

ABSTRACT
Human endogenous retroviruses (HERV) make up 8% of the human genome. While the youngest of these retroviruses, HERV-K(HML-2), termed HK2, is able to code for all viral proteins and produce virus-like particles, it is not known if these virus particles package and transmit HK2-related sequences. Here, we analyzed the capacity of HK2 for packaging and transmitting HK2 sequences. We created an HK2 probe, termed Bogota, which can be packaged into HK2 viruses, and transfected it into cells that make HK2 particles. Supernatants of the transfected cells, which contained HK2 viral particles, then were added to target cells, and the transmissibility of the HK2 Bogota reporter was tracked by G418 resistance. Our studies revealed that contemporary HK2 virions produced by some teratocarcinoma and breast cancer cell lines, as well as by peripheral blood lymphocytes from lymphoma patients, can package HK2 Bogota probes, and these viruses transmitted these probes to other cells. After transmission, HK2 Bogota transcripts undergo reverse transcription, a step impaired by antiretroviral agents or by introduction of mutations into the probe sequences required for reverse transcription. HK2 viruses were more efficiently transmitted in the presence of HK2 Rec or HIV-1 Tat and Vif. Transmitted Bogota probes formed episomes but did not integrate into the cellular genome. Resistance to integration might explain the relatively low number of HK2 insertions that were acquired during the last 25 million years of evolution. Whether transient transmission of modern HK2 sequences, which encode two putative oncoproteins, can lead to disease remains to be studied.
IMPORTANCE Retroviruses invaded the genome of human ancestors over the course of millions of years, yet these viruses generally have been inactivated during evolution, with only remnants of these infectious sequences remaining in the human genome. One of these viruses, termed HK2, still is capable of producing virus particles, although these particles have been regarded as being noninfectious. Using a genetic probe derived from HK2, we have discovered that HK2 viruses produced in modern humans can package HK2 sequences and transmit them to various other cells. Furthermore, the genetic sequences packaged in HK2 undergo reverse transcription. The transmitted probe circularized in the cell and failed to integrate into the cellular genome. These findings suggest that modern HK2 viruses can package viral RNA and transmit it to other cells. Contrary to previous views, we provide evidence of an extracellular viral phase of modern HK2 viruses. We have no evidence of sustained, spreading infection.
INTRODUCTION
Actively replicating retroviruses infected the germ line multiple times over the millions of years of human evolution. These sequences were transmitted vertically in a Mendelian fashion to the next generation; therefore, they became a stable part of the inherited genetic material. Relics of these retroviral infections presently make up approximately 8% of the modern human genome. These retroviral sequences have acquired multiple mutations, and sometimes deletions, leading to the widely held assumption that they are no longer competent to replicate (1–3). The most recent of those infections were attributed to the human endogenous retrovirus type K [HERV-K(HML-2)], termed HK2 in this work (4, 5). After infection, HK2 integrated into the germ line DNA to form proviruses consisting of four retroviral genes (gag, pro, pol, and env) and two regulatory genes, np9 and rec. Np9 and Rec proteins are alternative splice products that are expressed depending upon the presence or absence, respectively, of a 292-bp segment in the env gene. The coding region of HK2 is flanked by two long terminal repeats (LTRs). Approximately 3,000 proviral remnants of the HK2 group remain in the modern human genome (6, 7). About 85% of these HK2 elements exist as solitary LTRs (solo-LTRs), which originated by recombination between the 5′ and 3′ LTRs of full-length proviruses, removing the internal gag, pro, pol, and env genes (8). To date, approximately two hundred HK2 elements have been found in the DNA of the modern human as full-length proviruses (3, 9, 10). A few HK2 loci have been copied by segmental duplication. About one hundred of these elements copied by recombination into the centromeres of several human chromosomes. Upon integration, HK2 created identical 5- to 6-bp sequences, known as target site duplications, flanking each side of the provirus. Homologous recombination between some HK2, however, created hybrid proviruses with different flanking target site sequences (11).
Certain HK2 elements are present in the genome of humans but not in other primates; therefore, they represent the youngest retroviruses to enter the human genome (4). Further, the youngest 11 HK2 are polymorphic in the human population (3, 9, 12). As HK2 is the most recent retrovirus to enter the genome, it is not surprising that HK2 still is transcriptionally active (13–19). The expression of HK2 viral RNA, proteins, and virus-like particles (VLPs) is detectable in breast cancer, leukemia, melanoma, and teratocarcinoma cell lines. These viruses, however, are widely considered to be noninfectious (15, 20–23). In addition to the findings of HERV-K expression in cancer cell lines, we have found HK2 RNA, proteins, and VLPs in the blood of patients with HIV-1 infection, lymphoma, and breast cancer (9, 10, 24–27); the capacity of these viruses to package HK2 sequences and transmit them to other cells remains to be tested.
While no single human endogenous retrovirus sequence thus far identified in the genome encodes a complete infectious virus, the diverse HK2 loci in the human genome retain coding capacity for every functional retroviral protein (4, 10, 28–30): the Gag protein, necessary for particle formation and release (31–35); the Pro protein, necessary for cleavage and maturation of viral proteins (35–37); the Pol protein, necessary for RNA-dependent DNA replication and integration (38–42); the accessory protein Rec, which exports unspliced viral RNA from the nucleus to the cytoplasm (43, 44); and functional Env proteins (45). Recombination or trans-complementation between these proviruses could lead to the formation of viruses that would be able to package and transmit HK2 sequences (9, 46–48), as seen with endogenous retroviruses in mice (49–51).
Recently, several groups have begun to suspect that modern HK2 sequences still retain the capacity to replicate. By analyzing HK2 sequences, Belshaw et al. revealed that during millions of years of HK2 evolution, the env gene, which is necessary only for virus infectivity, has been under purifying selection, meaning that this gene has accumulated synonymous mutations to preserve infectious capacity and suggesting that a pool of replication-competent HK2 still are present in modern humans (6). Subsequently, two groups resurrected infectious viruses using existing HK2 fragments of the human genome (52, 53); molecular recombination among present-day HK2 proviruses produced viruses competent to replicate, providing proof of principle of the possibility of HK2 replication in ancient and perhaps modern humans (53). Our laboratory has recently demonstrated that HK2 viral genomes found in HK2 particles are able to proceed through reverse transcription and have utilized a life cycle with a degree of genomic flexibility in which both RNA- and DNA-containing viruses are capable of mediating infection (54). Finally, we looked for evidence of HK2 transmission in modern humans by analyzing the viral genome found in viruses produced by living patients (9). Sequence analysis of the viral env gene in HIV-1-infected patients, but not breast cancer patients, revealed evidence consistent with HK2 transmission: (i) active recombination, suggesting the viral RNA was packaged into the particles and went through reverse transcription, and (ii) accumulation of synonymous mutations, a sign of purifying selection that could result from the copying of loci within the individual. These observations suggest, but do not prove, that HK2 sequences still could be packaged into HK2 particles and transmitted to other cells under certain conditions (9, 25).
Proof that HK2 sequences can be packaged and transmitted to other cells in modern humans is challenging to obtain using standard virology techniques, as all human cells contain HK2. It is difficult to differentiate between viral DNA, RNA, and protein products generated by de novo transmission from the already existent endogenous HK2. We have devised an innovative strategy to assess packaging and transmission of HK2 viruses, based on experiments previously employed to demonstrate that L1 elements are mobile (55–58). We constructed a molecular probe, termed Bogota, which has a presumed packaging sequence, the minimal HK2 viral sequences necessary to undergo reverse transcription, and a neomycin resistance reporter cassette. The genome of Bogota, however, does not encode any retroviral protein. The Bogota probe was designed so that it could be packaged into HK2 virus and then used to track cell-to-cell transmission and reverse transcription of genetic material packaged within HK2 viruses produced in human cells under various conditions.
In the present study, we report that HK2 viruses produced under certain conditions indeed can package HK2-related sequences and transmit these sequences to other cells. Furthermore, these sequences undergo reverse transcription. Efficient transmission of the HK2 sequences requires conserved sequences necessary for reverse transcription and is inhibited by reverse transcriptase inhibitors. Production of transmission-competent HK2 viruses occurs in certain cancer cell lines and peripheral blood lymphocytes of lymphoma patients. The expression of transmissible HK2 virus increases with the overexpression of the HK2 accessory protein Rec and is further enhanced by the HIV-1 accessory proteins Tat and Vif. Therefore, we have developed a strategy to assess packaging and transmissibility of endogenous retrovirus particles and have shown that modern HK2 viruses are competent to package HK2 sequences and transmit them to other cells. Upon passaging, we have demonstrated that the packaged genetic material undergoes reverse transcription. The viral sequences, however, do not undergo genomic integration into chromosomal DNA.
MATERIALS AND METHODS
Ethics statement.
Blood samples were collected from lymphoma patients after receiving informed consent for a protocol approved by the Institutional Review Board of the University of Michigan Medical School (IRBMED).
Construction of HERV-K Bogota plasmids and mutants.
Bogota constructs were derived from the HERV-K113 provirus (9,472 bp; accession no. AY037928.1) present in the bacterial artificial chromosome (BAC) clone RP11-398B1 obtained from BacPac Resources (https://bacpac.chori.org). Bogota was engineered to contain sequences necessary for reverse transcription, including the LTRs, the primer binding site (PBS), and the polypurine tract (PPT), whereas all the essential and accessory genes, gag, pol, env, and rec, were mutated or deleted. The Bogota construct contains only a portion of the gag gene and lacks pol, most of the env gene, and the rec gene. Therefore, Bogota constructs retain K113 sequence from the 5′ LTR to the middle portion of gag (bases 366 to 2665 of the K113 genome) and the 3′ LTR with a final portion of the env sequence (bases 8265 to 9472 of the K113 genome). Either a neo cassette (HERV-KBogota neo) or a neo cassette in reverse orientation with an intron (HERV-KBogota oenR) replaced the internal K113 genes (55). Bogota constructs were cloned into the pCEP4 vector (Invitrogen). The expression of Bogota is driven by the cytomegalovirus (CMV) promoter, which replaced the U3 portion of the 5′ LTR sequence (bases 1 to 366). Bogota mutants were constructed by deleting either the PBS (TGG TGC CCA ACG TGG AGG; 971 to 988 bp), the PPT site (AAA AGA AAA GGG GGA; bp 8488 to 8502), or the last 220 bp of the 3′ LTR. Further description of the cloning strategies is available upon request.
Other plasmid constructs.
Paul Bieniasz kindly provided the plasmid CHKCP, coding for the infectious HERV-KCON, and the plasmid pCR3.1/K-Rec.HERV-KCON, which contains a puromycin reporter cassette inserted into the env gene; its expression is driven by the CMV promoter (52). The plasmid pCR3.1/K-Rec codes for the HERV-K108 Rec protein. The VSV-G plasmid codes for the vesicular stomatitis virus (VSV) envelope. The HIV-1 Tat-coding plasmid pcDNA3.1-Tat86 was derived from the parent vector pcDNA3.1+/Tat101-flag (Eric Verdin, NIH AIDS Research and Reference Reagent Program; http://www.aidsreagent.org). The plasmids pcDNA-HVif and pcDNA-Vphu encode the HIV-1 accessory proteins Vif and Vpu, respectively. The sequences of Vif and Vpu are codon optimized for expression in human cells and are further described elsewhere (Stephan Bour and Klaus Strebel, NIH AIDS Research and Reference Reagent Program). The pcDNA3.1 empty vector control was made by excising the Tat insert with BamHI, followed by religation of the backbone.
Culture of cell lines, peripheral blood lymphocytes, and transfection.
The human cell lines 293T, NCCIT, PA-1, and MCF-7, as well as hamster CHO, rat C6, and buffalo green monkey BGMK cell lines, were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin. 293T cells are derived from embryonic kidney cells. The HK2 particle-producing NCCIT cell line is derived from male teratocarcinoma cells. PA-1 is derived from female ovarian teratocarcinoma cells. MCF-7 is derived from breast adenocarcinoma cells. The feline G355 cells were cultured in modified McCoy's 5a medium with the same additives. The quail QT6 cells were cultured in Ham's F-12 medium supplemented with 10% tryptose phosphate broth and 5% bovine calf serum. Blood samples were collected from lymphoma patients after receiving informed consent for a protocol approved by the University of Michigan Institutional Review Board. Peripheral blood lymphocytes (PBLs) were isolated from whole blood by Ficoll-Hypaque gradient and separated from macrophages by plate adherence. PBLs were maintained in RPMI medium supplemented with 10% FBS, penicillin-streptomycin and stimulated with 5 μg/ml phytohemagglutinin (PHA-P; Sigma-Aldrich) and 10 U/ml interleukin-2 (Sigma-Aldrich). Cells were transiently transfected in 10-cm plates at 5 × 106 cells/plate or in six-well plates at 1 × 105 cells/well using FuGENE HD (Roche) according to the manufacturer's protocol. After 24 h, cells were washed 5 times with phosphate-buffered saline and medium was replaced. Virus particle-containing supernatants were collected after an additional 24 to 48 h. Control experiments included mock transfections.
Immunoprecipitation of HK2 particles.
