

ABSTRACT
Enterovirus 71 (EV71) infection causes severe mortality involving multiple possible mechanisms, including cytokine storm, brain stem encephalitis, and fulminant pulmonary edema. Gamma interferon (IFN-γ) may confer anti-EV71 activity; however, the claim that disease severity is highly correlated to an increase in IFN-γ is controversial and would indicate an immune escape initiated by EV71. This study, investigating the role of IFN-γ in EV71 infection using a murine model, showed that IFN-γ was elevated. Moreover, IFN-γ receptor-deficient mice showed higher mortality rates and more severe disease progression with slower viral clearance than wild-type mice. In vitro results showed that IFN-γ pretreatment reduced EV71 yield, whereas EV71 infection caused IFN-γ resistance with attenuated IFN-γ signaling in IFN regulatory factor 1 (IRF1) gene transactivation. To study the immunoediting ability of EV71 proteins in IFN-γ signaling, 11 viral proteins were stably expressed in cells without cytotoxicity; however, viral proteins 2A and 3D blocked IFN-γ-induced IRF1 transactivation following a loss of signal transducer and activator of transcription 1 (STAT1) nuclear translocation. Viral 3D attenuated IFN-γ signaling accompanied by a STAT1 decrease without interfering with IFN-γ receptor expression. Restoration of STAT1 or blocking 3D activity was able to rescue IFN-γ signaling. Interestingly, viral 2A attenuated IFN-γ signaling using another mechanism by reducing the serine phosphorylation of STAT1 following the inactivation of extracellular signal-regulated kinase without affecting STAT1 expression. These results demonstrate the anti-EV71 ability of IFN-γ and the immunoediting ability by EV71 2A and 3D, which attenuate IFN-γ signaling through different mechanisms.
IMPORTANCE Immunosurveillance by gamma interferon (IFN-γ) may confer anti-enterovirus 71 (anti-EV71) activity; however, the claim that disease severity is highly correlated to an increase in IFN-γ is controversial and would indicate an immune escape initiated by EV71. IFN-γ receptor-deficient mice showed higher mortality and more severe disease progression, indicating the anti-EV71 property of IFN-γ. However, EV71 infection caused cellular insusceptibility in response to IFN-γ stimulation. We used an in vitro system with viral protein expression to explore the novel IFN-γ inhibitory properties of the EV71 2A and 3D proteins through the different mechanisms. According to this study, targeting either 2A or 3D pharmacologically and/or genetically may sustain a cellular susceptibility in response to IFN-γ, particularly for IFN-γ-mediated anti-EV71 activity.
INTRODUCTION
Enterovirus 71 (EV71) is a single-stranded RNA virus in the Picornaviridae family. The EV71 genome encodes four structural proteins, VP1 to VP4, and seven nonstructural proteins, 2A to 2C and 3A to 3D (1). Numerous studies have investigated the functions of viral proteins in viral replication and virulence (2). During EV71 infection, capsid VP proteins mediate virus entry by binding to cellular receptors, human scavenger receptor class B and P-selectin glycoprotein ligand 1 (3). Additionally, VP proteins participate in the assembly of viral particles (4). The 3C protein, a chymotrypsin-like protease, reduces host cell transcription dramatically by inhibiting cell polyadenylation (5) and induces caspase-regulated neural cell apoptosis (6). To develop specific anti-EV71 drugs, a number of small molecules targeting viral proteins have been designed, such as the 3C inhibitor rupintrivir and the 3D inhibitor aurintricarboxylic acid (ATA) (7–9).
EV71 infection typically causes mild, self-limiting hand-foot-and-mouth disease; however, patients sometimes have significant morbidity and mortality resulting from hemorrhagic pulmonary edema following acute central nervous system-related cardiopulmonary failure and brain stem encephalitis (2, 10, 11). In addition to the direct cytotoxicity caused by EV71 infection (12–16) and the resultant virulence factors (6, 17), host factors such as the aberrant production of cytokines that is detected during EV71-associated pulmonary edema can also lead to disease. In infected patients with brain stem encephalitis, increased levels of interleukin 8 (IL-8), IL-10, IL-13, gamma interferon (IFN-γ), CXC chemokine ligand 10 (IFN-γ-inducible protein 10), and monokine induced by IFN-γ are observed in the serum (18, 19), and large amounts of IL-1β, IL-6, IL-8, IFN-γ, and chemokine (C-C motif) ligand 2 (monocyte chemotactic protein 1) are found in the cerebrospinal fluid (19, 20). Neutralization of IL-6 by antibody confers protection against severe complications in EV71-infected neonatal mice (21). Additionally, an increased frequency of Th17 cells in peripheral blood and increased serum levels of the Th17 cell-derived cytokines IL-17 and IL-23 are observed in EV71-infected patients (22). While immunopathogenesis of EV71 infection has been proposed (23), the protective versus the pathogenic effects of most cytokines have not been addressed.
Host-derived IFNs exert antiviral activity via several mechanisms, including interference with viral replication through inhibiting the transcription and translation of viral components (24). Studies have demonstrated the antiviral activity of type I IFNs against EV71 infection in vitro and in vivo (25, 26). Furthermore, treatment with an IFN-inducing agent (27) and synergistic inhibition of EV71 replication by IFN-α and rupintrivir (28) have been identified as effective treatment regimens. However, during EV71 infection, several immune escape strategies are elicited by EV71 to attenuate the type I IFN response. The viral 3C protein retards retinoid acid-inducible gene I-mediated IFN regulatory factor 3 (IRF3) activation (29) and inhibits Toll-like receptor 3 signaling by cleaving the adaptor proteins TRIF (30) and IRF7 (31) to reduce type I IFN production (29–32). Viral protein 2A decreases the type I IFN response by targeting the antiviral signaling protein in mitochondria (33) and by reducing the IFN-α/β receptor 1 (IFNAR1) level (34). In addition to type I IFNs, the role of type II IFN (IFN-γ), a proinflammatory cytokine that is produced by natural killer cells, T cells, and antigen-presenting cells for immune defense against microbial infections and tumorigenesis (35, 36), is controversial in EV71 infection, specifically with regard to its dual roles in both cellular protection and immunopathogenesis. Compared to mice with IFNAR deficiency alone, mice with both IFNAR and IFN-γ receptor (IFN-γR) deficiency display increased susceptibility to EV71 infection (37), indicating a protective role for IFN-γ. In contrast, expression and gene polymorphism of IFN-γ have been shown to associate with EV71-induced pulmonary edema and encephalitis in patients (18, 38), and exogenous administration of three cytokines (IL-6, IL-13, and IFN-γ) together exacerbates EV71-induced pulmonary abnormality with edema in mice (39). Infection by EV71 may alter IFN-γ responses, leading to escape from both antiviral strategies and immunopathogenesis. The present study was therefore designed to examine the role of IFN-γ in EV71 infection and to determine how EV71 amends IFN-γ signaling.
MATERIALS AND METHODS
Cell cultures and reagents.
