

ABSTRACT
Cytokines are a group of small secreted proteins that mediate a diverse range of immune and nonimmune responses to inflammatory and microbial stimuli. Only a few of these cytokines mount an antiviral response, including type I, II, and III interferons (IFNs). During viral infections and under inflammatory conditions, a number of cytokines and chemokines are coproduced with IFN; however, no systematic study exists on the interactions of the cytokine repertoire with the IFN response. Here, we performed the largest cytokine and chemokine screen (the human cytokinome, with >240 members) to investigate their modulation of type I and type II IFN responses in a cell line model. We evaluated the cytokine activities in both IFN-stimulated response element (ISRE) and IFN-γ activation sequence (GAS) reporter systems. Several cytokine clusters that augment either or both ISRE- and GAS-mediated responses to IFNs were derived from the screen. We identified novel modulators of IFN response—betacellulin (BTC), interleukin 11 (IL-11), and IL-17F—that caused time-dependent induction of the IFN response. The ability to induce endogenous IFN-β and IFN-stimulated genes varies among these cytokines and was largely dependent on Stat1, as assessed by Stat1 mutant fibroblasts. Certain cytokines appear to augment the IFN-β response through the NF-κB pathway. The novel IFN-like cytokines augmented the antiviral activity of IFN-α against several RNA viruses, including encephalomyocarditis virus, vesicular stomatitis virus, and influenza virus, in susceptible cell lines. Overall, the study represents a large-scale analysis of cytokines for enhancing the IFN response and identified cytokines capable of enhancing Stat1, IFN-induced gene expression, and antiviral activities.
IMPORTANCE Innate immunity to viruses is an early defense system to ward off viruses. One mediator is interferon (IFN), which activates a cascade of biochemical events that aim to control the virus life cycle. In our work, we examined more than 200 cytokines, soluble mediators produced within the body as a result of infection, for the ability to enhance IFN action. We identified enhanced interactions with specific IFNs and cytokines. We also revealed that betacellulin, IL-17, and IL-11 cytokines have the novel property of enhancing the antiviral action of IFN against several viruses. These results demonstrate that the human genome codes for previously unknown proteins with unrelated functions that can augment the innate immunity to viruses. Knowing these interactions not only helps our understanding of immunity to viruses and emerging diseases, but can also lead to devising possible new therapeutics by enhancing the mediator of antiviral action itself, IFN.
INTRODUCTION
The interferon (IFN) response plays an important role in innate immunity to viruses (1, 2). Many cell types produce IFN in response to a viral invasion, and IFN acts on neighboring healthy cells to ward off further viral attack. IFN stimulates the expression of a multitude of genes (IFN-stimulated genes [ISGs]) that collectively participate in the control of the virus life cycle (3–7). During viral infections, many cytokines and chemokines are coproduced and secreted in tissues and into the circulation, but only a few mediators have been investigated for their effects on the IFN response.
Type I IFN, which includes the multiple IFN-α subtypes, IFN-β, IFN-κ, IFN-ω, and IFN-ε, binds to the IFNAR1/IFNAR2 receptor complex, while IFN-γ (also called immune IFN) binds to the IFNGR1/IFNGR2 complex (reviewed in references 8 and 9). Type III IFN (interleukin 28A [IL-28A]/IFN-λ2/IFNL2, IL-28B/IFN-λ3/IFNL3, and IL-29/IFN-λ1/IFNL1) comprises distantly related proteins of the IL-10 cytokine family and binds a different receptor complex, IFN-λR1/IL10R2 (10, 11), in which IL10R2 is common to other IL-10 members (12). This heterogeneous group of IFNs shares signaling via the JAK/Stat pathway (e.g., Stat1 phosphorylation), although each type has distinct regulatory components. Type I and type III IFNs activate similar signaling pathways but with different kinetics that lead to IFN-stimulated response element (ISRE)-mediated transcription (13). IFN-γ leads to IFN-γ activation sequence (GAS)-mediated transcription (14).
Despite the fact that a plethora of cytokines are coproduced with IFN during viral infections, there is no study that investigates the cytokinome effect on the IFN response. As “ome” refers to an entire set of objects, e.g., kinome, the cytokinome is also defined as the totality of these proteins and their interactions in and around cells (15). Using an ISRE/GAS reporter system in human hepatoma Huh7 cells, a well-established model of IFN biology, we performed a cytokinome screen targeting 244 cytokines and analyzed cytokine synergy in the IFN response. We identified positive regulators and interactions of the cytokines with the IFN response. We pinpointed those that affect the IFN response either directly or in combination with IFN, which included both known and unknown mediators. Finally, we show that the cytokines betacellulin (BTC) and IL-17 possess novel characteristics in that they augment the antiviral action of IFN.
MATERIALS AND METHODS
Cell culture, small interfering RNAs (siRNAs), cytokines, and reagents.
U3A (a Stat1-deficient cell line) generated from HT1080 was obtained from George Stark (Cleveland Clinic Foundation, Cleveland, OH). The Huh-7 cell line was obtained from the JCRB Cell Bank, Japan. The human epithelial amnion cell line WISH (HeLa markers) was obtained from the ATCC. Huh-7 and U3A cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics. WISH cells were maintained in minimal essential medium (MEM) (Invitrogen) supplemented with 10% FBS and antibiotics. The recombinant human IFN-α2a (rIFN-α2a) (Roferon; Hoffman-LaRoche, Basel, Switzerland) used had a specific activity of 2 × 108 IU/mg. Recombinant human IFN-γ (1 × 107 U/ml) was obtained from R&D Systems. The cytokines were all human recombinant forms obtained from Peprotech and R&D Systems and were stored as a stock solution at −20°C in 0.1% bovine serum albumin (BSA)-phosphate-buffered saline (PBS). Based on the manufacturers' information sheets, all the cytokines were endotoxin free. The NF-κB inhibitor is (E)-2-fluoro-4ʹ-methoxystilbene (Calbiochem; San Diego, CA).
Plasmids, IFN response, and NF-κB reporter assay.