Virus-containing supernatant of NCCIT cells transfected with HERV-KBogota constructs was cleared of cellular debris with two centrifugation steps at 1,000 × g at 4°C, followed by filtration through a 0.45-μm Puradisc filter (Whatman). Viral particles were concentrated by ultracentrifugation through a 20% sucrose cushion, resuspended in 600 μl of NH buffer (0.8% NaCl, 10 mM HEPES, pH 7.4), and DNase treated using a Turbo DNA-free kit (Ambion). Viral particles were precleared with 50 μl of protein A/G Plus agarose beads (Santa Cruz Biotech) for 30 min with rocking. The beads then were removed by centrifugation for 3 min at 2,000 × g, and the supernatant was split into two equal volumes for immunoprecipitation (IP). To one tube, 6 μg of control mouse IgG2a was added. To the second, 6 μg of mouse anti-HERV-K Env (HERM-1811-5; Austral Biologicals) was added. Both samples were incubated for 90 min with rocking, after which 50 μl of protein A/G plus agarose beads was added to each tube. The samples then were incubated for an additional 2 h. The samples were centrifuged for 3 min at 2,000 × g to pellet the beads, and the supernatant was removed and washed three times with 300 μl NH buffer with centrifugation. The samples once again were split into two tubes, one for Western blot analysis and one for quantitative reverse transcription-PCR (qRT-PCR) analysis.
Western blot analysis following immunoprecipitation.
To prepare Western blot samples, the bound protein A/G beads were resuspended in 40 μl of NH buffer and 10 μl of 5× Laemmli sample buffer with dithiothreitol. Samples were boiled for 10 min to lyse and denature viral proteins and loaded onto an SDS-PAGE gel. HK2 capsid expression was detected using a rabbit anti-HERV-K Gag antibody, developed by the group of Norbert Bannert (36), and a mouse anti-rabbit IgG antibody conjugated to horseradish peroxidase. SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) was added, and protein bands were detected with X-ray film.
Viral and cellular RNA extraction.
Viral RNA was extracted from purified and DNase-treated viral particles, from cell-free supernatants, or from HK2 Env-immunoprecipitated samples using a QIAamp viral RNA minikit (Qiagen) by following the manufacturer's protocol. For immunoprecipitated samples, the protein A/G beads were removed by centrifugation after viral lysis. Viral RNA was eluted from the QIAamp columns in equal volumes and stored at −80°C. Cellular RNA was extracted with the QIAmp RNA blood minikit by following the manufacturer's instructions (Qiagen) and treated with RNase-free DNase as recommend by the manufacturer. The elimination of DNA was confirmed by PCR without RT using primers that bind HERV-K gag and Bogota neo. No PCR amplification product was detected in the cellular or viral RNA samples, confirming the absence of genomic or plasmid DNA.
Quantitative RT-PCR.
Viral and cellular RNA was analyzed for gene expression of the endogenous retrovirus HK2 np9, rec, gag, and env, HERV-W gag and env, HERV-H env, and HERV-KBogota neo, as well as housekeeping genes (gapdh, cox-2, and β-actin), by real-time RT-PCR using the primers described in Table 1. Quantitative RT-PCR was done using the Brilliant III ultrafast SYBR green qRT-PCR kit (Agilent Technologies) in a StepOne Plus real-time PCR system (Applied Biosystems). Quantitation analyses were performed using the ΔΔCT method (where CT is threshold cycle).
TABLE 1.
List of primers
| Name | Sequence |
|---|---|
| Knp9F | 5′-CCA ACG TGG AGG CTT TTC TCT AG-3′ |
| Knp9R | 5′-GTA CAC CTG CAG TCT CCG TCT CC-3′ |
| KrecF | 5′-GAG GCT GGC GGG ATC CTC-3′ |
| KrecR | 5′-ACA AAG CTT CCT ACG TCA TCA TGG CCC G-3′ |
| KgagRTF | 5′-AGC AGG TCA GGT GCC TGT AAC ATT-3′ |
| KgagRTR | 5′-TGG TGC CGT AGG ATT AAG TCT CCT-3′ |
| Kenvtype2F | 5′-AGA CAC CGC AAT CGA GCA CCG TTG A-3′ |
| Kenvtype2R | 5′-ATC AAG GCT GCA AGC AGC ATA CTC-3′ |
| BogotaneoF | 5′-ATG TTT CGC TTG GTG GTC GAA TGG-3′ |
| BogotaneoR | 5′-ACC TTG CTC CTG CCG AGA AAG TAT-3′ |
| HERV-WenvF | 5′-TCA TAT CTA AGC CCC GCA AC-3′ |
| HERV-WenvR | 5′-CGT TCC ATG TCC CCA TTT AG-3′ |
| HERV-WgagF | 5′-TCA GGT CAA CAA TAG GAT GAC AAC A-3′ |
| HERV-WgagR | 5′-CAA TGA GGG TCT ACA CTG GGA ACT-3′ |
| HERV-HenvF | 5′-GTC GGT TTA GGA CTT TCT GC-3′ |
| HERV-HenvR | 5′-TGT GGG AAC CTA GAG CGG GA-3′ |
| GAPDHF | 5′-TGC ACC ACC AAC TGC TTA GCA CCC-3′ |
| GAPDHR | 5′-CTT GAT GAC ATC ATA TTT GGC AGG-3′ |
| Cox-2F | 5′-TGA CTA TGG CTA CAA AAG CTG GG-3′ |
| Cox-2R | 5′-GCA AAC ATC ATG TTT GAG CCC T-3′ |
| B-actinF | 5′-GTG GGG CGC CCC AGG CAC CA-3′ |
| B-actinR | 5′-CTC CTT AAT GTC ACG CAC GAT TTC-3′ |
| E1 NEO1720F | 5′-TGC GCT GAC AGC CGG AAC ACG-3′ |
| E2 NEO210R | 5′-GAC CGC TTC CTC GTG CTT TAC G-3′ |
| E3 NEO437F | 5′-GAG CCC CTG ATG CTC TTC GTC C-3′ |
| E4 NEO1808R | 5′-CAT TGA ACA AGA TGG ATT GCA CGC-3′ |
Virus transmission.
Cells that may or may not produce transmissible HERV-K endogenous virus particles were transfected with HERV-K Bogota probe constructs and cotransfected with plasmids expressing HERV-K Rec or HIV-1 regulatory proteins where indicated. After 24 h, the transfection medium was removed and cells were washed at least five times with phosphate-buffered saline in order to eliminate any residual plasmid. Transfected cells were incubated in serum-free media for an additional 24 to 48 h. The production of infectious HERV-KCON viruses was performed as described previously (52). Briefly, plasmid CHKCP, encoding HERV-KCON, was transfected into 293T cells with or without plasmids expressing HERV-K Rec and VSV-G. Virus-containing supernatant was treated with DNase and cleared of cellular debris with two centrifugations at 1,000 × g at 4°C, followed by filtration through a 0.45-μm Whatman Puradisc filter (Whatman). Infection was performed by exposing the filtered virus-containing supernatant onto 60 to 70% confluent target cells seeded in 6-well or 10-cm plates along with fresh media in the presence of 5 μg/ml of Polybrene. In some experiments, target cells were incubated in the presence or absence of nucleoside and nonnucleoside reverse transcriptase inhibitors (RTIs). Cells transduced with HERV-K particles containing Bogota probes were selected in medium containing 250 to 500 μg/ml of neomycin for 2 to 4 weeks. Cells transduced with HERV-KCON were selected in 2.5 μg/ml puromycin for approximately 10 days. Colonies then were stained with Coomassie blue for visualization. The number of resistant colonies appearing as plaques was scored, and where indicated, the frequency of transmission was determined by calculating the percentage of resistant colonies formed by either RTI-treated cells or Bogota mutants in relation to the number of colonies observed in the untreated, Bogota-transmitted cells.
Detection of Bogota integration.
Identification of Bogota integration into the target cell genome was performed on genomic DNA isolated from G418 cells. The existence of Bogota DNA, either in episomal form or integrated into chromosomes, was assessed by several methodologies.
(i) PCR.
DNA was screened for Bogota integration by PCR using primers that bind to the neo gene, NEO437F and NEO1808R (Table 1). PCRs were performed using fast-start Taq DNA polymerase (Roche Applied Science) using the following conditions: one cycle at 95°C for 2 min, followed by 35 cycles at 94°C for 15 s, 55°C for 20 s, and 72°C for 30 s. PCR products were purified using the QIAquick PCR purification kit (Qiagen) and sequenced.
(ii) Deep-sequencing analyses.
Paired-end libraries were prepared from the genomic DNA samples, and Bogota elements were enriched for by hybridization with a biotinylated probe set spanning a consensus sequence of the HK2 LTR. The hybridized DNA then was captured with streptavidin beads, washed, and reamplified prior to deep sequencing using the Illumina HiSeq 2000 system (59). Potential integration sites were determined by PCR with primers that bind to the HK2 LTR and their potential flanking genomic sequences.
(iii) Inverse PCR.
Five micrograms of genomic DNA was digested with SpeI or HindIII (New England BioLabs). The digested DNA was ligated using T4 DNA ligase (3,200 U; New England BioLabs) in a volume of 600 μl at 16°C for 16 h. The ligated DNA was ethanol precipitated and dissolved in 50 μl of distilled water. The inverse PCR was performed using the Expand Long-Range dNTPack PCR kit (Roche Applied Science). Two microliters of DNA was used in a primary PCR in a 50-μl volume containing 2.5 mM MgCl2, 500 μM deoxynucleoside triphosphates (dNTPs), 300 nM each primer (NEO1720F and NEO210R; Table 1), and 3.5 U of Expand Long-Range enzyme mix. PCR was performed in one cycle of 95°C for 2 min and then 30 cycles of 94°C for 10 s, 63°C for 30 s, and 68°C for 15 min, followed by a final extension step at 68°C for 30 min. One μl of the resultant product was subjected to a secondary PCR using the same conditions, except that we used primers NEO1808F and NEO173R (Table 1). PCR products from the second amplification were purified with a QIAquick PCR purification kit (Qiagen) and sequenced (60).
(iv) Alu and B2 Ran PCR.
We performed Alu PCR and B2 Ran PCR to account for posttransmission Bogota insertion into areas of highly abundant species-specific repetitive elements. PCR was performed using primers that bind to human Alu (61) or B2 rodent (62) sequences and complementary primers that bind the HK2 LTR of Bogota using the conditions described in the citations (61, 62).
(v) ATLAS suppression PCR.
We adapted a protocol for the amplification typing of L1 active subfamilies (ATLAS) that enables the selective amplification of DNA fragments containing the ends of human-specific L1s and flanking sequence as described previously (63), using primers specific for the Bogota virus instead of L1 primers.
In silico sequence analysis.
The env sequences obtained in supernatants from HK2 particle-producing cell lines were subjected to BLAST searches with the HERVd and NCBI databases. The sequences were aligned in BioEdit and exported to the MEGA 6 matrix. Viral RNA sequences were aligned to known HK2 proviruses (3, 9). Phylogenetic trees were constructed and corroborated by the neighbor-joining method, using the statistical bootstrap test (10,000 replicates) of inferred phylogeny and the Kimura 2-parameter model (64, 65). Open reading frames (ORFs) were calculated using translated-BLAST in the NCBI database. Highlighter plots were generated using the highlighter tool of the Los Alamos HIV sequence database (http://www.hiv.lanl.gov).
Test for recombination.
We evaluated sequences for potential recombination events using several methods. We first inspected the neighbor-joining tree for each data set. Recombination of large portions of different elements may lead to branches with unresolved topology, resulting in taxonomic units (TUs) that either protrude far beyond the other taxa or fall far short in comparison. The potential recombinant sequences were verified and the parent sequences identified using RIP 3.0 (http://www.hiv.lanl.gov). This program used a sliding window (200 bp in this study) that moves over an alignment containing the query sequence and all of the possible parental proviruses. Best matches are marked if they are significant by using an internal statistical test. Recombination analyses were corroborated using the recombination analysis tool (RAT) program (66). We verified the sequence similarity between the putative parent and query sequence on each side of the recombination spot. On several occasions, recombinants were more than 99% similar to each parental sequence.
RESULTS
Design of a strategy to test HK2 packaging and transmission: the Bogota system.
To examine whether present-day HK2 viruses can package and transmit HK2-related transcripts, we engineered a molecular HK2 probe, termed Bogota, which uses the transmission of resistance to the aminoglycoside neomycin (G418) to detect transmission into target cells. Similar approaches that use neomycin resistance as a surrogate marker have been created to successfully demonstrate the retrotransposition of L1 elements in human cells (55–58). Therefore, we cloned a neomycin (neo) resistance reporter cassette consisting of the simian virus 40 (SV40) promoter, the neomycin resistance gene, and a 3′ polyadenylation signal into a defective HERV-K clone derived from provirus K113 (12). This clone, HERV-KBogota neo, also contains the sequences necessary to fully undergo reverse transcription: the primer binding site (PBS), the polypurine tract (PPT), and the R and U5 segments of the LTRs. This construct contains only partial segments of the gag and env TM genes; therefore, Bogota is incompetent to make any retroviral protein itself (Fig. 1). The Bogota construct, HERV-KBogota neo, was designed to contain a sense neo reporter cassette (Fig. 1). Expression of the Bogota construct is driven by the CMV promoter, which replaced the U3 portion of the 5′ LTR, in order to achieve high levels of expression and avoid the variable expression of HERV-K seen in different cell types (14, 67). Transfection of Bogota into HK2 particle-producing cells results in production of Bogota retroviral transcripts that were predicted to be copackaged into HK2 VLPs, as Bogota contains the putative HK2 packaging signal. When these VLPs are used for transmission, they can confer G418 resistance onto the target cell only when the Bogota genome undergoes reverse transcription and forms episomal forms (1LTR and 2LTR) or integrates into chromosomal DNA (Fig. 1). However, if small traces of contamination of the HERV-KBogota neo construct are inadvertently transfected to a detector cell, G418 resistance can be created without the need for reverse transcription. Therefore, a second construct, HERV-KBogota oenR, was designed to contain an antisense neo resistance indicator gene (oenR) disrupted by the intron IVS 2 of the λ-globin gene in the opposite transcriptional orientation (Fig. 1). The arrangement of HERV-KBogota oenR ensures that G418-resistant target cells arise only when transmitted HERV-KBogota oenR transcripts are spliced, packaged into VLPs, reverse transcribed, and expressed by their own promoter (55–58). Cells will remain sensitive to neomycin (G418) if Bogota transcripts cannot be spliced and reverse transcribed; therefore, the neo resistance product was not synthetized.
