

ABSTRACT
The discovery that measles virus (MV) uses the adherens junction protein nectin-4 as its epithelial receptor provides a new vantage point from which to characterize its rapid spread in the airway epithelium. We show here that in well-differentiated primary cultures of airway epithelial cells from human donors (HAE), MV infectious centers form rapidly and become larger than those of other respiratory pathogens: human respiratory syncytial virus, parainfluenza virus 5, and Sendai virus. While visible syncytia do not form after MV infection of HAE, the cytoplasm of an infected cell suddenly flows into an adjacent cell, as visualized through wild-type MV-expressed cytoplasmic green fluorescent protein (GFP). High-resolution video microscopy documents that GFP flows through openings that form on the lateral surfaces between columnar epithelial cells. To assess the relevance of the protein afadin, which connects nectin-4 to the actin cytoskeleton, we knocked down its mRNA. This resulted in more-limited infectious-center formation. We also generated a nectin-4 mutant without the afadin-binding site in its cytoplasmic tail. This mutant was less effective than wild-type human nectin-4 at promoting MV infection in primary cultures of porcine airway epithelia. Thus, in airway epithelial cells, MV spread requires the nectin-4/afadin complex and is based on cytoplasm transfer between columnar cells. Since the viral membrane fusion apparatus may open the passages that allow cytoplasm transfer, we refer to them as intercellular membrane pores. Virus-induced intercellular pores may contribute to extremely efficient measles contagion by promoting the rapid spread of the virus through the upper respiratory epithelium.
IMPORTANCE Measles virus (MV), while targeted for eradication, still causes about 120,000 deaths per year worldwide. The recent reemergence of measles in insufficiently vaccinated populations in Europe and North America reminds us that measles is extremely contagious, but the processes favoring its spread in the respiratory epithelium remain poorly defined. Here we characterize wild-type MV spread in well-differentiated primary cultures of human airway epithelial cells. We observed that viral infection promotes the flow of cytoplasmic contents from infected to proximal uninfected columnar epithelial cells. Cytoplasm flows through openings that form on the lateral surfaces. Infectious-center growth is facilitated by afadin, a protein connecting the adherens junction and the actin cytoskeleton. The viral fusion apparatus may open intercellular pores, and the cytoskeleton may stabilize them. Rapid homogenization of cytoplasmic contents in epithelial infectious centers may favor rapid spread and contribute to the extremely contagious nature of measles.
INTRODUCTION
Measles virus (MV) is a highly contagious aerosol-transmitted virus that affects more than 10 million children each year and accounted for approximately 120,000 deaths in 2012 (1). The reemergence of measles in insufficiently vaccinated populations in Europe and North America (2) reminds us that MV is extremely contagious, but the mechanisms favoring its spread remain incompletely understood. Once inhaled, MV bypasses the respiratory epithelium by infecting alveolar macrophages or dendritic cells that express the immune cell-specific protein signaling lymphocyte activation molecule (SLAM) (3–5). After extensive replication in SLAM-expressing cells in lymphatic organs, epithelial invasion occurs from the basolateral side (6); the virus is likely delivered by circulating SLAM-expressing immune cells (7, 8) to epithelial cells expressing the adherens junction (AJ) protein nectin-4 (9, 10). Rapid MV spread in the upper airway epithelium may contribute to the extremely contagious nature of MV; however, these processes are incompletely characterized.
One of the critical challenges for the characterization of MV spread is the identification of model systems that mirror the way in which viruses spread in living organisms. Primary cultures of well-differentiated airway epithelial cells from human donors (HAE) recapitulate the surface cells of the conducting airways. HAE are pseudostratified and include multiple cell types, such as ciliated cells, nonciliated cells, goblet cells, and basal cells. Unlike infection of immortalized cells, infection of HAE with a vaccine (6) or wild-type (11, 12) virus is noncytopathic. MV-infected HAE form infectious centers that retain individual nuclei, plasma membranes, and transepithelial resistance (6). A great deal of evidence demonstrates that this culture model is highly representative of the in vivo airways (13, 14). Moreover, tissue collected at the peak of acute disease in experimentally MV infected macaques had large infectious centers similar to those of the primary-culture model (7), confirming its relevance. In these studies, MV was delivered as free virus to the basolateral surface of HAE. During the course of a viral infection in vivo, circulating immune cells are likely the vehicles of delivery to epithelial cells (7, 8). Studies to compare the efficiencies of free and immune cell MV delivery in the HAE model system are ongoing.
