

ABSTRACT
Latent Kaposi's sarcoma-associated herpesvirus (KSHV) genomes encode a homolog of cellular FLICE-inhibitory proteins (termed v-FLIP) that activates NF-κB and can trigger important proinflammatory and antiapoptotic changes in latently infected cells. The protein is present at very low levels in infection and has generally been difficult to efficiently express in recombinant vectors. Here we show that codon usage in the v-FLIP gene is strikingly suboptimal. Optimization of codon use in expression vectors, as expected, restores efficient protein expression. Surprisingly, however, it also dramatically increases the steady-state level of v-FLIP mRNA, at least in part by increasing mRNA stability. When codon-optimized v-FLIP sequences are reintroduced into intact KSHV genomes, the resulting virus expresses readily detectable monocistronic v-FLIP mRNAs that are undetectable in wild-type (WT) infection by blot hybridization, suggesting that such RNAs are in fact transcribed in WT infection but fail to accumulate. The overexpression of v-FLIP by codon-optimized latent genomes results in a 5- to 7-fold decrement in virus production following lytic induction, indicating that maximizing NF-κB signaling is deleterious to induction. These studies provide a clear explanation for the evolution of inefficient codon usage in this gene and point to a strong connection between translational efficiency and RNA accumulation in mammalian cells.
IMPORTANCE This study reports that inefficient codon usage in a herpesviral gene is strikingly correlated with the inability of its mRNA to accumulate in cells; correction of efficient translatability restores RNA abundance. A similar correlation has been reported in yeast species, but the mechanisms operating in mammalian cells appear substantially different.
INTRODUCTION
The major latency locus (MLL) of the Kaposi's sarcoma-associated herpesvirus (KSHV) expresses several important proteins (1) and approximately 20 microRNAs (2, 3). The latent proteins include (i) the latency-associated nuclear antigen (LANA), which plays a central role in stable maintenance of the viral genome in latency (4), (ii) a virally encoded cyclin D homolog (5), (iii) kaposins, a family of proteins affecting cell signaling (6, 7), and (iv) a viral homolog of cellular FLICE-inhibitory proteins (v-FLIP) (8).
Like cellular FLIPs, the v-FLIP contains death effector domains (DEDs) (9), but unlike its host homologs, it does not appear to directly modulate caspase activation from the plasma membrane. Rather, it interacts with cytosolic IKK-γ (10), leading to phosphorylation and proteasomal degradation of IκB, the inhibitor of the transcription factor NF-κB. As a result, NF-κB undergoes constitutive activation, leading to the expression of a proinflammatory (11) and antiapoptotic (12) program. The antiapoptotic aspect of this program is essential for B-cell survival in primary effusion lymphoma (PEL) (13), and the proinflammatory signaling is speculated to play a role in the inflammatory phenotype of Kaposi's sarcoma (KS). NF-κB activation in human primary endothelial cells is also responsible for the spindle-like elongation characteristic of the infected endothelial cells of KS (14). Finally, v-FLIP-mediated NF-κB activation helps to stabilize the latent state by impairing lytic reactivation (15, 16), and inhibition of NF-κB triggers enhanced lytic induction (14). Therefore, v-FLIP expression is considered to be an important feature of KSHV latency in most lineages examined to date.
Despite this, how v-FLIP is expressed in infection has been difficult to rigorously define. Expression of the protein appears to be extremely inefficient; it is difficult to detect by immunoblotting in latently infected cells, and attempts to express it from potent recombinant vectors generally result in unexpectedly low levels of protein accumulation (see reference 17 and Fig. 3 below). Northern blots of latently infected cells do not show the presence of a monocistronic mRNA for v-FLIP; rather, v-FLIP sequences detected by this method are found as the 3′-most open reading frame (ORF) in bi- or tricistronic transcripts directed from the LANA promoter (a map of the known transcripts from the MLL is shown in Fig. 1) (18–22). This seeming paradox led to the identification of sequences in the v-cyclin gene that can function as an internal ribosome entry site (IRES) element in recombinant vectors (17, 23), and since that time, it has been presumed that v-FLIP is expressed from polycistronic mRNAs via IRES-mediated internal translational initiation. Direct evidence for this mechanism in infected cells has been lacking, however. In fact, reverse transcription (RT)-PCR-based studies have detected low-level expression of a spliced monocistronic v-FLIP RNA (Fig. 1) in some latently infected cells (17). Moreover, the reasons for the difficulty in achieving efficient expression of the protein in recombinant vectors have remained largely unexplored.
FIG 3.

Codon optimization of v-FLIP gene sequence increases abundance of protein and RNA. (A) Western blot of proteins from 293 cells that were transfected with recombinant FLAG-tagged v-FLIP. Western blotting was done using an anti-FLAG monoclonal antibody. (B) Northern blot of increasing amounts of RNA (1 μg, 5 μg, and 10 μg of total RNA), denoted by the number of plus signs, is shown. v-FLIP RNA is detected with a biotin-labeled oligonucleotide probe complementary to the FLAG tag sequences of v-FLIP; neo transcripts are detected with a probe against the neo gene present on the vector encoding recombinant v-FLIP.
FIG 1.

Structure of KSHV major latency locus transcripts. Arrows depict mRNA structures. Transcripts of 5.4 to 5.8 kb (tricistronic) encode LANA; 1.7-kb (bicistronic) mRNAs encode v-cyclin and possibly v-FLIP (via an IRES-mediated mechanism). The 1.1-kb (monocistronic) mRNA, previously only detected by RT-PCR, can encode only v-FLIP. Abbreviations: LTc, constitutive latency promoter; LTi, inducible latency promoter; LANA, latency-associated nuclear antigen; v-CYC, virally encoded cyclin homolog; v-FLIP, viral homolog of FLICE-inhibitory protein.
Here we show that a major reason underlying the difficulty expressing v-FLIP has to do with its strikingly abnormal codon usage. Importantly, however, this abnormal codon use not only impairs its efficient translation but also imposes a dramatic block to the accumulation of v-FLIP mRNA. Restoration of efficient codon use in recombinant expression vectors not only improves protein expression but also strikingly increases steady-state levels of the corresponding mRNA. When the codon-optimized gene is recombined back into the viral genome and progeny virus used to infect cells, the resulting cells display strong accumulation of a monocistronic v-FLIP mRNA, suggesting that such a transcript is indeed produced by wild-type (WT) viral infection but fails to accumulate. When latently infected cells bearing the codon-optimized v-FLIP gene are induced, the resulting viral yields are diminished 5- to 7-fold, as are many important viral lytic transcripts. These data (i) resolve many of the extant questions concerning v-FLIP expression, (ii) affirm that efficient v-FLIP expression is deleterious to lytic viral growth, and (iii) point to strong connections between translational elongation efficiency and RNA accumulation in mammalian cells. Such connections have been previously observed in yeast species (24), but to our knowledge this is the first report of such effects in mammalian cells, and their determinants appear significantly different from those described in yeast.