A human muscle rhabdomyosarcoma (RD) cell line (ATCC CCL-136) and murine neuroblastoma (Neuro-2a) cell line (ATCC CCL-131) were maintained in media according to the instructions of the ATCC. Wild-type mouse embryonic fibroblasts (MEFs), a gift from Hsiao-Sheng Liu, Department of Microbiology and Immunology, National Cheng Kung University, Taiwan, were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum, 50 U/ml of penicillin, and 50 μg/ml of streptomycin in a humidified atmosphere with 5% CO2 and 95% air. The reagents and antibodies used were 3D inhibitor ATA (Sigma-Aldrich); recombinant mouse IFN-γ (PeproTech or R&D Systems); recombinant mouse IFN-β (R&D Systems); fluorescein isothiocyanate- or phycoerythrin-conjugated control antibodies or antibodies against mouse CD4 (clone GK 1.5; eBioscience), CD8a (clone 53-6.7; eBioscience), F4/80 (clone BM8; eBioscience), phospho-STAT1α/β at Tyr701 and Ser727, STAT1α/β, phospho-Janus-associated kinase 2 (phospho-Jak2) at Tyr1007/1008, Jak2, phospho-extracellular signal-regulated kinase 1/2 (phospho-ERK1/2) at Thr202/Tyr204, ERK1/2, phospho-MEK1/2 at Ser217/221, MEK1/2, phospho-c-Raf at Ser338, and c-Raf (Cell Signaling Technology), V5 tag, IFN-γR1, and IFN-γR2 (Abcam), and β-actin (Chemicon International); and Alexa Fluor 488- and horseradish peroxidase-conjugated goat anti-mouse, goat anti-rabbit, and donkey anti-goat IgG (Invitrogen Life Technologies).
The virus and mice.
EV71 strain M2 was propagated and titrated on RD cell monolayers and was used to infect mice as previously described (40). C57BL6/J mice and C57BL6/J-derived mice deficient in IFN-γR1 (B6.129S7-Ifngr1tm1Agt/J) were purchased from The Jackson Laboratory and maintained under specific-pathogen-free conditions in the laboratory animal center of our college.
Infection of mice.
Animal work in this study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals (41), the Animal Welfare Act, and U.S. federal law. All mouse experiment protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of National Cheng Kung University (IACUC approval no. 98012). Fourteen-day-old mice were mock infected or infected with 3 × 105 PFU per mouse of EV71 strain M2 by intraperitoneal injection and monitored for survival rate and disease progression for 30 days. The disease severity of infected mice was graded from 0 to 5 as follows: 0, health; 1, ruffled hair; 2, weakness in hind limbs; 3, paralysis in single hind limb; 4, paralysis in both hind limbs; and 5, death. In separate experiments, mice were anesthetized, and blood was collected. After perfusion with ice-cold phosphate-buffered saline (PBS) by intracardiac injection, mouse tissues were harvested, frozen, homogenized, frozen, and sonicated. The resulting samples were assayed for viral titers by plaque assay using RD cell monolayers.
IFN-γ measurement.
Mouse blood was processed into sera. Mouse brains were frozen and homogenized in ice-cold PBS containing a protease inhibitor cocktail (Sigma-Aldrich). The brain homogenates were centrifuged to obtain supernatants. The supernatants and sera were subjected to enzyme-linked immunosorbent assay to measure mouse IFN-γ (R&D Systems) by using the commercially available kit according to the manufacturer's instructions.
Flow cytometry.
Spleens and brains were harvested from mice perfused with PBS at day 9 postinfection, and leukocytes were isolated from the organs and stained with antibodies specific for mouse leukocytes or control antibodies as previously described (42). To detect the expression of viral proteins and IFN-γ receptors, cells were stained with antibodies against V5 tag, IFN-γR1, or IFN-γR2 and then incubated with a mixture of Alexa Fluor 488-conjugated goat anti-rabbit IgG antibodies. Stained cells were analyzed using flow cytometry (FACSCalibur; BD Biosciences) with excitation set at 488 nm; emission was detected with the FL-1 (515 to 545 nm) and FL-2 (564 to 606 nm) channels. Samples were analyzed using CellQuest Pro 4.0.2 software (BD Biosciences), and quantification was performed using WinMDI 2.8 software (The Scripps Institute). Small cell debris was excluded by gating on a forward scatter plot.
IFN treatment.
Neuro-2a cells were pretreated with 25 or 100 U/ml of recombinant mouse IFN-β or IFN-γ for 24 h and infected with EV71 strain M2. Cultures were collected 24 and 36 h postinfection to determine viral titers by plaque assay.
Plasmid construction and transfection.
To construct plasmids expressing each of the EV71 proteins, the full-length cDNAs encoding EV71 VP4, VP3, VP2, VP1, 2A, 2B, 2C, 3A, 3B, 3C, or 3D protein were amplified from an EV71/MP4 infectious clone, pEV71/MP4/y-5, by PCR with the corresponding viral gene-specific primer pairs (32, 43). PCR products were cloned into the pcDNA3.1/V5-His-TOPO vector using a pcDNA3.1/V5-His TOPO TA expression kit (Invitrogen) according to the manufacturer's instructions. Viral protein expression plasmids and a control plasmid, pcDNA3.1(−), were isolated and purified using an EndoFree Plasmid Maxi kit (Qiagen) to eliminate endotoxin contamination. MEFs were transfected with the plasmids using Lipofectamine 2000 (Invitrogen Life Technologies) and cultured for 3 days. Stable cell lines were selected by culturing transfected cells in medium containing 1.8 to 2.0 μg/ml of Geneticin (Invitrogen Life Technologies) for 14 days. The expression of viral genes and proteins in stable cell lines was confirmed using reverse transcription-PCR (RT-PCR) with the gene-specific primer pairs (32, 43) and immunoblotting with V5 tag, respectively. For STAT1 transfection, transient transfection was performed using an MP-100 Microporator (Digital Biotechnology, South Korea) according to the manufacturer's instructions for optimization and usage. The pSG5 construct expressing STAT1 was provided by Chien-Kuo Lee (National Taiwan University).
RT-PCR analysis.
Total RNA was extracted from mouse brain stems or plasmid-transfected cells using TRIzol reagent (Invitrogen Life Technologies), and 5 μg of total RNA was converted to cDNA using Moloney murine leukemia virus reverse transcriptase (Promega). PCR amplification was performed on 4 μl of cDNA sample. Numbers of amplification cycles and annealing temperatures were optimized for the specific primer pairs for viral genes and mouse IFN-γ and β-actin cDNA sequences (32, 43, 44). PCR products were electrophoresed on 1.2% agarose gels. The gels were analyzed, and the intensity of the IFN-γ band was quantified using ImageJ software and normalized to the intensity of the β-actin band in the sample.
Cytotoxicity assay.
To evaluate cell damage, the activity of lactate dehydrogenase released in culture supernatants was assayed using a colorimetric assay with cytotoxicity detection kit (Roche Diagnostics, United Kingdom) according to the manufacturer's instructions. A microplate reader (SpectraMax 340PC; Molecular Devices) was used to measure the absorbance at 620 nm with a reference wavelength of 450 nm, and data were analyzed with Softmax Pro software (Molecular Devices).
Luciferase reporter assay.
For the luciferase reporter assay, the cells were transiently cotransfected with the IRF1 promoter-driven firefly luciferase reporter (0.2 μg) and 0.01 μg of Renilla luciferase-expressing plasmid (pRL-TK; Promega) using the GeneJammer transfection reagent (Stratagene). Twenty-four hours after the transfection, the cells were treated with IFN-γ for 0.5 h, lysed, and then harvested to measure firefly and Renilla luciferase activities using a luciferase assay system (Dual-Glo; Promega). For each lysate, the firefly luciferase activity was calculated as relative light units (RLU).