MV and other paramyxoviruses are enveloped negative-strand RNA viruses that spread via aerosolization and cause a wide range of diseases in humans and other mammals. While MV, which belongs to the genus Morbillivirus, preferentially infects the basolateral surface of HAE and forms large infectious centers (6, 11), respiratory syncytial virus (RSV), of the genus Pneumovirus, preferentially infects at the apical surface of HAE, and infections remain limited to a single cell or a few contiguous cells (15). Parainfluenza virus 5 (PIV5), of the genus Rubulavirus, can infect HAE from the basolateral surface, but with less efficiency than from the apical surface (16). It has also been observed that Sendai virus (SeV), of the genus Respirovirus, infects airway epithelial cells preferentially from the apical surface (17). As with PIV5 and RSV, SeV infection of airway cells results in a pattern of discrete cellular infection, not infectious centers.
While these studies suggested significant differences in the ways in which these four paramyxoviruses spread in primary respiratory epithelial cells, to our knowledge the spreading patterns of these viruses have not been directly compared with MV spread under the same culture conditions. All four paramyxoviruses considered here enter cells by fusion at the plasma membrane at a neutral pH; however, the attachment proteins of respiroviruses and rubulaviruses bind ubiquitous sialic acids, while the attachment proteins of morbilliviruses bind the receptors SLAM and nectin-4 (18). Tissue-specific expression of SLAM and nectin-4 appears to be a main determinant of MV tropism (18, 19), and the biological properties of these receptors may also facilitate steps of the pathogenic process.
We asked here how nectin-4, also known as poliovirus receptor-like-4 (PVRL4), could facilitate rapid viral spread in respiratory epithelia. Nectins are immunoglobulin superfamily glycoproteins that initiate cell-cell adhesion by their trans-interactions and recruit other proteins to establish first AJs and then tight junctions (TJs) (20). Recruitment of these proteins is mediated by afadin, an actin filament binding protein that connects nectins to the cytoskeleton (21). The terminal 4 amino acids of nectin bind afadin, which tethers nectin to F-actin (22, 23).
Here we provide experimental proof that the nectin-4–afadin protein connection is necessary for efficient MV infectious-center formation in HAE. In addition, we document how the cytoplasm of infected columnar epithelial cells suddenly flows into an adjacent cell through openings that form on the lateral surfaces. Rapid homogenization of cytoplasmic contents in epithelial infectious centers may favor rapid spread and thus contribute to the extremely contagious nature of measles.
MATERIALS AND METHODS
Cell culture.
Primary cultures of human and pig airway epithelia were prepared from tracheae and bronchi by enzymatic dispersion using established methods (13). Briefly, epithelial cells were dissociated and were seeded onto semipermeable collagen-coated membranes with a pore size of 0.4 μm (Millicell HA; surface area, 0.6 cm2; Millipore Corp., Bedford, MA). Human and pig airway epithelial cultures were maintained in Ultroser G (USG) medium at 37°C under 5% CO2. Millicell inserts were placed in 24-well plastic cell culture plates (Costar, Cambridge, MA). Twenty-four hours after seeding, the mucosal medium was removed, and the cells were allowed to grow at the air-liquid interface as reported previously (13). Unless otherwise indicated, only well-differentiated cultures (>3 weeks old) were used in these studies. The presence of TJs was confirmed by measuring transepithelial resistance using a volt-ohm meter (resistance, >500 Ω · cm2; World Precision Instruments, Sarasota, FL).
Virus production and determination of titers.
MV-GFP was produced as described previously (11, 24). MV-GFP is a derivative of wild-type strain ICB-323 (12) expressing green fluorescent protein (GFP) from an additional transcription unit inserted upstream of the nucleocapsid gene (11). Briefly, Vero cells stably expressing the MV receptor CD150 and cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific, Waltham, MA) containing 8% newborn calf serum (NCS; Thermo Fisher Scientific, Waltham, MA) and penicillin-streptomycin (100 μg/ml; Thermo Fisher Scientific, Waltham, MA) were used to produce MV-GFP. MV-GFP titers of approximately 1 × 107 50% tissue culture infective doses (TCID50)/ml were obtained. Recombinant Sendai virus (SeV) expressing GFP was kindly provided by Ultan Power (Queens University, Belfast, United Kingdom), and working stocks were prepared in specific-pathogen-free eggs. GFP expressing recombinant respiratory syncytial virus (RSV) was kindly provided by Mark Peeples (Nationwide Children's Hospital, Columbus, OH). The working stock of RSV was prepared in HEp-2 cells (ATCC, Manassas, VA). Recombinant parainfluenza virus 5 (PIV5) expressing mCherry was provided by Biao He (University of Georgia, Atlanta, GA) and was propagated in MDBK cells as described previously (25). All studies described in this article received Institutional Review Board (IRB) approval.
Infection of primary airway epithelial cells.