The constructs utilized expression cassettes that contained a minimal cytomegalovirus (CMV) promoter linked to a strongly expressed TT/TA-reduced enhanced green fluorescent protein (EGFP) coding region (16) and a stable 3′ untranslated region (UTR) (17). The ISRE expression cassette contained the sequence GATTCTGTTTCAGTTTCCCCTCAGTTTCACTTTCCTTTCCCCTTTCAGCAGTTTCACTTTCCTTTCCCCTTTCC and was cloned into the expression vector using SalI and EcoRV. The GAS reporter construct contained the promoter sequence AGCCTGATTTCCCCGAAATGACGGCAGCCTGATTTCCCCGAAATGACGGCC. The minimal IFNB1 promoter sequence is GTAAATGACATAGGAAAACTGAAAGGGAGAAGTGAAAGTGGGAAATTCCTCTGAATAGAGAGAGGACCATCTCATATAAAT. We previously described the ISRE and mutant ISRE constructs (17). The NF-κB response element-linked reporter was constructed as previously described (17); the NF-κB element sequence is as follows: CCAAGGGAAACCCCAGGGAAGTACCGGGAAGGACTTTCCAGTTCTGGAAATTCCCA GAAATCCCCGGTAGGCGTGTACGGTG. Transfection of these constructs was performed as follows. Huh-7 cells were seeded in a 96-well microplate at a density of 5 ×105 cells/ml (0.1 ml/well) and incubated for 6 h. The cells were transfected with 50 ng/well of either ISRE or GAS reporters. After 24 h of incubation, the cells were treated with 10 IU/ml IFN-α2a or 1 nM the recombinant cytokines or their combinations, unless otherwise indicated. The fluorescence intensity was measured using a high-throughput BD-Pathway 435 imager (BD Biosciences, San Jose, CA). Automated image acquisition and quantification were performed using the ProXcell algorithm (18). Transfection efficiency was monitored by fluorescence before the addition of cytokines, and the coefficient of variation was <10% among replicate wells. The final results are presented as normalized fold changes. All data were collected at least in triplicate, and the experiments were independently repeated at least twice.
Reverse transcription-quantitative PCR.
Huh-7 cells were seeded in 6-well plates at a density of 3 × 105 cells/ml (0.1 ml/well). After 24 h of incubation, the media were replaced with fresh media, the selected cytokines, and/or 10 IU/ml of IFN-α2a. To assess the role of the NF-κB pathway, Huh-7 cells were treated with 10 μM NF-κB small-molecule inhibitor (Calbiochem) or dimethyl sulfoxide (DMSO) for 1 h before adding 1 nM cytokine and were incubated for a further 6 h. Total RNA was then extracted and subjected to reverse transcription (RT) to generate cDNA. TaqMan expression quantitative PCR (qPCR) was then performed using a TaqMan primer pair/hybridization probe (Applied Biosystems) with a 6-carboxyfluorescein (FAM) probe specific for human IFIT3, IFN-β, and OAS1 and normalized using a GAPDH (glyceraldehyde-3-phosphate dehydrogenase) TaqMan primer (as a VIC probe). For quantitation of viral RNA replication, EvaGreen Real-Time PCR was performed using 5× EvaGreen master mix (Al-Bio, Riyadh, Saudi Arabia) with a primer pair specific for vesicular stomatitis virus (VSV) rhabdovirus nucleocapsid protein (P) (forward, CGCCAGAGGGTTTAAGTGGA, and reverse, TATTTGACTCTCGCCTGATTGT) and primers specific for the influenza virus H1N1 strain Puerto Rico/8/34 nonstructural protein NS1 (forward, TAAGGGCTTTCACCGAAGAG, and reverse, TTACTGCTTCTCCAAGCGAA). Both reactions were normalized using the endogenous human GAPDH primers (forward, CACCATCTTCCAGGAGCGAG, and reverse, TCACGCCACAGTTTCCCGGA). The final results were converted to ratios ± standard errors of the mean (SEM) of the gene-specific mRNA levels to housekeeping gene mRNA levels. The qPCR was performed in multiplex with Chromo4 DNA engine cycler (Bio-Rad).
Western blotting.
Western blotting was used to assess the effects of the selected panel of cytokines on IFN-stimulated genes. Huh-7 cells were plated in 6-well plates at a density of 6 × 105 cells/well and incubated for 24 h. The cells were treated with 1 nM cytokines with or without 10 IU/ml of IFN-α2a and incubated for an additional 24 h. An equal amount of protein samples (50 μg) was resolved into sodium dodecyl sulfate-10% polyacrylamide gels (NuPAGE; Invitrogen), followed by gel transfer to nitrocellulose membranes (Hybond ECL; Amersham Biosciences, United Kingdom). The membranes were blotted with primary mouse monoclonal anti-human OAS1 antibody (1/500) or rabbit polyclonal anti-human IFIT3 antibody (1/500) (Santa Cruz Biotechnology, CA), followed by secondary horseradish peroxidase (HRP)-conjugated antibody. The blots were reprobed with an anti-human β-actin antibody as a control. Protein signals were detected using enhanced chemiluminescence (ECL)-Western blotting detection reagents (Amersham, NJ). Protein molecular weight markers were included for each blot.
Viruses and CPE assays.
Encephalomyocarditis virus (EMCV), VSV (strain Indiana), human respiratory syncytial virus (HRSV), and human influenza virus (H1N1 strain A/Puerto Rico/8/34) were obtained from the ATCC. Virus preparations were propagated and titrated in their appropriate host cells (18)—Vero cells (African green monkey kidney cell line; ATCC) for EMCV and VSV, HEp-2 cells (human epidermoid carcinoma cell line; ATCC) for HRSV, and embryonated eggs for H1N1—followed by clarification and filtration through 0.22-μm membranes for sterilization. The titers of the viruses were as follows: EMCV, 1.5 × 108 PFU/ml; VSV, 8 × 108 PFU/ml; RSV, 107 50% tissue culture infective doses (TCID50)/ml; and H1N1, 5,120 hemagglutinin (HA) units/ml. The virus stocks were aliquoted and stored at −70°C until use. The crystal violet stain assay was used to assess the virus-induced cytopathic effect (CPE) and to establish correlation with the real-time label-free electronic biosensor system (see below). Cells were seeded in 96-well plates at a density of 3 × 104 cells per well for 16 h. Then, the cells were replaced with appropriate concentrations of the cytokines alone or in combination with IFN-α prepared in 2% FBS medium. After overnight incubation, the medium was replaced with 0.1% BSA containing MEM and an appropriate virus dilution. Depending on the virus, there was a 16- to 40-h incubation, after which significant CPE was observed (∼80 to 90%), and the cells were fixed with 5% formalin-0.9% saline solution. The cells were then stained with 0.5% crystal violet (Fisher, Inc., NJ) solution, washed extensively, and air dried.
Label-free continuous monitoring of virus-induced changes.
A label-free cell-based assay (ACEA Biosystems, San Diego, CA) was employed to monitor virus-induced cellular changes in real time (19, 20). Cells were seeded in electrode strip-coated 96-well electrode-integrated microplates at a density of 3 × 105 cells per well. After overnight incubation, the cytokine-IFN combinations were added, and the cultures were further incubated for 16 h. Viruses were added at the appropriate dilution in reduced-serum MEM, and the CPE was continuously monitored at 15-min intervals for 48 h. The cell index (CI) parameter was derived to represent the cell status based on the change in the electrode impedance due to cell growth and attachment relative to that of background, i.e., without cells. The percent CPE inhibition was calculated using the following formula: [(experiment CI − virus control CI)/(cell control CI − virus control CI)] × 100. Both the percent inhibition and the time to CPE could be deduced continuously at any time point.