Western blotting.
Both procedures used were described previously (45). In brief, total cell lysates were extracted, and proteins were separated using SDS-polyacrylamide gel electrophoresis and then transferred to a polyvinylidene difluoride membrane (Millipore Corporation). After blocking with 5% bovine serum albumin (Sigma-Aldrich), blots were incubated with the desired antibodies and developed using an ECL Western blot detection kit (Millipore Corporation) according to the manufacturer's instructions. The relative signal intensity was quantified using ImageJ software (version 1.41o) from W. Rasband (National Institutes of Health, Bethesda, MD).
Immunostaining.
To detect the expression of STAT1, cells were fixed, stained, and analyzed as previously described (46). For confocal microscopy analysis, cells were stained with the STAT1 antibody and then with Alexa Fluor 488-conjugated goat anti-mouse IgG. The cells were then visualized using a confocal laser scanning microscope (Digital Eclipse C1si-ready; Nikon).
Statistical methods.
Values are presented as means ± standard errors (SE). The Kaplan-Meier survival curves were analyzed by the log rank test. The disease scores were analyzed by the Wilcoxon signed-rank test. The tissue viral loads were analyzed by the Mann-Whitney U test. The rest of results were analyzed by Student's two-tailed unpaired t test (SigmaPlot 8.0 for Windows; Systat Software). Statistical significance was set at a P value of <0.05.
RESULTS
EV71 infection induces IFN-γ in the brain, and absence of IFN-γR1 increases the mortality of EV71-infected mice.
In EV71-infected patients with brain stem encephalitis and pulmonary edema complications, IFN-γ levels in the plasma and cerebrospinal fluid are significantly elevated (18, 20). To investigate the induction of IFN-γ by EV71 infection in the host in vivo, we used a murine infection model that can reproduce neurological symptoms and death, analogous to those of infected patients (40). C57BL6/J mice were mock infected or infected with EV71 at a dose of 3 × 105 PFU/mouse, which induced signs of encephalitis, hunched posture, lethargy, hind limb paralysis, and ataxia in mice, with a death rate of 37% (42). Our previous report showed that infection of mice with this inoculum resulted in brain viral titers which increased from days 2 to 5 postinfection and then declined (42). In contrast to mock-infected mice, in which brain IFN-γ protein levels were below the detection level (Fig. 1A), EV71-infected mice showed significantly increased brain IFN-γ levels at day 6 postinfection (P < 0.001). We also found that EV71 infection increased IFN-γ mRNA levels in the mouse brain stem at days 4 and 6 postinfection, but not at day 8 postinfection, 1.2- and 1.8-fold, respectively, compared to those of mock-infected mice, as determined by RT-PCR analysis (data not shown). Serum IFN-γ protein levels were below the detection level in both mock-infected and infected mice from days 2 to 10 postinfection.
FIG 1.
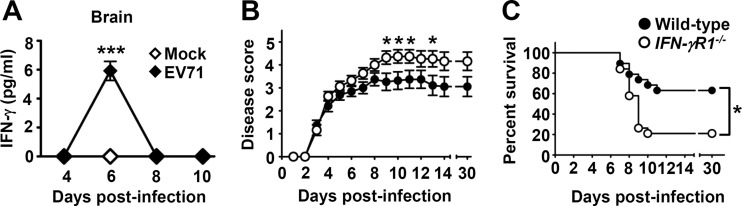
EV71 infection induces IFN-γ in the brain, and absence of IFN-γR1 increases the mortality of EV71-infected mice. (A) Brain IFN-γ levels in mice mock infected (Mock) or infected with EV71 were measured at the indicated days postinfection. Data are means ± SEs from at least 3 samples per data point, with samples derived from individual mice. ***, P < 0.001 compared with mock-infected mice. (B and C) The disease severity (B) and survival rates (C) of wild-type mice (n = 19) and IFN-γR1−/− mice (n = 19) infected with EV71 at the indicated days postinfection are shown. Data are means ± SEs in panel B. *, P < 0.05 compared with wild-type mice in panel B and between the indicated groups in panel C.
IFN-γR consists of the heterodimer of two chains: IFN-γR1 and IFN-γR2 (35, 36). To determine the role of IFN-γ signaling in EV71 infection, mice with a targeted disruption of the gene encoding IFN-γR1 were used. The growth of uninfected IFN-γR1-deficient (IFN-rR1−/−) mice appeared normal, without obvious abnormalities (47). After EV71 infection, IFN-γR1−/− mice displayed severe hind limb paralysis compared to wild-type mice, with significant differences in disease scores (P < 0.05) found at several days postinfection (Fig. 1B). The final survival rate of infected IFN-rR1−/− mice was 21%, which was significantly lower than that of infected wild-type mice, by 42% (Fig. 1C) (P < 0.05).
Absence of IFN-γR1 increases tissue viral loads of infected mice.
To investigate the high mortality rate of infected IFN-γR1−/− mice compared with that of infected wild-type mice, we harvested mouse central nervous system (brain without brain stem region, brain stem, and spinal cord) and peripheral tissues (spleen, kidney, lung, heart, intestine, and liver) to measure viral titers (Fig. 2A). In IFN-γR1−/− mice, the mean viral titers in all tissues examined were always higher than those of wild-type mice from days 5 to 10 postinfection, with significant differences found in the brain without brain stem region, brain stem, spinal cord, and spleen at day 10 postinfection and in the kidney at days 5 and 10 postinfection (P < 0.05). These results show that IFN-γR1 deficiency reduces EV71 clearance from infected mouse tissues.
FIG 2.

Effects of IFN-γR1 deficiency on tissue viral loads and leukocyte numbers. (A) Viral titers in the indicated tissues of wild-type and IFN-γR1−/− mice were determined at the indicated days postinfection. Wild-type and IFN-γR1−/− mice were mock infected (mock) or infected with EV71. (B and C) The numbers of CD4+, CD8+, and F4/80+ cells (B) and CD69+ CD4+ and CD69+ CD8+ cells (C) in spleens or brains at day 9 postinfection are shown. Data are means ± SEs from at least 3 samples per data point or group, with samples derived from individual mice. Asterisks show statistically significant differences compared with values from wild-type mice as follows: *, P < 0.05; **, P < 0.01.
We further investigated how IFN-γ could contribute to EV71 clearance. We previously found that CD4+ and CD8+ T lymphocytes protected mice from EV71 infection by reducing tissue viral loads (40). IFN-γ is known to enhance the proliferation of T lymphocytes and macrophages and activation of T lymphocytes (35). Therefore, we quantified CD4+ and CD8+ T lymphocytes and macrophages in infected organs, the spleen and brain, at day 9 postinfection by flow cytometric analysis. In spleens, the mean numbers of CD4+ and CD8+ T lymphocytes, F4/80+ macrophages, and activated T lymphocytes (CD69+ CD4+ and CD69+ CD8+ cells) in infected IFN-γR1−/− mice were slightly lower than those of infected wild-type mice (Fig. 2B and C). These results may partially explain the slow EV71 clearance found in IFN-γR1−/− mice. In brains, the mean numbers of CD4+ and CD8+ T lymphocytes and F4/80+ macrophages in infected wild-type and IFN-γR1−/− mice were not significantly different.