To infect airway epithelia with MV, RSV, PIV5, or SeV from the basolateral side, the Millicell culture insert containing the airway epithelial culture was turned over, and the virus was applied to the basolateral surface for 2 to 4 h in 100 μl of serum free medium. Following the 4-h infection, the virus was removed, and the culture was turned upright and was allowed to incubate at 37°C under 5% CO2 for the times indicated in the figure legends. For apical infection, MV, RSV, PIV5, or SeV was simply applied to the apical surface for 2 to 4 h in 100 μl of serum-free medium. After the incubation, the cells were rinsed with MEM three times to remove residual virus. Recombinant adenoviral vectors expressing either nectin-4 or a nectin-4 mutant were delivered to the basolateral surface using the same protocol. The nectin-4 ΔGHLV (N4ΔGHLV) vector was generated using site-directed mutagenesis to insert a premature stop codon, deleting the final 4 amino acids. The adenoviral vectors used in this study were produced at the University of Iowa Viral Vector Core (www.medicine.uiowa.edu/vectorcore).
Imaging.
Three days postinfection, the cells were initially imaged on an inverted UV fluorescence microscope and were then processed further for confocal microscopy. For processing, the cells were fixed in 2% paraformaldehyde, permeabilized in 0.2% Triton X-100, and blocked in 1% bovine serum albumin (BSA) for 1 h. For immunostaining, fixed and permeabilized HAE were stained with primary antibodies against human afadin (dilution, 1:200; R&D Systems, Minneapolis, MN), human nectin-4 (1:200; catalog no. AF2659; R&D Systems, Minneapolis, MN), ZO-1 (1:500; Thermo Fisher Scientific, Waltham, MA), or N protein (1:500; in-house-generated rabbit polyclonal antibody) overnight at 4°C, followed by a 1-h incubation with either an Alexa 488- or Alexa 568-labeled goat anti-sheep secondary antibody or an Alexa 568-labeled goat anti-rabbit secondary antibody (Life Technologies). The cells were then stained with the nuclear stain To-Pro-3 for 10 min. The filters containing the cells were removed from the culture insert by cutting the edges with a razor blade and were mounted on a slide with Vectashield (Vector Laboratories Inc., Burlingame, CA). The cells were visualized and photographed on a confocal microscope (Zeiss LSM 510; Zeiss USA, Thornwood, NY).
siRNA knockdown of the afadin gene in HAE.
The Dicer substrate short interfering RNAs (DsiRNAs) for human afadin and scrambled control sequences were designed (26, 27) and synthesized by Integrated DNA Technologies (Coralville, IA). For the human afadin DsiRNA, the sense strand (SS) is 5′-pCUGCAGCAUCGUAGCAAGGACAUGGCU and the antisense strand (AS) is 3′-GACGUCGUAGCAUCGUUCCUGUACC (DNA bases are shown in boldface). The scrambled sequences (negative control for DsiRNAs) are as follows: SS, 5′-pCGUUAAUCGCGUAUAAUACGCGUAT; AS, 3′-CAGCAAUUAGCGCAUAUUAUGCGCAUA. Lipofectamine RNAiMAX was purchased from Thermo Fisher Scientific, Waltham, MA.
RT-qPCR.
Total RNA (500 ng) was reverse transcribed by using a high-capacity reverse transcription (RT) kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. RT-quantitative PCRs (RT-qPCRs) were performed in an ABI Prism 7900 HT real-time PCR system (Thermo Fisher Scientific, Waltham, MA). The PCR conditions were as follows: 95°C for 10 min, 95°C for 15 s, and 60°C for 1 min for 40 cycles. The following primers were used in RT-qPCR analysis: for afadin, CTCTCAGCTCTTCCACTTCATC (forward) and CCCAACTGCTGACGGATAAT (reverse); for hsSFRS9, TGCGTAAACTGGATGACACC (forward) and CCTGCTTTGGTATGGAGAGTC (reverse). All the reactions were analyzed by the software (SDS, version 2.3) provided with the instrument. The relative expression of the genes was calculated by the 2−ΔΔCT formula using SFRS-9 as a normalizer. The values reported are means for at least three biological replicates, each with three technical replicates. Gene expression in samples is presented as the level of remaining target mRNA compared with the mRNA level in control samples (with scrambled control siRNA), which was normalized to 100%.
SDS-PAGE and Western blotting.
The primary human culture was washed with phosphate-buffered saline and was lysed in freshly prepared lysis buffer (1% Triton, 25 mmol/liter Tris [pH 7.4], 150 mmol/liter NaCl, protease inhibitors (cOmplete; mini, EDTA-free; Roche Biochemicals, Penzberg, Upper Bavaria, Germany) for 30 min at 4°C. Lysates were quantified by a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL). Fifty micrograms of protein per lane was separated on a 10% SDS-PAGE gel. Antibodies against human nectin-4 (1:200; R&D Systems, Minneapolis, MN) and α-tubulin (1:10,000; Sigma-Aldrich, St. Louis, MO) were used for Western blot analysis.