Data analysis.
For screening studies, fluorescence levels normalized to the control were converted to fold ratios and then used for centroid clustering using the JMP SAS program (SAS Institute, Cary, NC). The centroid method is a form of hierarchical clustering based on the distance between two clusters as the squared Euclidean distance between their means. Heat maps were also generated with JMP SAS. Student's t test was used to determine the significance of the difference when comparing two columns of data. One-way and two-way analysis of variance (ANOVA) was used in cases of multiple-column comparisons of one or two factors, respectively.
RESULTS
Cytokinome screen and IFN response.
Cytokines and chemokines are coproduced during viral infections and inflammatory diseases. Therefore, we first evaluated the abilities of 244 of these mediators representing almost the entire cytokine list (Gene Ontology) and Kyoto Encyclopedia of Genes and Genomes (KEGG) cytokine activity pathway (see Table S1 in the supplemental material). We used the human hepatoma cell line Huh-7 because it responds to all types of IFNs (i.e., types I, II, and III) and it expresses receptors for a multitude of cytokines and chemokines (21–25). It is also highly sensitive to IFN when using an IFN response reporter system (17, 26). We utilized ISRE/GAS response reporter cell-based assays, which respond to all types of IFNs, and generated large-scale data (see Table S2 in the supplemental material). A centroid clustering of fold changes in ISRE/GAS reporter fluorescence due to the combination of each cytokine with IFN-α (10 IU/ml) was generated. The gene cluster of cytokines that possessed the most upregulated reporter activity (Fig. 1) was also studied. This cytokine gene cluster included known IFN and IFN-like cytokines, in addition to a few non-IFN cytokines (Fig. 1). These cytokines boosted the IFN response from nearly 2-fold to 6-fold. The greatest ability to augment IFN-α-induced ISRE/GAS activity occurred with IFN-γ, IFN-λ1/IFNL1, and IFN-β (nearly 6-fold each; P < 0.001; n = 4). Both TNF-α (TNF) and TNF-β (LTA) also enhanced the IFN-α response. Oncostatin M (OSM) and cytokines not previously known for their ability to elicit an IFN response, namely, BTC, IL-17F, IL-11, and neurotrophins 3 and 4 (NTF3/4), were able to augment IFN-α and induced further ISRE/GAS activity (Fig. 1).
FIG 1.
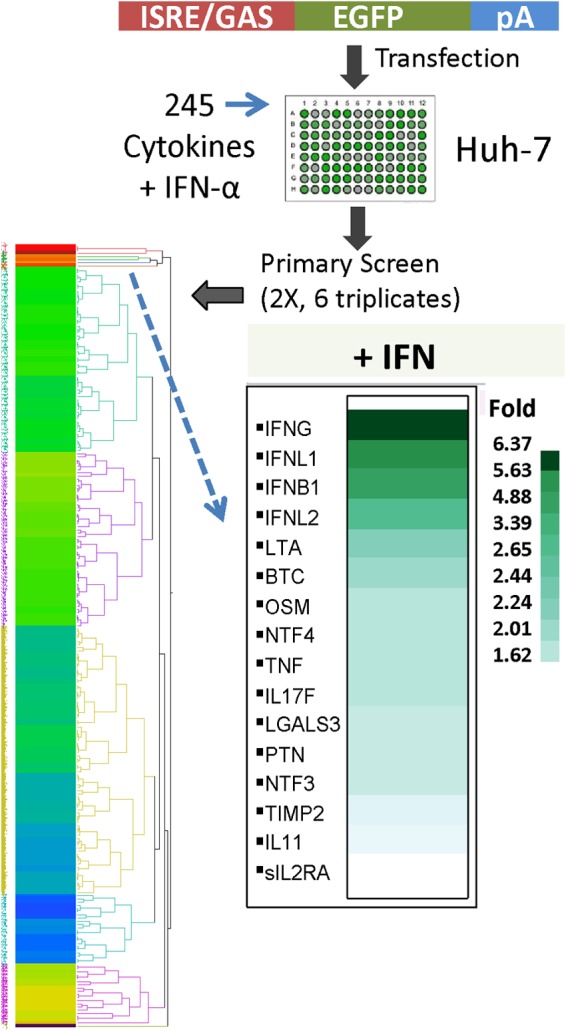
Cytokinome modulation of the IFN response. The Huh-7 liver cell line was transfected with ISRE/GAS-EGFP reporter constructs for 20 h. The cells were treated with a combination of IFN-α (10 IU/ml) and ∼1 nM each member of the cytokinome (>200 recombinant human cytokines [see Table S1 in the supplemental material]). The screening experiments (see Table S2 in the supplemental material) were performed twice independently in triplicate (total, 6 replicates). After 20 h of additional incubation, the fluorescence intensity was detected using a BD-Pathway cell imager and quantified with the ProXcell program. The heat plot represents the clustering of fold increases in fluorescence intensity due to IFN and cytokine combinations compared to IFN treatment alone. The inset shows the cytokine cluster that boosted the IFN response from nearly 2-fold to 6-fold.
ISRE- and GAS-dependent cytokine augmentation of the IFN response.
In order to determine whether ISRE or GAS was responsible for cytokine enhancement of IFN-α signaling, we examined the effector cytokine cluster obtained from the screen (Fig. 1) for either ISRE- or GAS-linked fluorescent reporter experiments in the presence of IFN-α and IFN-γ, respectively. The enhanced response to the ISRE reporter was generally lower (range, 1.5- to 3.5-fold) (Fig. 2A) than to the combined ISRE/GAS reporter (range, 1.6- to 6-fold) (Fig. 1). All types of IFNs (types I to III), as well as the cytokines BTC, IL-11, IL-17F, OSM, and TNF, were able to enhance the IFN response through ISRE, but not with the mutant ISRE reporter activity (Fig. 2A). The highest modulators of the ISRE response were the type III IFNL1 (3.5-fold), IFNB1, BTC, IL-11, and IFN-γ, with each cytokine eliciting an approximately 2.0-fold increase in reporter activity compared to IFN-α alone.
FIG 2.