IFN-γ pretreatment reduces EV71 yield, whereas EV71 infection causes IFN-γ resistance with attenuated IFN-γ signaling in infected cells.
IFN-γ has been shown to reduce viral infection by inhibiting viral replication (48, 49). To examine the anti-EV71 activity of IFN-γ, a mouse neuronal cell line (Neuro-2a) was treated or not with IFN-γ (25 or 100 U/ml) 24 h before EV71 infection at a multiplicity of infection (MOI) of 0.0003. To compare the anti-EV71 activities of type I and II IFNs, we also included IFN-β for study. Pretreatment with 25 U/ml of IFN-β or IFN-γ significantly reduced the mean viral titers (P ≤ 0.05) in Neuro-2a cells at both 24 and 36 h postinfection (Fig. 3A), and pretreatment with 100 U/ml of IFN slightly increased reductions of viral titers compared to 25 U/ml of IFN at 36 h postinfection. IFN-β reduced viral titers more efficiently than IFN-γ. However, treatment of Neuro-2a cells with IFN-γ after infection failed to reduce viral titers. We next investigated the possible immunoediting effects of EV71 infection on IFN-γ signaling. The transactivation of IRF1, which encodes an important transcription factor for IFN-γ-targeted genes, is typically induced by IFN-γ (35, 36). Our results showed that either IFN-γ or IFN-β treatment increased IRF1 promoter activity (P < 0.01) in Neuro-2a cells (Fig. 3B) as determined by a luciferase-based reporter assay (46). More importantly, EV71 infection significantly attenuated IFN-γ- and IFN-β-induced IRF1 transactivation (Fig. 3B) (P < 0.01). These results show IFN-γ with anti-EV71 activity and an immune escape strategy by EV71 infection.
FIG 3.

IFN-γ pretreatment reduces EV71 yield, whereas EV71 infection attenuates IFN-γ signaling in infected cells. (A) Neuro-2a cells were treated or not (−) with the indicated concentrations of type I IFN (IFN-β) or type II IFN (IFN-γ) for 24 h before infection with EV71 (MOI = 0.0003). Cultures were harvested to determine viral titers 24 and 36 h postinfection. Data are means plus SEs from 3 samples per group. (B) Neuro-2a cells were mock infected (MOCK) or infected with EV71 (MOI = 0.0003) for 24 h and then treated with PBS, IFN-γ (100 U/ml), or IFN-β (100 U/ml) for 30 min. A luciferase reporter assay showed the transactivation of IRF-1 as relative light units (RLU). Data are means plus SEs from three individual experiments of triplicate tests. *, P ≤ 0.05; **, P ≤ 0.01.
EV71 protein 2A or 3D abolishes IFN-γ-induced STAT1 activation and IRF1 transactivation.
EV71 infection diminishes IFN-γ antiviral activity. EV71 proteins may possibly possess immunoediting functions in cells to amend IFN-γ signaling. To ascertain which, if any, of the EV71 proteins were involved in the EV71 inhibitory effect, recombinant plasmids expressing each of the 11 viral proteins, including 4 structural proteins and 7 nonstructural proteins, were constructed (32). Reverse transcription-PCR (RT-PCR) analysis (Fig. 4A, left), Western blot analysis (Fig. 4A, right), and immunostaining-based flow cytometric assay (Fig. 4B) confirmed the stable expression of the genes encoding the 11 viral proteins in mouse embryonic fibroblasts (MEFs) after Lipofectamine delivery of the plasmids on day 3 posttransfection. The protein expression profile was similar to that reported in a previous study (32). Furthermore, there were no cytotoxic effects resulting from these viral proteins in MEFs as monitored by assaying the release of lactate dehydrogenase at day 3 posttransfection (Fig. 4C). The EV71 protein-expressing cells were further examined to determine whether and which viral proteins cause inhibitory effects on IFN-γ signaling.
FIG 4.

Effects of EV71 proteins on IFN-γ-induced transactivation of IRF1 promoter and STAT1 nuclear translocation. MEFs were transfected with the plasmid without (Vector) or with the indicated viral gene. (A) RT-PCR and Western blot analysis were performed with stable MEFs with viral gene transfection as indicated. Lane M, molecular weight markers. (B) Representative histogram of flow cytometric analysis showing the percentage of viral protein expression as indicated in the transfected MEFs. (C) The cytotoxicity, as determined by lactate dehydrogenase (LDH) release, was determined 3 days after transfection. ns, not significant. (D) A luciferase reporter assay showed the transactivation of IRF-1 in the transfected MEFs treated with PBS or IFN-γ for 30 min. Data are means plus SEs from triplicate tests. **, P < 0.01. (E) The transfected MEFs were treated with PBS or IFN-γ for 30 min. Immunostaining, analyzed by fluorescence microscopy, was performed to determine STAT1 expression. Arrowheads indicate the positive cells with STAT1 nuclear translocation. Scale bar: 20 μm. The data shown are representative of three individual experiments. *, P < 0.05 compared with PBS; #, P < 0.05 compared with IFN-γ.
In IFN-γ signaling pathway, the binding of IFN-γ initially leads to IFN-γR dimerization followed by activation of Janus-associated kinase 1/2 (Jak1/2) and recruitment of signal transducers and activators of transcription 1 (STAT1). Jak2 mediates STAT1 phosphorylation, leading to a STAT1 homodimer complex that translocates into the nucleus and turns on a variety of IRFs (35, 50). After cells were treated with 10 ng/ml of IFN-γ, IFN-γ-induced IRF1 transactivation was significantly reduced in cells expressing the EV71 2A or 3D protein but not other viral proteins compared with a vector-transfected control group (Fig. 4D) (P ≤ 0.01). We next examined whether these proteins have inhibitory effects on the activation (nuclear translocation) of STAT1, an upstream transcriptional factor of IRF1 (35, 50, 51). Immunostaining analysis showed a significant inhibitory effect of EV71 2A or 3D expression on IFN-γ-induced STAT1 nuclear translocation (Fig. 4E) (P < 0.05), while 3D expression caused a decrease in STAT1 expression. These results provide evidence that the IFN-γ inhibitory effect of EV71 was attributable to the 2A and 3D proteins.
EV71 3D, but not 2A, decreases IFN-γ-induced STAT1 tyrosine phosphorylation.
To investigate the possible mechanisms underlying insensible IFN-γ signaling in EV71 2A- and 3D-expressing cells, we evaluated the activity of the Jak2/STAT1 signaling axis, which is required for IFN-γ-induced signal transduction (35, 50). Notably, Western blotting showed that IFN-γ failed to cause STAT1 phosphorylation at a specific tyrosine residue (Tyr701) only in 3D-expressing cells (Fig. 5A), without affecting the phosphorylation of Jak2 (Tyr1007/1008), the upstream tyrosine kinase that phosphorylates STAT1 (35, 50) (Fig. 5B). Notably, the expression of STAT1 was decreased in 3D-expressing cells not only at the protein level (Fig. 5A) but also at the mRNA level (data not shown). These results demonstrate that the EV71 3D protein caused defective IFN-γ signaling by attenuating STAT1 tyrosine phosphorylation independent of Jak2 inactivation.
FIG 5.