Time lapse microscopy and 3D rendering.
Confocal time lapse microscopy was performed on an LSM 510 microscope (Carl Zeiss AG, Oberkochen, Germany) in the Central Microscopy Research Core Facility (CMRF) at the University of Iowa. Three-dimensional (3D) stack images were acquired using a 40× objective with Zeiss Zen 2010 software. Throughout the time lapse, the dish was maintained at a constant 37°C temperature by a heating sample holder with a continuous supply of 5% CO2 to ensure optimal growth conditions. Single MV-infected cells were selected on the basis of GFP expression. The top and bottom points of the infected cell were defined, and 20 to 30 z-stacks with a step size of 1 μm per time point were acquired as a series of .tif files with 1,024 × 1,024 pixels. To avoid photobleaching, no more than 1 set of z-stacks was collected per 10 min. The raw .tif series data sets were imported into Imaris software, version 7.3 (Bitplane AG, Zurich, Switzerland), and were reconstructed in 4D (3D plus time) data sets.
Statistical analysis.
Data are presented as means ± standard errors of individual data points. The statistical significance of differences between groups was determined by Student's t test. A P value of <0.01 was considered significant.
RESULTS
Comparative spread of four paramyxoviruses in HAE.
While MV infectious centers form rapidly (6, 11), their growth has not yet been compared “head-to-head” with that of other paramyxoviruses. Figure 1 directly compares the spread of wild-type MV with that of three paramyxoviruses of different genera 3 days after apical (Fig. 1, top) or basolateral (Fig. 1, center) inoculation of HAE at equivalent multiplicities of infection. All the recombinant viruses expressed fluorescent proteins reporting infection: SeV, RSV, and MV expressed GFP, while PIV5 expressed mCherry.
FIG 1.
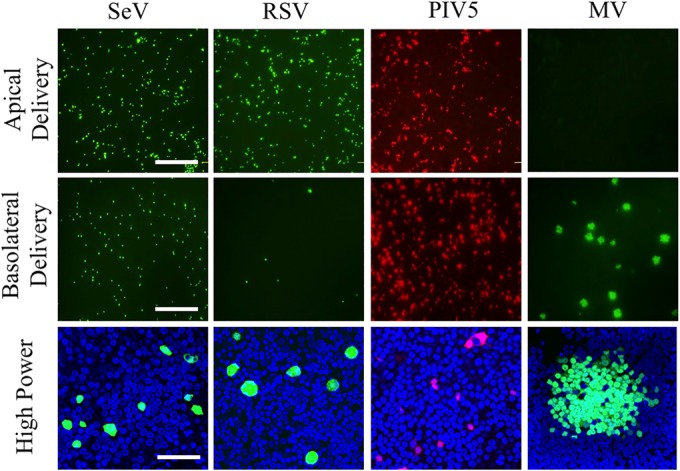
Paramyxovirus infection of HAE. (Top and center) Recombinant Sendai virus (SeV) expressing GFP, respiratory syncytial virus (RSV) expressing GFP, parainfluenza virus 5 (PIV5) expressing mCherry, and measles virus (MV) expressing GFP were applied to the apical (top row) or basolateral (center row) surface of HAE for 2 h at a multiplicity of infection of 1. Three days later, low-power images of live cells were collected with an inverted fluorescence microscope. Bar, 200 μm. (Bottom) High-power images of fixed cells collected by confocal microscopy. Bar, 50 μm.
Comparison of the two top rows of images in Fig. 1 indicates that RSV entered HAE with a clear apical preference, SeV with some apical preference, and PIV5 with almost equivalent efficiencies from both sides. The high-magnification analyses (Fig. 1, bottom) document that at this early stage, SeV and RSV infections remained restricted to single cells and PIV5 infections were limited to two or three contiguous cells. On the other hand, large MV infectious centers formed.
Noncytopathic MV spread.
In contrast to what is observed in transformed cells, MV infection of HAE does not result in cell-cell fusion or visible syncytium formation (6, 11). To assess whether the plasma membranes of individual cells remain intact in MV infectious centers, we used red fluorophore-conjugated wheat germ agglutinin (WGA) to stain sialic acid and N-acetylglucosaminyl residues of plasma membrane proteins. Figure 2A to D document that WGA stains the plasma membranes of MV-infected cells (Fig. 2A, green) as efficiently as the membranes of noninfected cells. Interestingly, similar amounts of GFP are detected in most cells of the same infectious center. Thus, while the transepithelial resistance of MV-infected HAE remains constant and the plasma membranes of individual cells appear largely intact, the cytoplasmic contents of infectious centers appear to equilibrate.