MATERIALS AND METHODS
Cell culture.
SLK, SLK.Bac16KSHV, and 293 cells were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco), l-glutamine (2 mM; Invitrogen), penicillin (100 IU/ml; Gibco), and streptomycin (100 μg/ml; Gibco). SLK.Bac16KSHV cells were grown in the presence of 300 μg/ml hygromycin B to maintain selection of the viral episome. Lymphatic endothelial cells (LECs) and blood endothelial cells (BECs) were purchased from Lonza and cultured in EBM-2 medium and supplemented with EGM-2 MV (Lonza). All cells were grown at 37°C under a 5% CO2 atmosphere.
Codon optimization of v-FLIP and eGFP (enhanced green fluorescent protein) genes.
The codon optimization of each gene was done using the polymerase cycling assembly (PCA) procedure adapted from published work (25, 26). Polyacrylamide gel- or high-performance liquid chromatography (HPLC)-purified 60-nucleotide-long overlapping oligonucleotides were designed to span both strands of the desired sequence. (Sequences of the oligonucleotides are available upon request.) These were mixed at a final concentration of 200 nM to a 50-μl final volume. The PCA reaction was performed using Expand high-fidelity polymerase (Roche) according to the manufacturer's instructions. The PCR was done at an initial 95°C for 2 min, followed by a chain of 95°C for 30 s, 55°C for 30 min, and 72°C for 60 s for 35 cycles, followed by a final 72°C for 5 min. Two microliters of the PCA reaction mixture was used as a template for amplifying the full-length assembled product using short forward and reverse terminal primers.
Transcription inhibition experiments.
Two methods were used to shut off transcription. In one method, actinomycin D at a concentration of 5 μg/ml was added to 293 cells that had been transfected with recombinant v-FLIP (completely or partially optimized clones in pcR3.1). Treatment was done 48 h after transfection for up to 7.5 h, and total RNA was extracted at different time points and then analyzed by Northern analyses. In another method, the mutant v-FLIP was cloned into pTRE2 vector and then transfected into Tet-Off cells, which express a doxycycline-dependent repressor. After 2 days of constitutive FLIP expression, doxycycline was added at various time points and RNA was extracted from cells and analyzed for v-FLIP mRNA by Northern analysis.
BAC mutagenesis.
Mutagenesis of the Bac16 clone (27) was done by bacterial artificial chromosome (BAC) recombineering using galK selection as described previously (28) with Escherichia coli SW102 cells. In this procedure, a GalK gene bearing flanking v-FLIP sequences is first inserted into the WT v-FLIP locus; in a second step, the insert is then replaced with that for a mutated v-FLIP gene, again relying on homologous recombination in the flanking v-FLIP sequences. After each step of recombination, the location of the insert and the junctions of the insert and the neighboring bacmid sequence were confirmed by PCR. The primers used for amplifying and then recombining the GalK gene into the location of the v-FLIP gene are as follows: Fwd, 5′AAATCAGATACATACATTCTACGGACCAAAAATTAGCAACAGCTTGTTATCCTGTTGACAATTAATCATCGGCA3′; Rev, 5′AATAAATTTTCCTTTGTTTTTCCACATCGGTGCCTTCACATATACAAGCCGGCACCTCAGCACTGTCCTGCTCCT3′. The primers used for amplifying and then recombining the mutated sequence in order to replace the GalK gene were as follows: Fwd, 5′CTTCAAACGTACCCTGGTTGGC3′; Rev, 5′GCTGGCAAGCATATTGGGATGC3′. The primers used to check the presence and location of the optimized sequence of v-FLIP were as follows: Fwd, 5′CCGCGACATCCGCAGCC3′; Rev, 5′GCCAATAACCCGCCCTCGGG3′.
Infection of human cell lines bearing the mutated Bac16-derived virus. (i) Transfection into 293 cells.
For infections through coculture, 293T cells were transfected with 6 μg of Bac16KSHV (WT or mutant) DNA with 15 μl of Lipofectamine 2000 (Life Technologies) in accordance with the manufacturer's instructions. Twenty-four to 48 h after transfection, when some cells were clearly GFP positive, the culture was split into a larger plate and grown in medium that contained 50 μg/ml of hygromycin B until a stably infected 293T cell population was obtained.
(ii) Coculture of infected 293T cells with iSLK-Puro.
A total of 5 × 106 293T cells in 10 ml of DMEM was mixed with 5 × 106 iSLK-Puro cells in 10 ml of DMEM, 1.2 mM valproic acid was added, and the mixture was incubated for 72 h. After this, the cells were treated with 5 μg/ml of puromycin (to deplete 293T cells) and 300 μg/ml of hygromycin B for 3 to 4 days. After this step, the cells were grown in only hygromycin B (to enrich for infected SLK cells) until a healthy, fully GFP-positive population of latently infected iSLK-Puro cells was obtained.
Infections using virus stock.
KSHV virus stocks bearing wild-type or mutant v-FLIP were made by induction of latently infected cells as previously described (29). Precipitated virus was resuspended into complete DMEM to make an ∼75× stock of concentrated virus. Infection into cell lines was done by spinning the virus on cells at 2,000 rpm for 1 h, after which unbound virus was removed and replaced with new medium. Following this, cells were grown for 48 h and then either treated with hygromycin B containing medium (to select for the episome) or analyzed directly by Northern analyses, Western analyses, flow cytometry, PCR quantification, or sequencing.
For quantitation of viral infectivity, cells were infected with serial dilutions of the virus stock; at 48 h postinfection, the total cell count was determined and the proportion of GFP-positive cells was determined by flow cytometry. From this, the number of GFP focus-forming (i.e., infectious) units in each dilution was determined, and the number of infectious units per microliter in the parental virus stock was determined by back-calculation.
Viral genome sequencing.
The WT and mutated genomes of viral DNA in Bac16 were purified using a maxiprep kit (Qiagen). Different concentrations of purified bacmid were used as inputs into the Nextera XT DNA sample preparation protocol, in accordance with the manufacturer's instructions. The PCR products were analyzed using a high-sensitivity DNA kit on a bioanalyzer (Agilent Technologies). PCR products were then size-selected to enrich for fragments between 450 and 650 bp using a LabChip XT DNA kit (Caliper Life Sciences.) This was done to enhance efficient clustering during sequencing. The library prepared was indexed, and each library was quantified using a double-stranded DNA (dsDNA) HS assay, on a Qubit 2.0 fluorometer (Life Technologies). An 8 pM concentration of each library was loaded onto the MiSeq flow cell. The reads obtained were aligned by use of a MiSeq reporter to the reference Bac16KSHV genome sequence (GenBank accession number GQ994935.1) to obtain the depth of coverage for each position and assess the quality of the reads and the frequency of single-nucleotide polymorphisms, deletions, or insertions at each position.