EV71 3D attenuates IFN-γ-induced tyrosine phosphorylation of STAT1 accompanied by a STAT1 decrease. Western blotting was used to detect the expression of phospho-STAT1α/β Tyr701 (pY STAT1) and STAT1α/β (STAT1) (A) or phospho-Jak2 Tyr1007/1008 (pY Jak2) and Jak2 (B) in MEFs transfected with the plasmid without (Vector) or with the indicated viral gene and treated or not (−) with IFN-γ for the indicated times. β-Actin was used as an internal control. The ratios of phosphorylated protein to total protein are shown. The data shown are representative of three individual experiments.
EV71 2A decreases IFN-γ-induced STAT1 serine phosphorylation.
Although the activation of STAT1 and IRF1 was defective in EV71 2A-expressing cells, IFN-γ-induced tyrosine phosphorylation of STAT1 was not affected (Fig. 5A). In addition to tyrosine phosphorylation, which initially activates STAT1, the next serine phosphorylation site (Ser727) is required for the dimerization and stability of the STAT1/STAT1 complex, leading to nuclear translocation followed by DNA binding (35, 50). We found that EV71 2A expression effectively diminished IFN-γ-induced serine phosphorylation of STAT1 at Ser727 (Fig. 6A). The signaling of the mitogen-activated protein kinase, extracellular signal-regulated kinase (ERK), which is the primary kinase for STAT1 serine phosphorylation (35, 50), was therefore examined next. Western blotting showed a decrease in the phosphorylation of ERK (Thr202/Tyr204), MEK1/2 (Ser217/221), and c-Raf (Ser338) (Fig. 6B). The expression of Ras was not affected by 2A expression (data not shown). These findings show a novel inhibitory effect of EV71 2A on the serine phosphorylation of STAT1 through a mechanism involving dephosphorylation of the c-Raf/MEK1/2/ERK signaling axis pathway.
FIG 6.

EV71 2A decreases IFN-γ-induced serine phosphorylation of STAT1 by inhibiting ERK signaling. Western blotting was used to detect the expression of phospho-STAT1α/β Ser727 (pS STAT1) and STAT1α/β (STAT1) (A) or phospho-ERK1/2 Thr202/Tyr204 (pT/Y ERK1/2), ERK1/2, phospho-MEK1/2 Ser217/221 (pS MEK1/2), MEK1/2, phospho-c-Raf Ser338 (pS c-Raf), and c-Raf (B) in MEFs transfected with the plasmid without (Vector) or with 2A and treated or not (−) with IFN-γ for the indicated times. β-Actin was used as an internal control. The ratios of phosphorylated protein to total protein are shown. The data shown are representative of three individual experiments.
EV71 3D decreases STAT1 expression and activation.
3D-expressing cells were defective in IFN-γ-induced nuclear translocation of STAT1, as demonstrated by immunostaining (Fig. 4E), and displayed a decrease in STAT1 expression, as demonstrated by Western blotting (Fig. 5A). However, the phosphorylation of Jak2 (Tyr1007/1008) was not affected by 3D expression (Fig. 5B), suggesting that 3D expression should not affect IFN-γR expression. Flow cytometric analysis showed that the surface expression of IFN-γR1 and IFN-γR2 was not different between the control vector and the 3D-expressing cells (Fig. 7A). Complementary expression of STAT1 significantly reversed the decrease of IFN-γ-induced IRF1 transactivation in 3D-expressing cells (Fig. 7B) (P ≤ 0.01). While EV71 3D protein exhibits polymerase activity (2), administration of the 3D inhibitor ATA reversed the decreased expression of STAT1 in 3D-expressing cells (Fig. 7C) as well as 3D-inhibited IRF1 transactivation in response to IFN-γ (Fig. 7D) (P < 0.01). These results demonstrate that defects in endogenous STAT1 expression attenuated IFN-γ signaling in 3D-expressing cells.
FIG 7.

EV71 3D facilitates a STAT1 decrease. MEFs were transfected with the plasmid without (Vector) or with 3D or STAT1. (A) Immunostaining followed by flow cytometric analysis was used to detect IFN-γR1 and IFN-γR2 expression on the cell surface. (B and D) The transfected MEFs were transfected with the plasmid without (Vector) or with STAT1 (STAT1) for 24 h and then treated with PBS or IFN-γ for 30 min (B). The transfected MEFs were treated with PBS or ATA for 6 h and then with PBS or IFN-γ for 30 min (D). Luciferase reporter assay was used to detect the transactivation ratio of IRF-1 to Renilla compared to normalized control (fold increase). Data are means plus SEs of three individual experiments with triplicate tests. **, P ≤ 0.01. (C) Western blotting was used to detect STAT1 expression in 3D-transfected MEFs treated or not (−) with a 3D inhibitor, ATA, for the indicated times. The ratios of STAT1 to β-actin are shown. In panels A and C, the data shown are representative of three individual experiments.
DISCUSSION
A previous study indicated that IFN-γ might have anti-EV71 activity, as mice deficient in both IFNAR and IFN-γR were more susceptible to EV71 infection than mice deficient only in IFNAR (37). Here we provide direct evidence that IFN-γ indeed protects mice from EV71 infection, with decreases in tissue viral loads and slight increases in spleen leukocyte numbers. To search the protective mechanism of IFN-γ in EV71 infection in mice, we performed in vitro studies and found that treatment of a mouse neuronal cell line with a low dose (25 U/ml) of IFN-γ before infection significantly reduces virus yields, despite the fact that the anti-EV71 efficacy of IFN-γ is less than that of type I IFN. The capacities of IFN-γ to increase spleen leukocytes and, more importantly, to reduce virus yield may contribute to the decreases of tissue viral loads and lethality detected in wild-type mice compared to IFN-γR1−/− mice after EV71 infection. As the major targets of IFN-γ are leukocytes, not neuronal cells, the response of neuronal cells might be limited and can be easily saturated. This may explain our result showing that the high dose (100 U/ml) of IFN-γ treatment fails to further inhibit viral replication compared to 25 U/ml of IFN-γ. Interestingly, EV71 infection causes IFN-γ resistance. Our investigation on how EV71 facilitates immune evasion from IFN-γ antiviral surveillance sheds light on the possible inhibitory roles of EV71 proteins 2A and 3D on IFN-γ activity. Our results showed that 2A and 3D attenuated IFN-γ signaling by different mechanisms. Questions that remain following this study are whether these viral proteins cause the identified IFN-γ resistance during infection and whether such IFN-γ resistance has benefits for the virus. Further studies are needed to clarify the mechanisms through which viral proteins perform the observed immunoediting of the host response.