FIG 1.

HK2 packaging and transmission assay. (A) Organization of HK2 proviruses. ORFs coded by the gag, prt, pol, and env genes, and the accessory rec gene, are indicated by closed rectangles. The 5′ and 3′ LTRs (light gray boxes) at each side of the provirus and the surface (SU) and transmembrane (TM) domains of the encoded Env protein are shown. (B) An overview of the packaging and transmission assay of HERV-KBogota reporters. The constructs HERV-KBogota neo and HERV-KBogota oenR were derived from the K113 provirus by replacing the retroviral genes with a neomycin (neo) resistance indicator gene to tag the retroviral transcripts. An SV40 promoter and a 3′ polyadenylation signal (A′) regulate the expression of neo. Although Bogota transcripts do not code for retroviral proteins, the transcripts retain sequences necessary for reverse transcription (the primer binding site [PBS], the polypurine tract [PPT], and the U3, R, and U5 parts of the LTRs). As the LTR of HK2 is not functional in every cell line, a CMV promoter replaced the U3 portion of the 5′ LTR. Bogota retroviral transcripts contain the packaging signal of HK2; thus, they can be packaged into HK2 viral particles. Upon cell transmission, these transcripts confer G418 resistance only when they are reverse transcribed and then integrated into chromosomal DNA or form episomes (1LTR and 2LTR). HERV-KBogota oenR (right) contains an antisense copy of the neo gene disrupted by intron 2 of the γ-globin gene in the sense direction. The splice donor (SD) and splice acceptor (SA) sites of the intron are indicated. Transcripts originating from the HERV-KBogota oenR promoter can splice out the intron but contain only an antisense copy of the neo gene. G418-resistant colonies should arise only when, upon packaging and transmission, these transcripts are reverse transcribed, producing a sense copy of the neo resistance gene, which could be integrated into the genome or into episomal forms, and expressed from their own promoter.
Evaluation of cell lines that might produce transmissible HK2 viruses.
We first evaluated several cancer cell lines that might produce HK2 VLPs that can package and transmit HK2 sequences. While some breast cancer, teratocarcinoma, leukemia, and melanoma cell lines produce HK2 VLPs, these viruses appear to be noninfectious (15, 20–23). Bieda et al. characterized the phenotype of HK2 VLPs produced by the teratocarcinoma cell lines GH, Tera-1, NCCIT, 2102Ep, and Tera-2; one additional cell line, PA-1, served as a virus-negative control (20). Some VLPs released by NCCIT cells appeared to have morphological and biochemical properties of transmissible viruses: (i) by electron microscopy, free virus particles are seen with processed cores, suggesting mature viruses; (ii) Western blot analysis detected processed Gag protein in those particles. However, Env protein was not detected by Western blotting in those particles with the anti-HK2 sera used in this study, suggesting that the particles cannot be infectious (20). In experiments conducted in our laboratory, however, by electron microscopy we saw that the VLPs derived from NCCIT cells can be immunostained with anti-HERV-K Env antibody. This antibody further detected processed Env protein in VLP preparations by Western blotting (Fig. 2), suggesting that VLPs produced from NCCIT cells are promising candidates for transmissible HK2 viruses. The transmembrane (TM) band observed in the Western blot is very weak compared to the full Env bands, suggesting that the HERV-K Env protein is hardly cleaved and not fusogenic. However, the small portion of HERV-K viruses with processed or cleaved TM protein could be infectious.
FIG 2.
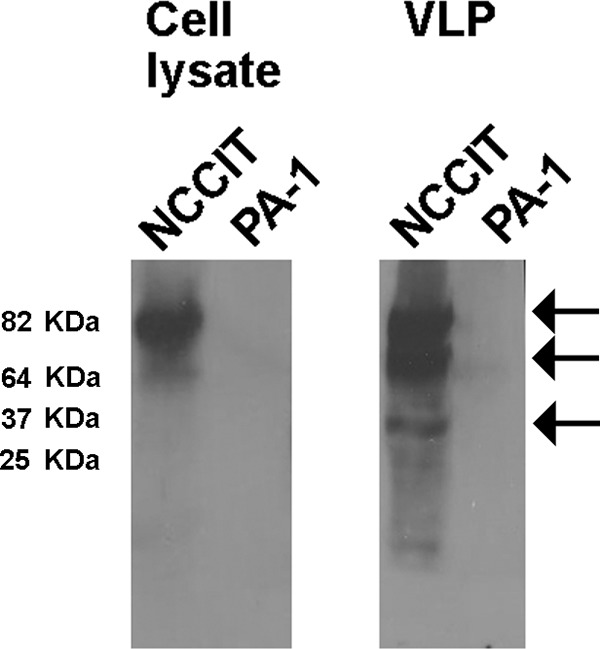
Western blot analysis of HK2 Env protein found in the cell-associated lysate and cell-free VLPs released from NCCIT cells. Cell lysates and VLPs contained in the supernatants of human NCCIT and PA-1 teratocarcinoma cells were assessed for the presence of the HK2 Env protein using the anti-HK2 Env antibody HERM1811-5 (Austral Biologicals, San Ramon, CA) as previously described (9). Two bands of 90 and 80 kb were detected in the cell lysate of NCCIT but not PA-1 cells. These bands are consistent with the unprocessed precursor Env protein, with or without the signal peptide. These bands also were detected in cell-free VLPs of NCCIT cells together with a band of ∼37 kb that corresponds to the TM subunit cleaved after Env processing (100).
In a further step, we analyzed the viral env genome sequence contained in HK2 VLPs produced by breast cancer, melanoma, and teratocarcinoma cell lines, looking for evidence of viral RNA replication by reverse transcription, as demonstrated by sequence diversification and recombination. Analysis of HK2 env sequences found in the supernatants of the NCCIT cell line, but not other cell lines, revealed evidence of virus diversity and recombination, suggesting that the HK2 genome replicated through reverse transcription (unpublished data). It is possible that ex vivo RT-PCR recombination artificially created these recombinant HK2 env sequences detected in our study (68). However, the detection of recombinant env sequences (45% of the sequences) only in the supernatants of NCCIT cells, but not other cell lines, argues that these recombinant env sequences arose by HK2 replication through reverse transcription in the NCCIT cells and not artifactually. It also is important to clarify that many more HK2 proviruses are expressed in NCCIT cells than in other cell lines studied (unpublished data); therefore, the occurrence of recombination might be related to the diversity of HK2 proviruses that are expressed.
Packaging of Bogota into HK2 viruses.
Having determined that the HK2 VLPs produced by the NCCIT cell line have the potential to be transmissible, we next addressed whether the Bogota probe can be packaged into these VLPs by transfecting the Bogota construct into NCCIT cells and analyzing the released HK2 VLPs for copackaging of Bogota transcripts. The packaging of Bogota transcripts in HK2 VLPs was evaluated using two different methodologies. In the first approach, the enveloped HK2 VLPs were immunoprecipitated with anti-HK-2 Env antibody, and in the second approach, total VLPs were pelleted by ultracentrifugation; the amount of Bogota transcripts packaged into those VLPs was measured by real-time RT-PCR. For the immunoprecipitation studies, VLPs produced were concentrated from NCCIT supernatants by ultracentrifugation and immunoprecipitated using anti-HERV-K Env antibody or control nonspecific IgG. VLPs immunoprecipitated with anti-HERV-K Env antibody reacted with anti-HERV-K Gag antibody in Western blot analysis, confirming the precipitation of HK2 VLPs using this methodology (Fig. 3A). Immunoprecipitation of HK2 VLPs with nonspecific IgG was detected only at a low level. We next measured the abundance of the HK2 gag and Bogota neo RNA transcripts contained in immunoprecipitated HK2 VLPs, using glyceraldehyde-3-phosphate dehydrogenase (gapdh) as a control. Bogota neo and HK2 gag RNA transcripts were enriched in VLPs immunoprecipitated with HERV-K Env antibody compared to the VLPs precipitated with nonspecific IgG (Fig. 3B). The levels of gapdh transcripts decreased in IP samples compared to those of the raw supernatant. The residual low levels of gapdh in IP samples is expected, as this has been reported in studies of other retroviruses (32, 69–76), which can package different nonviral RNA sequences at low levels. Further, enrichment of HK2 gag and Bogota neo transcripts found in HK2 VLPs was 5 to 20 times greater than that of gapdh transcripts (Fig. 3B), suggesting that Bogota transcripts are efficiently packaged into HK2 viral particles.
FIG 3.
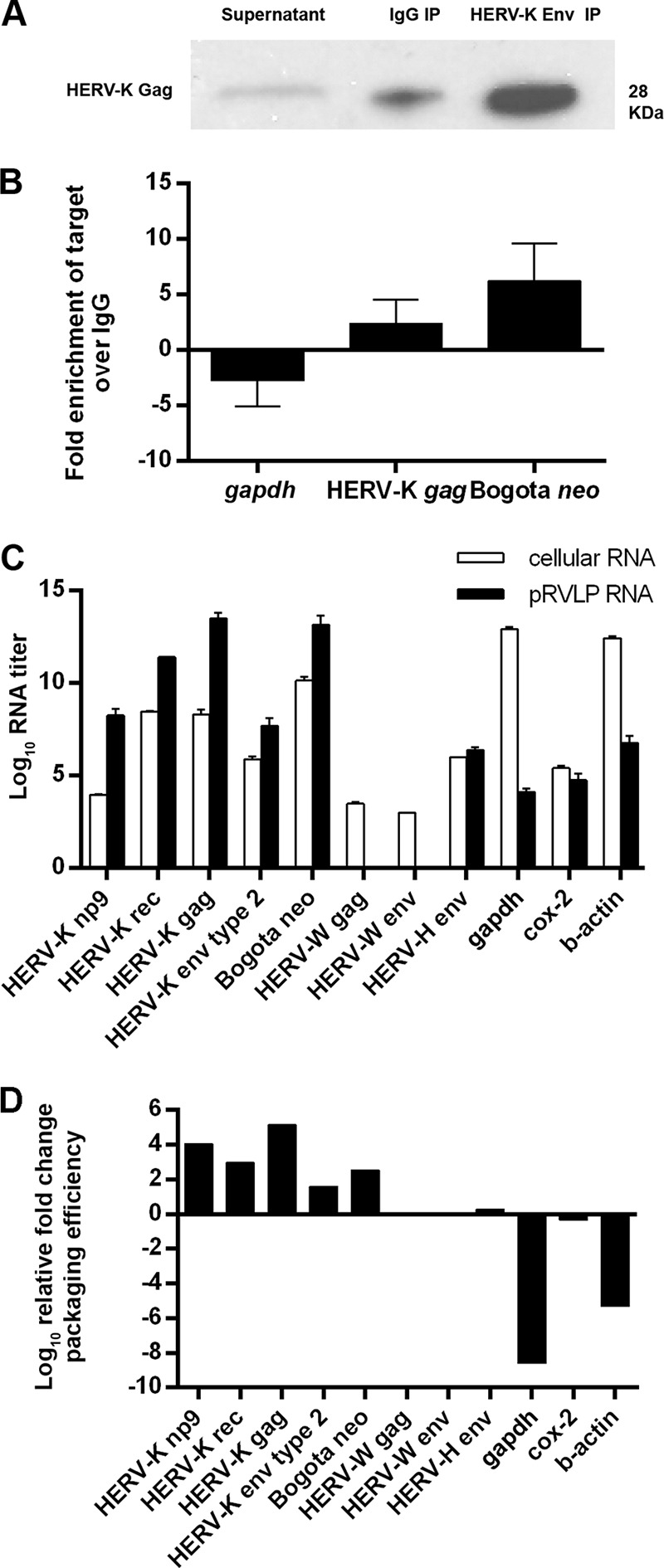
Packaging of Bogota transcripts in HK2 viral particles. (A and B) Immunoprecipitation of HERV-K virions containing Bogota transcripts. NCCIT cells were transfected with HERV-KBogota neo, and viral particles were collected 48 h after. Viral particles were pelleted by ultracentrifugation prior to immunoprecipitation with either an anti-HERV-K Env antibody or a control anti-mouse IgG2a. (A) Cleared supernatant and the immunoprecipitated samples were analyzed for HERV-K capsid enrichment by Western blotting with an anti-HERV-K Gag antibody. The untreated supernatant is about 5-fold diluted compared to the volumes obtained in the concentrated and immunoprecipitated samples; therefore, equal amounts of the supernatants could not be added in the gel due to well-volume constraints. The low background level detected with the anti-IgG2a immunoprecipitation may be nonspecific. (B) RNA from immunoprecipitated particles was quantitated for HERV-K gag, Bogota neo, or nonviral gapdh by qRT-PCR. The average fold enrichment for each target in particles precipitated with anti-HERV-K Env compared to those precipitated by control anti-mouse IgG2a is shown. (C) RNA expression levels of HERV-K (np9, rec, gag, and env type 2), Bogota neo, HERV-W (gag and env), HERV-H env, gapdh, cox-2, and β-actin were analyzed by qRT-PCR in Bogota-transfected NCCIT cells (white bars) and pelleted retrovirus-like particles (pRVLPs; black bars). The log10 RNA titers show the average quantitations ± standard deviations (SD) from at least three independent experiments expressed in arbitrary units. (D) Packaging efficiency of Bogota transcripts in NCCIT-produced viral particles. The relative fold change of packaging efficiency was calculated by dividing expression levels of target RNA copies in NCCIT pRVLPs by their expression levels in total cellular RNA. Values are presented as the means ± SD from at least three independent experiments. A value of 0 is given for HERV-W gag and env due to the absence of these transcripts from NCCIT pRVLPs.
