

ABSTRACT
Influenza A virus (IAV) infection provokes an antiviral response involving the expression of type I and III interferons (IFN) and IFN-stimulated genes (ISGs) in infected cell cultures. However, the spatiotemporal dynamics of the IFN reaction are incompletely understood, as previous studies investigated mainly the population responses of virus-infected cultures, although substantial cell-to-cell variability has been documented. We devised a fluorescence-activated cell sorting-based assay to simultaneously quantify expression of viral antigens and ISGs, such as ISG15, MxA, and IFIT1, in IAV-infected cell cultures at the single-cell level. This approach revealed that seasonal IAV triggers an unexpected asymmetric response, as the major cell populations expressed either viral antigen or ISG, but rarely both. Further investigations identified a role of the viral NS1 protein in blocking ISG expression in infected cells, which surprisingly did not reduce paracrine IFN signaling to noninfected cells. Interestingly, viral ISG control was impaired in cultures infected with avian-origin IAV, including the H7N9 virus from eastern China. This phenotype was traced back to polymorphic NS1 amino acids known to be important for stable binding of the polyadenylation factor CPSF30 and concomitant suppression of host cell gene expression. Most significantly, mutation of two amino acids within the CPSF30 attachment site of NS1 from seasonal IAV diminished the strict control of ISG expression in infected cells and substantially attenuated virus replication. In conclusion, our approach revealed an asymmetric, NS1-dependent ISG induction in cultures infected with seasonal IAV, which appears to be essential for efficient virus propagation.
IMPORTANCE Interferons are expressed by infected cells in response to IAV infection and play important roles in the antiviral immune response by inducing hundreds of interferon-stimulated genes (ISGs). Unlike many previous studies, we investigated the ISG response at the single-cell level, enabling novel insights into this virus-host interaction. Hence, cell cultures infected with seasonal IAV displayed an asymmetric ISG induction that was confined almost exclusively to noninfected cells. In comparison, ISG expression was observed in larger cell populations infected with avian-origin IAV, suggesting a more resolute antiviral response to these strains. Strict control of ISG expression by seasonal IAV was explained by the binding of the viral NS1 protein to the polyadenylation factor CPSF30, which reduces host cell gene expression. Mutational disruption of CPSF30 binding within NS1 concomitantly attenuated ISG control and replication of seasonal IAV, illustrating the importance of maintaining an asymmetric ISG response for efficient virus propagation.
INTRODUCTION
Influenza A viruses (IAVs) are prototypic members of the Orthomyxoviridae family, featuring a segmented RNA genome composed of eight single-stranded RNAs that have negative polarity (1). IAVs circulate in the human population, causing periodic epidemic outbreaks and occasional pandemic waves of respiratory disease (2). Moreover, there is a large natural IAV host reservoir in wild aquatic birds, such as ducks and geese, in which the viruses cause mainly mild or no apparent symptoms. IAV strains are usually well adapted to their particular host species, which is reflected not only in the existence of stable virus lineages but also in polymorphic amino acid positions in viral proteins distinctively found in human or avian strains (3).
IAVs target the epithelial cell layers lining the human respiratory tract, in which they are subject to immune control in infected cells, mediated by the antiviral type I interferon (IFN) response (4). Many of the key events and factors driving the IFN response have been identified and involve initial recognition of the viral genomic 5′-triphosphorylated RNA by the intracellular RNA helicase RIG-I, which governs a signaling module culminating in the activation of transcription factors, such as IRF-3 and NF-κB, thereby inducing the transcription of type I IFN genes (5, 6). Type I IFNs comprise 14 subtypes of IFN-α and one IFN-β that are secreted from virus-infected cells and exert antiviral effects against many virus families, including IAV (4, 7). Type I IFNs secreted by infected cells act by para- and autocrine signaling and can activate surrounding as well as originally infected cells by ligation to the ubiquitously expressed dimeric IFN-α/β receptor. This key event activates the JAK-STAT pathway through the receptor-associated kinases JAK1 and TYK2, which phosphorylate the cytosolic transcription factors STAT1 and STAT2, resulting in their dimerization, subsequent nuclear translocation, and binding of IRF9, which generates the trimeric ISGF3 complex (8). Nuclear ISGF3 triggers transcriptional upregulation of more than 350 IFN-stimulated genes (ISGs) generally associated with the establishment of an antiviral state (9, 10). Some ISGs can also be upregulated directly by activated IRF3 (11). The type I IFN system has evolved to include positive-feedback activation, as several factors involved in the signaling events themselves are ISGs, such as STAT1 and IRF9. The more recently identified type III IFN (IFN-λ) family, whose expression appears to depend particularly on NF-κB, also signals through the JAK-STAT pathway and thereby activates ISG upregulation, but it utilizes a dedicated IFN-λ receptor (12).
ISGs encode different gene products with diverse biochemical or enzymatic functions that are expressed to inhibit establishment or continuation of an ongoing infection (13, 14). Some ISG products detect viral molecules, others are transcription factors that amplify interferon transcript synthesis, and some ISGs encode proteins with direct antiviral effector functions. Antiviral functions include the induction of apoptosis and the regulation of posttranscriptional events or posttranslational modifications (13). (Ortho)myxovirus resistance protein 1 (Mx1 in mice and MxA in humans) was the first ISG product shown to confer resistance to IAV infection in mice (15). Other genes that are strongly induced by IFNs and inhibit IAV replication are ISG54 and ISG56, which encode the p54/IFIT2 and p56/IFIT1 proteins, respectively (16). One of the most prominently induced proteins upon IFN stimulation is the ISG15 gene product, which has potent antiviral activity against a variety of DNA and RNA viruses, such as influenza A and B viruses and herpesviruses (17, 18).
Pathogenic viruses have acquired gene products that sabotage the induction or execution phase of the IFN response at various stages (19). The best-studied IFN antagonist expressed by IAV is the viral NS1 protein, which has evolved multiple mechanisms to inhibit the type I IFN response of the host cell (20, 21). NS1 interacts not only with RNA but also with several cellular proteins, such as PKR, OAS, TRIM25, RIG-I, CPSF30, PABII, NS1-BP, NS1-I, and phosphatidylinositol 3-kinase (PI3K), in order to control maturation or translation of mRNA, suppress the cellular immune response to infection, and inhibit apoptosis of the cell (20, 22–26). Furthermore, strain-specific differences have been reported for the ability of NS1 proteins to inhibit IFN-β induction via targeting of the RIG-I signaling module and/or the 30-kDa subunit of the cleavage and polyadenylation specificity factor (CPSF30) (27). More recent evidence showed that additional IAV gene products can modulate the cellular IFN response, including the viral PB2 and PB1-F2 proteins (28, 29). Despite this multifactorial viral antagonism, low levels of IFN and ISG expression can still be detected upon infection with wild-type (WT) IAV (30, 31). Silencing of the induction arm of type I IFN expression resulted in increased IAV growth, implying that this residual ISG induction has antiviral activity (32). However, it is much less clear whether this residual IFN expression slows virus replication directly at the level of the infected cell or by enhanced paracrine signaling to neighboring noninfected cells.
Despite considerable insights into the processes activating the IFN system in response to IAV infection and its blockade by viral gene products, we have only a limited understanding of the spatiotemporal dynamics of the IFN alarm response within an infected tissue or cell layer. One important challenge that needs to be considered to overcome this limitation is the considerable variation among cells in their capability to respond to biological stimuli (33), which also includes IFN and ISG expression. In fact, only a fraction of cells stimulated with a RIG-I ligand upregulate IFN gene transcription due to stochastic events (34–36), and cells within one culture differ widely in the minimal type I IFN concentration required to upregulate ISGs (37). This suggests that it is important to determine the activation and execution of the antiviral IFN response at the level of single cells. However, most studies in this field have employed averaging methods, such as Northern blot or immunoblot analyses, that do not allow allocation of ISG expression to distinct populations.
In this study, we scrutinized ISG expression in response to IAV infection in a fluorescence-activated cell sorting (FACS)-based assay enabling us to trace ISG signatures in single cells. This analysis revealed a striking bimodal response in epithelial cell populations infected with seasonal human IAVs, in which cells were found either to be infected or to express ISGs, but rarely both. This polarization between infected and noninfected cells was not observed in infections with Newcastle disease virus (NDV) and was less pronounced among avian-origin IAV strains. Further analysis traced the phenotypes back to the viral NS1 protein, in particular to polymorphic amino acid positions 103 and 106, which govern NS1 binding to the polyadenylation factor CPSF30 and the general inhibition of host cell gene expression (38, 39). The mutation-driven abrogation of NS1-dependent ISG control in seasonal IAV strongly attenuated viral growth, demonstrating that asymmetric ISG expression is associated with efficient viral propagation.