STAT1 and IRF1, the transcription factors required for gene regulation of antiviral type I and II IFNs, are required for the host response against viral infection by promoting the expression of cellular factors, including antiviral RNA-activated protein kinase, the 2-5A system, Mx protein, and inflammatory cytokines (24). Our recent report showed that an increase in CXC chemokine ligand 10 in EV71-infected mice and a deficiency in this chemokine led to increased susceptibility to EV71 infection (42). Another report indicates that IFN-γ might reduce EV71 infection (37). The antiviral activity of IFN-γ is therefore thought to be important for an anti-EV71 response; however, it is possible that EV71 may develop strategies to circumvent IFN-γ for immunoevasion. For IFN-γ homeostasis, suppressor of cytokine signaling 1 (SOCS1) and SOCS3, which can bind to IFN-γRs to attenuate Jak/STAT signaling (52, 53), and Src homology 2 (SH2) domain-containing phosphatase 2, which can dephosphorylate Jak2 and STAT1 (54, 55), are negative regulators and may be abnormally utilized by pathogens for IFN-γ-mediated immune response escape (56). However, a role for these proteins in EV71 infection has not been documented until now. On the other hand, it is well established that viral proteins 2A and 3C are involved in altering the type I IFN response to facilitate viral replication (29–33). To our knowledge, this work identifies, for the first time, a novel strategy utilized by EV71 against IFN-γ via different 2A- and 3D-mediated mechanisms. Immune escape by EV71 against IFN-γ may also represent a strategy to maintain viral replication.
EV71 2A protease, a cysteine protease, has multiple functions that permit viral replication as well as evasion of the host response during infection (2). Previous studies showed that the proapoptotic effects of 2A in Vero and HeLa-229 cells are mediated via cleavage of the eukaryotic initiation factor 4GI, a key factor required for host protein synthesis (17). However, in our study, no cytotoxicity was observed in 2A-expressing MEFs. This is consistent with recent studies with HeLa and RD cells and yeasts (33, 57), in which overexpression of EV71 2A did not affect the growth of these cells. In 2A-expressing HeLa cells and yeasts, the newly identified transcriptional activity of 2A protease may also regulate some cellular genes to benefit viral replication (57); however, the targets of 2A protease in host cells require further investigation. Wang et al. (33) revealed a novel strategy employed by EV71 2A to inhibit the production of type I IFN by targeting the mitochondrial antiviral signaling protein, a unique adaptor molecule activated upon retinoic acid-induced gene I and melanoma differentiation-associated gene viral recognition receptor signaling. EV71 2A also blocks the IFN-β-mediated phosphorylation of STAT1, STAT2, Jak1, and Tyk2 by reducing IFNAR1 (34). In this study, we also discovered an additional EV71 2A-mediated immune evasion strategy in which the virus alters type II IFN-γ signal transduction. Without affecting the protein expression of IFN-γR and STAT1 and tyrosine phosphorylation of STAT1, a novel IFN-γ inhibitory mechanism by the 2A protein is exhibited by attenuating STAT1 phosphorylation at a specific serine residue. While the upstream signaling cascade consisting of c-Raf/MEK1/2/ERK was also abolished by 2A, it is speculated that the 2A protein may directly regulate c-Raf autophosphorylation (58) and/or indirectly regulate upstream host factors such as Ras, phosphatidylinositol 3-kinase, Cdc42/Rac, p21-activated kinase 1, and phosphatases related to c-Raf activation (59–61) to cause STAT1 inactivation. Further studies are needed to address this issue. For EV71 infection, the 2A protease is therefore required for viral escape of the host antiviral innate immune response for both type I and type II IFNs.
Certain inhibitors, such as ATA, and RNA interference (RNAi) against 3D have been used as antiviral strategies to suppress EV71 transcription and replication (8, 9), as the 3D protein serves as an RNA-dependent RNA polymerase for initial de novo transcription with a poly(C) template and genomic RNA through EV71 VP protein binding and uridylylation (62, 63). To our knowledge, there has been no report describing a host interaction with 3D during EV71 infection. In this study, we unexpectedly found that 3D-expressing cells demonstrate defects in IFN-γ signal transduction. Without affecting Jak2 activation or IFN-γR expression, an unknown mechanism led to 3D-mediated STAT1 downregulation. Either restoring STAT1 or inhibiting 3D activity effectively reversed IFN-γ-induced IRF1 transactivation. It is still unknown how the 3D protein regulates STAT1 activation and expression. Previous studies showed that STAT1 is upregulated by IFN-γ-activated Jak/STAT1/IRF1 signaling (64). The specific causes of the decrease in STAT1 transcriptional and/or posttranslational levels by the 3D protein require further investigation. During EV71 infection, 3D may also contribute to IFN-γ resistance, perhaps in analogy to the role of 2A as a viral factor for immunoediting.
In conclusion, this work used an in vitro system with viral protein expression to explore the novel IFN-γ inhibitory properties of the EV71 2A and 3D proteins. Although IFN-γ is upregulated for immunopathogenesis during EV71 pulmonary disorders, the blockade of IFN-γ may cause severe infection and disease progression as demonstrated by studies using two different murine models. According to this study, targeting either 2A or 3D pharmacologically and/or genetically may sustain a cellular susceptibility in response to IFN-γ, particularly for IFN-γ-mediated anti-EV71 activity.
ACKNOWLEDGMENTS
This work was supported by grant NHRI-EX102-9917NC from the National Health Research Institutes and grants MOST100-2320-B-006-009-MY3 and 104-2321-B-006-015 from the Ministry of Science and Technology in Taiwan.
We thank Robert Anderson for critical reading of the manuscript and the Immunobiology Core of the Research Center of Clinical Medicine in National Cheng Kung University Hospital for providing services that include training, technical support, and assistance with experimental designs and data analyses using the flow cytometry core facilities.
REFERENCES
- 1.Brown BA, Pallansch MA. 1995. Complete nucleotide sequence of enterovirus 71 is distinct from poliovirus. Virus Res 39:195–205. doi: 10.1016/0168-1702(95)00087-9. [DOI] [PubMed] [Google Scholar]
- 2.Yi L, Lu J, Kung HF, He ML. 2011. The virology and developments toward control of human enterovirus 71. Crit Rev Microbiol 37:313–327. doi: 10.3109/1040841X.2011.580723. [DOI] [PubMed] [Google Scholar]