Review of the sequence of the recombinant v-FLIP codon-optimized virus revealed possible polymorphisms in the ORF32, RTA, MTA, and LANA loci. The ORF32 polymorphism was silent and therefore was not further investigated. The remaining potential polymorphisms were investigated by PCR amplification and resequencing by the Sanger method. This revealed that the base changes in the Illumina sequencing of RTA and MTA were artifactual, as were 3 of the 6 of the base change calls in LANA. The three remaining changes that deep sequencing called were all low-frequency variants (frequency, 0.14 to 0.19) and were in the highly repetitive region of the LANA gene (nucleotides [nt] 125339 to 125360); the majority reads in this region were WT, suggesting that these were sequencing artifacts. In accord with this, one of these changes would generate a stop codon, but the LANA encoded by this genome was full length on immunoblotting (data not shown). Moreover, the LANA produced was fully functional, since it allowed selection of stable 293 cell transductants that maintained functional viral genomes. Our attempts to resequence this region after PCR amplification failed owing to its highly repetitive nature, but the above evidence points strongly to all three of these putative minor polymorphisms being sequencing errors.
Flow cytometry.
Cells were trypsinized, washed (with medium and then phosphate-buffered saline [PBS]), and then fixed with 0.5% paraformaldehyde for 5 min. Cells were then washed twice with fluorescence-activate cell sorter (FACS) buffer containing 4% FBS and 0.09% sodium azide and analyzed on an LSRII flow cytometer (BD Biosciences) for GFP-positive (infected) cells.
Northern blotting.
Total RNA was harvested from cells using RNA-BEE (AMSBIO catalog no. Cs-104B) in accordance with the manufacturer's instructions; poly(A) mRNA was further purified using an Oligotex mRNA minikit (Qiagen). Ten micrograms of total RNA or 500 ng of poly(A) mRNA was separated by 1% agarose-formaldehyde gel electrophoresis and transferred onto a Nytran membrane. For Northern blotting, a DNA oligonucleotide probe complementary to the FLAG sequence was labeled with the BrightStar Psoralen biotin kit (Life Technologies catalog no. AM1480) in accordance with the manufacturer's guidelines. Gene and sense-specific riboprobes used for detecting the v-FLIP and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNAs were synthesized from PCR products using the MAXIscript T7-T3 transcription kit (Ambion catalog no. AM1326) according to the manufacturer's instructions. The primers used for PCR amplification of gene-specific probes are as follows: for the WT v-FLIP probe (nt 122588 to 122940), 5′TAATACGACTCACTATAGGGCTATGGTGTATGGCGATAGTGTTGGG3′ (FwdT7) and 5′TGCTTCACTTAGACCCGCGTTTTTTAG3′ (Rev); for the HS v-FLIP probe (nt 122686 to 123017), 5′TAATACGACTCACTATAGGGTGCGCAGCATGCTCATCAGGGCGTCC3′ (FwdT7) and 5′GGAGGAGGGCCGCCTGACCTTC3′ (Rev).
Western blotting.
Medium was removed from the cells, and the cells were washed with PBS and then lysed in radioimmunoprecipitation assay (RIPA) buffer on the plate at 4°C, vortexed in a tube for 5 s, incubated on ice for 10 min, and then spun at 14,000 × g for 15 min at 4°C. The protein content of supernatant was quantified using a bicinchoninic acid (BCA) protein assay, 20 μg of total protein was loaded on a Bis-Tris gel (Invitrogen), and Western blotting was performed. Antibodies against ORF8 (Abcam), K8alpha (Abcam), ORF45 (Abcam), gpK8.1 (Advanced), ORF57 (Santa Cruz), ORFKbZIP (Santa Cruz), and v-FLIP (a rabbit polyclonal antibody made by Adam Grundhoff) were detected with horseradish peroxidase (HRP)-conjugated secondary antibodies using enhanced chemiluminescence (ECL; GE).
Real-time PCR quantification.
To estimate amounts of virus in the supernatant of cells, the virus DNA was purified as described by Grossman et al. (30). Quantitative PCR was performed using an Applied Biosystems 7300 real-time PCR system and SYBR green real-time PCR master mix (Life Technologies) according to the manufacturer's instructions. The promoter sequences in the K2 gene were quantified using the following primers: K2Fwd, GACCTGCAAACTTTTCCATTGC; K2Rev, GCTGACTAAGACGCACTACA. A foreign DNA sequence was added as an internal control to the samples and was measured as described previously (14), except that the primers used were as follows: Fwd, 5′AGGACCCGATCAACAACATC3′; Rev, 5′ATCGCGTTCTTGTTCAGCTT3′.
RESULTS
Inefficient codon usage and expression of v-FLIP.
Having encountered difficulties in the efficient expression of recombinant v-FLIP, we first inspected the codon usage in the v-FLIP coding region, using the Graphical Codon Usage Analyzer (GCUA) program. As shown in Fig. 2 (top panel), codon usage in the wild-type (WT) KSHV v-FLIP gene is markedly suboptimal, with numerous positions across the entire coding region displaying use of extremely inefficient codons; for comparison, codon usage in the ß-actin gene, which is efficiently expressed, is depicted in Fig. 2 (center panel). Therefore, we constructed a synthetic v-FLIP gene in which every codon position was optimized for efficient translation in human cells; the predicted translational efficiency of codons in this construct, which we denote v-FLIP HS (for homo sapiens optimized), is shown in the bottom panel of Fig. 2.
FIG 2.

Codon usage of the β-actin and wild-type and codon-optimized KSHV-encoded FLIP genes. The codon usage of the above-mentioned genes are represented using the Graphical Codon Usage Analyzer (GCUA) available online at http://gcua.schoedl.de/. Codon usage in a sequence is shown from the 5′ end to the 3′ end of the mRNA (along the horizontal axis). It is represented using the codon adaptation index that is derived from the codon frequency in the Homo sapiens codon usage table and also takes into account the number of codons that code for each amino acid. Codon efficiency of less than 10% is shown by red bars, that between 10% and 20% by gray bars, and that above 20% by black bars.