FIG 2.

MV does not form syncytia in HAE. HAE were infected from the basolateral surface with MV-GFP. Three days later, GFP expression was documented (A), the cell surface was stained with wheat germ agglutinin (WGA) (B), and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (C). A merged image is shown (D). MV N protein colocalizes with GFP in infectious centers. Three days postinfection with MV-GFP, HAE were stained with anti-N protein antibody. GFP expression (E) and N protein staining (F) were observed in almost every MV-infected cell with few exceptions (white arrow). Panels E and F each represent a digitally compressed z-stack; panel G is a merged image of a single z-slice from panels E and F. Bars, 50 μm.
In our recombinant wild-type MV, GFP is expressed from an additional transcriptional unit inserted upstream of the nucleoprotein (N) gene (11). To confirm that GFP expression is a suitable proxy for MV in infectious centers, we infected HAE with MV-GFP, allowed infectious centers to form over 3 days, and then stained the HAE with an anti-N protein antibody. Figure 2E to G document that MV N was observed in almost every infected cell of an infectious center, except for two cells at its periphery that were GFP positive but not N positive (Fig. 2E, white arrows). This observation is consistent with the cell-to-cell spread of cytoplasmic GFP, followed by the spread of ribonucleocapsids. We next investigated this process of cell-to-cell spread in greater detail.
Mechanism of MV spread in HAE: cytoplasm flows from infected cells into adjacent cells.
Cytoplasmic content equilibration suggests that in HAE fusion, pores open between infected and adjacent cells, as happens in nonpolarized monolayers of stable cell lines (19). Remarkably, while in transformed cell lines, membrane fusion is rapidly followed by syncytium formation and cell death, in HAE, no syncytia are observed, and transepithelial resistance is maintained (11).
To gain insight into the mechanism of cytoplasm equilibration without cytopathic effect, we sought to follow the development of MV infections over time. Movie S1 in the supplemental material documents the development of HAE infectious centers over 3 days. Figure 3A presents 20 frames of this analysis, taken at 2-h intervals starting at 39 h postinoculation. Individual GFP-positive cells are first observed 24 to 36 h after basolateral MV-GFP inoculation. Typically, ∼8 h later, GFP expression is observed in a neighboring cell. From this point, viral spread occurs rapidly, and by 2 to 3 days postinfection, an infectious center reaches a 50- to 100-infected-cell stage. Most infectious centers grow slowly if at all from day 4 onward, and occasionally, progressive regression is observed during the 2nd week of infection (6).
FIG 3.

Time lapse microscopy analysis of MV infectious-center formation in HAE. HAE were infected from the basolateral surface with MV-GFP. (A) Growth of one infectious center shown at 2-h intervals starting at 39 h postinfection. An arrow in the 39-h panel indicates a single infected cell that infects another cell (at 47 h) and then a third cell (at 49 h). During the entire observation period, there was no visible cytopathic effect, and syncytium formation did not occur. (B and C) Higher-magnification, 10-min interval analyses of intercellular cytoplasmic GFP transfer at ∼48 h postinfection. (B) An inverted fluorescence microscope was used. (C) 3D reconstruction by confocal microscopy. The arrow indicates the point of initial GFP leakage.
We then sought to follow GFP transfer at a higher magnification. Movie S2 in the supplemental material documents the flow of GFP from an infected cell to an adjacent cell over 30 min. Figure 3B presents three frames of this movie, taken at 10-min intervals. The flow into the receiving cell begins at ∼8 min, and the level of fluorescence of this cell grows linearly over the next 12 to 15 min, reaching saturation/maximum level at minute 20 to 30. Movie S3 in the supplemental material shows similar cytoplasm transfer kinetics in multiple events.
We also followed GFP transfer using confocal microscopy and 3D reconstruction. Movie S4 in the supplemental material is an 18-h time lapse analysis of MV-GFP infection in HAE starting at ∼36 h after inoculation. Initially, only a single infected cell is visualized (green fluorescence). For more than 2 h, the infected cell maintains an unchanging shape, but then the cell contracts vertically, and within minutes, GFP diffuses to an adjacent columnar cell. Figure 3C presents three frames of this movie, taken at 10-min intervals. These data indicate that the cytoplasm leaked from the lateral surface of the MV-infected columnar cell. Interestingly, the leak originated at a near-apical location. We note that the apical junction complex that contains the TJ and AJ is usually located approximately at the position where the passage opened. Since the MV epithelial receptor nectin-4 is concentrated at the AJs of columnar cells, we postulate that it triggered the viral fusion apparatus expressed on the surfaces of infected cells to open an intercellular pore.