- 3.Yamayoshi S, Ohka S, Fujii K, Koike S. 2013. Functional comparison of SCARB2 and PSGL1 as receptors for enterovirus 71. J Virol 87:3335–3347. doi: 10.1128/JVI.02070-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curry S, Fry E, Blakemore W, Abu-Ghazaleh R, Jackson T, King A, Lea S, Newman J, Stuart D. 1997. Dissecting the roles of VP0 cleavage and RNA packaging in picornavirus capsid stabilization: the structure of empty capsids of foot-and-mouth disease virus. J Virol 71:9743–9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weng KF, Li ML, Hung CT, Shih SR. 2009. Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoS Pathog 5:e1000593. doi: 10.1371/journal.ppat.1000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li ML, Hsu TA, Chen TC, Chang SC, Lee JC, Chen CC, Stollar V, Shih SR. 2002. The 3C protease activity of enterovirus 71 induces human neural cell apoptosis. Virology 293:386–395. doi: 10.1006/viro.2001.1310. [DOI] [PubMed] [Google Scholar]
- 7.Zhang XN, Song ZG, Jiang T, Shi BS, Hu YW, Yuan ZH. 2010. Rupintrivir is a promising candidate for treating severe cases of enterovirus-71 infection. World J Gastroenterol 16:201–209. doi: 10.3748/wjg.v16.i2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hung HC, Chen TC, Fang MY, Yen KJ, Shih SR, Hsu JT, Tseng CP. 2010. Inhibition of enterovirus 71 replication and the viral 3D polymerase by aurintricarboxylic acid. J Antimicrob Chemother 65:676–683. doi: 10.1093/jac/dkp502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thibaut HJ, De Palma AM, Neyts J. 2012. Combating enterovirus replication: state-of-the-art on antiviral research. Biochem Pharmacol 83:185–192. doi: 10.1016/j.bcp.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 10.McMinn PC. 2002. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol Rev 26:91–107. doi: 10.1111/j.1574-6976.2002.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 11.Wang SM, Liu CC. 2009. Enterovirus 71: epidemiology, pathogenesis and management. Expert Rev Anti Infect Ther 7:735–742. doi: 10.1586/eri.09.45. [DOI] [PubMed] [Google Scholar]
- 12.Chan YF, Abubakar S. 2003. Enterovirus 71 infection induces apoptosis in Vero cells. Malays J Pathol 25:29–35. http://www.mjpath.org.my/past_issue/MJP2003.1/EV71%2003.pdf. [PubMed] [Google Scholar]
- 13.Chang SC, Lin JY, Lo LY, Li ML, Shih SR. 2004. Diverse apoptotic pathways in enterovirus 71-infected cells. J Neurovirol 10:338–349. doi: 10.1080/13550280490521032. [DOI] [PubMed] [Google Scholar]
- 14.Liang CC, Sun MJ, Lei HY, Chen SH, Yu CK, Liu CC, Wang JR, Yeh TM. 2004. Human endothelial cell activation and apoptosis induced by enterovirus 71 infection. J Med Virol 74:597–603. doi: 10.1002/jmv.20216. [DOI] [PubMed] [Google Scholar]
- 15.Chen TC, Lai YK, Yu CK, Juang JL. 2007. Enterovirus 71 triggering of neuronal apoptosis through activation of Abl-Cdk5 signalling. Cell Microbiol 9:2676–2688. doi: 10.1111/j.1462-5822.2007.00988.x. [DOI] [PubMed] [Google Scholar]
- 16.Shih SR, Weng KF, Stollar V, Li ML. 2008. Viral protein synthesis is required for enterovirus 71 to induce apoptosis in human glioblastoma cells. J Neurovirol 14:53–61. doi: 10.1080/13550280701798980. [DOI] [PubMed] [Google Scholar]
- 17.Kuo RL, Kung SH, Hsu YY, Liu WT. 2002. Infection with enterovirus 71 or expression of its 2A protease induces apoptotic cell death. J Gen Virol 83:1367–1376. http://vir.sgmjournals.org/content/83/6/1367.long. [DOI] [PubMed] [Google Scholar]
- 18.Wang SM, Lei HY, Huang KJ, Wu JM, Wang JR, Yu CK, Su IJ, Liu CC. 2003. Pathogenesis of enterovirus 71 brainstem encephalitis in pediatric patients: roles of cytokines and cellular immune activation in patients with pulmonary edema. J Infect Dis 188:564–570. doi: 10.1086/376998. [DOI] [PubMed] [Google Scholar]
- 19.Wang SM, Lei HY, Yu CK, Wang JR, Su IJ, Liu CC. 2008. Acute chemokine response in the blood and cerebrospinal fluid of children with enterovirus 71-associated brainstem encephalitis. J Infect Dis 198:1002–1006. doi: 10.1086/591462. [DOI] [PubMed] [Google Scholar]
- 20.Wang SM, Lei HY, Su LY, Wu JM, Yu CK, Wang JR, Liu CC. 2007. Cerebrospinal fluid cytokines in enterovirus 71 brain stem encephalitis and echovirus meningitis infections of varying severity. Clin Microbiol Infect 13:677–682. doi: 10.1111/j.1469-0691.2007.01729.x. [DOI] [PubMed] [Google Scholar]
- 21.Khong WX, Foo DG, Trasti SL, Tan EL, Alonso S. 2011. Sustained high levels of interleukin-6 contribute to the pathogenesis of enterovirus 71 in a neonate mouse model. J Virol 85:3067–3076. doi: 10.1128/JVI.01779-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Tong J, Liu H, Liu Y, Su Z, Wang S, Shi Y, Zheng D, Sandoghchian S, Geng J, Xu H. 2012. Increased frequency of Th17 cells in the peripheral blood of children infected with enterovirus 71. J Med Virol 84:763–767. doi: 10.1002/jmv.23254. [DOI] [PubMed] [Google Scholar]
- 23.Wang SM, Lei HY, Liu CC. 2012. Cytokine immunopathogenesis of enterovirus 71 brain stem encephalitis. Clin Dev Immunol 2012:876241. doi: 10.1155/2012/876241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malmgaard L. 2004. Induction and regulation of IFNs during viral infections. J Interferon Cytokine Res 24:439–454. doi: 10.1089/1079990041689665. [DOI] [PubMed] [Google Scholar]
- 25.Liu ML, Lee YP, Wang YF, Lei HY, Liu CC, Wang SM, Su IJ, Wang JR, Yeh TM, Chen SH, Yu CK. 2005. Type I interferons protect mice against enterovirus 71 infection. J Gen Virol 86:3263–3269. doi: 10.1099/vir.0.81195-0. [DOI] [PubMed] [Google Scholar]
- 26.Yi L, He Y, Chen Y, Kung HF, He ML. 2011. Potent inhibition of human enterovirus 71 replication by type I interferon subtypes. Antivir Ther 16:51–58. doi: 10.3851/IMP1720. [DOI] [PubMed] [Google Scholar]
- 27.Lin CW, Wu CF, Hsiao NW, Chang CY, Li SW, Wan L, Lin YJ, Lin WY. 2008. Aloe-emodin is an interferon-inducing agent with antiviral activity against Japanese encephalitis virus and enterovirus 71. Int J Antimicrob Agents 32:355–359. doi: 10.1016/j.ijantimicag.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hung HC, Wang HC, Shih SR, Teng IF, Tseng CP, Hsu JT. 2011. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J Infect Dis 203:1784–1790. doi: 10.1093/infdis/jir174. [DOI] [PubMed] [Google Scholar]
- 29.Lei X, Liu X, Ma Y, Sun Z, Yang Y, Jin Q, He B, Wang J. 2010. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J Virol 84:8051–8061. doi: 10.1128/JVI.02491-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. 2011. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J Virol 85:8811–8818. doi: 10.1128/JVI.00447-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lei X, Xiao X, Xue Q, Jin Q, He B, Wang J. 2013. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J Virol 87:1690–1698. doi: 10.1128/JVI.01855-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee YP, Wang YF, Wang JR, Huang SW, Yu CK. 2012. Enterovirus 71 blocks selectively type I interferon production through the 3C viral protein in mice. J Med Virol 84:1779–1789. doi: 10.1002/jmv.23377. [DOI] [PubMed] [Google Scholar]