To determine if codon optimization improved v-FLIP expression, the WT and HS versions of the gene were cloned into the pcDNA3.1 expression vector, in which transcription is driven by the strong cytomegalovirus (CMV) immediate early (IE) promoter; each construct was FLAG epitope-tagged at its N terminus. Two days posttransfection into 293T cells, extracts were prepared and immunoblotted with anti-FLAG monoclonal antibody (MAb). As shown in Fig. 3A, no v-FLIP protein was observed in cells transfected with the empty vector (left lane), and a barely detectable signal was present in the lane transfected with the WT v-FLIP gene (center lane); in contrast, a strong signal at the expected molecular weight (MW) of v-FLIP was obtained with the codon-optimized HS variant (right lane). Although this result was anticipated, analysis of v-FLIP mRNA levels by Northern blotting (Fig. 3B) revealed a major surprise. Wild-type v-FLIP mRNA was nearly undetectable by this method, while the codon-optimized HS variant showed abundant accumulation of correctly sized v-FLIP transcripts. This result was not due to poor transfection of (or faulty RNA preparation from) the WT cells, since pcDNA-encoded neo transcripts accumulated identically in all transfected cells (Fig. 3B). qRT-PCR quantitation of the relative levels of v-FLIP in HS and WT-transfected cells indicated that, by 48 h posttransfection, HS RNA accumulation was ca. 70- to 140-fold that of the WT transcripts.
The HS variant is the result of multiple base changes throughout the length of the v-FLIP coding region. Although these lesions do not alter the amino acid sequence of the protein, they might have created an RNA element that promotes mRNA accumulation or (more likely) disrupted an element that impairs accumulation. To examine this possibility, and to map any putative accumulation-influencing element, we constructed a series of chimeras between the WT and HS versions of v-FLIP (Fig. 4A). Each chimera was transfected into 293T cells and assayed for transcript accumulation by Northern blotting. The results in Fig. 4B show that no single region could be identified as uniquely associated with RNA accumulation. Overall, the v-FLIP transcript levels were roughly proportional to the length of sequence derived from the codon-optimized HS variant, although position effects are also likely playing a role (as suggested by constructs 4 and 5 [lanes 4 and 5], which each contain 50% WT sequences in reciprocal locations but accumulate to modestly different levels).
FIG 4.
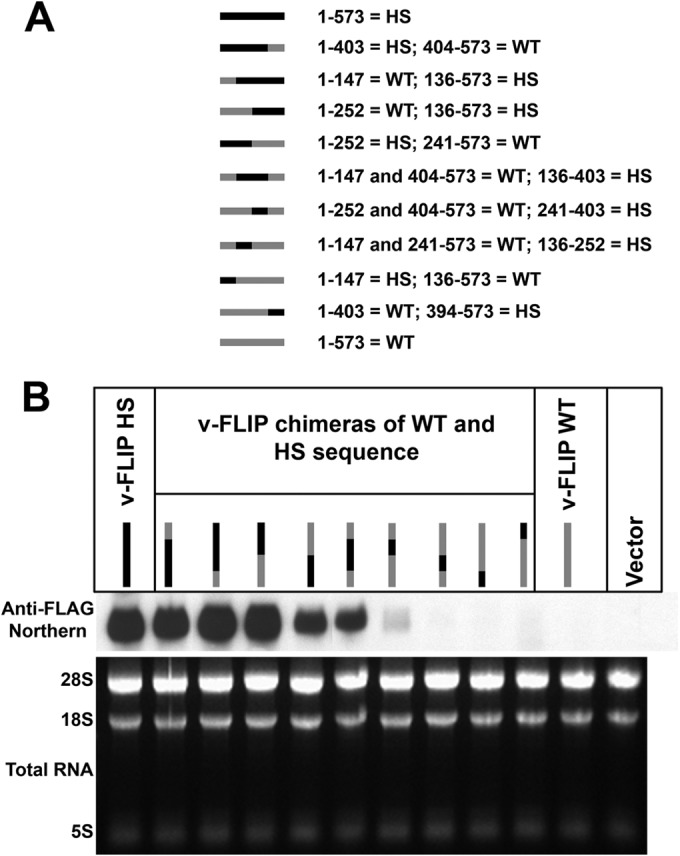
Chimeras between wild-type and optimized sequences of the v-FLIP gene show correlation between amount of optimized sequence and abundance of RNA. (A) The chimeras are represented by gray (for wild-type sequences) and black (for optimized sequences), and the nucleotide positions of these regions are indicated alongside. Note that the regions between nt 136 and 147, nt 241 and 252, and nt 394 and 403 of the sequence are the same in wild-type and optimized v-FLIP genes. (B) Northern blot probing for the invariant 5′ FLAG tag on the chimeras using labeled probe against FLAG (upper panel). The lower panel depicts ethidium bromide staining of total RNA as a loading control.
To determine if the effect of codon usage on RNA accumulation was peculiar to the KSHV v-FLIP gene, we asked if similar effects could be seen on a nonviral reporter gene, the eGFP gene. As shown in Fig. 5A (top), this gene's codon usage has previously been optimized for efficient mammalian expression. We constructed two variants of this gene, both of which encode GFPs identical in amino acid sequence to the parental cistron: one (Fig. 5A, middle panel) in which only the first portion of the coding region was deoptimized, and a second (bottom panel) in which codon usage was deoptimized across the entire gene. The probe used in this experiment was against the invariant 5′ FLAG tag. As shown in Fig. 5B, transfection of 293T cells with these constructs revealed that the fully deoptimized GFP gene showed a dramatic decrease in mRNA accumulation compared to the optimized gene; the partially deoptimized variant showed a modest reduction in accumulation.
FIG 5.

Codon usage deoptimization in enhanced green fluorescent protein (eGFP) decreases the abundance of its mRNA. (A) The codon usage is represented using the Graphical Codon Usage Analyzer (GCUA) available online at http://gcua.schoedl.de/, as was done for v-FLIP in Fig. 2. The upper panel shows the (optimized) eGFP, the middle panel shows the mutant with deoptimization of the 5′ 180 nt, and the bottom panel shows eGFP, whose codon usage has been completely deoptimized. (B) Northern blot (using probe against FLAG tag) of total RNA from HEK293 cells transfected with recombinant FLAG-tagged eGFP and mutants.
Inefficient codon use in the 3′ gene does not prevent accumulation of bicistronic RNAs.