MATERIALS AND METHODS
Viruses and cells.
A549 and 293T cells were cultivated in Dulbecco's modified Eagle's medium (DMEM), MDCKII cells in minimal essential medium (MEM) (with both media containing 10% fetal calf serum, 2 mM l-glutamine, and antibiotics), and Vero cells in serum-free OptiPro medium at 37°C and 5% CO2. The viruses used for this study included the recently emerged virus strain A/Anhui/1/2013 [Anhui/2013 (H7N9)], the seasonal viruses A/Panama/2007/1999 [Pan/99 (H3N2)] and A/New Caledonia/20/1999 [NewC/99 (H1N1)], a mutant variant of A/Panama/2007/1999 lacking the gene encoding NS1 (Pan/99ΔNS1) (40), the lowly pathogenic avian virus A/mallard/Germany/439/2004 [mallard/Ger (H3N2)], the laboratory-adapted A/PR/8/1934 virus [PR/8 (H1N1)], and the seasonal influenza B/Thüringen/02/2006 virus. Newcastle disease virus was kindly provided by Hans-Dieter Klenk (University of Marburg). The human isolates NewC/99 and Anhui/2013 were grown in MDCK cells, Pan/99 and Pan/99ΔNS1 were grown in Vero cells, and the mallard/Ger and PR/8 viruses were propagated in 11-day-old embryonated chicken eggs. Virus stocks were aliquoted, stored at −80°C, and titrated on MDCK cells by standard plaque assay. The recombinant viruses PR/8 NS1 S103 I106 and PR/8 NS1 S103F I106M were kindly provided by Georg Kochs (Freiburg, Germany) (41). An established reverse genetic system was employed to generate Pan99-derived recombinant wild-type and NS1 mutant IAVs (40). All experiments with the H7N9 virus were performed in a biosafety level 3 containment laboratory approved for work with this virus by local authorities. Infection was done for 30 min at 33°C for influenza B virus (IBV) and at 37°C for all other viruses. Infected cells were incubated in DMEM (containing 2% bovine serum albumin [BSA], 2 mM l-glutamine, and antibiotics) at 37°C (33°C in the case of influenza B virus) until further use. Where indicated, cells were preincubated with JAK inhibitor I (Calbiochem, Darmstadt, Germany) or BAY 11-7085 (Enzo Life Sciences, Lörrach, Germany), to block IFN signaling and NF-κB activation, respectively, for 1 h in infection medium. The inhibitor-containing medium was collected prior to infection, and the same medium was re-added to the cells when infection was completed. Blockade of the IFN-α/β receptor was conducted by incubation of A549 cells with 15 μg/ml anti-IFNAR2 antibody (PBL Assay Science) for 8 h before and 16 h after infection.
Transfection.
Transfection of 293T cells with plasmid DNA was done using Lipofectamine 2000 following the manufacturer's instructions. If not stated otherwise, cells were incubated for 24 h before they were either analyzed or stimulated with IFN-α. For the RNP reconstitution assay, 6 × 105 293T cells were transfected with plasmids expressing the A/Panama/2007/1999 PB1, PB2, PA (50 ng each), and NP (100 ng) genes, together with a pPol-I-GFP reporter plasmid (500 ng). To analyze whether other viral proteins influence ISG15 expression, plasmids encoding the HA, M, NA, and NS segments or a ΔNS1 control plasmid (50 ng each) was added separately, as indicated. After transfection, 293T cells were incubated in DMEM containing 10% fetal calf serum and 2 mM l-glutamine for 48 h at 37°C until FACS analysis was performed.
Confocal laser scanning microscopy.
For indirect immunofluorescence staining, cells were seeded on coverslips on the day before infection. After infection for 16 h, cells were fixed with 2.5% formaldehyde for 20 min, permeabilized with 0.2% Triton X-100 solution for 10 min, and blocked with 3% BSA solution for 1 h. Primary antibodies were diluted in 3% BSA solution (rabbit anti-human ISG15 serum [1:1,000] and monoclonal mouse anti-IAV NP IgG [1:500]; Serotec) and used to stain cells for 1 h. After washing, cells were stained with suitable secondary fluorescent antibodies (1:1,000; Molecular Probes) for 45 min in the dark. In order to stain the nuclei, DAPI (4′,6-diamidino-2-phenylindole) was added to the secondary antibody solution. Finally, cells were mounted in Mowiol and analyzed using confocal laser scanning microscopy (LSM 510 Meta confocal laser scanning microscope; Zeiss).
FACS assays.
FACS analysis was conducted to analyze ISG expression in A549 cells after infection or in 293T cells after transfection. Cells were washed with phosphate-buffered saline (PBS) and detached from the surface with trypsin (100 μl/well in a 12-well plate), and trypsinization was stopped by the addition of DMEM (500 μl/well in a 12-well plate). For FACS analysis, 1 × 105 to 5 × 105 cells were transferred to a FACS tube, 600 μl PBS was added, and cells were centrifuged for 5 min at 4°C and 800 rpm. The supernatants were removed, cells were resuspended in the remaining fluid in the tube, and the washing step was repeated. Next, cells were fixed in 500 μl 4% paraformaldehyde for at least 20 min at 4°C. The fixation period was extended to at least 16 h for cells infected with the H7N9 virus. Cells were washed twice with PBS, and then 500 μl Triton buffer (0.5% BSA, 0.02% sodium azide, 0.1% Triton X-100 in PBS) was added. Permeabilization of cellular membranes with Triton X-100 was done for 10 min at room temperature (RT). Primary antibodies were diluted in Triton buffer, and the following antibody dilutions were used: rabbit anti-ISG15 serum, 1:1,000 to 1:2,000; Acris mouse anti-IAV NP–fluorescein isothiocyanate (FITC), 1:250 to 1:500; the monoclonal anti-IAV NS1 antibody IA7 (kindly provided by Jonathan Yewdell, NIH), 1:250 to 1:500; anti-IAV NS1 (24), 1:500; Serotec mouse anti-IAV NP, 1:500; Serotec mouse anti-IBV NP, 1:500; anti-MxA (kindly provided by Georg Kochs), 1:500; Abcam mouse anti-ISG56, 1:500; and Serotec anti-NDV HN, 1:500. After centrifugation of the permeabilized cells, supernatants were removed, and 30 μl antibody staining solution was added. Cells were mixed with the antibody solution and incubated for at least 1 h in the dark at 4°C, followed by two washings with Triton buffer. Subsequently, supernatants were removed and cells were incubated in 30 μl secondary antibody staining solution (Alexa 488-conjugated goat anti-mouse IgG and Alexa 647-conjugated donkey anti-rabbit IgG [Molecular Probes], diluted 1:2,000 in Triton buffer) for at least 45 min in the dark, followed by two washings with Triton buffer and one with buffer without Triton X-100. After the last washing step, cell pellets were resuspended in 150 to 200 μl buffer without Triton X-100 and kept in the dark at 4°C until measurement. All samples were measured using a BD FACSCalibur cytometer and BD Cell Quest Pro software (version 6.0). FACS data were analyzed using FlowJo (version 9.5.1).
GST coprecipitation assay.
A fusion protein consisting of bovine CPSF30 and glutathione S-transferase (GST) (a kind gift of Georges Martin, University of Basel, Switzerland) was expressed in Escherichia coli BL26 bacterial cells, purified, and adsorbed to glutathione (Glu) Sepharose beads according to standard procedures (25). Bovine CPSF30 is identical to its human homolog in the region of the NS1 binding domain (42). As a control, the unmodified GST protein was expressed. In vitro translation of NS1 from 1 μg plasmid DNA was conducted in the presence of 4 μl l-[35S]methionine (10 mCi/ml) according to the manufacturer's instructions (TNT T7 coupled reticulocyte lysate kit; Promega). To determine binding to CPSF30, 10 μl of in vitro-translated NS1 protein was mixed with 30 μl loaded Glu beads in Tris binding buffer (10 mM Tris-HCl, pH 8.0, 0.075% IGEPAL, 150 mM NaCl, 0.01% BSA), followed by rotation for 1 to 2 h at 4°C. Samples were centrifuged for 3 min at 3,000 rpm (4°C) and washed 4 times with cold Tris washing buffer (10 mM Tris-HCl, pH 8.0, 0.075% IGEPAL, 500 mM NaCl) on ice before 20 μl 2× SDS sample buffer was added to each sample. Next, samples were boiled and stored at −20°C before separation by SDS-PAGE and Coomassie staining. Input and bound NS1 proteins were visualized by autoradiography. Band intensity was quantified using ImageJ software.