We next pelleted retroviral VLPs by ultracentrifugation to examine Bogota packaging in total, as opposed to immunoprecipitated, HK2 VLPs. We quantitated the abundance of Bogota neo and HK2 np9, rec, gag, and env type 2 transcripts. We then compared the amounts of these transcripts in relation to the levels of expression of the cellular housekeeping genes gapdh and β-actin and the mitochondrial gene cytochrome c oxidase II (cox-2), as well as transcripts from the endogenous retrovirus families HERV-W and HERV-H (Fig. 3C). In order to determine the packaging efficiency of these transcripts into the pelleted retroviral VLPs, we compared the levels of transcripts found in the pelleted retroviral VLPs to the ones found in the cellular RNA using a previously validated methodology (32, 77, 78). Of these transcripts, HK2 genes and Bogota HK2 appear to be the most abundant retroviral transcripts in NCCIT cellular RNA. A small degree of expression of the endogenous retroviruses HERV-W and HERV-H was detected in the cellular RNA as well, as opposed to the high levels of expression of the housekeeping genes gapdh and β-actin. In the pelleted retroviral VLPs, however, we detected larger amounts of HK2 and Bogota transcripts compared to the levels of expression of the housekeeping genes. Transcripts of the HERV-H family also were detected in the pelleted retroviral VLPs but at a low level, and transcripts of the HERV-W family were not detected (Fig. 3C).
We calculated the relative packaging efficiency of each RNA transcript by dividing the expression levels of each transcript found in pelleted retroviral VLPs by its expression level found in cellular RNA and normalizing these numbers to the packaging efficiency level of HERV-H env, which was arbitrarily set to 1. The relative packaging efficiencies of HK2 and Bogota transcripts were 2 to 5 logs higher than the packaging efficiency of HERV-H env. The relative packaging efficiency could not be calculated for HERV-W transcripts, as they were not detectable in pelleted retroviral VLPs from NCCIT cells (Fig. 3D). Calculation of the packaging efficiencies of cellular and mitochondrial housekeeping genes produced negative values, suggesting they are not selectively packaged into pelleted retroviral VLPs. These results strongly suggest that HK2 and Bogota transcripts are selectively packaged into pelleted retroviral VLPs from NCCIT cells transfected with the Bogota construct. In contrast, transcripts from housekeeping genes, as well as from other endogenous retroviral families, seem not to be selectively packaged into pelleted retroviral VLPs, arguing that these HK2 RNAs are packaged into structures other than HK2 VLPs, such as exosomes, that would coexist in these pellets.
Infectivity of HK2 viruses containing Bogota transcripts.
Having determined that Bogota transcripts are efficiently packaged into HK2 VLPs, we next determined whether these particles could transmit Bogota from one cell to another. In order to accomplish this, we tested whether HK2 VLPs from NCCIT containing the Bogota genome, which uses neomycin resistance as a surrogate marker for transmissibility, can transmit neomycin (G418) resistance to target cells (Fig. 4A). As a negative control, we transfected 293T cells, which do not produce detectable HK2 VLPs and should not package Bogota and transmit neomycin (G418) resistance to target cells. As a positive control for the transmissibility assay, we artificially produced infectious HK2 VLPs in 293T cells by transfecting the cells with the molecular clone CHKCP, a plasmid that produces the infectious HK2 virus HERV-KCON. This virus uses the puromycin resistance marker for infectivity (52), and HERV-KCON-infected cells were selected by their resistance to puromycin. As infectivity of HERV-KCON is improved by expression in trans of HK2 Rec and the vesicular stomatitis virus envelope protein (VSV-G), we also cotransfected CHKCP with Rec and VSV-G (Fig. 4B) constructs. As expected, HERV-KCON expression in 293T cells in the presence of Rec and VSV-G produced VLPs that infected target cells grown in puromycin as previously reported (52) (Fig. 4B). Expression of Bogota in 293T cells, however, did not produce transmissible VLPs, even in the presence of Rec and VSV-G (Fig. 4B). These results demonstrate that cell lines that do not express transmissible HK2 particles are not able to transmit Bogota into target cells.
FIG 4.
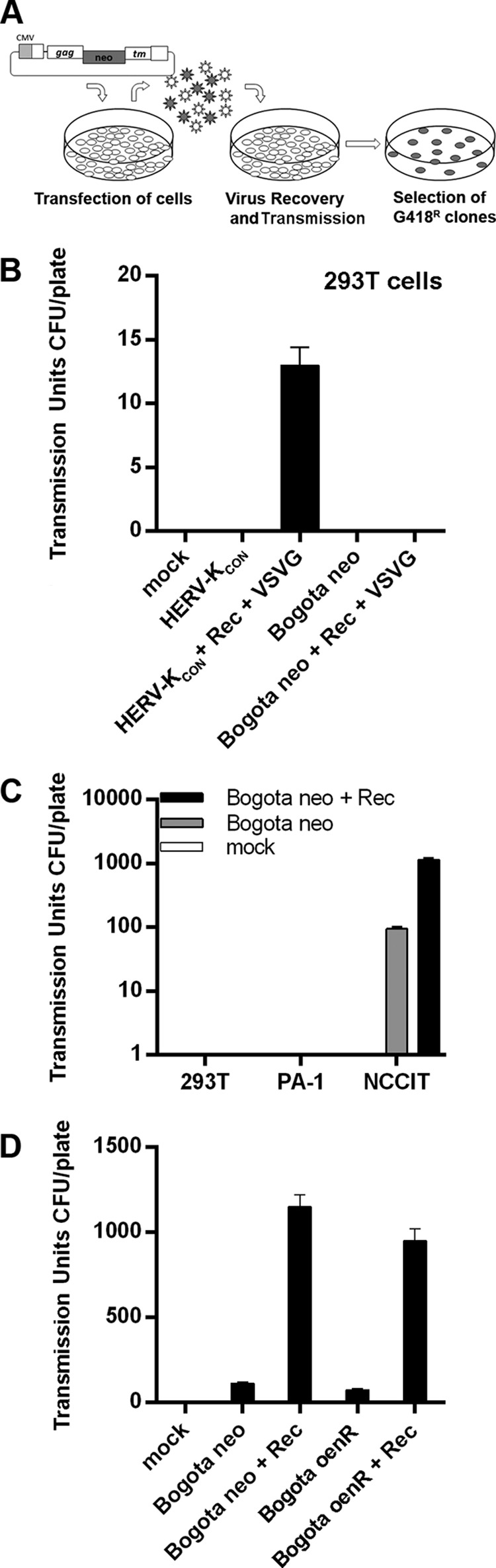
Transmission of HK2 viral particles. (A) Methodology of the transmissibility assay. Cells were transfected with Bogota constructs or the indicated plasmid mixtures. Supernatants were collected 48 h posttransfection, and the particles were incubated with target 293T cells. Cells exposed to HK2 VLPs were selected for G418 resistance and the apparent transmissibility units calculated by CFU per plate. (B) The infectivity of the resurrected HERV-KCON, produced by the CHKCP plasmid, following transfection into 293T cells was used as a positive control. HERV-KCON (VSV-G)-pseudotyped virions were produced in 293T cells in the presence or absence of Rec. HERV-KCON-infected cells were selected with puromycin. A similar transmissibility assay using HERV-KBogota neo did not reveal production of transmissible HK2 particles in 293T cells, which was predicted, as 293T cells do not make HK2 particles. (C) Transmissible units of HK2 particles containing Bogota transcripts produced by 293T and the germ cell tumor cell lines PA-1 and NCCIT, in the presence or absence of Rec. The target cells were 293T cells. (D) Transmissible units of NCCIT-producing HK2 particles containing Bogota transcripts with neo markers (the neo gene is displayed in sense orientation or antisense orientation and disrupted with an intron [see the text, particularly Materials and Methods, for further explanation]) in the presence or absence of Rec. The target cells were 293T cells. Values represent the means ± SD from at least three independent experiments.
We then addressed whether Bogota transcripts produced in HK2 VLP-producing cell lines can be transmitted to target cells. We transfected HERV-KBogota neo in the presence or absence of a Rec construct into the HK2 particle-producing NCCIT cells and the non-virus-producing cell lines 293T and PA-1. Cotransfection with Rec has been shown to increase the production of transmissible particles (52). VLPs were collected and used to transmit Bogota onto target cells, and these cells were selected with G418 resistance. The Bogota probe packaged into VLPs produced by the NCCIT cells was transmitted into target cells (Fig. 4C). Notably, expression in trans of the Rec protein increased the production of transmissible HK2 VLPs by ∼10-fold. The expression of the Bogota probe in non-virus-producing 293T and PA-1 cells, either in the presence or absence of Rec, did not lead to the production of transmissible HK2 VLPs. G418-resistant cells were never obtained when target cells were exposed to VLPs of cells transfected with the backbone vector pCEP4, the expression vector used to clone the Bogota probe (data not shown).
Reverse transcription of transmitted Bogota HK2 sequences.
We next determined whether transmission of the Bogota genome into target cells followed a reverse transcription step. We transfected NCCIT cells with HERV-KBogota oenR, which contains an antisense copy of the neo gene disrupted by an intron, and assessed the transmissibility of this genome into target cells. We observed that HK2 VLPs produced by the NCCIT cell line transmit the HERV-KBogota oenR probe into target cells, suggesting that the HERV-KBogota oenR genome was spliced and reverse transcribed in order to generate G418 resistance in target cells (Fig. 4D). The production of transmissible HK2 VLPs containing the HERV-KBogota oenR probe also was enhanced in the presence of Rec. G418-resistant cells could have arisen only if a spliced HERV-KBogota oenR genome is packaged into VLPs, transmitted into the target cell, replicated through reverse transcription to a sense strand encoding the neo resistance gene, integrated into chromosomal DNA or formed episomal forms, and then transcribed to produce G418 resistance.
Thus, transmission of HK2 Bogota into target cells appears to require a reverse transcription step. As Bogota itself does not encode a reverse transcriptase, the RT activity is hypothesized to come from the HK2 VLP in which it is packaged. Thus, we next studied in further detail whether the reverse transcriptase enzyme indeed modulates the reverse transcription of the Bogota sequence. We tested the capacity of the HK2 Bogota genome to undergo reverse transcription in the presence of reverse transcriptase inhibitors, molecules known to terminate the reverse transcription of a wide variety of retroviruses (79, 80). Transmission of Bogota into target cells was reduced by ∼80% when target cells were subjected to treatment with the nucleoside analogue reverse transcriptase inhibitors zidovudine (AZT) and lamivudine (3TC). Transmission of the Bogota genome was not inhibited when the experiments were conducted in the presence of the HIV-1-specific reverse transcriptase inhibitor efavirenz (Fig. 5A), suggesting that the reverse transcription of the Bogota genome is modulated by an HK2-specific reverse transcriptase. We also determined whether reverse transcription of the HK2 Bogota genome depends on substrate sequences necessary for reverse transcription, notably the primer-binding site (PBS), the polypurine tract (PPT), and the U5 LTR (80). We deleted these sequences in the HK2 Bogota genome (Fig. 5B) and tested the reverse transcription efficiency of Bogota. Deletion of PBS, PPT, and U5 LTR sequences reduced the transmission of Bogota into target cells by more than 80%, suggesting that reverse transcription of the Bogota genome required these substrate sequences (Fig. 5C). Thus, deletions introduced into key elements necessary for reverse transcription of the HK2 Bogota probe significantly reduced transmissibility. However, transmission was not abolished, suggesting that remaining nucleotide sequences/leaky mutations allow for residual reverse transcription.
FIG 5.