ISRE-dependent cytokine augmentation of the IFN response. (A and B) Huh-7 cells were transfected with either ISRE-linked or ISRE-mutant (Mut)-linked EGFP reporter constructs. After 20 h of incubation, the cells were treated with 1 nM each selected cluster cytokine in the presence (A) or absence (B) of 10 U/ml IFN-α. pA denotes 3′ UTR poly(A) signal. (C) Cytokine modulation of the IFN response. Huh-7 cells were transfected with ISRE-linked reporter constructs for 20 h. The cells were then treated with increasing log0.5 doses of IFN-α in the absence or presence of the indicated cytokines (1 nM each). The data shown are mean (±SEM) fold increases in fluorescence activities (pixels), compared to IFN alone, from one representative experiment out of two. The P values were determined by two-curve comparison by F test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Because the above-described experiments focused on the abilities of combinations of various IFNs and cytokines to augment IFN-α or IFN-λ responses, we examined the ability of one cytokine to directly activate ISRE or GAS responses. As expected, type I IFNs were able to directly activate ISRE by more than 20-fold (Fig. 2B). The combined effects of the cytokines IL-11, IL-17F, and BTC on ISRE activation were tested with increasing doses of IFN-α (Fig. 2C). The cytokines themselves were far less potent alone than with the different doses of IFN-α (approximately 2-fold over mock treatment). Although most of the prior experiments were performed with a fixed dose of IFN (e.g., 10 IU/ml), the enhanced ISRE-mediated responses appeared to be operative across the linear dose-response curves (P < 0.001; F test) (Fig. 2C).
The response to GAS-mediated activity demonstrated a restricted pattern: OSM augmented IFN-γ-mediated activation of GAS from 9-fold to 17-fold in the absence or presence of IFN-γ, respectively (P < 0.001) (Fig. 3A). This appears to be lower than ISRE-mediated activation by IFN-α (7- to 12-fold; P < 0.01) (Fig. 2A). Other cytokines, specifically, BTC and TNF, interacted slightly with IFN-γ in GAS-linked reporter activity but with only a 40% increase (1.4-fold).
FIG 3.

GAS-dependent cytokine augmentation of the IFN response. (A and B) Huh-7 cells were transfected with GAS-linked EGFP reporter constructs. After 20 h of incubation, the cells were treated with 1 nM each selected cytokine cluster in the presence (A) or absence (B) of 1 nM IFN-γ. (C and D) Huh-7 cells were transfected with GAS-linked reporter constructs for 20 h and then were treated with increasing doses of IFN-γ in the absence or presence of 1 nM OSM (C) or BTC (D). CC, cell control. The data shown are mean (and SEM) fold increases in fluorescence activities (pixels) from three independent experiments; P < 0.0001 as determined by two-way ANOVA. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
For GAS (type II) responses, IFN-α had little effect, but IFN-β had a modest effect (Fig. 3B). The highest direct GAS activation was triggered by IFN-γ or OSM treatment (Fig. 3B). Moderate GAS induction was also achieved with BTC, IL-11, and TNF (Fig. 3B). The cytokines IL-11, BTC, and IL-17F had the ability to directly trigger ISRE by at least 4-fold but did not affect the mutant ISRE. IFNL2, which has a strong effect on the dual ISRE/GAS reporter, did not activate GAS reporter activity. Alone, OSM, but not BTC, displayed dose-response characteristics, but both enhanced GAS activation in combination with different doses of IFN-γ (Fig. 3C and D).
Induction of IFN-stimulated gene expression by the cytokine cluster.
Next, we examined the abilities of all types of IFN plus the newly identified IFN response modulators to induce ISGs. We predicted that ISG mRNA expression would be stronger than that of the transcriptional reporter system. There was strong induction of mRNA levels for IFIT3, one of the highly inducible ISGs in Huh-7 cells (17), by the various IFN and cytokine combinations. When the cytokines were used alone, the highest IFIT3 mRNA levels were induced with all types of IFN (100 to >1,000-fold; P < 0.001), and IFNL2 was the most potent, followed by IFNB, IFN-γ, and IFNL1. The cytokines BTC, IL-17F, and OSM were able to increase this ISG only up to 2-fold (P < 0.01) (Fig. 4A). However, these cytokines, and also TNF, LTA, TIMP2, and NTF3, potently upregulated IFIT3 mRNA expression when combined with IFN-α at nearly 15-fold (Fig. 4B). Among the IFNs, IFNB caused a notable 1,000-fold enhancement of IFN-α-induced IFIT3 mRNA, followed by IFN-γ, which caused a 200-fold enhancement of IFN-α-induced IFIT3 mRNA (Fig. 4B). We also queried another ISG mRNA, OAS1, which showed that these cytokines caused a 3-fold enhancement of IFN-α-induced OAS1 mRNA (Fig. 4C). The effect of combining cytokines with IFN-α on OAS1 mRNA induction was weaker than that on IFIT3 mRNA. There was virtually no induced expression of OAS1 by other cytokines individually (data not shown). The ability to induce MX1, another ISG, was also studied. The most potent inducers were type I IFN and type III IFNs (300- to 1,000-fold enhancement), followed by IFN-γ and OSM (Fig. 4D). The other cytokines did not individually trigger MX1 mRNA expression, similar to mRNA OAS1 expression patterns. The IFN combinations led to enhanced MX1 mRNA expression, and the most potent was when type III IFNs (IFNL1/L2) were combined with IFN-α (Fig. 4D). When cytokines were combined with IFN-α, BTC had the most potent activity, with 5-fold enhancement over IFN-α alone, followed by TNF and LTA and, to a lesser extent, the rest of the cytokine cluster (Fig. 4D). The protein levels of OAS confirmed the enhanced expression of IFN and cytokine combinations (Fig. 4E and F).
FIG 4.

Induction of ISG expression by the cytokine cluster. (A) Huh-7 cells were pretreated with selected cytokines (∼1 nM) or a combination for 6 h. Quantitative RT-PCR using TaqMan primers specific for human IFIT3 and control GAPDH was performed. (B and C) Huh-7 cells were treated with IFN-α (10 IU/ml) with or without selected cytokines (∼1 nM) for 6 h. Quantitative RT-PCR using TaqMan primers specific for human IFIT3 (B) or for human OAS1 (C) was performed, while GAPDH primers were used for GAPDH housekeeping control. (D) Huh-7 cells were treated with cytokines individually or in combination with IFN-α (10 IU/ml) for 6 h. Quantitative RT-PCR using TaqMan primers specific for human MX1 and GAPDH mRNA levels was performed. The values shown represent the mean ISG/GADPH mRNA ratio plus SEM from two independent experiments. **, P < 0.01. (E and F) Huh-7 cells were treated with IFN-α (10 IU/ml) with or without selected cytokines (∼1 nM). The Huh-7 cells were treated with the indicated cytokine combinations for 20 h, and then lysates (50 μg) were obtained and resolved by SDS-PAGE. Western blots were performed using an OAS1 antibody. The blots were reprobed with an anti-β-actin antibody for loading control. The images are from a representative experiment out of two. *, P < 0.01; **, P < 0.001.
Roles of endogenous IFN-β and Stat1 in cytokine-IFN interactions.