RESULTS
A FACS-based assay reveals distinct ISG expression patterns in IAV-infected epithelial cell cultures.
To assess the spatiotemporal aspects of the antiviral immune response toward IAV infection in single cells, we established a FACS-based assay to quantify the presence of viral antigen and IFN-stimulated genes in human A549 epithelial cell cultures. Using ISG15 as a sensitive marker of ISG expression, this assay revealed a surprising bimodal response toward infection with the seasonal H3N2 strain A/Panama/2007/1999 (Pan/99), which was robustly observed at various multiplicities of infection (MOIs), ranging from 0.1 to 5, and across different time points of infection (Fig. 1A and data not shown). More than 90% of the cells expressed either the viral NP (NP+) or ISG15 (ISG15+), but both antigens were rarely (<4%) detectable within the same cell, even after infection with a high dose of virus. The rest of the cells did not show a signal for either of the two markers and were not taken into account any further. Immunofluorescence staining of infected human epithelial cell layers confirmed these observations, as cells showed signals for either the viral NP or ISG15 (Fig. 1B). Notably, we did not add trypsin to the infected cultures to prevent reinfection by progeny virions, allowing us to focus on ISG induction by the primary infection.
FIG 1.

ISG15 is predominantly induced in noninfected cells upon IAV infection. (A) A549 cells were infected with the seasonal Pan/99 strain at the indicated MOI. At 24 h postinfection (hpi) cells were stained for ISG15 (y axis) and NP (x axis) and analyzed via FACS. Numbers indicate the percentages of cells in each gate. One representative dot plot is shown for each MOI (n > 3). Control staining is shown for cells after mock infection or treatment of cells with 500 U/ml IFN-α. (B) A549 cells were infected with Pan/99 at an MOI of 1. At 16 hpi, cells were stained for ISG15 (red channel) and NP (green channel) and analyzed via indirect immunofluorescence assay. One representative image is shown in each panel (n = 2). Bar, 10 μm. (C) Percentages of ISG15- and NP-double-positive cells at 24 hpi with Pan/99 (influenza A virus; light gray), Thü/2006 (influenza B virus; black), or NDV (Newcastle disease virus; dark gray) when more than 80% of the A549 cells in the respective experiment were positive for viral antigen. Data are means + standard errors of the means (SEM) (n = 4), *, P < 0.05 (Mann-Whitney U test).
To examine whether the distinct ISG expression phenotypes were also provoked by other negative-strand RNA viruses, we carried out similar FACS analyses using the related viruses influenza B virus (IBV) and Newcastle disease virus (NDV), an avian negative-strand RNA virus considered unable to antagonize the human type I IFN system (43). Interestingly, Fig. 1C shows that both IBV and NDV generated up to 10-fold more abundant populations of infected, ISG15-expressing positive cells than those with seasonal IAV (Fig. 1C). These results suggested that a poor ISG15 expression phenotype in infected cells is a distinctive feature of IAV rather than a general hallmark of negative-strand RNA viruses. Moreover, questions arose regarding whether the lack of ISG15 expression in IAV-infected cells was due to an intrinsic block of gene activation or a consequence of viral inhibition of the JAK-STAT pathway and which viral gene product(s) would have a role in this scenario.
ISG15 induction in noninfected cells requires the JAK/STAT and NF-κB pathways.
We next examined the prediction that ISG15 gene expression in noninfected cells of our cultures was driven by IFNs secreted from infected cells by using inhibitors blocking regulatory steps in viral ISG15 induction. This included BAY 11-7085, a substance that inhibits NF-κB activation, as well as JAK inhibitor I, which reduces the expression of interferon-stimulated genes via blockade of the receptor-associated kinases JAK1 and TYK2. Cells were treated with the inhibitors for 1 h before and throughout the infection with Pan/99 or the laboratory-adapted A/PR/8/34 [PR/8 (H1N1)] strain. Subsequent FACS analyses pointed out that either inhibitor reduced the proportion of ISG15-positive cells induced by Pan/99 or PR/8 in a dose-dependent manner (Fig. 2A and B). As expected, both inhibitors concomitantly decreased type I and type III IFN secretion in these cultures (Fig. 2C and D). Interestingly, antibody-mediated neutralization of IFNAR only weakly affected virus-induced ISG15 expression, indicating that type III IFN is sufficient to stimulate ISG induction, although we cannot rule out that type I also contributes in the noninhibited situation (Fig. 2E). These results confirmed that ISG15 expression upon IAV infection depends on viral IFN induction and active JAK-STAT signaling.
FIG 2.

ISG15 induction during IAV infection depends on NF-κB and JAK1/TYK2 signaling. A549 cells were pretreated with BAY 11-7085 to inhibit NF-κB (A) or with JAK inhibitor I to inhibit JAK1/TYK2 (B to D) for 1 h prior to mock treatment or infection with Pan/99 or PR/8 IAV at an MOI of 0.5 for 16 h. The inhibitors were present throughout the experiments, and their concentrations are indicated. Dimethyl sulfoxide (DMSO) was used as a control. (A and B) Bars represent percentages of total ISG15-positive cells as assessed by FACS. Data are means + SEM for experiments conducted in duplicate (n = 3). IFN-β (C) and IFN-λ2 (D) levels in supernatants taken 16 h after infection with the different viruses were assessed by enzyme-linked immunosorbent assay (ELISA). Data are means + SEM for experiments conducted in duplicate (n = 3). (E) To examine the role of type I IFN signaling, A549 cells were not treated or pretreated with 15 μg/ml anti-IFNAR2 neutralizing antibody for 8 h. Subsequently, the cells were mock treated, stimulated with 50 U/ml IFN-α, or infected with Pan/99 at an MOI of 1 for 16 h. The antibody was present or absent throughout the experiments, as indicated. Bars represent percentages of total ISG15-positive cells as assessed by FACS. Data are means + SEM (n = 4). *, P < 0.05 (Mann-Whitney U test).
IAVs of avian origin are debilitated in controlling ISG expression in infected cells.
The experiments shown so far involved two human-derived IAV strains. To determine whether poor ISG15 expression in infected cells was a conserved feature, we analyzed additional IAV strains (Fig. 3). To facilitate direct comparisons between strains, cell cultures were analyzed under conditions in which the overall proportions of infected cells were equivalent for the different viruses. Interestingly, the seasonal A/New Caledonia/20/1999 (H1N1) virus (NewC/99) provoked a low ISG15 response in infected cells, similarly to Pan/99 (H3N2) (Fig. 3A, black and dark gray bars). In contrast, a significant increase in the proportion of double-positive cells was observed upon infection with a lowly pathogenic avian H3N2 virus (A/mallard/Germany/439/2004 [mallard/Ger]), as well as the prototypic A/Anhui/1/2013 isolate of H7N9 subtype viruses that recently emerged in humans in eastern China (44) (Fig. 3A, medium gray and light gray bars). Nevertheless, we detected comparable populations of ISG15-positive noninfected cells for all analyzed strains (Fig. 3B). These findings suggested that the minimal ISG15 expression phenotype in infected cells is a feature of seasonal IAVs.
FIG 3.