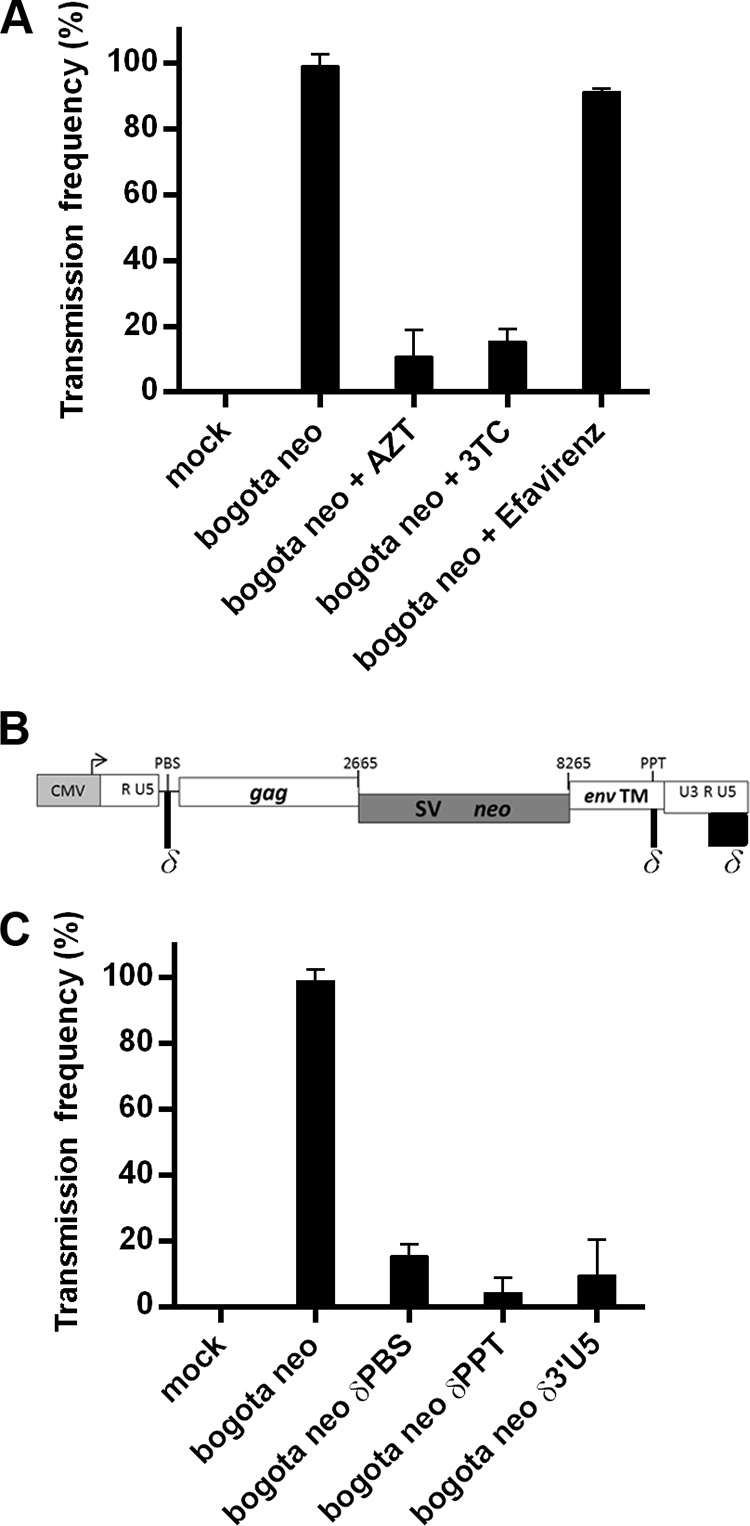
Transmission of HK2 particles into 293T target cells requires reverse transcription. (A) Inhibition of transmission of HK2 particles containing Bogota transcripts by treatment of the target cells with nucleoside reverse transcriptase inhibitors (NRTIs). The frequency of transmission of HK2 particles containing Bogota transcripts in the presence or absence of 50 μM AZT, 3TC, or efavirenz. (B) Genomic position of sequences required for reverse transcription modified in Bogota mutant constructs. The primer binding site (PBS), necessary to initiate reverse transcription, the polypurine tract (PPT), which resists digestion by the RNase H activity of the reverse transcriptase (RT) enzyme and primes plus-strand DNA synthesis, and the last 220 bp of the 3′ LTR containing a portion of R and U5, required for template switching during reverse transcription, were deleted by site-directed mutagenesis to test the effect of these sequences during reverse transcription of Bogota transcripts. (C) Transmission of HK2 particles containing Bogota transcripts and reverse transcription-associated mutant transcripts. Frequency of infection of HK2 particles containing Bogota or mutant δPBS, δPPT, or δ3′RU5 transcripts. Values represent the means ± SD from at least three independent experiments.
Lack of integration of transmitted HK2 Bogota reporters.
Having determined that Bogota genomes contained in HK2 VLPs produced by NCCIT cells were transmitted and reverse transcribed in target cells, we tested whether Bogota could integrate into chromosomal DNA or form episomes (1LTR or 2LTR episomes) (Fig. 6A). We further verified whether or not HK2 Bogota DNA is detected in transmitted G418-resistant cell clones by amplifying the Bogota surrogate antibiotic marker, the neo resistance gene. PCR detected neo DNA in Bogota G418-resistant cells (human NCCIT or hamster CHO cells) but not in G418-susceptible cells (Fig. 6A and B).
FIG 6.
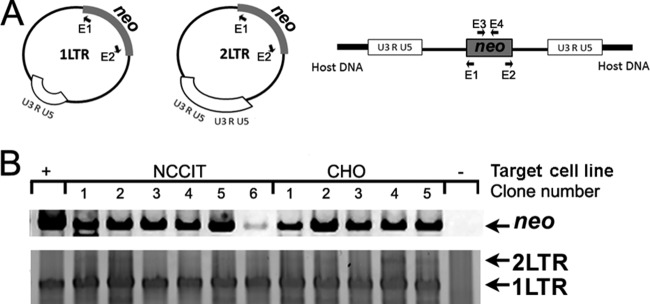
Bogota reverse-transcribed DNA sequences are found in episomal forms in target cells. (A) Schematic representation of the possible integration outcomes of Bogota reverse-transcribed cDNAs. After reverse transcription, Bogota cDNA may integrate into chromosomal DNA or form episomes (1LTR or 2LTR forms). Bogota integration forms can be detected with primers E1 and E2 by inverse PCR (Table 1). Primers E3 and E4 can detect Bogota neo DNA, which can be present in any of the integration forms. (B) Detection of Bogota DNA and Bogota episomal forms in recipient cells. DNA was extracted from target cells with Bogota-transmitted neomycin resistance. DNA of the neo resistance gene, as well as Bogota 1LTRs and 2LTRs, were detected by PCR and inverse PCR, respectively, using the primers described above. Target cells included the human NCCIT cell line and the hamster CHO cell line. DNA from Bogota neo was detected in all of the neomycin-resistant clones but not in untreated, neomycin-susceptible cells. The episomal 1LTR Bogota form was detected in all target cells, yet the 2LTR form was detected only in NCCIT clones 1 and 2 and the CHO clones 4 and 5. Sequencing confirmed the presence of episomal forms. The HERV-KBogota neo plasmid served as a positive control for neo DNA, and a clone containing a 1LTR sequence obtained in one of the experiments performed in this investigation served as a positive control for 1LTR amplification.
Having detected Bogota DNA in G418-resistant cells, we aimed to detect the possible Bogota integration forms. After completion of reverse transcription, retroviral cDNA could be integrated into the host chromosomal DNA or be circularized into episomal forms, i.e., the 1LTR or 2LTR circles. 2LTRs can be formed upon autointegration, sometimes called suicidal integration or circularization of the unintegrated provirus, by the viral integrase. Homologous recombination among the LTRs of linear unintegrated proviruses can further generate 1LTR episomes. Episomal integration forms have been detected for infectious HIV-1 and resurrected HK2 viruses (59, 81–83). We assessed the existence of Bogota episomal forms in DNA isolated from G418-resistant cells by inverse PCR using primers that bind to the neo resistance gene in the outward orientation (Fig. 6A). We detected 1LTR forms of the inferred Bogota construct in all G418-resistant cells exposed to HK2 viruses containing Bogota but not in untreated cells (Fig. 6B); the DNA sequence of these episomal forms was confirmed by sequencing to be Bogota. Less frequently, the inverse PCR amplified the 2LTR circles, suggesting preferential formation or amplification of 1LTR over 2LTR episomes (Fig. 6B). As 1LTR forms arise by recombination between the 5′ and 3′ LTRs of unintegrated viral cDNA (81–83), we analyzed the 1LTR sequence for signs of recombination. Sequence analysis revealed that the LTR produced in the 1LTR episome arose by recombination between the 5′ and 3′ LTR of Bogota (data not shown). These data suggest that Bogota DNA recircularized into episomal forms. Attempts to detect chromosomal integration of Bogota into the genome of the target G418-resistant cells was unsuccessful despite the use of multiple methodologies: (i) deep-sequencing analysis (59), (ii) inverse PCR in DNA digested with restriction enzymes and recircularized with DNA ligase (60), (iii) Alu and B2 Ran PCR (61, 62), and (iv) ATLAS suppression PCR (63) (see Materials and Methods).
Production of transmissible HK2 viruses by cell lines other than NCCIT.
As the Bogota probe packaged into HK2 particles proved to be a useful indicator of HK2 transmission, we used it next to assess whether or not cell lines other than NCCIT produce transmissible HK2 VLPs. We tested the capacity of the transmission of putative HK2 VLPs produced by the breast cancer cell line MCF-7 and the peripheral blood lymphocytes (PBLs) from patients with lymphoma, in which some recombinant HK2 viral genomes are detected, suggesting the presence of transmissible HK2 viruses (25, 84). Compared to the standard particle-producing NCCIT cells, transfection of HK2 Bogota into MCF-7 and PBLs from patients with lymphoma results in the production of a small number of transmissible HK2 particles that conferred G418 resistance onto target 293T cells (Fig. 7A). Transmission of these HK2 VLPs again was reduced by ∼75% when sequences necessary for reverse transcription, the PBS, the PPT, and the U5 LTR, were deleted from the Bogota genome (Fig. 7A). No transmission was seen with the empty vector. While it is possible that the small number of transmissible units detected in our studies reflects differences in the efficiency of transfection of MCF-7 and PBLs compared to that of NCCIT cells, the more limited production may be due to the lower titer of HK2 produced by the MCF-7 cell line and PBLs of patients with lymphoma.
FIG 7.
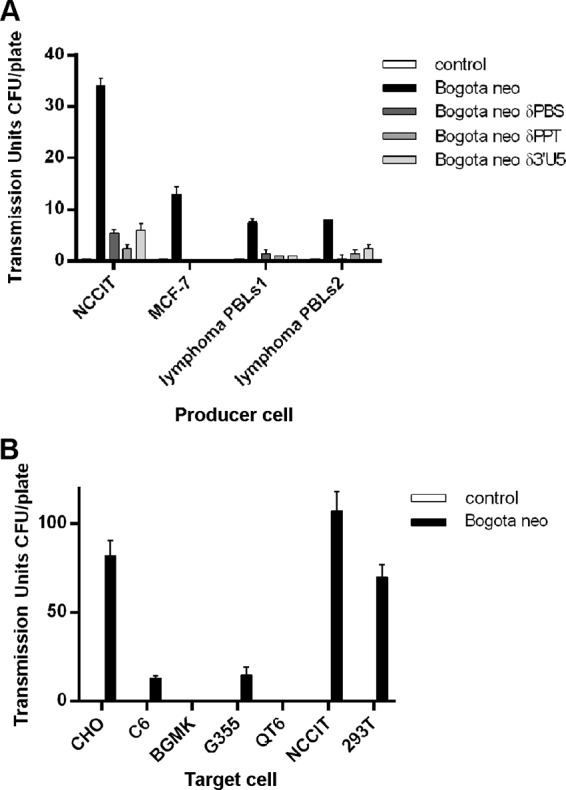
Transmissibility of HK2 particles produced from cancer cells and virus tropism. (A) Transmission of HK2 particles produced from cancer cell lines and lymphocytes from lymphoma patients to target 293T cells. The frequency of transmissibility of HK2 particles containing Bogota and reverse transcription-associated δPBS, δPPT, or δ3′RU5 mutant transcripts produced in NCCIT cells, the breast cancer MCF-7 cell line, and the peripheral blood lymphocytes (PBLs) of lymphoma patients (PBLs1 corresponds to a patient with large B-cell lymphoma, and PBLs2 corresponds to a patient with follicular lymphoma) is shown. (B) Tropism of HK2 particles produced in NCCIT cells containing Bogota transcripts. HK2 particles containing Bogota transcripts were incubated with hamster CHO, rat C6, buffalo green monkey BGMK, feline G355, quail QT6, and human NCCIT and 293T cell lines and selected for G418. Transmissible units per plate represent the means ± SD from at least three independent experiments.
Tropism of HK2 viruses.
We determined the tropism of HK2 VLPs produced by NCCIT cells using the Bogota system. The capacity of transmission of HK2 VLPs was evaluated in target human (NCCIT and 293T), hamster CHO, rat C6, buffalo green monkey BGMK, feline G355, and quail QT6 cells (Fig. 7B). Cells were selected by G418 resistance. No transmission was seen with the empty vector. We observed that HK2 VLPs were efficiently transmitted to human and hamster cells. Rat and feline cells were less susceptible to transmission. HK2 could not be transmitted to buffalo green monkey and quail cells (Fig. 7B). These results suggest that human and hamster cells are permissive to transmission of HK2 VLPs. Similar results were reported when investigators assessed the infectious capacity of resurrected HK2 in human, hamster, and feline cell lines (52, 53), further suggesting that Bogota is being transmitted in an HK2 virion.
Expression of HIV-1 Tat and Vif increases HK2 transmission.
We next asked whether expression of HIV-1 viral proteins increases the production of transmissible HK2 VLPs. The rationale behind this approach is that HIV infection, specifically the expression of the HIV-1 Tat and Vif proteins, has been found in previous studies to enhance expression of HK2 (9, 10, 24, 26, 27, 85, 86). Expression of HIV-1 Tat and Vif, but not Vpu, in NCCIT cells increased the production of transmissible HK2 VLPs containing the Bogota probe, as determined by measuring the number of G418-resistant clones (Fig. 8). A synergistic effect on the production of transmissible HK2 VLPs was observed when HIV-1 Tat and Vif were expressed together. Furthermore, production of transmissible HK2 VLPs increased further when the expression of HIV-1 Tat or Vif took place in the presence of the HK2 Rec protein, and increased transmission of HK2 VLP resulted when the cells were stimulated with the combination of all of these proteins (Fig. 8). These results suggest that the HIV-1 accessory proteins Tat and Vif enhance the production and, as a result, the transmission of HK2 VLPs produced in NCCIT cells.
FIG 8.