Afadin knockdown reduces MV infection levels.
The contraction of the infected cell before cytoplasm extrusion suggested the involvement of the apical actin and myosin belt in the passage-opening process. Since nectins are tethered to filamentous actin through afadin, we sought to document the location of these molecules in HAE.
The top panel of Fig. 4A presents an en face view of HAE. En face, afadin (green) colocalizes with the TJ protein ZO-1 (red); however, vertical sections indicate that ZO-1 is slightly apical of afadin (red and green arrows, respectively). This observation is expected in polarized epithelia, because TJs and AJs occur in tight conjunction (28). Figure 4B (top, white arrows) documents the colocalization of afadin (red) and nectin-4 (green). The afadin signal is focused at the AJ; however, the nectin-4 signal is more broadly distributed on the basolateral surface (Fig. 4B, bottom, yellow arrows).
FIG 4.

Localization of AJ proteins in HAE. Fixed and permeabilized HAE were immunostained for the TJ marker ZO-1 (red) or afadin (green) (A) and for afadin (red) or nectin-4 (green) (B). (Top) xy en face views; (bottom) xz vertical views. Red and green arrows indicate the localizations of ZO-1 and afadin, respectively. White arrows indicate the colocalization of afadin and nectin-4, and yellow arrows indicate nectin-4 expression. Cell nuclei were visualized using DAPI (blue). Images were acquired with a confocal laser scanning microscope (Zeiss LSM 510).
To assess whether afadin is a cofactor for MV infection in HAE, we transiently knocked it down using siRNA. Since well-differentiated HAE are very difficult to transfect, in order to achieve acceptable knockdown efficacy we delivered siRNA to freshly seeded, poorly differentiated HAE (29, 30). Using this method, we obtained 60% knockdown efficiency for an afadin-specific siRNA relative to expression with a scrambled siRNA negative control (Fig. 5A).
FIG 5.

Afadin knockdown reduces the efficiency of MV infection in HAE. Afadin was silenced in primary human airway epithelia by using a reverse transfection technique (35). (A) Afadin mRNA abundance following transfection with scrambled siRNA or afadin siRNA, as determined by RT-qPCR 24 h posttransfection. Twenty-four hours after siRNA transfection, MV-GFP was delivered to the basolateral surfaces of epithelial cells. Results are means for 3 experiments, each with samples from 3 donors; *, P < 0.001. (B) The mean fluorescence intensity (MFI) was measured using bioluminescence imaging 3 days after infection. (C and D) Representative images of MV spread, as monitored by GFP expression, in epithelia transfected with scrambled or afadin siRNA. Bars, 100 μm.
Following afadin knockdown, HAE were infected with MV-GFP, and 3 days later, MV infection efficiency was quantified using the mean GFP fluorescence intensity of the culture. Figure 5B documents that infection efficiency was reduced by ∼60% in the afadin siRNA-transfected cells. In addition, fewer and slightly smaller infectious centers were documented (Fig. 5C and D). Thus, afadin knockdown reduces MV infection levels. AJs contribute to TJ formation and the differentiation of polarized epithelia. Typically, up to 3 weeks is required for this model to fully differentiate. The transient knockdown of afadin may alter or delay the differentiation process; therefore, we next sought to examine the contribution of afadin in fully differentiated epithelia.
A nectin-4 mutant without the afadin-binding site inefficiently promotes viral spread.
The four C-terminal amino acids of nectin-4 (GHLV) bind afadin. Afadin, in turn, tethers nectin-4 to F-actin (31). We asked whether the deletion of these C-terminal residues results in inefficient MV spread in airway epithelial cells. To avoid the confounding effects of endogenous human nectin-4 expression in HAE, we used well-differentiated primary pig airway epithelia (PAE). We demonstrated recently that PAE are nonpermissive for MV replication because of a glutamic acid-to-glycine substitution at position 62 of pig nectin-4 (19). To deliver the human nectin-4 constructs into PAEs, we generated recombinant adenoviruses (Ad) expressing either human nectin-4 or a nectin-4 mutant with a truncation of four C-terminal residues (N4ΔGHLV).
We first confirmed the expression patterns of human nectin-4 and N4ΔGHLV. PAE were transduced with an adenovirus expressing human nectin-4 or the N4ΔGHLV mutant; 72 h postransduction, the expression pattern was determined by confocal microscopy. Figure 6A and B document similar expression patterns of human nectin-4 and N4ΔGHLV after PAE transduction. No human nectin-4 was detected in naïve PAE, confirming antibody specificity (Fig. 6C). Western blot analysis documented equivalent levels of protein production following transduction of CHO cells (Fig. 6D). Using an mCherry-expressing adenoviral vector and the identical delivery protocol in matched donors, we observed >90% transduction efficiency; however, the percentage of transduction as determined by human nectin-4 immunostaining was lower. Nectin-4 immunostaining of primary airway epithelia likely does not provide a fair estimate of the percentage of transduction.