One of the features of the transcripts of the KSHV major latency locus is the abundant accumulation of bicistronic and (to a lesser extent) tricistronic RNAs in which the v-FLIP gene is in the 3′ position (Fig. 1). This suggests that, if our transient-transfection system faithfully reproduces the regulation in the viral genome, engineering the WT v-FLIP gene into the 3′ position of a bicistronic mRNA (where it might be expected to be poorly translated) should not impair accumulation of the bicistronic transcript. To examine this issue, we constructed (in pcDNA3.1) bicistronic constructs bearing eGFP in the 5′ position and v-FLIP (WT or HS) in the 3′ position. As shown in Fig. 6A, such constructs either contained (top line) or lacked (middle two lines) a functional poliovirus IRES between the two ORFs; a fourth construct (bottom line) fused eGFP and v-FLIP in-frame, creating a single ORF from the two elements. In all constructs, v-FLIP was FLAG tagged, and transcripts were detected using a probe recognizing FLAG coding sequences. Each construct was transfected into 293T cells, and RNA was prepared at 48 h posttransfection. Figure 6B shows that transcripts corresponding in size and sequence to the expected bicistronic RNAs were readily detected from all of these constructs, in roughly equal abundances. Paradoxically, there did not appear to be a large effect of manipulating the translation of the downstream v-FLIP gene: whether an IRES was present (Fig. 6, lanes 1 and 2 or 9 and 10) or absent in the mRNA (which should have strongly affected v-FLIP translation) (Fig. 6, lanes 3 to 6 or 11 to 14) seemed to have little influence upon transcript accumulation. Similarly, entry of ribosomes into the (poorly translatable) v-FLIP sequences in the fusion construct should be very efficient, yet this construct displays abundant accumulation of its transcript (compare lanes 7 and 8 with lanes 15 and 16). Thus, the dramatic effect of suboptimal codon usage on RNA levels appears to depend upon position in the transcript. We do not yet understand the basis of this position dependence; however, these results affirm that our transient-transfection assay faithfully reproduces this aspect of gene expression from the intact KSHV genome in virally infected cells.
FIG 6.
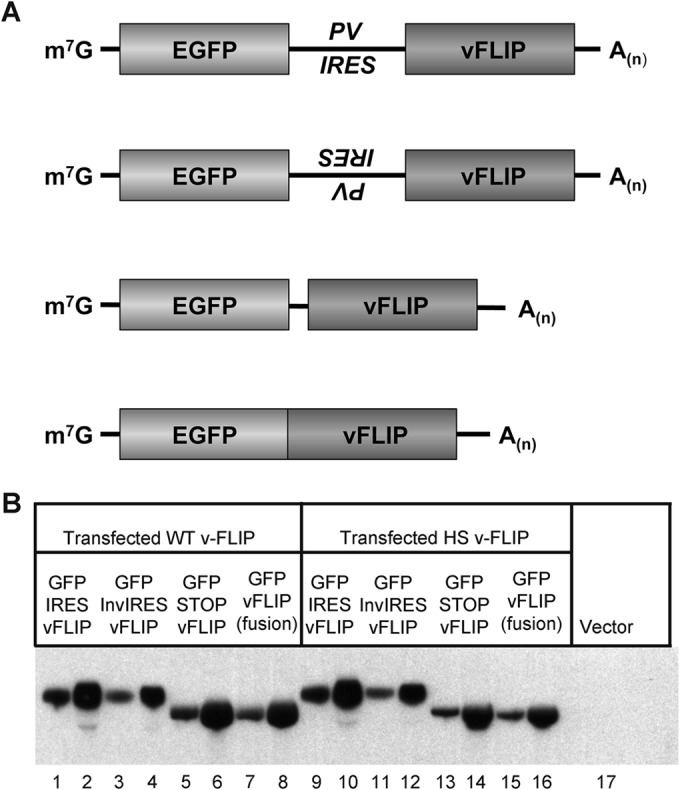
Transcripts of bicistronic constructs accumulate efficiently in transfected cells even when the 3′ gene has inefficient codon usage. (A) Diagrams of plasmids used in this study. First diagram, eGFP–IRES–v-FLIP; second diagram, eGFP–inverted IRES–v-FLIP; third diagram, eGFP–Stop–v-FLIP; fourth diagram, eGFP–v-FLIP fusion. (B) Northern blot of 293 cells transfected with the indicated constructs; shown for each construct are blots examining 3 μg and 10 μg of total RNA. Left panel (lanes 1 to 8), the FLIP gene in each construct is WT; right panel (lanes 9 to 16), the FLIP gene in each construct has been codon optimized.
Transcripts with suboptimal codon usage are destabilized.
Given that translation is a cytosolic process, we speculated that the failure of WT (codon-deoptimized) v-FLIP transcripts to accumulate was likely due to destabilization of the mRNA. To examine this, we treated v-FLIP-expressing cells with actinomycin D to shut off additional transcription and then examined the accumulation of transcripts at various times thereafter. Because WT v-FLIP mRNA is virtually undetectable at steady state, we chose to study the chimera (here termed construct 7) depicted in lane 7 of Fig. 4B; this transcript accumulates to lower but detectable levels, thereby facilitating the analysis. For a control, we examined the stability of codon-optimized v-FLIP-HS transcripts in parallel; RNA was examined by Northern blotting with a probe directed at the FLAG sequences common to both constructs. As shown in Fig. 7A, codon-optimized mRNAs are strikingly stable, with no visible decrement over the 7.5-h chase period. In contrast, construct 7, in which roughly 75% of the gene displays inefficient codon use, shows substantial degradation beginning at 40 to 60 min of the actinomycin D chase. However, we also note that there is very substantially less accumulation of construct 7 mRNA at the zero time point. We speculate that this may be due to extremely rapid (cotranscriptional?) degradation, since such a process would result in failure to generate the full-length RNA being measured in the Northern analysis. Alternatively, it could reflect additional effects on RNA synthesis, processing, or cytoplasmic transport. In all of these cases, however, it is unclear how such intranuclear events would be affected by base changes designed to affect translation, a cytosolic process (see Discussion).
FIG 7.

Reduced stability of partially optimized (construct 7) RNA compared to completely optimized (HS) RNA. (A) RNA from 293 cells that had been transfected with construct 7 or HS v-FLIP was purified after treatment with actinomycin D for the indicated times. Northern blotting was done using labeled probe against the invariant 5′ FLAG tag. (B) Doxycycline-induced repression of transfected v-FLIP genes (construct 7 or HS) was instituted at 48 h posttransfection (here designated 0 h), and RNA was harvested at the indicated times thereafter and examined by Northern blotting against the invariant FLAG tag of v-FLIP.