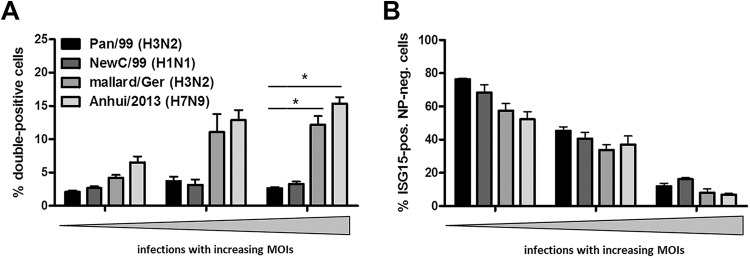
ISG15 is induced more prominently in infected cells in response to avian-origin IAV. A549 cells were infected with the indicated viruses at increasing MOIs. At 24 hpi, cells were stained for ISG15 and NP and analyzed via FACS. Percentages of double-positive cells (A) or ISG15-positive noninfected cells (B) are represented by bars. Proportions of the respective cell populations were compared in groups containing 15 to 25%, 45 to 55%, and 75 to 85% infected cells in total, shown as increasing MOIs. (A) Comparison of the proportions of infected cells expressing ISG15 upon infection with Pan/99 (black bars), NewC/99 (dark gray bars), mallard/Ger (medium gray bars), and Anhui/2013 (light gray bars). Data are means + SEM (n ≥ 3). *, P < 0.05 (Mann-Whitney U test). (B) Comparison of the proportions of ISG15-expressing noninfected cells after infection with Pan/99 (black bars), NewC/99 (dark gray bars), mallard/Ger (medium gray bars), and Anhui/2013 (light gray bars). Data are means + SEM (n = 4). *, P < 0.05 (Mann-Whitney U test).
The viral NS1 protein controls virus- and type I IFN-induced ISG15 expression in a strain-dependent manner.
Next, we investigated whether a viral protein would directly control ISG expression in cells expressing an active viral RNA polymerase. Hence, we reconstituted the viral ribonucleoprotein of the Pan/99 strain by transfecting 293T cells with expression plasmids for the viral polymerase and NP. In addition, we coexpressed a viral RNA (vRNA)-like molecule encompassing the open reading frame for green fluorescent protein (GFP) flanked by conserved noncoding regions of IAV gene segments with negative polarity. Cells containing all necessary components of an active viral ribonucleoprotein complex expressed GFP and were detected by FACS (Fig. 4). As predicted (45), expression of this viral minigenome was sufficient to induce ISG15 expression in these cells, and this depended on the presence of all three polymerase subunits (Fig. 4A). To screen for modulatory proteins, we coexpressed single viral gene segments of the Pan/99 strain together with the reconstituted viral minigenomes. Addition of the M, NA, or HA gene did not alter the proportion of transfected ISG15-positive cells (Fig. 4A). On the contrary, expression of the NS segment, encoding the NS1 and NEP polypeptides, or a combination of the M, NA, HA, and NS vectors resulted in a significant decrease of the transfected ISG15-positive cell population. In a control experiment in which an NS segment with deleted NS1 but an intact NEP open reading frame was expressed, ISG15 levels were not affected, pointing to a regulatory activity of the viral NS1 protein (Fig. 4A). This conclusion was confirmed in the context of a virus infection. A549 cell cultures infected with a recombinant Pan/99 wild-type virus contained infected ISG15-positive cells at a much lower abundance than that for infections with an NS1-deleted mutant virus (Fig. 4B). Interestingly, the ΔNS1 virus stimulated ISG15 expression in noninfected cells, to a proportion similar to that with the wild type (Fig. 4C). This result indicates that NS1 does not reduce IFN secretion from wild-type virus-infected cells below a threshold sufficient to lower ISG activation in neighboring cells. In conclusion, these data demonstrate that NS1 is responsible for inhibiting ISG15 expression in both infected and transfected cells.
FIG 4.
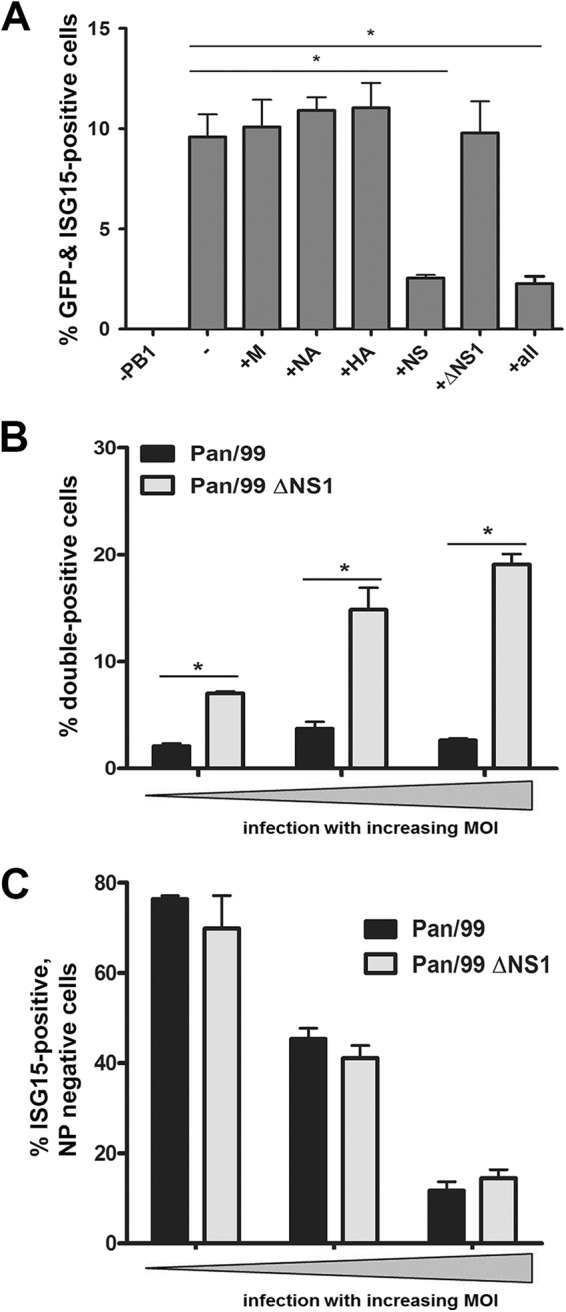
NS1 inhibits ISG15 expression in infected cells. (A) 293T cells were transfected with plasmids encoding the following viral segments from the seasonal H3N2 strain Pan/99: PB1, PB2, PA, and NP. The cells were cotransfected with a pPol-I-GFP plasmid (2nd bar). On the x axis, the plasmids that were left out (1st bar) or added (bars 3 to 8) are indicated. Percentages of transfected cells (assessed by their GFP signals) that were positive for ISG15 are shown. Cells were transfected for 48 h and then analyzed via FACS. Data are means + SEM for experiments performed in duplicate (n ≥ 3). *, P < 0.05 (Mann-Whitney U test). (B and C) A549 cells were infected with Pan/99 (black bars) or Pan/99 ΔNS1 (light gray bars) at increasing MOIs. At 24 hpi, cells were stained for ISG15 and NP and analyzed via FACS. Percentages of infected (B) and noninfected (C) ISG15-positive cells are represented by bars. Proportions of double-positive cells were compared in groups containing 15 to 25%, 45 to 55%, and 75 to 85% infected cells in total, shown here as increasing MOIs. Data are means + SEM (n = 4). *, P < 0.05 (Mann-Whitney U test).
The previous findings suggested that NS1-mediated restriction of ISG15 expression by seasonal IAV was less stringent in other IAV strains, raising questions about the activity of the corresponding NS1 proteins. To address this aspect, we transfected cells with plasmids expressing the NS1 proteins of the respective viruses and added exogenous type I interferon to characterize ISG15 expression by FACS. By using the viral NP as a control, we determined that approximately 40% of the cells were transfected and positive for ISG15 (Fig. 5A, first bar). This population was decreased to below 10% by expression of the Pan99 NS1 protein (Fig. 5A), which is consistent with a previously described activity of inhibiting maturation of ISG mRNAs (31). A similar strong decrease in ISG15 expression was observed for the NS1 proteins of the seasonal NewC/99 (H1N1) virus and the pandemic Brevig Mission/1/1918 [BrevM/18 (H1N1)] virus (Fig. 5). In contrast, the proportion of ISG15-expressing transfected cells remained unaltered with NS1 proteins encoded by the avian mallard/Ger virus, the highly pathogenic avian A/Hong Kong/157/1997 [HK/97 (H5N1)] virus, and the PR/8 virus (Fig. 5A). Expression of A/Anhui/1/2013 NS1 indicated a low suppressive activity compared to those of the seasonal and pandemic strains. Collectively, these findings revealed remarkable differences in the ability of NS1 proteins to inhibit ISG15 expression upon type I IFN stimulation.
FIG 5.