FIG 6.

Relevance of the afadin-binding sequence in the nectin-4 cytoplasmic tail for MV epithelial infection. (A to C) Localization of human nectin-4 in PAE either transduced from the basolateral surface with an adenoviral vector expressing human nectin-4 (A) or human N4ΔGHLV (B) or mock transduced (C). Two days later, cells were fixed with 2% paraformaldehyde, permeabilized with 0.2% Triton X-100, and incubated overnight with rabbit polyclonal antibodies against human nectin-4. Nectin-4 was visualized with a secondary antibody conjugated to Alexa Fluor 546 (red). Cell nuclei were visualized using DAPI (blue). For each panel, both an xy en face view (top) and an xz vertical view (bottom) are shown. Images were acquired with a confocal laser scanning microscope. White arrows indicate basolateral localization; yellow arrows indicate junctional expression. (D) Analysis of Ad-nectin-4 and Ad-N4ΔGHLV expression levels in CHO cells. β-tub., β-tubulin. (E) PAE transduced with an adenoviral vector expressing either human nectin-4 or N4ΔGHLV were infected from either the apical or the basolateral surface with MV-GFP, and the infectious centers were counted. An adenoviral vector expressing mCherry served as a negative control. Results are means for 3 experiments, each with samples from 3 donors; *, P < 0.01.
We next tested whether expression of human nectin-4 in PAE is sufficient to confer MV infection. PAE were inoculated with an Ad expressing human nectin-4, N4ΔGHLV, or an irrelevant negative control (mCherry) for 4 h. Two days following Ad delivery, MV-GFP was applied to the apical or basolateral surface for 4 h. Infectious centers were counted 3 days following MV-GFP delivery. A recombinant adenovirus expressing human nectin-4 conferred basolateral entry and lateral spread of MV-GFP in PAE (Fig. 6E). N4ΔGHLV also conferred basolateral entry and lateral spread, but the efficacy of MV infection was reduced to ∼25% of that with human nectin-4. Thus, the binding of nectin-4 to afadin is necessary to achieve maximum levels of infection in airway epithelial cells. Because deletion of the nectin-4–afadin interaction did not reduce MV infection to background levels, we must conclude that other cellular proteins play a role. Taken together, these data suggest that in well-differentiated airway epithelial cells, the nectin-4/afadin complex contributes to MV infection efficiency.
DISCUSSION
We document how in HAE the cytoplasm of MV-infected columnar epithelial cells suddenly flows into adjacent cells through openings that form on their lateral surfaces. It appears likely that viral attachment proteins expressed on the surfaces of infected cells bind nectin-4 molecules located on the lateral sides of adjacent columnar cells. This interaction may trigger the unfolding of viral fusion protein trimers, opening intercellular pores.
In an effort to estimate the diameters of the intercellular pores, we quantified GFP fluorescence transfer from 20 infected donor cells to receiving cells. We analyzed these data with a 2-cell computational model of GFP diffusion. In this way, fluorescence intensity measurements are converted to an estimate of pore diameter by using publicly available VCell software (www.vcell.org) (32). Our preliminary analysis predicts that the MV-induced pores are wider than 250 nm, a size sufficient for the transit of viral ribonucleocapsids. Using transmission electron microscopy, the average distance between the cytoplasms of two adjacent columnar airway cells in the HAE model was estimated to be 10 to 15 μm. The diffusion coefficient of GFP in cytoplasm was previously determined to be 25 μm2 s−1 (33). The images in our time lapse movies were collected at 10-min intervals. Because the transfer process was completed within 30 min, only 2 to 3 data points were collected per cellular transfer (see movies S2 and S3 in the supplemental material). Ideally, 10 to 15 data points should provide a better estimate of the pore size. In future studies, we intend to improve the temporal resolution by collecting live-cell images at shorter intervals (1 to 2 min) and also by using MV expressing a photoactivatable variant of GFP that is more suitable for studying protein diffusion (34).
Interestingly, in cell lines, membrane fusion is rapidly followed by syncytium formation and cell death, but in HAE, no cytopathic effect is observed. Moreover, infected HAE maintain high transepithelial resistance. Thus, while the cytoplasmic contents of infected HAE mix, as documented by homogeneous GFP distribution, apical TJs remain functional. Like GFP, viral components may rapidly spread laterally. Since large infectious centers but no syncytia are detected in the tracheae of experimentally inoculated monkeys (7), rapid infection spread through intercellular pores may also occur in the airways of naturally infected hosts. It seems likely that eventually infectious centers may detach, provoking coughing and sneezing, which would favor aerosolization and facilitate efficient contagion.