- 33.Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, Jin Q, Zhao Z. 2013. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog 9:e1003231. doi: 10.1371/journal.ppat.1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu J, Yi L, Zhao J, Yu J, Chen Y, Lin MC, Kung HF, He ML. 2012. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J Virol 86:3767–3776. doi: 10.1128/JVI.06687-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schroder K, Hertzog PJ, Ravasi T, Hume DA. 2004. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 36.Schoenborn JR, Wilson CB. 2007. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol 96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 37.Caine EA, Partidos CD, Santangelo JD, Osorio JE. 2013. Adaptation of enterovirus 71 to adult interferon deficient mice. PLoS One 8:e59501. doi: 10.1371/journal.pone.0059501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J, Zhao N, Su NL, Sun JL, Lv TG, Chen ZB. 2012. Association of interleukin 10 and interferon gamma gene polymorphisms with enterovirus 71 encephalitis in patients with hand, foot and mouth disease. Scand J Infect Dis 44:465–469. doi: 10.3109/00365548.2011.649490. [DOI] [PubMed] [Google Scholar]
- 39.Huang SW, Lee YP, Hung YT, Lin CH, Chuang JI, Lei HY, Su IJ, Yu CK. 2011. Exogenous interleukin-6, interleukin-13, and interferon-gamma provoke pulmonary abnormality with mild edema in enterovirus 71-infected mice. Respir Res 12:147. doi: 10.1186/1465-9921-12-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin YW, Chang KC, Kao CM, Chang SP, Tung YY, Chen SH. 2009. Lymphocyte and antibody responses reduce enterovirus 71 lethality in mice by decreasing tissue viral loads. J Virol 83:6477–6483. doi: 10.1128/JVI.00434-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed U.S. National Academies Press, Washington, DC. [Google Scholar]
- 42.Shen FH, Tsai CC, Wang LC, Chang KC, Tung YY, Su IJ, Chen SH. 2013. Enterovirus 71 infection increases expression of interferon-gamma-inducible protein 10 which protects mice by reducing viral burden in multiple tissues. J Gen Virol 94:1019–1027. doi: 10.1099/vir.0.046383-0. [DOI] [PubMed] [Google Scholar]
- 43.Kung YH, Huang SW, Kuo PH, Kiang D, Ho MS, Liu CC, Yu CK, Su IJ, Wang JR. 2010. Introduction of a strong temperature-sensitive phenotype into enterovirus 71 by altering an amino acid of virus 3D polymerase. Virology 396:1–9. doi: 10.1016/j.virol.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 44.Chen SH, Garber DA, Schaffer PA, Knipe DM, Coen DM. 2000. Persistent elevated expression of cytokine transcripts in ganglia latently infected with herpes simplex virus in the absence of ganglionic replication or reactivation. Virology 278:207–216. doi: 10.1006/viro.2000.0643. [DOI] [PubMed] [Google Scholar]
- 45.Tsai CC, Kai JI, Huang WC, Wang CY, Wang Y, Chen CL, Fang YT, Lin YS, Anderson R, Chen SH, Tsao CW, Lin CF. 2009. Glycogen synthase kinase-3beta facilitates IFN-gamma-induced STAT1 activation by regulating Src homology-2 domain-containing phosphatase 2. J Immunol 183:856–864. doi: 10.4049/jimmunol.0804033. [DOI] [PubMed] [Google Scholar]
- 46.Tseng PC, Huang WC, Chen CL, Sheu BS, Shan YS, Tsai CC, Wang CY, Chen SO, Hsieh CY, Lin CF. 2012. Regulation of SHP2 by PTEN/AKT/GSK-3beta signaling facilitates IFN-gamma resistance in hyperproliferating gastric cancer. Immunobiology 217:926–934. doi: 10.1016/j.imbio.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 47.Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. 1993. Immune response in mice that lack the interferon-gamma receptor. Science 259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- 48.Stubblefield Park SR, Widness M, Levine AD, Patterson CE. 2011. T cell-, interleukin-12-, and gamma interferon-driven viral clearance in measles virus-infected brain tissue. J Virol 85:3664–3676. doi: 10.1128/JVI.01496-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burdeinick-Kerr R, Govindarajan D, Griffin DE. 2009. Noncytolytic clearance of sindbis virus infection from neurons by gamma interferon is dependent on Jak/STAT signaling. J Virol 83:3429–3435. doi: 10.1128/JVI.02381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 51.Coccia EM, Marziali G, Stellacci E, Perrotti E, Ilari R, Orsatti R, Battistini A. 1995. Cells resistant to interferon-beta respond to interferon-gamma via the Stat1-IRF-1 pathway. Virology 211:113–122. doi: 10.1006/viro.1995.1384. [DOI] [PubMed] [Google Scholar]
- 52.Song MM, Shuai K. 1998. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J Biol Chem 273:35056–35062. doi: 10.1074/jbc.273.52.35056. [DOI] [PubMed] [Google Scholar]
- 53.Yoshimura A, Naka T, Kubo M. 2007. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 54.You M, Yu DH, Feng GS. 1999. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol Cell Biol 19:2416–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu TR, Hong YK, Wang XD, Ling MY, Dragoi AM, Chung AS, Campbell AG, Han ZY, Feng GS, Chin YE. 2002. SHP-2 is a dual-specificity phosphatase involved in Stat1 dephosphorylation at both tyrosine and serine residues in nuclei. J Biol Chem 277:47572–47580. doi: 10.1074/jbc.M207536200. [DOI] [PubMed] [Google Scholar]
- 56.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 57.Yang CH, Li HC, Jiang JG, Hsu CF, Wang YJ, Lai MJ, Juang YL, Lo SY. 2010. Enterovirus type 71 2A protease functions as a transcriptional activator in yeast. J Biomed Sci 17:65. doi: 10.1186/1423-0127-17-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zang M, Gong J, Luo L, Zhou J, Xiang X, Huang W, Huang Q, Luo X, Olbrot M, Peng Y, Chen C, Luo Z. 2008. Characterization of Ser338 phosphorylation for Raf-1 activation. J Biol Chem 283:31429–31437. doi: 10.1074/jbc.M802855200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun H, King AJ, Diaz HB, Marshall MS. 2000. Regulation of the protein kinase Raf-1 by oncogenic Ras through phosphatidylinositol 3-kinase, Cdc42/Rac and Pak. Curr Biol 10:281–284. doi: 10.1016/S0960-9822(00)00359-6. [DOI] [PubMed] [Google Scholar]
- 60.Zang M, Waelde CA, Xiang X, Rana A, Wen R, Luo Z. 2001. Microtubule integrity regulates Pak leading to Ras-independent activation of Raf-1. Insights into mechanisms of Raf-1 activation. J Biol Chem 276:25157–25165. doi: 10.1074/jbc.M100152200. [DOI] [PubMed] [Google Scholar]
- 61.Zang M, Hayne C, Luo Z. 2002. Interaction between active Pak1 and Raf-1 is necessary for phosphorylation and activation of Raf-1. J Biol Chem 277:4395–4405. doi: 10.1074/jbc.M110000200. [DOI] [PubMed] [Google Scholar]
- 62.Jiang H, Weng L, Zhang N, Arita M, Li R, Chen L, Toyoda T. 2011. Biochemical characterization of enterovirus 71 3D RNA polymerase. Biochim Biophys Acta 1809:211–219. doi: 10.1016/j.bbagrm.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 63.Sun Y, Wang Y, Shan C, Chen C, Xu P, Song M, Zhou H, Yang C, Xu W, Shi PY, Zhang B, Lou Z. 2012. Enterovirus 71 VPg uridylation uses a two-molecular mechanism of 3D polymerase. J Virol 86:13662–13671. doi: 10.1128/JVI.01712-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lehtonen A, Matikainen S, Julkunen I. 1997. Interferons up-regulate STAT1, STAT2, and IRF family transcription factor gene expression in human peripheral blood mononuclear cells and macrophages. J Immunol 159:794–803. [PubMed] [Google Scholar]