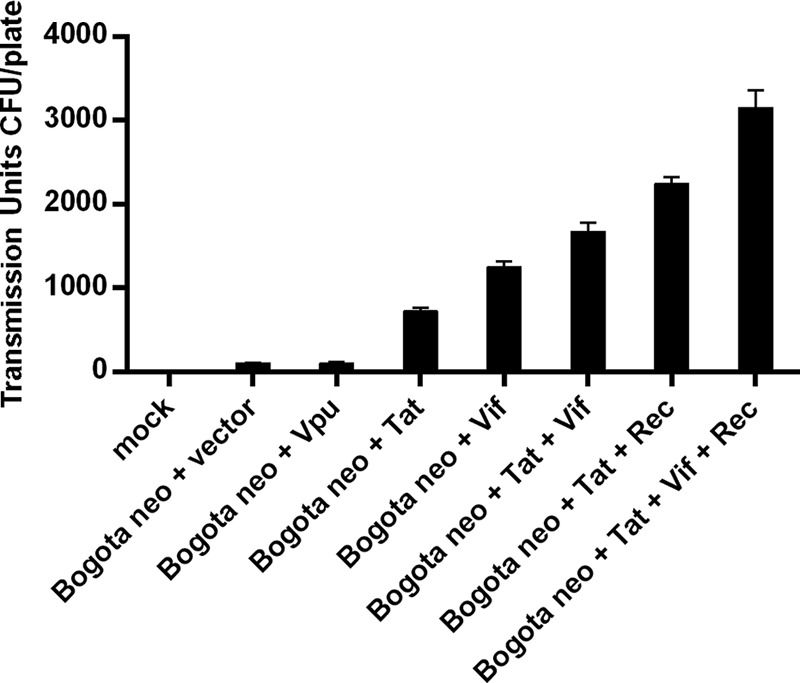
Expression of HIV-1 accessory proteins increases the production of transmissible HK2 particles. NCCIT cells were transfected with the indicator Bogota construct and plasmids expressing the HIV-1 Vpu, Tat, and Vif proteins. HK2 particles produced in treated cells were collected and assessed for transmissibility to 293T target cells by using the Bogota neomycin selection assay. The graph shows the transmissible units of HK2 particles containing Bogota transcripts stimulated by overexpression of HIV-1 proteins in the presence or absence of Rec. Expression of HIV-1 Tat or Vif, but not Vpu, increases the production of transmissible HK2 particles. Furthermore, an additive effect in the transmissible units of HK2 particles is seen when HIV-1 Tat and Vif, together with HK2 Rec, are expressed in NCCIT cells. Values represent the means ± SD from at least three independent experiments.
DISCUSSION
The prevailing wisdom for many years has been that no endogenous retrovirus is capable of infection in modern humans due to mutations in their viral sequences. Beyond this assumption, experimental approaches to examine the transmission capacity of endogenous retroviruses in humans have been hampered by the inability to distinguish de novo infection from the proviruses already present in the genome. Recently, two groups reconstructed what appeared to be the most recent HK2 infectious retrovirus to have endogenized in the human species by correcting the mutations found in present-day HK2 sequences to make these viruses infectious (52, 53). To assess the transmissibility of these ancient HK2 viruses, these investigators introduced surrogate markers (antibiotic resistance or fluorescent tags) to trace their mobility in modern human cells. Although these viruses were transmitted to other human cells and are a very helpful tool to examine endogenous retrovirus biology, it has remained unclear whether contemporary endogenous retroviruses, those that are encoded by the modern human genome, can be transmitted. We have devised a molecular system that traces the transmissibility of modern HK2 viruses using the molecular probe Bogota, which uses a surrogate marker to indicate transmission of HK2 viruses. Moreover, transmission of the Bogota probe is dependent on sequences necessary to undergo reverse transcription. Therefore, this probe can assess not only transmissibility but also reverse transcription of modern HK2 viral genomes.
In the present study, we have shown that HK2 viruses from cell lines that produce HK2 viruses with mature cores and processed Gag and Env proteins were capable of transmitting the Bogota marker to other cells (20). Bieda et al. reported that, in contrast to other teratocarcinoma cell lines producing HK2 viruses, the cell line NCCIT produced free mature viral particles with processed Gag protein, despite Env protein being undetectable in these studies (20). However, we have detected the Env protein in HK2 particles released by NCCIT cells and have found evidence of transmissibility of those particles using the Bogota probe. Transfer of Bogota cannot take place if this probe is expressed in non-HK2 particle-producing cells. Our studies indicate that HK2 viruses produced by NCCIT cells and MCF-7 cells and the peripheral blood lymphocytes from lymphoma patients produce transmission-competent HK2 viral particles, and that the Bogota sequences contained in these particles underwent further reverse transcription. Furthermore, HK2 viruses could be transmitted, to some extent, to certain nonhuman cells, suggesting that the tropism of HK2 viruses is not limited to human cells, an observation compatible with those of other groups (52, 53). We cannot rule out the possibility that the broad tropism of HK2 particles depends on other human retroviral envelopes able to pseudotype HK2 particles. At least an Env protein would be required for virus transmission, since other studies have failed to transmit unenveloped HK2 particles, such as unenveloped HERV-KCON (52). Even though our studies indicate HK2 RNA is packaged selectively into HK2 enveloped VLPs, particles that allow for the transmission of RNA to target cells, the possibility exists that transmission of some RNA was mediated by exosomes (87).
The transmission of Bogota packaged into HK2 particles was enhanced in the presence of retroviral regulatory proteins. Expression in trans of HK2 Rec, a protein that facilitates the export of incompletely spliced RNA from the nucleus to the cytoplasm (43, 44), increased the transmission of Bogota by ∼10-fold. Similar results were obtained when the resurrected infectious HERV-KCON virus was expressed in the presence of HK2 Rec (53). In addition, expression in trans of the HIV-1 accessory proteins Tat and Vif increased the production of transmissible HK2 Bogota. The HIV-1 Tat protein likely activates expression of HK2 viruses through transactivation of the HK2 viral promoter; we previously reported that HIV-1 Tat upregulated HK2 expression by modulating the binding of the transcription factors NF-κB and NFAT in the HK2 LTR (85, 86). Expression of the HIV-1 protein Vif could facilitate the transmissibility of HK2 virus by interfering with cytidine deaminases of the APOBEC family; these proteins introduce mutations into the viral genome by deaminating minus-strand cytidines during reverse transcription (88–90). APOBEC deaminases have been shown to inhibit infection with HK2 viruses (52, 91); therefore, the expression of HIV-1 Vif may counteract this effect. These findings are consistent with those from our group and others, who have found increased expression of HK2 viruses in the setting of HIV-1 infection (9, 10, 24–27, 92). The consequence of this increased expression of transmissible HK2 viruses, for both HIV-1-infected cells and the HIV-1-infected individual, remains to be elucidated. However, it is important to note that expression of HK2 epitopes, and perhaps transmissible HK2 viruses, on HIV-1-infected cells render these cells susceptible to lysis by immune T cells and antibody responses directed against HK2, creating a tool that conceivably could be used to eradicate HIV-1 infection (93–95).
Our studies indicate that modern HK2 viruses indeed remain transmissible and that genetic material packaged in these viral particles can undergo reverse transcription. Several lines of evidence suggest that expression and transmission of Bogota required reverse transcription: (i) transmission of BogotaoenR, a probe that uses a neo resistance gene disrupted by an intron and in an antisense orientation, could have generated G418 resistance in target cells only by removing the intron through splicing and positioning the neo gene in the sense orientation after reverse transcription (Fig. 4D); (ii) transmission of Bogota was impaired by introducing genetic mutations in sequences required for reverse transcription, i.e., the PBS, the PPT, and the U5 LTR; (iii) transfer of Bogota was reduced substantially in the presence of nucleoside analogue reverse transcriptase inhibitors; and (iv) detection of Bogota episomal integration (1LTR and 2LTR).
We also examined the fate of Bogota cDNA in target cells and whether it integrated into chromosomal DNA and/or formed 1LTR and 2LTR episomes (59, 81–83). While we detected abundant Bogota integration in episomal forms, we were unable to detect Bogota integration into chromosomal DNA using multiple methodological approaches. The inability of Bogota to integrate into cellular DNA could be attributed to properties inherent to the Bogota system. Bogota may have lacked unique sequences necessary for integration, or perhaps the viral cDNA product was so short that it favored autointegration and not chromosomal integration. Replication of the Bogota episomes might be attributed to the presence of an SV40 origin of replication upstream of the neo resistance gene. The SV40 large T antigen, an antigen expressed in the cell line 293T used as the target cell in some of our transmission experiments, is known to promote episomal DNA replication in the nucleus of plasmids with the SV40 origin of replication (96). Interestingly, however, it also has been shown that plasmids containing the SV40 origin of replication can replicate in hamster CHO cells (cells we also have used as the target cells in the transmission studies), even though they lack the expression of the SV40 large T antigen, suggesting that Bogota episomes containing the SV40 origin of replication similarly are able to replicate autonomously in CHO cells (97). It appears, however, that the replication of these SV40 origin of replication-containing episomes required the expression of the scaffold/matrix attachment region (S/MAR) protein from the human beta interferon gene cluster as well as transcription of sequences upstream of the S/MAR gene (98). In any case, we have very consistently detected Bogota episomes in several target cell lines that apparently do not express the SV40 large T antigen (the human NCCIT, the hamster CHO, the rat C6, and the feline G355 lines), suggesting that the replication of the Bogota episomes is not dependent on the SV40 large T antigen. How these cells maintain the replication of the Bogota episome remains unclear, but it is possible that trans-acting factors other than the large T antigen modulate the SV40 origin of replication in the Bogota plasmid.
Although the Bogota probe seems to favor episomal replication when transmitted in an autonomous form, infection and retroviral integration of HK2 into the genome of an infected cell has been demonstrated using the resurrected HK2 viruses HERV-KCON and Phoenix (53, 54), as well as the autointegration of HERV-KCON, to form episomes (59). Thus, HK2 is, in principle, able to integrate into the genome of human cells, and it is surprising that the majority of HK2 viruses led to transmissible viral genomes that resulted in circular forms rather than genomic integrations. Perhaps, unlike the integrase enzyme encoded by the consensus HERV-KCON and Phoenix viruses, which are functional and allow integration, none of the HK2 elements in the cells tested in our studies contribute a functional integrase protein to the VLPs. It is likely that HK2 episomes can be transcribed and translated into proteins able to be packaged into new particles, as the Bogota episomes were able to produce a protein that confers G418 resistance. It is still unclear to us whether the recombinant viral genomes found in the VLPs produced by NCCIT come from these episomal forms or whether they are derived from a recombinant integrated provirus. Our data suggest that cellular or viral factors prevent transmission of HK2 virus from integrating in the human genome; this could explain why only about 3,000 HK2 sequences integrated in the last 25 million years of human evolution (3, 9). This may be due to the previously described cell restriction factors or to even more potent restriction mechanisms in the germ cells of primates. Thus, our inability to detect chromosomal integration may well reflect a biological reality, in that passage of truly infectious HK2 is a rare event in modern humans. It is important to note that the transmission of HK2 particles was seen most impressively with particles produced by the NCCIT cell line, in which a great portion of the repertoire of HK2 particle-associated sequences are recombinant and the expression profile of HK2 proviruses was more diverse. As HIV-1 viral proteins further increased the transmission of HK2 viruses produced by NCCIT, it is likely that expression of more HK2 proviruses enhances the production of transmissible HK2 viruses. Whether transmissible HK2 viruses are produced in HIV-1 patients remains to be further explored. However, the findings in this study suggest that the increased expression, occurrence of recombination, and evidence of purifying selection of HK2 found in the HIV-1 setting (9, 10, 24, 26, 27, 85, 91) are indicative of the production of transmissible HK2 viruses in HIV-1-infected individuals.
What pathogenic effect a modern transmissible HK2 virus may have needs to be investigated. HK2 expression, and production of HK2 particles, has been linked to cancers, in particular, teratocarcinoma, breast cancer, melanoma, and lymphoma (15, 20–23, 25). Our data have revealed that HK2 virus released from certain cell lines can be transmitted from one cell to the next; therefore, transmissible HK2 viruses might contribute to the pathogenesis of cancer. It is conceivable that with viral recombination and sequence diversification, more transmissible particles can be created. As we could not find passaged HK2 integrated into chromosomes, a mechanism of insertional mutagenesis mediated by HK2 seems unlikely to contribute to the pathogenesis of cancer. Nonetheless, transmission of HK2 viral sequences could enhance the burden of HK2 proteins, such as Rec, Np9, and Env, which have been associated with cancer development (reviewed in reference 99). The data presented here suggest that, contrary to previous views, certain modern HERVs retain the ability to be reverse transcribed and transmitted to new cells. Furthermore, the transmitted HK2 sequences circularize preferentially in the target cells, generating episomes in abundance. However, as no chromosomal integration was seen, the potential for true replication of modern HERVs with ongoing, continuous viral spread has not yet been demonstrated.
ACKNOWLEDGMENTS
We thank Joseph Zahn and Anjan K. Saha for their thoughtful insights into the manuscript and Paul Bieniasz (Rockefeller University) for providing us with the plasmids HERV-KCON CHKCP and pCR3.1/K-Rec. The plasmids pcDNA3.1-Tat86, pcDNA-HVif, and pcDNA-Vphu were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS. We thank Norbert Bannert for supplying the rabbit anti-HERV-K Gag antibody. We thank José L. García-Pérez and John M. Moran for providing the pCEP4, pBSKS, and pINT plasmids.