Because endogenous IFN-β may play a role in the induction of the IFN response by cytokines, such as TNF-α (27), we examined IFNB1 mRNA expression in response to the various types of IFNs and cytokines (Fig. 5A). BTC and IFNL2 displayed the strongest triggering of IFNB1 mRNA expression, whereas other IFNs and cytokines had a modest 2-fold effect (Fig. 5A). Several cytokines were examined for the kinetics of induction of IFNB1 mRNA. IFNL2 and IL-17 displayed transient induction of IFNB1 mRNA expression, which peaked at 3 to 5 h after induction, while BTC-induced IFNB1 mRNA levels displayed sustained production over the 24-h period (Fig. 5B). The requirement for Stat1 was also explored using U3A cells, which lack Stat1 and do not respond to IFNs (28). Stat1 was required for all of the effector cytokines (BTC, IL-11, IL-17F, and IFNL2) except TNF-α (Fig. 5C). The cytokine cluster induced ISG expression (IFIT3) in a Stat1-dependent fashion (Fig. 5D).
FIG 5.

Roles of endogenous IFN-β and Stat1 in the activity of the cytokine cluster. (A) Huh-7 cells were treated with selected cytokines (1 nM) for 6 h. Quantitative RT-PCR using TaqMan primers specific for human IFN-β (and GAPDH as a control) was performed. (B) Time-dependent expression of IFN-β mRNA. Huh-7 cells were treated with IL-17F, BTC, and IFNL2 (1 nM) for the indicated times. Quantitative RT-PCR using TaqMan primers specific for human IFN-β and GAPDH was performed. (C) Wild-type (WT) and U3A (Stat1-deficient) fibroblasts were treated with the selected cytokine panel (1 nM) for 6 h. Quantitative RT-PCR using TaqMan primers specific for human IFN-β was performed, and the results were normalized to human GAPDH levels. (D) WT and U3A (Stat1-deficient) cells were treated with a selected cytokine panel (1 nM) for 6 h. Quantitative RT-PCR using TaqMan primers specific for human IFIT3 was performed, and the results were normalized to human GAPDH levels. (E) Huh-7 cells were transfected with nanoluciferase (NanoLuc) reporter fused to a minimal IFNB1 promoter and then subjected to the indicated IFN and cytokines for 6 h. Cells were lysed, and luciferase activity was quantitated and presented as fold increase over the control (no cytokine treatment). The data represent means and SEM of triplicate readings from two independent experiments. (F) Huh-7 cells were transfected with nanoluciferase reporter fused to NF-κB response elements and then subjected to the indicated IFN and cytokines for 6 h. The cells were lysed, and the luciferase activity was quantitated and presented as fold increase over the control (no cytokine treatment). (G) Huh-7 cells were pretreated with 10 μM NF-κB inhibitor or DMSO for 1 h before adding 1 nM each of the cytokines and incubating for a further 6 h. Quantitative RT-PCR using TaqMan primers specific for human IFN-β (and GAPDH as a control) was performed. The data are means and SEM from two independent experiments. *, P < 0.01; **, P < 0.001.
The ability of the cytokine cluster to induce the transcription of the IFNB1 gene was assessed using the IFNB1 minimal promoter-linked luciferase reporter (Fig. 5E). Three of the cytokines, BTC, OSM, and TNF, but not others, were able to induce IFNB1 promoter activity by 2-fold (Fig. 5E), suggesting that other posttranscriptional regulatory mechanisms may exist. To explore the involvement of the IFN-independent pathway, the NF-κB reporter cell assay and NF-κB inhibitor experiments were utilized. Except for IL-17F and IFNL2, the cluster cytokines were able to induce NF-κB element-linked reporter activity by at least 2-fold (Fig. 5F). The NF-κB inhibitor was also able to reduce the ability of the cluster cytokines to induce IFNB1 mRNA by ≥25% (Fig. 5G), except IFNL2.
Antiviral activity of the cytokine cluster.
We developed a real-time label-free IFN bioassay based on the existing electric impedance technology, which comprises microwells in culture plates coated with micro-electronic chips. We used the WISH cell line, which is susceptible to EMCV and sensitive to IFN. As cells are grown or destroyed by a virus (CPE), the change in the ionic chip environment is registered, and an electric impedance cellular index curve is generated. There was a clear cellular change due to the virus-induced CPE, with a distinct peak in cellular changes at approximately 14 h postinfection (Fig. 6A, red arrow), which likely corresponded to the virus being released. Subsequently, there was a sharp decline in the cell index within a few hours to complete cellular destruction. IFN, in a dose-dependent manner, was able to delay the onset of cellular destruction and to protect the cells from the virus-induced CPE. Interestingly, notable IFN-induced delay and flattening of the virus release/shedding curve peaks were noted in this technology (Fig. 6A, arrows). At the maximum dose of IFN-α used (10 U/ml), there was a dramatic change in the onset of virus effect and curve kinetics with a smoother decline in the cellular index than in the virus. As a validation of this method, we found the data obtained with the method comparable to the data obtained with the traditional crystal violet CPE assay and superbly correlated (Fig. 6B). Using the standard assay first, we evaluated several combinations; among the novel candidates, IL-17F and BTC were able to protect the cells from EMCV-induced CPE both directly and in combination with IFN-α (Fig. 6C). Representative images showing the protection of the cells in response to IFN and cytokines were obtained (Fig. 6D).
FIG 6.

Development of a real-time IFN bioassay and antiviral screen. (A) WISH cells were seeded in microwell plates precoated overnight with micro-electronic chips. The cells were treated with IFN-α at the indicated doses for 16 h and then challenged with EMCV at a multiplicity of infection (MOI) of 0.1. The CPE was monitored in 15-min windows in real time. The cell index (plus SEM), representing live-cell activity in relation to time postinfection, is shown. (B) WISH cells were seeded in 96-well microplates overnight. The cells were treated with IFN-α at the indicated doses for 16 h and then challenged with EMCV at an MOI of 0.1. The cells were fixed and stained as shown. (C) WISH cells were seeded in a 96-well microplate and then treated with the indicated IFN and cytokine combinations for 16 h, followed by EMCV challenge at an MOI of 0.1. The cells were fixed, stained with crystal violet, and then eluted for optical density (OD) quantitation. The data are percent CPE from triplicate readings from one representative experiment out of two. VC, virus control. (D) Representative images of WISH-EMCV experiments performed using the classical crystal violet assay for visual monitoring. *, P < 0.01; ***, P < 0.001.