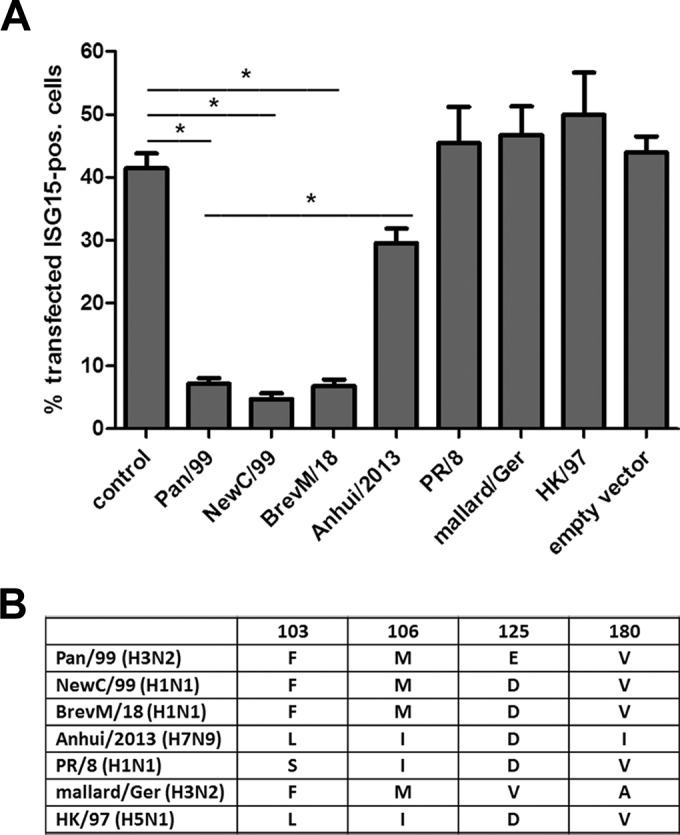
NS1-dependent inhibition of ISG15 expression is strain specific. (A) 293T cells were transfected with NS segments of the indicated viruses or with the Pan/99 NP segment as a control. At 24 h posttransfection, cells were treated with 500 U/ml IFN-α. At 18 h poststimulation, cells were stained for ISG15 and NS1 or NP and analyzed via FACS. Percentages of transfected ISG15-positive cells are represented by bars. Data are means + SEM (n ≥ 3). *, P < 0.05 (Mann-Whitney U test). (B) Overview of the main differences in amino acid sequence between the investigated NS1 proteins. Comparisons were performed for NS1 amino acid positions 103, 106, 125, and 180 between Pan/99 (GenBank accession no. ABE73108), NewC/99 (accession no. DQ508861), BrevM/18 (accession no. AAK14368), Anhui/2013 (accession no. EPI439510 [GISAID database]), PR/8 (accession no. AAM75163), mallard/Ger (accession no. KM364561), and HK/97 (accession no. CAC04091).
Reverse genetics identifies CPSF30 binding as a key determinant for strain-dependent regulation of ISG expression by the viral NS1 protein.
NS1 proteins from human and animal strains can differ by more than 30% of the amino acid sequence (46). Therefore, we asked whether the distinct activities of the examined NS1 proteins in limiting ISG15 induction were reflected in a specific amino acid motif. In fact, sequence comparisons revealed differences for a set of four NS1 amino acid positions (positions 103, 106, 125, and 180) (Fig. 5B). The NS1 proteins of ISG15-suppressing strains (Pan/99, NewC/99, and BrevM/18) followed the consensus F103, M106, D/E125, and V180, whereas the NS1 proteins of the second, nonsuppressing group (Anhui/2013, HK/97, mallard/Ger, and PR/8) differed from this consensus at two or more positions (Fig. 5B). Significantly, these four NS1 amino acids were previously shown to engage in binding of the 30-kDa subunit of the cleavage and polyadenylation specificity factor (38, 47), suggesting a direct tie between this association and ISG15 suppression.
To investigate this relationship in more detail, we analyzed the variable amino acids at positions 103 and 106 between NS1 proteins within two different strain backgrounds. On the one hand, we introduced the amino acid changes F103L and M106I, which were previously shown to diminish CPSF30 binding of the NS1 protein expressed by an H5N1 strain, into Pan/99 NS1 (48). On the other hand, we tested NS1 of the PR/8 strain, which does not bind CPSF30, and a derivative in which the natural S103 and I106 positions were reverted to the consensus amino acids F and M, respectively (27). The first analysis showed that mutation of the consensus sequence suspended the ability of transfected Pan/99 NS1 to suppress IFN-driven ISG15 expression, whereas the two amino acid substitutions restored ISG15 suppression within the PR8 NS1 protein (Fig. 6A). To validate these findings, we studied recombinant viruses expressing the modified NS1 proteins (Fig. 6B and C). In line with the prior results, infection of A549 cultures with the Pan/99 NS1 F103L M106I mutant virus generated a significant increase in the population of double-positive cells compared to the wild type, whereas the proportion of double-positive cells was drastically reduced in infections with the PR/8 NS1 S103F I106M mutant virus (Fig. 6B and C). These data confirmed the elemental role of the NS1 amino acids F103 and M106 in the inhibition of ISG15 expression in transfected as well as IAV-infected cells.
FIG 6.

NS1 proteins with amino acid residues F103 and M106 inhibit expression of ISG15 in transfected and infected cells. (A) 293T cells were transfected with NS segments of Pan/99 and PR/8 encoding NS1 proteins with the indicated amino acids at positions 103 and 106. At 24 h posttransfection, cells were treated with 500 U/ml IFN-α. At 18 h poststimulation, cells were stained for ISG15 and NS1 and analyzed via FACS. Percentages of double-positive cells are represented by bars. Data are means + SEM (n = 3). *, P < 0.05 (Mann-Whitney U test). (B and C) A549 cells were infected with the indicated viruses at increasing MOIs. At 24 hpi, cells were stained for ISG15 and NP and analyzed via FACS. Percentages of double-positive cells are represented by bars. (B) Proportions of double-positive cells were compared in groups of 5 to 15%, 16 to 25%, and 26 to 35% infected cells. Cells were infected with the Pan/99 wild type (black bars) or the mutant variant (light gray bars). (C) Populations of double-positive cells were compared in groups containing 15 to 25%, 45 to 55%, and 75 to 85% infected cells. Cells were infected with the PR/8 wild type (light gray bars) or the mutant variant (black bars). Data are means + SEM (n ≥ 3). *, P < 0.05 (Mann-Whitney U test).
To characterize the role of a physical interaction in the observed NS1 phenotypes, we investigated the binding of NS1 proteins to CPSF30 in a coprecipitation assay. This analysis demonstrated an association of CPSF30 with the NS1 proteins expressed by the seasonal viruses Pan/99 and NewC/99 (Fig. 7A and B), whereas the NS1 proteins of mallard/Ger and Anhui/2013, as well as the mutant Pan/99 NS1, interacted only poorly (Fig. 7). We did not investigate CPSF30 binding of the NS1 proteins from PR/8, HK/97, and BrevM/18, as previous studies showed that BrevM/18 NS1 binds to CPSF30, whereas the NS1 proteins of PR/8 and HK/97 cannot (27, 49). These results established a direct and, most likely, causal link between the capacities of IAV NS1 proteins from seasonal strains to bind to CPSF30 and limit ISG15 expression in infected cells.
FIG 7.
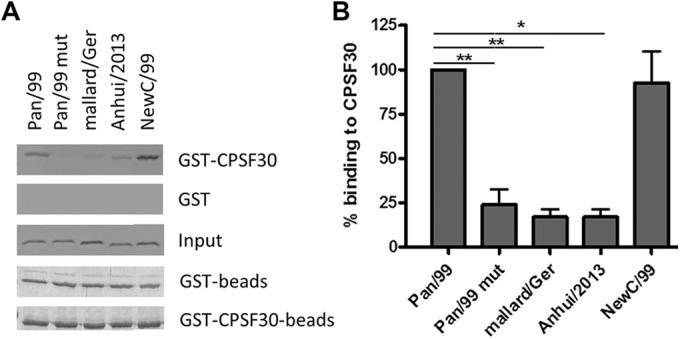
NS1 proteins that inhibit ISG15 expression bind to CPSF30. NS1 proteins from Pan/99, Pan/99 mut (with amino acid exchanges F103L and M106I), mallard/Ger, Anhui/2013, and NewC/99 were radioactively labeled via in vitro translation in the presence of [35S]methionine. (A) The proteins were incubated with GST-CPSF30 or GST linked to glutathione Sepharose, and bands on the film represent NS1 bound to CPSF30 or to the GST control. The input bands represent 1% of the in vitro-translated NS1 proteins. The bands in the Coomassie-stained gels (“beads”) confirm that equal amounts of beads were used. Data from one representative experiment of six are shown. (B) Quantitative analysis of CPSF30 binding, with the level for Pan/99 NS1 set to 100% binding. Data are means + SEM (n = 6). *, P < 0.05; **, P < 0.01 (Mann-Whitney U test).