This work was supported by grant RO1 CA144043 from the National Institutes of Health to D.M.M., grant K22 CA177824 from the National Cancer Institute and a Research Supplement to Promote Diversity in Health-Related Research (3R01CA144043-03S1) from the National Institutes of Health to R.C.-G., and grant 05-5089 from the Concerned Parents for AIDS Research (CPFA) to M.H.K. D.D. and M.J.G.-H. were supported by a Molecular Mechanisms of Microbial Pathogenesis Training Grant from the University of Michigan (5T32AI007528-13). D.D. was also supported by an NIH Ruth L. Kirschstein NRSA Individual Postdoctoral Fellowship (1 F32 AI106189-01). M.J.G.-H. was also supported by a Rackham Merit Fellowship and by NIH Ruth L. Kirschstein NRSA Individual Predoctoral Fellowship to Promote Diversity in Health-Related Research grant 1F31CA150523-01. G.S.O. acknowledges support from NIH grants RM-08-029, P30U54ES017885, U54DA021519, and UL1RR24986.
REFERENCES
- 1.Jern P, Coffin JM. 2008. Effects of retroviruses on host genome function. Annu Rev Genet 42:709–732. doi: 10.1146/annurev.genet.42.110807.091501. [DOI] [PubMed] [Google Scholar]
- 2.Nelson PN, Carnegie PR, Martin J, Davari Ejtehadi H, Hooley P, Roden D, Rowland-Jones S, Warren P, Astley J, Murray PG. 2003. Demystified. Human endogenous retroviruses. Mol Pathol 56:11–18. doi: 10.1136/mp.56.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Subramanian RP, Wildschutte JH, Russo C, Coffin JM. 2011. Identification, characterization, and comparative genomic distribution of the HK2 group of human endogenous retroviruses. Retrovirology 8:90. doi: 10.1186/1742-4690-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbulescu M, Turner G, Seaman MI, Deinard AS, Kidd KK, Lenz J. 1999. Many human endogenous retrovirus K (HERV-K) proviruses are unique to humans. Curr Biol 9:861–868. doi: 10.1016/S0960-9822(99)80390-X. [DOI] [PubMed] [Google Scholar]
- 5.Okahara G, Matsubara S, Oda T, Sugimoto J, Jinno Y, Kanaya F. 2004. Expression analyses of human endogenous retroviruses (HERVs): tissue-specific and developmental stage-dependent expression of HERVs. Genomics 84:982–990. doi: 10.1016/j.ygeno.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Belshaw R, Pereira V, Katzourakis A, Talbot G, Paces J, Burt A, Tristem M. 2004. Long-term reinfection of the human genome by endogenous retroviruses. Proc Natl Acad Sci U S A 101:4894–4899. doi: 10.1073/pnas.0307800101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paces J, Pavlícek A, Zika R, Kapitonov VV, Jurka J, Paces V. 2004. HERVd: the Human Endogenous RetroViruses Database: update. Nucleic Acids Res 32:D50. doi: 10.1093/nar/gkh075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hughes JF, Coffin JM. 2004. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution. Proc Natl Acad Sci U S A 101:1668–1672. doi: 10.1073/pnas.0307885100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contreras-Galindo R, Kaplan MH, Contreras-Galindo AC, Gonzalez-Hernandez MJ, Ferlenghi I, Giusti F, Lorenzo E, Gitlin SD, Dosik MH, Yamamura Y, Markovitz DM. 2012. Characterization of human endogenous retroviral elements in the blood of HIV-1-infected individuals. J Virol 86:262–276. doi: 10.1128/JVI.00602-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Contreras-Galindo R, Kaplan MH, He S, Contreras-Galindo AC, Gonzalez-Hernandez MJ, Kappes F, Dube D, Chan SM, Robinson D, Meng F, Dai M, Gitlin SD, Chinnaiyan AM, Omenn GS, Markovitz DM. 2013. HIV infection reveals widespread expansion of novel centromeric human endogenous retroviruses. Genome Res 23:1505–1513. doi: 10.1101/gr.144303.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes JF, Coffin JM. 2005. Human endogenous retroviral elements as indicators of ectopic recombination events in the primate genome. Genetics 171:1183–1194. doi: 10.1534/genetics.105.043976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner G, Barbulescu M, Su M, Jensen-Seaman MI, Kidd KK, Lenz J. 2001. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr Biol 11:1531–1535. doi: 10.1016/S0960-9822(01)00455-9. [DOI] [PubMed] [Google Scholar]
- 13.Johnston JB, Silva C, Holden J, Warren KG, Clark AW, Power C. 2001. Monocyte activation and differentiation augment human endogenous retrovirus expression: implications for inflammatory brain diseases. Ann Neurol 50:434–442. doi: 10.1002/ana.1131. [DOI] [PubMed] [Google Scholar]
- 14.Ruda VM, Akopov SB, Trubetskoy DO, Manuylov NL, Vetchinova AS, Zavalova LL, Nikolaev LG, Sverdlov ED. 2004. Tissue specificity of enhancer and promoter activities of a HERV-K(HML-2) LTR. Virus Res 104:11–16. doi: 10.1016/j.virusres.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 15.Seifarth W, Baust C, Murr A, Skladny H, Krieg-Schneider F, Blusch J, Werner T, Hehlmann R, Leib-Mösch C. 1998. Proviral structure, chromosomal location, and expression of HERV-K-T47D, a novel human endogenous retrovirus derived from T47D particles. J Virol 72:8384–8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugimoto J, Matsuura N, Kinjo Y, Takasu N, Oda T, Jinno Y. 2001. Transcriptionally active HERV-K genes: identification, isolation, and chromosomal mapping. Genomics 72:137–144. doi: 10.1006/geno.2001.6473. [DOI] [PubMed] [Google Scholar]
- 17.Tönjes RR, Löwer R, Boller K, Denner J, Hasenmaier B, Kirsch H, König H, Korbmacher C, Limbach C, Lugert R, Phelps RC, Scherer J, Thelen K, Löwer J, Kurth R. 1996. HERV-K: the biologically most active human endogenous retrovirus family. J Acquir Immune Defic Syndr Hum Retrovirol 1:S261–S267. [DOI] [PubMed] [Google Scholar]
- 18.Wang-Johanning F, Frost AR, Johanning GL, Khazaeli MB, LoBuglio AF, Shaw DR, Strong TV. 2001. Expression of human endogenous retrovirus K envelope transcripts in human breast cancer. Clin Cancer Res 7:1553–1560. [PubMed] [Google Scholar]
- 19.Yi JM, Kim HM, Kim HS. 2001. Molecular cloning and phylogenetic analysis of the human endogenous retrovirus HERV-K long terminal repeat elements in various cancer cells. Mol Cells 12:137–141. [PubMed] [Google Scholar]
- 20.Bieda K, Hoffmann A, Boller K. 2001. Phenotypic heterogeneity of human endogenous retrovirus particles produced by teratocarcinoma cell lines. J Gen Virol 82:591–596. [DOI] [PubMed] [Google Scholar]
- 21.Büscher K, Trefzer U, Hofmann M, Sterry W, Kurth R, Denner J. 2005. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res 65:4172–4180. doi: 10.1158/0008-5472.CAN-04-2983. [DOI] [PubMed] [Google Scholar]
- 22.Löwer R, Boller K, Hasenmaier B, Korbmacher C, Müller-Lantzsch N, Löwer J, Kurth R. 1993. Identification of human endogenous retroviruses with complex mRNA expression and particle formation. Proc Natl Acad Sci U S A 90:4480–4484. doi: 10.1073/pnas.90.10.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muster T, Waltenberger A, Grassauer A, Hirschl S, Caucig P, Romirer I, Födinger D, Seppele H, Schanab O, Magin-Lachmann C, Löwer R, Jansen B, Pehamberger H, Wolff K. 2003. An endogenous retrovirus derived from human melanoma cells. Cancer Res 63:8735–8741. [PubMed] [Google Scholar]
- 24.Contreras-Galindo R, Kaplan MH, Markovitz DM, Lorenzo E, Yamamura Y. 2006. Detection of HERV-K(HML-2) viral RNA in plasma of HIV type 1-infected individuals. AIDS Res Hum Retrovir 22:979–984. doi: 10.1089/aid.2006.22.979. [DOI] [PubMed] [Google Scholar]
- 25.Contreras-Galindo R, Kaplan MH, Leissner P, Verjat T, Ferlenghi I, Bagnoli F, Giusti F, Dosik MH, Hayes DF, Gitlin SD, Markovitz DM. 2008. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J Virol 82:9329–9336. doi: 10.1128/JVI.00646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Contreras-Galindo R, Almodóvar-Camacho S, González-Ramírez S, Lorenzo E, Yamamura Y. 2007. Comparative longitudinal studies of HERV-K and HIV-1 RNA titers in HIV-1-infected patients receiving successful versus unsuccessful highly active antiretroviral therapy. AIDS Res Hum Retrovir 23:1083–1086. doi: 10.1089/aid.2007.0054. [DOI] [PubMed] [Google Scholar]
- 27.Contreras-Galindo R, López P, Vélez R, Yamamura Y. 2007. HIV-1 infection increases the expression of human endogenous retroviruses type K (HERV-K) in vitro. AIDS Res Hum Retrovir 23:116–122. doi: 10.1089/aid.2006.0117. [DOI] [PubMed] [Google Scholar]
- 28.Mayer J, Sauter M, Racz A, Scherer D, Mueller-Lantzsch N, Meese E. 1999. An almost-intact human endogenous retrovirus K on human chromosome 7. Nat Genet 21:257–258. doi: 10.1038/6766. [DOI] [PubMed] [Google Scholar]
- 29.Ono M, Yasunaga T, Miyata T, Ushikubo H. 1986. Nucleotide sequence of human endogenous retrovirus genome related to the mouse mammary tumor virus genome. J Virol 60:589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ono M. 1986. Molecular cloning and long terminal repeat sequences of human endogenous retrovirus genes related to types A and B retrovirus genes. J Virol 58:937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayer J, Meese E, Mueller-Lantzsch N. 1997. Multiple human endogenous retrovirus (HERV-K) loci with gag open reading frames in the human genome. Cytogenet Cell Genet 78:1–5. [DOI] [PubMed] [Google Scholar]
- 32.Ruprecht K, Ferreira H, Flockerzi A, Wahl S, Sauter M, Mayer J, Mueller-Lantzsch N. 2008. Human endogenous retrovirus family HERV-K(HML-2) RNA transcripts are selectively packaged into retroviral particles produced by the human germ cell tumor line Tera-1 and originate mainly from a provirus on chromosome 22q11.21. J Virol 82:10008–10016. doi: 10.1128/JVI.01016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tönjes RR, Czauderna F, Kurth R. 1999. Genome-wide screening, cloning, chromosomal assignment, and expression of full-length human endogenous retrovirus type K. J Virol 73:9187–9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heslin DJ, Murcia P, Arnaud F, Van Doorslaer K, Palmarini M, Lenz J. 2009. A single amino acid substitution in a segment of the CA protein within Gag that has similarity to human immunodeficiency virus type 1 blocks infectivity of a human endogenous retrovirus K provirus in the human genome. J Virol 83:1105–1114. doi: 10.1128/JVI.01439-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraus B, Boller K, Reuter A, Schnierle BS. 2011. Characterization of the human endogenous retrovirus K Gag protein: identification of protease cleavage sites. Retrovirology 8:21. doi: 10.1186/1742-4690-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.George M, Schwecke T, Beimforde N, Hohn O, Chudak C, Zimmermann A, Kurth R, Naumann D, Bannert N. 2011. Identification of the protease cleavage sites in a reconstituted Gag polyprotein of an HERV-K(HML-2) element. Retrovirology 8:30. doi: 10.1186/1742-4690-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhelj R, Rizzo CJ, Chang CH, Jadhav PK, Towler EM, Korant BD. 2001. Inhibition of human endogenous retrovirus-K10 protease in cell-free and cell-based assays. J Biol Chem 276:16674–16682. doi: 10.1074/jbc.M008763200. [DOI] [PubMed] [Google Scholar]
- 38.Boyd MT, Foley B, Brodsky I. 1997. Evidence for copurification of HERV-K-related transcripts and a reverse transcriptase activity in human platelets from patients with essential thrombocythemia. Blood 90:4022–4030. [PubMed] [Google Scholar]
- 39.Tönjes RR, Boller K, Limbach C, Lugert R, Kurth R. 1997. Characterization of human endogenous retrovirus type K virus-like particles generated from recombinant baculoviruses. Virology 233:280–291. doi: 10.1006/viro.1997.8614. [DOI] [PubMed] [Google Scholar]
- 40.Berkhout B, Jebbink M, Zsíros J. 1999. Identification of an active reverse transcriptase enzyme encoded by a human endogenous HERV-K retrovirus. J Virol 73:2365–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golan M, Hizi A, Resau JH, Yaal-Hahoshen N, Reichman H, Keydar I, Tsarfaty I. 2008. Human endogenous retrovirus (HERV-K) reverse transcriptase as a breast cancer prognostic marker. Neoplasia 10:521–533. doi: 10.1593/neo.07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kitamura Y, Ayukawa T, Ishikawa T, Kanda T, Yoshiike K. 1996. Human endogenous retrovirus K10 encodes a functional integrase. J Virol 70:3302–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magin C, Löwer R, Löwer J. 1999. cORF and RcRE, the Rev/Rex and RRE/RxRE homologues of the human endogenous retrovirus family HTDV/HERV-K. J Virol 73:9496–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J, Bogerd HP, Peng S, Wiegand H, Truant R, Cullen BR. 1999. An ancient family of human endogenous retroviruses encodes a functional homolog of the HIV-1 Rev protein. Proc Natl Acad Sci U S A 96:13404–13408. doi: 10.1073/pnas.96.23.13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dewannieux M, Blaise S, Heidmann T. 2005. Identification of a functional envelope protein from the HERV-K family of human endogenous retroviruses. J Virol 79:15573–15577. doi: 10.1128/JVI.79.24.15573-15577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belshaw R, Katzourakis A, Paces J, Burt A, Tristem M. 2005. High copy number in human endogenous retrovirus families is associated with copying mechanisms in addition to reinfection. Mol Biol Evol 22:814–817. doi: 10.1093/molbev/msi088. [DOI] [PubMed] [Google Scholar]