Compared to control noninfected cells, the negative-strand RNA virus VSV induced CPE in Huh-7 cells, recorded as a reduced cell index by the electronic biosensor (Fig. 7A). Addition of a low dose of IFN-α (3 IU/ml) caused partial protection from virus-induced CPE. Cytokines alone did not cause a change in the cell index in the absence of the virus (data not shown). Although the cytokines exerted little or no antiviral action, the combination of IFN-α and either BTC or IL-17F resulted in enhanced antiviral action of IFN-α as measured by a reduction in the 50% effective dose (ED50) and delayed time to CPE (Fig. 7A). The IFN–IL-11 combination led to a modest effect compared to IFN-α activity alone. Using qPCR for VSV rhabdovirus nucleocapsid P mRNA assessment, we also found that the combinations of IFN-BTC and IFN–IL-17F were the most effective in inhibiting VSV P mRNA (Fig. 7B). IL-17F was most potent against VSV infection of Huh-7 cells, while BTC was most potent against EMCV infection of WISH cells (Fig. 7A and C). Unlike EMCV, there was no activity of the novel cytokines in enhancing IFN activity against VSV in WISH cells (data not shown) as opposed to Huh-7 cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
FIG 7.

Antiviral activities of cytokines and IFN combinations. (A) Huh-7 cells were seeded for overnight incubation in microwell plates precoated with micro-electronic chip electrodes. The cells were treated with the indicated cytokines and combinations with IFN-α and challenged with VSV at an MOI of 1. The CPE was monitored at 15-min intervals in real time. The cell index, representing live-cell activity in relation to time postinfection is presented as means ± SEM from triplicate wells. (B) Huh-7 cells were treated with the indicated cytokines in the presence or absence of IFN-α (10 IU/ml) for 16 h and then infected with VSV at an MOI of 1 for 6 h. Total RNA was extracted, and VSV nucleocapsid P RNA levels were assayed by RT-qPCR with primers specific for VSV P RNA. GAPDH mRNA was used as a control. UD, undetectable. (C) WISH cells were seeded in microwell plates precoated with micro-electronic chip electrodes. The cells were treated with the indicated cytokines and/or combinations with IFN-α and challenged with EMCV at an MOI of 0.1. The CPE was monitored at 15-min intervals in real time. The cell index, representing live-cell activity in relation to time postinfection, is presented as means ± SEM from triplicate wells. The results are from one representative experiment out of two. The horizontal line shows ED50 readings. (D) Confluent A549 lung cells were treated with cytokines (3 nM) in the presence or absence of IFN-α (100 IU/ml) for 16 h. The cells were infected with the human influenza virus H1N1 for 6 h, and then the RNA was extracted and assayed for H1N1 NS1 RNA using RT-qPCR with primers specific for H1N1 NS1. GAPDH was also measured as a control. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
A human respiratory disease virus, influenza virus H1N1, was evaluated here using a relevant cellular model, the A549 lung cell line. We looked at the effect of the cytokine-IFN combination on human H1N1 replication using a viral-RNA assay; we chose a sensitive viral-RNA measurement rather than a cell-based assay because IFN has weak activity against H1N1 due to its IFN-antagonistic NS1-mediated activity, mostly seen as post-mRNA transcriptional/posttranslational effects (29, 30). The BTC and IL-11 treatment itself reduced H1N1 NS1 mRNA and enhanced IFN-suppressing activity against H1N1 NS1 mRNA (Fig. 7D).
DISCUSSION
IFNs are part of the cytokine family, a large group of small secreted proteins that comprise a complex and highly interactive network that orchestrates innate and adaptive immunity. Viruses and other stimuli trigger the biosynthesis and release of IFNs from many cell types, with a plethora of coexpressed cytokines. The number of cytokines that make up the cytokinome is now more than 200. In this study, we used a systems biology approach for analyzing the potential of each molecule of the currently known cytokinome to modulate the IFN response in Huh-7 liver cells. We studied different combinations of cytokines and IFNs and focused on the resulting cluster, which contains the classical IFNs and a number of cytokines, including molecules with putative novel antiviral effects.
We used the Huh-7 cell line for several reasons. It is a well-established human hepatocyte-derived liver cell line model that responds to all types of IFNs and to many cytokines and chemokines (21–25) and is susceptible to several viruses, including liver and respiratory viruses (7, 31). It is important to note that despite the choice of Huh-7 liver cells based on their broad response to a wide variety of IFNs and cytokines, our study is limited by the repertoire of responsive receptors in the liver cells. Despite this limitation, we were still able to identify multiple combinatorial interactions between IFNs and cytokines. To broaden the scope for detecting responses, three different reporters, the dual ISRE/GAS-linked reporter, the ISRE-linked reporter, and the GAS-linked reporter, were utilized. The screen showed that certain IFNs and cytokines respond to both ISRE and GAS reporters, but with different potencies, while others do not respond. Notably, the response is higher to the ISRE/GAS composite reporter than to the individual reporters. There are many ISGs that respond to both IFN-α and IFN-γ (32), and thus, they harbor both ISRE and GAS.
Our data demonstrate the ability of the cytokine cluster (BTC, IL-11, IL-17, and IFNL2), except TNF-α, to induce IFN-β through Stat1 signaling. Although Stat1 signaling is mainly required for IFN activity rather than for IFN production, the data indicate that Stat1 signaling is also important in IFN-β gene expression, at least for these cytokines. IFN-β gene induction by viruses also requires coordinated combinations of ATF-2/c-Jun and IRF-3/IRF-7 (33). Thus, it is also possible that autocrine IFN-β upon binding to IFNR requires Stat1 and activates IRF7, leading to augmentation of IFN-β gene expression, although this usually happens in dendritic cells (34). In general, the ability of the candidate cytokine cluster to directly activate the IFNB1 promoter was weaker than endogenous IFNB gene expression and occurred only with BTC and TNF. This is not unexpected, because reporter studies address only one regulatory component—transcription. Another signaling factor that may explain other regulatory events in IFN-β induction by the cytokines is the NF-κB pathway. NF-κB signaling is an important signaling pathway in cytokine action, and it is an accessory factor for IFN-β induction, as it was previously known that IFN-β induction by double-stranded RNA and viruses requires NF-κB signaling (35, 36). Our data demonstrated a role of the NF-κB-dependent pathway in the induction of IFNB by this candidate cytokine cluster. However, knowledge of further mechanisms of the synergy will require detailed experimental studies in the future.
The IFNs, and the IFN-like cytokines identified here, comprise a functional (IFN-modulating) cluster; however, it belongs to various structurally unrelated subfamilies: the interferon family (type I IFNs), the hematopoietin family (OSM and IL-11), the TNF family (TNF-α and lymphotoxin), the IL-10 family (IL-28 and IL-29), the IL-17 family (IL-17F), and the epidermal growth factor (EGF) family (betacellulin). Among these are the less widely known (OSM) and previously unrecognized IFN-like (BTC, IL-11, and IL-17F) cytokines. OSM is a cytokine that is primarily released from dendritic cells and other antigen-presenting cells (37). It has been shown to possess anti-hepatitis C virus (HCV) activity in liver cells and enhances IFN-α action (38, 39). Our cytokinome study extended these findings by showing OSM individual and combined effects on ISRE- and GAS-linked expression, interaction with IFN-γ in a dose-response manner, and partial involvement of NF-κB-mediated IFN-β expression.