We suspected that the NS1-dependent inhibition of ISG15 expression reflects a more general effect that also applies to other ISGs. To examine this hypothesis, we studied the expression of other ISG products, including MxA and IFIT1, in cells infected with the recombinant Pan/99 and PR8 WT and mutant viruses. In confirmation of our assumption, we found that only viruses containing NS1 proteins displaying amino acids F103 and M106 (Pan/99 and PR8mut) inhibited the expression of IFIT1 (Fig. 8A and C) and MxA (Fig. 8B and D) in infected cells. Conversely, viruses containing NS1 proteins with amino acid changes at these positions led to the appearance of considerably larger proportions of double-positive cells. Finally, we conducted growth curve analyses to elucidate the impact of differential antiviral ISG activation on propagation of recombinant Pan/99 viruses. Figure 8E demonstrates retarded and decreased (>1.5 log) multicyclic replication in A549 cells for the Pan/99 NS1 F103L M106I mutant virus compared to the wild type, illustrating a substantial contribution of asymmetric ISG expression to the outcome of infection. Intriguingly, WT and mutant viruses replicated equally well in Vero cells that are devoid of functional type I IFN genes (50), suggesting that NS1-mediated control of ISG activation is beneficial for efficient virus propagation in IFN-competent hosts.
FIG 8.

NS1 amino acids F103 and M106 regulate expression of IFIT1 and MxA and viral propagation in human cells. Human A549 cells were infected with the indicated viruses at increasing MOIs. At 24 hpi, cells were stained for IFIT1 and NS1 (A and C) or MxA and NS1 (B and D) and analyzed via FACS. Percentages of double-positive cells are represented by bars. (A and B) Cells were infected with the Pan/99 wild-type virus (black bars) or its mutant variant (light gray bars). (C and D) Cells were infected with the PR/8 wild-type virus (light gray bars) or its mutant variant (black bars). Data are means + SEM (n ≥ 3). (A) Proportions of double-positive cells were compared in groups containing totals of 16 to 25% and 26 to 35% infected cells. (B) Proportions of double-positive cells were compared in groups containing totals of 5 to 10% and 16 to 20% infected cells. (C and D) Proportions of double-positive cells were compared in groups containing 15 to 25% and 75 to 85% total infected cells. (E and F) A549 (E) or Vero (F) cells were infected with isogenic Pan/99 viruses expressing either wild-type NS1 or the NS1 F103L M106I mutant protein at an MOI of 0.01. Supernatants were collected at the indicated time points and titrated on MDCK cells. Data are means ± standard deviations for experiments performed in duplicate (n = 3). *, P < 0.05 (Mann-Whitney U test).
DISCUSSION
A large body of our knowledge of IFN induction and antagonism in IAV-infected cells has been shaped by investigations on the population level. However, there is considerable variability in the ability of cells to upregulate IFN genes and to respond to these cytokines (36, 37). This implies that the complex scenario of the type I IFN reaction to virus infection also needs to be studied at the level of single cells to fully understand the dynamic forces within a given cell population. Our FACS-based approach captured several new insights into the differential expression of ISGs and viral antigens in single cells, which would not have been detected otherwise. First, cell cultures responded to infections with seasonal IAV in a bimodal fashion, in which ISG15 as well as other ISGs, such as MxA and IFIT1, were expressed almost exclusively in noninfected cells and hardly detected in infected cells. This polarization of ISG expression in noninfected cells was not influenced by the multiplicity of infection and most likely depended on paracrine signaling through the JAK-STAT pathway. Follow-up analysis demonstrated that the viral NS1 protein decisively restricts ISG expression in infected cells. Cultures infected with an NS1-deficient virus contained 3- to 5-fold increases in the population of infected cells expressing ISG15 in comparison to wild-type infection, suggesting effective autocrine IFN signaling in those cells. This conclusion was confirmed in a transfection-based setting, as the NS1 proteins of seasonal IAV strongly suppressed ISG expression in cells transfected with a viral minigenome.
The NS1-deficient Pan/99 variant has previously been shown to trigger secretion of >10-fold higher levels of IFN-β than those with the wild type, and similar findings exist for other strain backgrounds (40, 51, 52). It was therefore not unexpected that ISG expression was strongly increased in ΔNS1 virus-infected cells compared to wild-type-infected cells, but it came as a surprise that the populations of noninfected cells with an ISG signature were of comparable sizes for both viruses (Fig. 4B and C). This shows that the low level of IFN produced in wild-type-infected cultures, most likely before substantial amounts of intracellular NS1 have accumulated, is sufficient to alert large numbers of surrounding cells. Hence, the IFN antagonism of the NS1 protein functions mainly on the level of the initially infected cell to prevent expression of antiviral genes, but it does not decrease IFN secretion sufficiently to block effective paracrine signaling. Consequently, progeny viruses will face large numbers of ISG-expressing cells in a subsequent round of infection, which is expected to slow down virus propagation. Interestingly, this scenario is supported by independent evidence, as the ablation of IFN expression stimulated multicyclic IAV replication in A549 cells (32). Finally, we were intrigued by the finding that no more than 20% of the infected cells expressed an ISG signature even in the absence of the NS1 protein (Fig. 4B). Possible reasons include the possibility that other, less-well-characterized viral factors, such as the PA-X protein, compromise the cell's capacity to upregulate ISGs (53) or simply that not all cells are able to respond to virus-induced IFN secretion due to stochastic events (37). The observation that only about 30% of the infected cells in NDV-infected culture showed an ISG signature is compatible with the latter explanation, but further experiments will be required to confirm this.
In a comparative analysis, we observed that IAVs of avian origin triggered up to 5-fold larger populations of infected cells expressing ISGs than those with human seasonal H3N2 and H1N1 strains, although overall ISG expression levels in noninfected cells were similar (Fig. 3). This suggested that NS1 proteins of avian-origin strains were less capable of effectively antagonizing autocrine IFN signaling than human IAVs, and this conclusion was confirmed in a reporter setting with transfected NS1 genes.
To reconcile those findings, we considered the two major mechanisms by which NS1 proteins can reduce IFN and/or ISG induction: by silencing the RIG-I pathway or by a more general inhibition of host cell gene expression via binding of CPSF30 (20). The targeting of the two pathways has been linked to polymorphic NS1 amino acid positions 103 and 106, among others, for CPSF30 binding, and position 196, for suppressing RIG-I signaling (20). A sequence comparison correlated the asymmetric ISG control by human strains in infected and transfected cells with signature amino acids previously described to govern stable binding of NS1 to CPSF30. In fact, the present and previous reports (27, 31) showed that NS1 complex formation with CPSF30 plays a decisive role in this control. NS1 proteins expressed by seasonal H1N1 and H3N2 strains efficiently interacted with CPSF30. In contrast, the mallard/Ger (H3N2) and Anhui/2013 (H7N9) NS1 proteins were poor interactors, and others had previously determined similar failures for the NS1 proteins of highly and lowly pathogenic avian H5 subtype viruses (49, 54).
The elemental role of the NS1-CPSF30 interaction in viral replication and the control of ISG expression by the seasonal H3N2 virus were finally supported by a reverse genetic analysis (Fig. 6 to 8). The two amino acid substitutions, F103L and M106I, within the CPSF30 binding element of the Pan/99 NS1 protein strongly increased the proportion of ISG-expressing infected cells, and this was paralleled by a strong decrease in CPSF30 binding. Significantly, we showed that mutation of the CPSF30 binding element substantially reduced multicyclic replication of seasonal IAV in human A549 cells, by about 1.5 log, demonstrating that the tight control of ISG expression is decisive for efficient replication in such IFN-competent hosts, at least for seasonal strains. This conclusion is supported by the almost identical growth of WT and mutant viruses in Vero cells that are defective in type I IFN expression. The F103L and M106I exchanges have been shown previously not to alter NS1-dependent activation of c-Jun N-terminal kinase, and therefore we expect that this activity did not add to the attenuated phenotype of the mutant virus in A549 cells (55). Significantly, our study showed that the emerging H7N9 virus, which, to date, displays a case fatality rate of >35% in humans (56), cannot strictly control ISG expression in infected cells. We recently documented that the NS1 protein of the prototypic Anhui/1/2013 (H7N9) strain efficiently suppresses IFN-β induction in human cells, indicating that this activity is sufficient to antagonize IFN action (44). Interestingly, Ayllon and colleagues recently reported that an I106M mutation within the H7N9 NS1 protein concomitantly confers CPSF30 binding and increases viral replication and virulence in mice (57). We expect that this mutation will also strengthen ISG control in H7N9-infected cultures. Notably, the contribution of NS1-CPSF30 interaction to virus propagation appears not to be as limiting for all IAVs. Mutational restoration of CPSF30 binding in the NS1 protein of the pandemic H1N1-2009 IAV, which is a bona fide poor interactor, did not increase replication and virulence despite reduced IFN and ISG induction (54, 58).