- 47.Stoye JP. 2001. Endogenous retroviruses: still active after all these years? Curr Biol 11:R914–R916. doi: 10.1016/S0960-9822(01)00553-X. [DOI] [PubMed] [Google Scholar]
- 48.Magiorkinis G, Belshaw R, Katzourakis A. 2013. “There and back again”: revisiting the pathophysiological roles of human endogenous retroviruses in the post-genomic era. Philos Trans R Soc Lond B Biol Sci 368:20120504. doi: 10.1098/rstb.2012.0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ribet D, Dewannieux M, Heidmann T. 2004. An active murine transposon family pair: retrotransposition of “master” MusD copies and ETn trans-mobilization. Genome Res 14:2261–2267. doi: 10.1101/gr.2924904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans LH, Alamgir AS, Owens N, Weber N, Virtaneva K, Barbian K, Babar A, Malik F, Rosenke K. 2009. Mobilization of endogenous retroviruses in mice after infection with an exogenous retrovirus. J Virol 83:2429–2435. doi: 10.1128/JVI.01926-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Young GR, Eksmond U, Salcedo R, Alexopoulou L, Stoye JP, Kassiotis G. 2012. Resurrection of endogenous retroviruses in antibody-deficient mice. Nature 491:774–778. doi: 10.1038/nature11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee YN, Bieniasz PD. 2007. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog 3:e10. doi: 10.1371/journal.ppat.0030010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dewannieux M, Harper F, Richaud A, Letzelter C, Ribet D, Pierron G, Heidmann T. 2006. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res 16:1548–1556. doi: 10.1101/gr.5565706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dube D, Contreras-Galindo R, He S, King SR, Gonzalez-Hernandez MJ, Gitlin SD, Kaplan MH, Markovitz DM. 2014. Genomic flexibility of human endogenous retrovirus type K. J Virol 88:9673–9682. doi: 10.1128/JVI.01147-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH Jr. 1996. High frequency retrotransposition in cultured mammalian cells. Cell 87:917–927. doi: 10.1016/S0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- 56.Moran JV, DeBerardinis RJ, Kazazian HH Jr. 1999. Exon shuffling by L1 retrotransposition. Science 283:1530–1534. doi: 10.1126/science.283.5407.1530. [DOI] [PubMed] [Google Scholar]
- 57.Gilbert N, Lutz-Prigge S, Moran JV. 2002. Genomic deletions created upon LINE-1 retrotransposition. Cell 110:315–325. doi: 10.1016/S0092-8674(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 58.Garcia-Perez JL, Marchetto MC, Muotri AR, Coufal NG, Gage FH, O'Shea KS, Moran JV. 2007. LINE-1 retrotransposition in human embryonic stem cells. Hum Mol Genet 16:1569–1577. doi: 10.1093/hmg/ddm105. [DOI] [PubMed] [Google Scholar]
- 59.Brady T, Lee YN, Ronen K, Malani N, Berry CC, Bieniasz PD, Bushman FD. 2009. Integration target site selection by a resurrected human endogenous retrovirus. Genes Dev 23:633–642. doi: 10.1101/gad.1762309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O'Shea KS, Moran JV, Gage FH. 2009. LINE-1 retrotransposition in human neural progenitor cells. Nature 460:1127–1131. doi: 10.1038/nature08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nelson DL, Ledbetter SA, Corbo L, Victoria MF, Ramírez-Solis R, Webster TD, Ledbetter DH, Caskey CT. 1989. Alu polymerase chain reaction: a method for rapid isolation of human-specific sequences from complex DNA sources. Proc Natl Acad Sci U S A 86:6686–6690. doi: 10.1073/pnas.86.17.6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gentile A, D'Alessandro L, Lazzari L, Martinoglio B, Bertotti A, Mira A, Lanzetti L, Comoglio PM, Medico E. 2008. Met-driven invasive growth involves transcriptional regulation of Arhgap12. Oncogene 27:5590–5598. doi: 10.1038/onc.2008.173. [DOI] [PubMed] [Google Scholar]
- 63.Badge RM, Alisch RS, Moran JV. 2003. ATLAS: a system to selectively identify human-specific L1 insertions. Am J Hum Genet 72:823–838. doi: 10.1086/373939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- 65.Hills D, Bull J. 1993. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst Biol 42:182–192. doi: 10.1093/sysbio/42.2.182. [DOI] [Google Scholar]
- 66.Etherington GJ, Dicks J, Roberts IN. 2005. Recombination Analysis Tool (RAT): a program for the high-throughput detection of recombination. Bioinformatics 21:278–281. doi: 10.1093/bioinformatics/bth500. [DOI] [PubMed] [Google Scholar]
- 67.Lavie L, Kitova M, Maldener E, Meese E, Mayer J. 2005. CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERV-K(HML-2). J Virol 79:876–883. doi: 10.1128/JVI.79.2.876-883.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Flockerzi A, Maydt J, Frank O, Ruggieri A, Maldener E, Seifarth W, Medstrand P, Lengauer T, Meyerhans A, Leib-Mösch C, Meese E, Mayer J. 2007. Expression pattern analysis of transcribed HERV sequences is complicated by ex vivo recombination. Retrovirology 4:39. doi: 10.1186/1742-4690-4-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adkins B, Hunter T. 1981. Identification of a packaged cellular mRNA in virions of Rous sarcoma virus. J Virol 39:471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aronoff R, Linial M. 1991. Specificity of retroviral RNA packaging. J Virol 65:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berkowitz R, Fisher J, Goff SP. 1996. RNA packaging. Curr Top Microbiol Immunol 214:177–218. [DOI] [PubMed] [Google Scholar]
- 72.Onafuwa-Nuga AA, King SR, Telesnitsky A. 2005. Nonrandom packaging of host RNAs in Moloney murine leukemia virus. J Virol 79:13528–13537. doi: 10.1128/JVI.79.21.13528-13537.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Onafuwa-Nuga AA, Telesnitsky A, King SR. 2006. 7SL RNA, but not the 54-kd signal recognition particle protein, is an abundant component of both infectious HIV-1 and minimal virus-like particles. RNA 12:542–546. doi: 10.1261/rna.2306306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Muriaux D, Mirro J, Harvin D, Rein A. 2001. RNA is a structural element in retrovirus particles. Proc Natl Acad Sci U S A 98:5246–5251. doi: 10.1073/pnas.091000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jiang M, Mak J, Ladha A, Cohen E, Klein M, Rovinski B, Kleiman L. 1993. Identification of tRNAs incorporated into wild-type and mutant human immunodeficiency virus type 1. J Virol 67:3246–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rulli SJ Jr, Hibbert CS, Mirro J, Pederson T, Biswal S, Rein A. 2007. Selective and nonselective packaging of cellular RNAs in retrovirus particles. J Virol 81:6623–6631. doi: 10.1128/JVI.02833-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.D'Souza V, Summers MF. 2005. How retroviruses select their genomes. Nat Rev Microbiol 3:643–655. doi: 10.1038/nrmicro1210. [DOI] [PubMed] [Google Scholar]
- 78.Jouvenet N, Lainé S, Pessel-Vivares L, Mougel M. 2011. Cell biology of retroviral RNA packaging. RNA Biol 8:572–580. doi: 10.4161/rna.8.4.16030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rosenblum LL, Patton G, Grigg AR, Frater AJ, Cain D, Erlwein O, Hill CL, Clarke JR, McClure MO. 2001. Differential susceptibility of retroviruses to nucleoside analogues. Antivir Chem Chemother 12:91–97. doi: 10.1177/095632020101200202. [DOI] [PubMed] [Google Scholar]
- 80.Coffin JM, Hughes SH, Varmus HE (ed). 1997. Retroviruses. Overview of Reverse Transcription. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 81.Sharkey ME, Stevenson M. 2001. Two long terminal repeat circles and persistent HIV-1 replication. Curr Opin Infect Dis 14:5–11. doi: 10.1097/00001432-200102000-00002. [DOI] [PubMed] [Google Scholar]
- 82.Pierson TC, Kieffer TL, Ruff CT, Buck C, Gange SJ, Siliciano RF. 2002. Intrinsic stability of episomal circles formed during human immunodeficiency virus type 1 replication. J Virol 76:4138–4144. doi: 10.1128/JVI.76.8.4138-4144.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sloan RD, Wainberg MA. 2011. The role of unintegrated DNA in HIV infection. Retrovirology 8:52. doi: 10.1186/1742-4690-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ejthadi HD, Martin JH, Junying J, Roden DA, Lahiri M, Warren P, Murray PG, Nelson PN. 2005. A novel multiplex RT-PCR system detects human endogenous retrovirus-K in breast cancer. Arch Virol 150:177–184. doi: 10.1007/s00705-004-0378-8. [DOI] [PubMed] [Google Scholar]
- 85.Gonzalez-Hernandez MJ, Swanson MD, Contreras-Galindo R, Cookinham S, King SR, Noel RJ Jr, Kaplan MH, Markovitz DM. 2012. Expression of human endogenous retrovirus type K (HML-2) is activated by the Tat protein of HIV-1. J Virol 86:7790–7805. doi: 10.1128/JVI.07215-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gonzalez-Hernandez MJ, Cavalcoli JD, Sartor MA, Contreras-Galindo R, Meng F, Dai M, Dube D, Saha AK, Gitlin SD, Omenn GS, Kaplan MH, Markovitz DM. 2014. Regulation of the human endogenous retrovirus K (HML-2) transcriptome by the HIV-1 Tat protein. J Virol 88:8924–8935. doi: 10.1128/JVI.00556-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lokossou AG, Toudic C, Barbeau B. 2014. Implication of human endogenous retrovirus envelope proteins in placental functions. Viruses 6:4609–4627. doi: 10.3390/v6114609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 89.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809. doi: 10.1016/S0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 90.Lecossier D, Bouchonnet F, Clavel F, Hance AJ. 2003. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 91.Armitage AE, Katzourakis A, de Oliveira T, Welch JJ, Belshaw R, Bishop KN, Kramer B, McMichael AJ, Rambaut A, Iversen AK. 2008. Conserved footprints of APOBEC3G on hypermutated human immunodeficiency virus type 1 and human endogenous retrovirus HERV-K(HML2) sequences. J Virol 82:8743–8761. doi: 10.1128/JVI.00584-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bhardwaj N, Maldarelli F, Mellors J, Coffin JM. 2014. HIV-1 infection leads to increased transcription of human endogenous retrovirus HERV-K (HML-2) proviruses in vivo but not to increased virion production. J Virol 88:11108–11120. doi: 10.1128/JVI.01623-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Garrison KE, Jones RB, Meiklejohn DA, Anwar N, Ndhlovu LC, Chapman JM, Erickson AL, Agrawal A, Spotts G, Hecht FM, Rakoff-Nahoum S, Lenz J, Ostrowski MA, Nixon DF. 2007. T cell responses to human endogenous retroviruses in HIV-1 infection. PLoS Pathog 3:e165. doi: 10.1371/journal.ppat.0030165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tandon R, SenGupta D, Ndhlovu LC, Vieira RG, Jones RB, York VA, Vieira VA, Sharp ER, Wiznia AA, Ostrowski MA, Rosenberg MG, Nixon DF. 2011. Identification of human endogenous retrovirus (HERV)-specific T cell responses in vertically HIV-1-infected subjects. J Virol 85:11526–11531. doi: 10.1128/JVI.05418-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jones RB, Garrison KE, Mujib S, Mihajlovic V, Aidarus N, Hunter DV, Martin E, John VM, Zhan W, Faruk NF, Gyenes G, Sheppard NC, Priumboom-Brees IM, Goodwin DA, Chen L, Rieger M, Muscat-King S, Loudon PT, Stanley C, Holditch SJ, Wong JC, Clayton K, Duan E, Song H, Xu Y, SenGupta D, Tandon R, Sacha JB, Brockman MA, Benko E, Kovacs C, Nixon DF, Ostrowski MA. 2012. HERV-K-specific T cells eliminate diverse HIV-1/2 and SIV primary isolates. J Clin Investig 122:4473–4489. doi: 10.1172/JCI64560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chittenden T, Frey A, Levine AJ. 1991. Regulated replication of an episomal simian virus 40 origin plasmid in COS7 cells. J Virol 65:5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Piechaczek C, Fetzer C, Baiker A, Bode J, Lipps HJ. 1999. A vector based on the SV40 origin of replication and chromosomal S/MARs replicates episomally in CHO cells. Nucleic Acids Res 27:426–428. doi: 10.1093/nar/27.2.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Glover DJ, Lipps HJ, Jans DA. 2005. Towards safe, non-viral therapeutic gene expression in humans. Nat Rev Genet 6:299–310. doi: 10.1038/nrg1577. [DOI] [PubMed] [Google Scholar]
- 99.Hohn O, Hanke K, Bannert N. 2013. HERV-K(HML-2), the best preserved family of HERVs: endogenization, expression, and implications in health and disease. Front Oncol 3:246. doi: 10.3389/fonc.2013.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brinzevich D, Young GR, Sebra R, Ayllon J, Maio SM, Deikus G, Chen BK, Fernandez-Sesma A, Simon V, Mulder LC. 2014. HIV-1 interacts with human endogenous retrovirus K (HML-2) envelopes derived from human primary lymphocytes. J Virol 88:6213–6223. doi: 10.1128/JVI.00669-14. [DOI] [PMC free article] [PubMed] [Google Scholar]