Actinomycin D is a cytotoxic drug with many secondary effects, since it is a global inhibitor of transcription. To examine transcript stability in the absence of this drug, we separately cloned FLAG-tagged codon-optimized and construct 7 v-FLIP genes into a Tet-Off vector plasmid in which their mRNA synthesis can be repressed by the addition of doxycycline to the medium (7). We then examined the accumulation of mRNA for each construct by Northern blotting (again using the common FLAG probe) at various times after doxycycline addition. This experiment (Fig. 7B) again affirmed the stability of the codon-optimized sequence (the seeming loss at 24 h after doxycycline addition is due to underloading of this lane). Construct 7 RNA again showed very rapid early loss (see 0- and 2-h time points) but also showed clear destabilization during the chase, here between 8 and 24 h after doxycycline-mediated repression. (The slower kinetics of loss in the doxycycline studies than in the actinomycin studies is almost certainly due to the cytotoxicity of actinomycin D.) We conclude that accelerated RNA turnover is at least partially responsible for the reduced accumulation that follows from inefficient codon use but cannot exclude the possibility of additional mechanisms that could contribute importantly to RNA loss (see below and Discussion).
Attempts to decipher the mechanism of impaired transcript accumulation in WT v-FLIP mRNA.
Several well-described pathways connect correct mRNA translation to RNA accumulation in eukaryotic cells. The best characterized of these is nonsense-mediated mRNA decay (NMD), in which RNA degradation follows the encounter of translating ribosomes with premature stop codons in mutant mRNAs. Another pathway (31), nonstop decay (NSD), operates on model transcripts that lack stop codons at the end of their ORFs—as a result, ski7, a homolog of the release factor eRF3, is recruited to the 3′ end of the RNA, where it recruits a complex of 3′-5′ exonucleases known as the exosome, leading to RNA turnover (32). Neither mechanism is likely to operate in v-FLIP, as the gene has a conventionally positioned stop codon.
In 2006, another process linking impaired translation to RNA turnover was described in yeast. This process, termed “no-go decay” (NGD), was triggered by runs of suboptimal codons (or by engineered hairpins in mRNA), processes that are expected to impede the progress of translating ribosomes on the mRNA (33). To our knowledge, this process has not previously been described in vertebrate cells, but we considered it likely that the KSHV v-FLIP gene could represent the first example of no-go decay in the mammalian context. However, it has been difficult to affirm that the processes at work in this case recapitulate the described yeast pathway. For example, if NGD acted in mammalian cells as it does in yeast, one might have expected the GFP–v-FLIP fusion protein of Fig. 6 to have triggered transcript turnover, but it does not (Fig. 6, compare lanes 7 and 8 with lanes 15 and 16). Moreover, when we engineered a strong RNA hairpin (or stem-loop [SL]) (Fig. 8) into the 5′ UTR of the WT v-FLIP gene (with or without second mutations in the v-FLIP initiator AUG codon, denoted as ΔAUG in Fig. 8), in order to decrease ribosome entry into the deoptimized coding region, no accumulation of v-FLIP mRNA was observed on Northern blots (Fig. 8, lanes 5 to 7). This was true despite a 90% decrement in v-FLIP translation, as judged by examining the ability of the WT and SL/ΔAUG versions of the HS homolog to activate an NF-κB reporter gene (data not shown). We found this result particularly surprising, since rescue of codon-deoptimized RNA accumulation by an upstream hairpin is one of the defining experimental features of yeast NGD (24).
FIG 8.
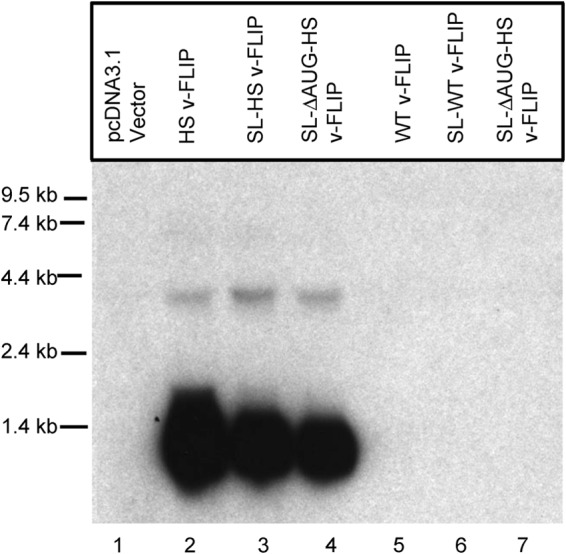
Cloning a stem-loop into the 5′ UTR and mutating the v-FLIP start codon does not enhance accumulation of v-FLIP mRNA. 293 cells were transfected with the indicated plasmid construct, and 48 h later, total RNA was prepared for Northern blotting. All constructs were tagged with a 5′ FLAG tag, and a probe complementary to this sequence was employed to detect the transcripts. Lanes 2 to 4, variants of the HS (codon-optimized) FLIP sequence; lanes 5 to 7, variants of the WT sequence. Abbreviations: SL, stem-loop sequence; ΔAUG, start codon deletion.
The described pathway of NGD in yeast is thought to involve dependence upon two genes, the Dom34 and Hbs1 genes (33, 34). Accordingly, we attempted to knock down expression of the mammalian homologs of these genes in 293 cells and to examine the effect of the knockdown on accumulation of WT or construct 7 v-FLIP RNA. Although we achieved substantial decrease in both proteins (as judged by immunoblotting) we generally did not observe reproducible enhancement of v-FLIP mRNA accumulation (data not shown). This is, of course, a negative result, and subject to all the caveats of the same; perhaps, for example, a more striking impairment of these functions than can be generated by small interfering RNA (siRNA) is required to augment RNA stability. Moreover, a recent review of yeast NGD has raised the possibility that there may be forms of NGD in yeast that do not display dependence on these genes (33), although none of the responsible mechanisms for such independence have been elucidated. For all these reasons, we do not conclude from these experiments that NGD is not operating in our system but simply that multiple first-generation tests designed to implicate this pathway in v-FLIP regulation do not unambiguously do so. The mechanism(s) underlying our observation remains to be rigorously defined, but it is certainly possible that entirely novel mechanisms are at work here.
Properties of KSHV virions bearing codon-optimized v-FLIP sequences.