All of the indicated members appear to mediate the IFN response through a Stat1-mediated pathway, except the TNF family members. Candidate novel cytokines that possess IFN-like activity, such as inducing ISRE-mediated and/or GAS-mediated transcription and inducing ISG mRNA expression, were further analyzed for their antiviral activities. Both IFNL2 and IL-17F caused transient activation of IFN-β, while the effect of BTC demonstrated sustained activation. The latter effect is similar to the effect of type III IFNs on ISG expression in Huh-7 cells (22, 40). The antiviral effects of IL-17F, IL-11, and BTC are not as broad as those of classical IFNs in terms of virus-cell-type combinations but appear to be highly selective in regard to virus type and cell type. IL-17F, originally named for and derived from Th-17-producing T cells, is primarily a proinflammatory cytokine that is involved in innate immunity and autoimmune diseases (reviewed in reference 41). The IL-17 receptor is widely expressed in immune cells, fibroblasts, and epithelial cells, including Huh-7 liver cells (42, 43). Our results show that IL-17F induced ISRE-mediated ISG expression, particularly in combination with IFN; these effects required Stat1 signaling and involved endogenous IFN-β. There is very little or no information on IL-17 and the other cytokines identified here (IL-11 and BTC) regarding virus-induced production, antiviral activity, or IFN response.
The IFN biosensor assay is based on real-time monitoring of the change in electric impedance allowing sensing of cellular activity and has been used in several studies in a virology setting (19, 20). It allowed us to develop an IFN assay that made detailed observations, such as the effective dose at any given time point, e.g., the ED30, ED50, and ED95, and the time until CPE at any desired percentage of intact cells. We found that assaying IFN antiviral action correlated with CPE in a dose-dependent manner, which allowed us to monitor the enhancement action of IL-17F and BTC on IFN-mediated antiviral activity at both early and later stages.
The IFN-cytokine combination is clearly superior to IFN alone in inhibiting viral mRNA and cytopathic effects, including those of human respiratory viruses. The enhancing effect of the cytokines on IFN is dependent not only on the virus but also on the cell type. Thus, more detailed work is needed to confirm the effects of these novel IFN-like cytokines on viral replication with different viruses and multiple cell types. Also, more work is required to confirm their suitability for preclinical experiments, including animal models. In addition to the novel findings of additional IFN-like cytokines, combinations, and ISRE/GAS selectivity, this work can be utilized as a resource. For example, it can be used for exploring IFNs, other cytokines, and their combinations in their selectivity toward ISRE, GAS, or both and for understanding the relative activities of the IFNs, cytokines, and their combinations in eliciting IFN activity.
Supplementary Material
ACKNOWLEDGMENTS
We thank Robert H. Silverman and George Stark at the Lerner Research Center in Cleveland, OH, for providing the HT1080 and U3A mutant cell lines. The technical assistance of Maher Al-Saif is greatly appreciated.
This project was supported by intramural funding and approval of the King Faisal Specialist Hospital and Research Centre.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03729-14.
REFERENCES
- 1.Van Boxel-Dezaire AH, Rani MR, Stark GR. 2006. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 2.Khabar KS, Young HA. 2007. Post-transcriptional control of the interferon system. Biochimie 89:761–769. doi: 10.1016/j.biochi.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Itsui Y, Sakamoto N, Kurosaki M, Kanazawa N, Tanabe Y, Koyama T, Takeda Y, Nakagawa M, Kakinuma S, Sekine Y, Maekawa S, Enomoto N, Watanabe M. 2006. Expressional screening of interferon-stimulated genes for antiviral activity against hepatitis C virus replication. J Viral Hepat 13:690–700. doi: 10.1111/j.1365-2893.2006.00732.x. [DOI] [PubMed] [Google Scholar]
- 5.Jiang D, Weidner JM, Qing M, Pan XB, Guo H, Xu C, Zhang X, Birk A, Chang J, Shi PY, Block TM, Guo JT. 2010. Identification of five interferon-induced cellular proteins that inhibit West Nile virus and dengue virus infections. J Virol 84:8332–8341. doi: 10.1128/JVI.02199-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. 2012. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A 109:4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metz P, Dazert E, Ruggieri A, Mazur J, Kaderali L, Kaul A, Zeuge U, Windisch MP, Trippler M, Lohmann V, Binder M, Frese M, Bartenschlager R. 2012. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology 56:2082–2093. doi: 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- 8.Gad HH, Hamming OJ, Hartmann R. 2010. The structure of human interferon lambda and what it has taught us. J Interferon Cytokine Res 30:565–571. doi: 10.1089/jir.2010.0062. [DOI] [PubMed] [Google Scholar]
- 9.Levy DE, Marie IJ, Durbin JE. 2011. Induction and function of type I and III interferon in response to viral infection. Curr Opin Virol 1:476–486. doi: 10.1016/j.coviro.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. 2003. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 11.Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, Ostrander C, Dong D, Shin J, Presnell S, Fox B, Haldeman B, Cooper E, Taft D, Gilbert T, Grant FJ, Tackett M, Krivan W, McKnight G, Clegg C, Foster D, Klucher KM. 2003. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 12.Donnelly RP, Sheikh F, Kotenko SV, Dickensheets H. 2004. The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J Leukocyte Biol 76:314–321. doi: 10.1189/jlb.0204117. [DOI] [PubMed] [Google Scholar]
- 13.Olagnier D, Hiscott J. 2014. Type I and type III interferon-induced immune response: it's a matter of kinetics and magnitude. Hepatology 59:1225–1228. doi: 10.1002/hep.26959. [DOI] [PubMed] [Google Scholar]