In conclusion, our analysis of sorted cells revealed that seasonal human IAVs provoke an asymmetric antiviral reaction in infected cultures, as they confine ISG expression to noninfected cells, which appears to be important for efficient viral growth. We show that this asymmetric response is mediated by the viral NS1 protein targeting the cellular polyadenylation factor CPSF30, which translates into an effective block of autocrine IFN signaling. We believe that these findings contribute to an improved understanding of IFN induction and control by IAVs. They will be valuable not only to explain strain-dependent differences in biological responses to IAV infection downstream of IFN signaling, such as inflammasome and TRAIL induction (59, 60), but also for interpretations of NS1 mutant phenotypes (61) and the refinement of mathematical models concerning viral IFN induction (37, 62).
ACKNOWLEDGMENTS
We thank Georg Kochs (Freiburg, Germany) for providing the MxA antibody as well as PR/8 plasmids and viruses, Jon Yewdell (NIH) for providing the anti-NS1 antibody IA7, and Georges Martin (Basel, Switzerland) for the bacterial CPSF30 expression plasmid. We thank Gudrun Heins for excellent technical assistance and Matthias Budt for critical comments on the manuscript.
This work was supported by grants from the German Ministry of Education and Research (FluResearchNet [grant 01 KI 1006] and Virosign [grant 0316180D]) and the German Research Foundation (SFB-TR84 project B2). J.V.R.-K. acknowledges support from the ZIBI Graduate School Berlin.
REFERENCES
- 1.Shaw ML, Palese P. 2013. Orthomyxoviridae. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed (electronic) Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Wright PF, Neumann G, Kawaoka Y. 2013. Orthomyxoviruses. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed (electronic) Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Reperant LA, Kuiken T, Osterhaus AD. 2012. Adaptive pathways of zoonotic influenza viruses: from exposure to establishment in humans. Vaccine 30:4419–4434. doi: 10.1016/j.vaccine.2012.04.049. [DOI] [PubMed] [Google Scholar]
- 4.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 5.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 6.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 7.Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat Rev Immunol 14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 9.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A 95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grandvaux N, Servant MJ, ten Oever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol 76:5532–5539. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durbin RK, Kotenko SV, Durbin JE. 2013. Interferon induction and function at the mucosal surface. Immunol Rev 255:25–39. doi: 10.1111/imr.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat Rev Immunol 8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horisberger MA, Staeheli P, Haller O. 1983. Interferon induces a unique protein in mouse cells bearing a gene for resistance to influenza virus. Proc Natl Acad Sci U S A 80:1910–1914. doi: 10.1073/pnas.80.7.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pichlmair A, Lassnig C, Eberle CA, Gorna MW, Baumann CL, Burkard TR, Burckstummer T, Stefanovic A, Krieger S, Bennett KL, Rulicke T, Weber F, Colinge J, Muller M, Superti-Furga G. 2011. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat Immunol 12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- 17.Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE, Garcia-Sastre A, Leib DA, Pekosz A, Knobeloch KP, Horak I, Virgin HW IV. 2007. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci U S A 104:1371–1376. doi: 10.1073/pnas.0607038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsiang TY, Zhao C, Krug RM. 2009. Interferon-induced ISG15 conjugation inhibits influenza A virus gene expression and replication in human cells. J Virol 83:5971–5977. doi: 10.1128/JVI.01667-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Versteeg GA, Garcia-Sastre A. 2010. Viral tricks to grid-lock the type I interferon system. Curr Opin Microbiol 13:508–516. doi: 10.1016/j.mib.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krug RM, Garcia-Sastre A. 2013. The NS1 protein: a master regulator of host and viral functions, p 114–132. In Webster RG, Monto AS, Braciale TJ, Lamb RA (ed), Textbook of influenza, 2nd ed John Wiley & Sons, Ltd, Oxford, United Kingdom. [Google Scholar]
- 21.Wolff T, Ludwig S. 2009. Influenza viruses control the vertebrate type I interferon system: factors, mechanisms, and consequences. J Interferon Cytokine Res 29:549–557. doi: 10.1089/jir.2009.0066. [DOI] [PubMed] [Google Scholar]
- 22.Ehrhardt C, Wolff T, Pleschka S, Planz O, Beermann W, Bode JG, Schmolke M, Ludwig S. 2007. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J Virol 81:3058–3067. doi: 10.1128/JVI.02082-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hale BG, Randall RE, Ortin J, Jackson D. 2008. The multifunctional NS1 protein of influenza A viruses. J Gen Virol 89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- 24.Opitz B, Rejaibi A, Dauber B, Eckhard J, Vinzing M, Schmeck B, Hippenstiel S, Suttorp N, Wolff T. 2007. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell Microbiol 9:930–938. doi: 10.1111/j.1462-5822.2006.00841.x. [DOI] [PubMed] [Google Scholar]
- 25.Wolff T, O'Neill RE, Palese P. 1996. Interaction cloning of NS1-I, a human protein that binds to the nonstructural NS1 proteins of influenza A and B viruses. J Virol 70:5363–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolff T, O'Neill RE, Palese P. 1998. NS1-binding protein (NS1-BP): a novel human protein that interacts with the influenza A virus nonstructural NS1 protein is relocalized in the nuclei of infected cells. J Virol 72:7170–7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kochs G, Garcia-Sastre A, Martinez-Sobrido L. 2007. Multiple anti-interferon actions of the influenza A virus NS1 protein. J Virol 81:7011–7021. doi: 10.1128/JVI.02581-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varga ZT, Grant A, Manicassamy B, Palese P. 2012. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol 86:8359–8366. doi: 10.1128/JVI.01122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Z, Yi C, Zhao L, Wang S, Zhou L, Hu Y, Zou W, Chen H, Jin M. 2014. PB2-588I enhances 2009 H1N1 pandemic influenza virus virulence by increasing viral replication and exacerbating PB2 inhibition of beta interferon expression. J Virol 88:2260–2267. doi: 10.1128/JVI.03024-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayman A, Comely S, Lackenby A, Hartgroves LC, Goodbourn S, McCauley JW, Barclay WS. 2007. NS1 proteins of avian influenza A viruses can act as antagonists of the human alpha/beta interferon response. J Virol 81:2318–2327. doi: 10.1128/JVI.01856-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noah DL, Twu KY, Krug RM. 2003. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAs. Virology 307:386–395. doi: 10.1016/S0042-6822(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 32.Chen S, Short JA, Young DF, Killip MJ, Schneider M, Goodbourn S, Randall RE. 2010. Heterocellular induction of interferon by negative-sense RNA viruses. Virology 407:247–255. doi: 10.1016/j.virol.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snijder B, Pelkmans L. 2011. Origins of regulated cell-to-cell variability. Nat Rev Mol Cell Biol 12:119–125. doi: 10.1038/nrm3044. [DOI] [PubMed] [Google Scholar]
- 34.Hu J, Nudelman G, Shimoni Y, Kumar M, Ding Y, Lopez C, Hayot F, Wetmur JG, Sealfon SC. 2011. Role of cell-to-cell variability in activating a positive feedback antiviral response in human dendritic cells. PLoS One 6:e16614. doi: 10.1371/journal.pone.0016614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hwang SY, Hur KY, Kim JR, Cho KH, Kim SH, Yoo JY. 2013. Biphasic RLR-IFN-beta response controls the balance between antiviral immunity and cell damage. J Immunol 190:1192–1200. doi: 10.4049/jimmunol.1202326. [DOI] [PubMed] [Google Scholar]