To explore the role of impaired codon usage in v-FLIP in the context of authentic viral infection, we constructed a mutant of KSHV bearing the codon-optimized v-FLIP gene in place of the WT gene. This was done using homologous recombination into a bacmid-derived KSHV genome (Bac16) in E. coli. Individual recombinant bacmids were subjected to Illumina sequencing to ensure that no secondary mutations were acquired during the recombination; this procedure obviates the need for construction of revertant viruses in which the WT gene is restored via recombination. The details of this analysis are reported in Materials and Methods, and the results validated that the recombinant bore no additional mutations, with the caveat that the repeat region within LANA (nt 125330 to 125360) could not be accurately resequenced owing to its repetitive nature. Once the appropriate recombinant was verified, bacmid DNA bearing either the WT or HS transgene was transfected into 293 cells, which were then induced with butyrate and cocultivated with iSLK cells, which harbor a doxycycline-inducible RTA gene. Following selection in puromycin to counterselect against 293 cells, the resulting latently infected iSLK mass culture was induced with doxycycline and butyrate, and a viral stock was prepared from each culture.
iSLK cells were then infected with virus bearing either the WT or optimized (HS) v-FLIP gene, and infected cells were selected in puromycin. The stable, latently infected cells were then induced with doxycycline plus butyrate, and RNA was prepared at 0 h (latent) and at 24, 48, and 72 h postinduction. After poly(A) selection, we examined each postinduction preparation for v-FLIP transcripts by Northern blotting with a probe corresponding to either WT or HS v-FLIP sequences. As shown in Fig. 9A, FLIP-containing transcripts in this SLK-based cell line have very low abundance in latency but are strongly induced during the lytic cycle. This inducible pattern, while not common in primary effusion lymphoma cell lines, has been described previously and is attributable to an alternative LANA promoter called LTi (inducible latency promoter) that is activated postinduction (35). As expected, in WT infection, v-FLIP-containing RNAs accumulate primarily as a bicistronic RNA (with v-cyclin) and a tricistronic RNA (with LANA and v-cyclin). In contrast, the codon-optimized v-FLIP genes show the clear presence of an additional band corresponding in size to a monocistronic v-FLIP mRNA.
FIG 9.

Codon optimization of the v-FLIP gene increases abundance of monocistronic v-FLIP in a KSHV infection and decreases lytic reactivation and virus production. (A) SLK cells were infected with recombinant KSHV bearing either WT or HS (codon-optimized) v-FLIP genes, and RNA was prepared from uninduced (latent) cells or 24, 48, or 72 h after lytic induction with Na butyrate plus doxycycline, as indicated. Shown are Northern blots of poly(A)-purified RNA from the indicated cultures, after probing with homologous v-FLIP sequences (top) or GAPDH probe (bottom). (B) The medium from infected, induced SLK cells bearing WT or codon-optimized (HS) FLIP recombinants was used to infect HEK293 cells, and 48 h later, 293 cells were analyzed for infection by quantifying GFP positivity by flow cytometry. (C) Infection of LEC (top) and BEC (bottom) is analyzed by GFP positivity. Infection was done using equal volumes of the viral stock, and in both lines diminished infectivity was produced by the codon-optimized (HS) virus. (D) Virions from medium of infected, lytic SLK cells were quantified by counting the number of DNase-resistant viral genomes per milliliter in the culture supernatant of induced cells. The amount of virus in both WT and HS v-FLIP medium was normalized to an internal control DNA (as done by Grossman et al. [30]) and then to the WT v-FLIP, and this is represented in the bar graph.
What is the impact of v-FLIP codon optimization on the biology of KSHV? Given what has previously been published about the role of NF-κB activation in stabilizing latency (15, 16, 30), we might expect v-FLIP overexpression to be associated with impaired lytic reactivation (though this can vary with cell type [14]). To examine this, latent iSLK mass cultures bearing WT and HS v-FLIP genes were induced with doxycycline plus butyrate, and supernatants were examined for (i) infectivity and (ii) viral particle counts, as judged by the presence of nuclease-resistant viral DNA quantified by qPCR. Figure 9B shows that infectivity in the medium was reduced ca. 5- to 7-fold in the codon-optimized virus, compared to WT, in both 293 and SLK recipient cells. Similar reductions were observed in primary endothelial cells as well (Fig. 9C). Figure 9D shows that the reductions in infectivity were associated with corresponding reductions in particle counts in the medium. These defects can be correlated with defects in the expression of several lytic proteins, as judged by immunoblotting of infected cell lysates (data not shown). These results are consistent with expectations based on prior work from our laboratory (30) and others (16).
DISCUSSION
In this study, we have addressed long-standing questions concerning the expression of v-FLIP, with an eye toward explaining why the v-FLIP gene appears to be so inefficiently expressed. Our results show that (i) poor expression of the v-FLIP gene is largely due to its profoundly inefficient codon usage, (ii) this translational inefficiency also leads to a dramatic block of RNA accumulation, (iii) RNA turnover accounts for at least part of this effect, and (iv) the prior failure to observe monocistronic v-FLIP mRNA by Northern blotting in WT infection is the result of this phenomenon. When codon-optimized v-FLIP sequences are restored to the viral genome, the resulting enhancement of NF-κB activity impairs lytic reactivation, which in itself can explain the evolutionary selection for inefficient translation of this gene. Given the known proinflammatory consequences of NF-κB activation, it is also possible that limitation of NF-κB activation by this mechanism provides a mechanism for limiting immune targeting of infected cells in the human host.
While the linkage of RNA turnover to inefficient translational elongation has been described in yeast (and in cultured insect cells [33]), we believe that this is the first report of this phenomenon in a mammalian cell. Our results with deoptimization of GFP translation indicate that this phenomenon is not limited to viral genes or virus-infected cells; it is likely a general observation, though it may require substantial deoptimization to produce the dramatic effects we see on RNA accumulation. However, whether a common mechanism (e.g., no-go decay [NGD]) underlies all of these examples remains to be seen. Traditional NGD, as defined by experiments in yeast, does not adequately explain the position dependence of the RNA loss; that is, why manipulating the translation of a codon-deoptimized 3′ ORF in a bicistronic transcript does not affect transcript accumulation. Similarly, traditional concepts of NGD in yeast suggest that impairing the entry of ribosomes into a codon-deoptimized ORF should improve RNA accumulation, but we did not observe this behavior in our system. As noted above, these negative results do not completely exclude NGD, but they do suggest that if NGD is operating here, it may be considerably more complex in mammalian cells than its yeast counterpart. Finally, we note that the chase experiments shown in Fig. 6 could indicate that manipulating codon usage could have effects on other steps of RNA maturation that operate extremely actively during or closely following transcription. Since many early steps in RNA maturation occur in the nucleus, it is not obvious how these would be influenced by translational efficiency. It may be that, as in NMD (31, 36), assembly of specific protein complexes on the pre-mRNA in the nucleus is a prerequisite for destabilization in the cytoplasm when ribosomes are stalled or paused at suboptimal codons. However this process works, it seems reasonable to view it as a form of quality control that ensures that poorly translatable RNAs are not made in abundance. We anticipate that further dissections of v-FLIP RNA regulation could shed considerable light on such quality assurance mechanisms.
ACKNOWLEDGMENTS
This work was funded by the Howard Hughes Medical Institute and the Novartis Institutes for Biomedical Research, whose support is gratefully acknowledged.