- 14.Ramana CV, Gil MP, Schreiber RD, Stark GR. 2002. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol 23:96–101. doi: 10.1016/S1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 15.Costantini S, Castello G, Colonna G. 2010. Human cytokinome: a new challenge for systems biology. Bioinformation 5:166–167. doi: 10.6026/97320630005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Al-Saif M, Khabar KS. 2012. UU/UA dinucleotide frequency reduction in coding regions results in increased mRNA stability and protein expression. Mol Ther 20:954–959. doi: 10.1038/mt.2012.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahmoud L, Al-Saif M, Amer HM, Sheikh M, Almajhdi FN, Khabar KS. 2011. Green fluorescent protein reporter system with transcriptional sequence heterogeneity for monitoring the interferon response. J Virol 85:9268–9275. doi: 10.1128/JVI.00772-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hitti E, Al-Yahya S, Al-Saif M, Mohideen P, Mahmoud L, Polyak SJ, Khabar KS. 2010. A versatile ribosomal protein promoter-based reporter system for selective assessment of RNA stability and post-transcriptional control. RNA 16:1245–1255. doi: 10.1261/rna.2026310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang Y, Ye P, Wang X, Xu X, Reisen W. 2011. Real-time monitoring of flavivirus induced cytopathogenesis using cell electric impedance technology. J Virol Methods 173:251–258. doi: 10.1016/j.jviromet.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian D, Zhang W, He J, Liu Y, Song Z, Zhou Z, Zheng M, Hu Y. 2012. Novel, real-time cell analysis for measuring viral cytopathogenesis and the efficacy of neutralizing antibodies to the 2009 influenza A (H1N1) virus. PLoS One 7:e31965. doi: 10.1371/journal.pone.0031965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Azuma Y, Murata M, Matsumoto K. 2000. Alteration of sugar chains on α1-acid glycoprotein secreted following cytokine stimulation of HuH-7 cells in vitro. Clin Chim Acta 294:93–103. doi: 10.1016/S0009-8981(99)00248-X. [DOI] [PubMed] [Google Scholar]
- 22.Bolen CR, Ding S, Robek MD, Kleinstein SH. 2014. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 59:1262–1272. doi: 10.1002/hep.26657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matak P, Chaston TB, Chung B, Srai SK, McKie AT, Sharp PA. 2009. Activated macrophages induce hepcidin expression in HuH7 hepatoma cells. Haematologica 94:773–780. doi: 10.3324/haematol.2008.003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sutton A, Friand V, Brule-Donneger S, Chaigneau T, Ziol M, Sainte-Catherine O, Poire A, Saffar L, Kraemer M, Vassy J, Nahon P, Salzmann JL, Gattegno L, Charnaux N. 2007. Stromal cell-derived factor-1/chemokine (C-X-C motif) ligand 12 stimulates human hepatoma cell growth, migration, and invasion. Mol Cancer Res 5:21–33. doi: 10.1158/1541-7786.MCR-06-0103. [DOI] [PubMed] [Google Scholar]
- 25.Bai H, Weng Y, Bai S, Jiang Y, Li B, He F, Zhang R, Yan S, Deng F, Wang J, Shi Q. 2014. CCL5 secreted from bone marrow stromal cells stimulates the migration and invasion of Huh7 hepatocellular carcinoma cells via the PI3K-Akt pathway. Int J Oncol 45:333–343. doi: 10.3892/ijo.2014.2421. [DOI] [PubMed] [Google Scholar]
- 26.al-Haj L, Al-Ahmadi W, Al-Saif M, Demirkaya O, Khabar KS. 2009. Cloning-free regulated monitoring of reporter and gene expression. BMC Mol Biol 10:20. doi: 10.1186/1471-2199-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB. 2008. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol 9:378–387. doi: 10.1038/ni1576. [DOI] [PubMed] [Google Scholar]
- 28.Pellegrini S, John J, Shearer M, Kerr IM, Stark GR. 1989. Use of a selectable marker regulated by alpha interferon to obtain mutations in the signaling pathway. Mol Cell Biol 9:4605–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khaperskyy DA, Emara MM, Johnston BP, Anderson P, Hatchette TF, McCormick C. 2014. Influenza a virus host shutoff disables antiviral stress-induced translation arrest. PLoS Pathog 10:e1004217. doi: 10.1371/journal.ppat.1004217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kochs G, Garcia-Sastre A, Martinez-Sobrido L. 2007. Multiple anti-interferon actions of the influenza A virus NS1 protein. J Virol 81:7011–7021. doi: 10.1128/JVI.02581-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freymuth F, Vabret A, Rozenberg F, Dina J, Petitjean J, Gouarin S, Legrand L, Corbet S, Brouard J, Lebon P. 2005. Replication of respiratory viruses, particularly influenza virus, rhinovirus, and coronavirus in HuH7 hepatocarcinoma cell line. J Med Virol 77:295–301. doi: 10.1002/jmv.20449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A 95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panne D, Maniatis T, Harrison SC. 2007. An atomic model of the interferon-beta enhanceosome. Cell 129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Honda K, Takaoka A, Taniguchi T. 2006. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 35.Thanos D, Maniatis T. 1995. Identification of the rel family members required for virus induction of the human beta interferon gene. Mol Cell Biol 15:152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lenardo MJ, Fan CM, Maniatis T, Baltimore D. 1989. The involvement of NF-kappa B in beta-interferon gene regulation reveals its role as widely inducible mediator of signal transduction. Cell 57:287–294. doi: 10.1016/0092-8674(89)90966-5. [DOI] [PubMed] [Google Scholar]
- 37.Suda T, Chida K, Todate A, Ide K, Asada K, Nakamura Y, Suzuki K, Kuwata H, Nakamura H. 2002. Oncostatin M production by human dendritic cells in response to bacterial products. Cytokine 17:335–340. doi: 10.1006/cyto.2002.1023. [DOI] [PubMed] [Google Scholar]
- 38.Larrea E, Aldabe R, Gonzalez I, Segura V, Sarobe P, Echeverria I, Prieto J. 2009. Oncostatin M enhances the antiviral effects of type I interferon and activates immunostimulatory functions in liver epithelial cells. J Virol 83:3298–3311. doi: 10.1128/JVI.02167-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikeda M, Mori K, Ariumi Y, Dansako H, Kato N. 2009. Oncostatin M synergistically inhibits HCV RNA replication in combination with interferon-α. FEBS Lett 583:1434–1438. doi: 10.1016/j.febslet.2009.03.054. [DOI] [PubMed] [Google Scholar]
- 40.Jilg N, Lin WY, Hong J, Schaefer EA, Wolski D, Meixong J, Goto K, Brisac C, Chusri P, Fusco DN, Chevaliez S, Luther J, Kumthip K, Urban TJ, Peng LF, Lauer GM, Chung RT. 2014. Kinetic differences in the induction of interferon stimulated genes by interferon-alpha and interleukin 28B are altered by infection with hepatitis C virus. Hepatology 59:1250–1261. doi: 10.1002/hep.26653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin W, Dong C. 2013. IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect 2:e60. doi: 10.1038/emi.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu FM, Li QL, Gao Q, Jiang JH, Zhu K, Huang XY, Pan JF, Yan J, Hu JH, Wang Z, Dai Z, Fan J, Zhou J. 2011. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol Cancer 10:150. doi: 10.1186/1476-4598-10-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lafdil F, Miller AM, Ki SH, Gao B. 2010. Th17 cells and their associated cytokines in liver diseases. Cell Mol Immunol 7:250–254. doi: 10.1038/cmi.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.