- 36.Zhao M, Zhang J, Phatnani H, Scheu S, Maniatis T. 2012. Stochastic expression of the interferon-beta gene. PLoS Biol 10:e1001249. doi: 10.1371/journal.pbio.1001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rand U, Rinas M, Schwerk J, Nohren G, Linnes M, Kroger A, Flossdorf M, Kaly-Kullai K, Hauser H, Hofer T, Koster M. 2012. Multi-layered stochasticity and paracrine signal propagation shape the type-I interferon response. Mol Syst Biol 8:584. doi: 10.1038/msb.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das K, Ma LC, Xiao R, Radvansky B, Aramini J, Zhao L, Marklund J, Kuo RL, Twu KY, Arnold E, Krug RM, Montelione GT. 2008. Structural basis for suppression of a host antiviral response by influenza A virus. Proc Natl Acad Sci U S A 105:13093–13098. doi: 10.1073/pnas.0805213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krug RM, Yuan W, Noah DL, Latham AG. 2003. Intracellular warfare between human influenza viruses and human cells: the roles of the viral NS1 protein. Virology 309:181–189. doi: 10.1016/S0042-6822(03)00119-3. [DOI] [PubMed] [Google Scholar]
- 40.Matthaei M, Budt M, Wolff T. 2013. Highly pathogenic H5N1 influenza A virus strains provoke heterogeneous IFN-alpha/beta responses that distinctively affect viral propagation in human cells. PLoS One 8:e56659. doi: 10.1371/journal.pone.0056659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steidle S, Martinez-Sobrido L, Mordstein M, Lienenklaus S, Garcia-Sastre A, Staheli P, Kochs G. 2010. Glycine 184 in nonstructural protein NS1 determines the virulence of influenza A virus strain PR8 without affecting the host interferon response. J Virol 84:12761–12770. doi: 10.1128/JVI.00701-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Twu KY, Noah DL, Rao P, Kuo RL, Krug RM. 2006. The CPSF30 binding site on the NS1A protein of influenza A virus is a potential antiviral target. J Virol 80:3957–3965. doi: 10.1128/JVI.80.8.3957-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park MS, Garcia-Sastre A, Cros JF, Basler CF, Palese P. 2003. Newcastle disease virus V protein is a determinant of host range restriction. J Virol 77:9522–9532. doi: 10.1128/JVI.77.17.9522-9532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knepper J, Schierhorn KL, Becher A, Budt M, Tonnies M, Bauer TT, Schneider P, Neudecker J, Ruckert JC, Gruber AD, Suttorp N, Schweiger B, Hippenstiel S, Hocke AC, Wolff T. 2013. The novel human influenza A(H7N9) virus is naturally adapted to efficient growth in human lung tissue. mBio 4:e00601-13. doi: 10.1128/mBio.00601-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, Reis e Sousa C. 2010. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140:397–408. doi: 10.1016/j.cell.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 46.Sevilla-Reyes EE, Chavaro-Perez DA, Piten-Isidro E, Gutierrez-Gonzalez LH, Santos-Mendoza T. 2013. Protein clustering and RNA phylogenetic reconstruction of the influenza A virus NS1 protein allow an update in classification and identification of motif conservation. PLoS One 8:e63098. doi: 10.1371/journal.pone.0063098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ping J, Keleta L, Forbes NE, Dankar S, Stecho W, Tyler S, Zhou Y, Babiuk L, Weingartl H, Halpin RA, Boyne A, Bera J, Hostetler J, Fedorova NB, Proudfoot K, Katzel DA, Stockwell TB, Ghedin E, Spiro DJ, Brown EG. 2011. Genomic and protein structural maps of adaptive evolution of human influenza A virus to increased virulence in the mouse. PLoS One 6:e21740. doi: 10.1371/journal.pone.0021740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Twu KY, Kuo RL, Marklund J, Krug RM. 2007. The H5N1 influenza virus NS genes selected after 1998 enhance virus replication in mammalian cells. J Virol 81:8112–8121. doi: 10.1128/JVI.00006-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dankar SK, Miranda E, Forbes NE, Pelchat M, Tavassoli A, Selman M, Ping J, Jia J, Brown EG. 2013. Influenza A/Hong Kong/156/1997(H5N1) virus NS1 gene mutations F103L and M106I both increase IFN antagonism, virulence and cytoplasmic localization but differ in binding to RIG-I and CPSF30. Virol J 10:243. doi: 10.1186/1743-422X-10-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Diaz MO, Ziemin S, Le Beau MM, Pitha P, Smith SD, Chilcote RR, Rowley JD. 1988. Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc Natl Acad Sci U S A 85:5259–5263. doi: 10.1073/pnas.85.14.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia-Sastre A. 2011. Induction and evasion of type I interferon responses by influenza viruses. Virus Res 162:12–18. doi: 10.1016/j.virusres.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kochs G, Koerner I, Thiel L, Kothlow S, Kaspers B, Ruggli N, Summerfield A, Pavlovic J, Stech J, Staeheli P. 2007. Properties of H7N7 influenza A virus strain SC35M lacking interferon antagonist NS1 in mice and chickens. J Gen Virol 88:1403–1409. doi: 10.1099/vir.0.82764-0. [DOI] [PubMed] [Google Scholar]
- 53.Khaperskyy DA, Emara MM, Johnston BP, Anderson P, Hatchette TF, McCormick C. 2014. Influenza A virus host shutoff disables antiviral stress-induced translation arrest. PLoS Pathog 10:e1004217. doi: 10.1371/journal.ppat.1004217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kainov DE, Muller KH, Theisen LL, Anastasina M, Kaloinen M, Muller CP. 2011. Differential effects of NS1 proteins of human pandemic H1N1/2009, avian highly pathogenic H5N1, and low pathogenic H5N2 influenza A viruses on cellular pre-mRNA polyadenylation and mRNA translation. J Biol Chem 286:7239–7247. doi: 10.1074/jbc.M110.203489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nacken W, Anhlan D, Hrincius ER, Mostafa A, Wolff T, Sadewasser A, Pleschka S, Ehrhardt C, Ludwig S. 2014. Activation of c-Jun N-terminal kinase upon influenza A virus (IAV) infection is independent of pathogen-related receptors but dependent on amino acid sequence variations of IAV NS1. J Virol 88:8843–8852. doi: 10.1128/JVI.00424-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.WHO. 23 February 2015, posting date Human infections with avian influenza A(H7N9) virus. WHO, Geneva, Switzerland: http://www.who.int/influenza/human_animal_interface/influenza_h7n9/RiskAssessment_H7N9_23Feb20115.pdf?ua=1. [Google Scholar]
- 57.Ayllon J, Domingues P, Rajsbaum R, Miorin L, Schmolke M, Hale BG, Garcia-Sastre A. 2014. A single amino acid substitution in the novel H7N9 influenza A virus NS1 protein increases CPSF30 binding and virulence. J Virol 88:12146–12151. doi: 10.1128/JVI.01567-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hale BG, Steel J, Medina RA, Manicassamy B, Ye J, Hickman D, Hai R, Schmolke M, Lowen AC, Perez DR, Garcia-Sastre A. 2010. Inefficient control of host gene expression by the 2009 pandemic H1N1 influenza A virus NS1 protein. J Virol 84:6909–6922. doi: 10.1128/JVI.00081-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hogner K, Wolff T, Pleschka S, Plog S, Gruber AD, Kalinke U, Walmrath HD, Bodner J, Gattenlohner S, Lewe-Schlosser P, Matrosovich M, Seeger W, Lohmeyer J, Herold S. 2013. Macrophage-expressed IFN-beta contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog 9:e1003188. doi: 10.1371/journal.ppat.1003188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, Vidal SM. 2013. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog 9:e1003256. doi: 10.1371/journal.ppat.1003256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perez-Cidoncha M, Killip MJ, Asensio VJ, Fernandez Y, Bengoechea JA, Randall RE, Ortin J. 2014. Generation of replication-proficient influenza virus NS1 point mutants with interferon-hyperinducer phenotype. PLoS One 9:e98668. doi: 10.1371/journal.pone.0098668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fribourg M, Hartmann B, Schmolke M, Marjanovic N, Albrecht RA, Garcia-Sastre A, Sealfon SC, Jayaprakash C, Hayot F. 2014. Model of influenza A virus infection: dynamics of viral antagonism and innate immune response. J Theor Biol 351:47–57. doi: 10.1016/j.jtbi.2014.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]