REFERENCES
- 1.Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 120:939–949. doi: 10.1172/JCI40567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. 2005. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci U S A 102:5570–5575. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu Y, Haecker I, Yang Y, Gao S-J, Renne R. 2013. γ-Herpesvirus-encoded miRNAs and their roles in viral biology and pathogenesis. Curr Opin Virol 3:266–275. doi: 10.1016/j.coviro.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballestas ME, Kaye KM. 2011. The latency-associated nuclear antigen, a multifunctional protein central to Kaposi's sarcoma-associated herpesvirus latency. Future Microbiol 6:1399–1413. doi: 10.2217/fmb.11.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Godden-Kent D, Talbot SJ, Boshoff C, Chang Y, Moore P, Weiss RA, Mittnacht S. 1997. The cyclin encoded by Kaposi's sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J Virol 71:4193–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadler R, Wu L, Forghani B, Renne R, Zhong W, Herndier B, Ganem D. 1999. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi's sarcoma-associated herpesvirus. J Virol 73:5722–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCormick C, Ganem D. 2005. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 307:739–741. doi: 10.1126/science.1105779. [DOI] [PubMed] [Google Scholar]
- 8.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schröter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. 1997. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhary PM, Jasmin A, Eby MT, Hood L. 1999. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 18:5738–5746. doi: 10.1038/sj.onc.1202976. [DOI] [PubMed] [Google Scholar]
- 10.Liu L, Eby MT, Rathore N, Sinha SK, Kumar A, Chaudhary PM. 2002. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J Biol Chem 277:13745–13751. doi: 10.1074/jbc.M110480200. [DOI] [PubMed] [Google Scholar]
- 11.An J, Sun Y, Sun R, Rettig MB. 2003. Kaposi's sarcoma-associated herpesvirus encoded vFLIP induces cellular IL-6 expression: the role of the NF-kappaB and JNK/AP1 pathways. Oncogene 22:3371–3385. doi: 10.1038/sj.onc.1206407. [DOI] [PubMed] [Google Scholar]
- 12.Sun Q, Zachariah S, Chaudhary PM. 2003. The human herpes virus 8-encoded viral FLICE-inhibitory protein induces cellular transformation via NF-kappaB activation. J Biol Chem 278:52437–52445. doi: 10.1074/jbc.M304199200. [DOI] [PubMed] [Google Scholar]
- 13.Guasparri I, Keller SA, Cesarman E. 2004. KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med 199:993–1003. doi: 10.1084/jem.20031467. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Grossmann C, Ganem D. 2008. Effects of NFkappaB activation on KSHV latency and lytic reactivation are complex and context-dependent. Virology 375:94–102. doi: 10.1016/j.virol.2007.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao J, Punj V, Matta H, Mazzacurati L, Schamus S, Yang Y, Yang T, Hong Y, Chaudhary PM. 2007. K13 blocks KSHV lytic replication and deregulates vIL6 and hIL6 expression: a model of lytic replication induced clonal selection in viral oncogenesis. PLoS One 2:e1067. doi: 10.1371/journal.pone.0001067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown HJ, Song MJ, Deng H, Wu T-T, Cheng G, Sun R. 2003. NF-kappaB inhibits gammaherpesvirus lytic replication. J Virol 77:8532–8540. doi: 10.1128/JVI.77.15.8532-8540.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grundhoff A, Ganem D. 2001. Mechanisms governing expression of the v-FLIP gene of Kaposi's sarcoma-associated herpesvirus. J Virol 75:1857–1863. doi: 10.1128/JVI.75.4.1857-1863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J Virol 72:8309–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J Virol 72:1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Talbot SJ, Weiss RA, Kellam P, Boshoff C. 1999. Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology 257:84–94. doi: 10.1006/viro.1999.9672. [DOI] [PubMed] [Google Scholar]
- 21.Pearce M, Matsumura S, Wilson AC. 2005. Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi's sarcoma-associated herpesvirus originate from a common promoter. J Virol 79:14457–14464. doi: 10.1128/JVI.79.22.14457-14464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai X, Cullen BR. 2006. Transcriptional origin of Kaposi's sarcoma-associated herpesvirus microRNAs. J Virol 80:2234–2242. doi: 10.1128/JVI.80.5.2234-2242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Low W, Harries M, Ye H, Du MQ, Boshoff C, Collins M. 2001. Internal ribosome entry site regulates translation of Kaposi's sarcoma-associated herpesvirus FLICE inhibitory protein. J Virol 75:2938–2945. doi: 10.1128/JVI.75.6.2938-2945.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doma MK, Parker R. 2006. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL. 1995. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164:49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]
- 26.Smith HO, Hutchison CA, Pfannkoch C, Venter JC. 2003. Generating a synthetic genome by whole genome assembly: phiX174 bacteriophage from synthetic oligonucleotides. Proc Natl Acad Sci U S A 100:15440–15445. doi: 10.1073/pnas.2237126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brulois KF, Chang H, Lee AS-Y, Ensser A, Wong L-Y, Toth Z, Lee SH, Lee H-R, Myoung J, Ganem D, Oh T-K, Kim JF, Gao S-J, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21. doi: 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grossmann C, Podgrabinska S, Skobe M, Ganem D. 2006. Activation of NF-kappaB by the latent vFLIP gene of Kaposi's sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J Virol 80:7179–7185. doi: 10.1128/JVI.01603-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Popp MW-L, Maquat LE. 2014. The dharma of nonsense-mediated mRNA decay in mammalian cells. Mol Cells 37:1–8. doi: 10.14348/molcells.2014.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Hoof A, Frischmeyer PA, Dietz HC, Parker R. 2002. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295:2262–2264. doi: 10.1126/science.1067272. [DOI] [PubMed] [Google Scholar]
- 33.Harigaya Y, Parker R. 2010. No-go decay: a quality control mechanism for RNA in translation. Wiley Interdiscip Rev RNA 1:132–141. [DOI] [PubMed] [Google Scholar]
- 34.Passos DO, Doma MK, Shoemaker CJ, Muhlrad D, Green R, Weissman J, Hollien J, Parker R. 2009. Analysis of Dom34 and its function in no-go decay. Mol Biol Cell 20:3025–3032. doi: 10.1091/mbc.E09-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 77:4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schoenberg DR, Maquat LE. 2012. Regulation of cytoplasmic mRNA decay. Nat Rev Genet 13:246–259. doi: 10.1038/ni.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glaunsinger B, Ganem D. 2004. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol Cell 13:713–723. doi: 10.1016/S1097-2765(04)00091-7. [DOI] [PubMed] [Google Scholar]