We provide experimental proof that the nectin-4–afadin protein connection is necessary for efficient MV infectious-center formation in HAE. Afadin connects the cytoplasmic tail of nectin to the actin cytoskeleton, which may help the cell contract while fusion pores form (see movie S4 in the supplemental material). Interestingly, F-actin contributes to particle assembly by several paramyxoviruses, including SeV, RSV, and Newcastle disease virus (35–39). F-actin, which contributes to the release of MV particles from HeLa cells (40), also accumulates at the cellular junctions during MV transmission from dendritic cells to lymphocytes (41). Interestingly, very large amounts of actin have been detected in MV particles (42–44), and inhibitors of actin polymerization can restrict MV replication (45, 46). Moreover, it has been shown recently that different forms of the viral matrix protein associate preferentially with either F-actin or the cytoplasmic tail of the viral attachment protein, processes involved in particle assembly (47). Finally, disruption of F-actin in Madin-Darby canine kidney epithelial cells affects MV particle maturation (48). While these observations are all consistent with a role for F-actin in MV spread, the mechanisms most relevant for the spread of MV infections in HAE remain to be determined.
Finally, different classes of viruses, including positive- and negative-strand RNA viruses (49) and double-strand DNA viruses (50, 51), exploit nectins or related proteins to spread in their hosts. In addition, other viruses use TJ proteins, such as occludins, claudins, and junctional adhesion molecules, as receptors (52, 53). This is remarkable, because junctional proteins are not readily accessible. Our observations suggest that the cytoskeletal connection of nectin-4 facilitates rapid MV spread and that a similar process could favor other viral infections, thereby contributing to the repeated selection of junctional proteins as viral receptors.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Ultan Power, Mark Peeples, Peter Collins, and Biao He for generating recombinant viruses expressing reporter proteins. We thank Paul McCray, Jr., and Steven Varga for providing stocks of RSV, PIV5, and SeV. We acknowledge the support of the University of Iowa Genomics Division, In Vitro Models and Cell Culture Core, Viral Vector Core, and Cell Morphology Core.
This work was partially supported by National Institutes of Health grant R01 HL-105821 (to P.L.S.). Core facilities at the University of Iowa were partially supported by the National Institutes of Health (P01 HL-51670, P01 HL-091842) and the Center for Gene Therapy for Cystic Fibrosis (P30 DK-54759).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00821-15.
REFERENCES
- 1.Mateo M, Navaratnarajah CK, Cattaneo R. 2014. Structural basis of efficient contagion: measles variations on a theme by parainfluenza viruses. Curr Opin Virol 5:16–23. doi: 10.1016/j.coviro.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen SY, Anderson S, Kutty PK, Lugo F, McDonald M, Rota PA, Ortega-Sanchez IR, Komatsu K, Armstrong GL, Sunenshine R, Seward JF. 2011. Health care-associated measles outbreak in the United States after an importation: challenges and economic impact. J Infect Dis 203:1517–1525. doi: 10.1093/infdis/jir115. [DOI] [PubMed] [Google Scholar]
- 3.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 4.Ferreira CS, Frenzke M, Leonard VH, Welstead GG, Richardson CD, Cattaneo R. 2010. Measles virus infection of alveolar macrophages and dendritic cells precedes spread to lymphatic organs in transgenic mice expressing human signaling lymphocytic activation molecule (SLAM, CD150). J Virol 84:3033–3042. doi: 10.1128/JVI.01559-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lemon K, de Vries RD, Mesman AW, McQuaid S, van Amerongen G, Yuksel S, Ludlow M, Rennick LJ, Kuiken T, Rima BK, Geijtenbeek TB, Osterhaus AD, Duprex WP, de Swart RL. 2011. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog 7:e1001263. doi: 10.1371/journal.ppat.1001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinn PL, Williams G, Vongpunsawad S, Cattaneo R, McCray PB Jr. 2002. Measles virus preferentially transduces the basolateral surface of well-differentiated human airway epithelia. J Virol 76:2403–2409. doi: 10.1128/jvi.76.5.2403-2409.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frenzke M, Sawatsky B, Wong XX, Delpeut S, Mateo M, Cattaneo R, von Messling V. 2013. Nectin-4-dependent measles virus spread to the cynomolgus monkey tracheal epithelium: role of infected immune cells infiltrating the lamina propria. J Virol 87:2526–2534. doi: 10.1128/JVI.03037-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ludlow M, Lemon K, de Vries RD, McQuaid S, Millar EL, van Amerongen G, Yuksel S, Verburgh RJ, Osterhaus AD, de Swart RL, Duprex WP. 2013. Measles virus infection of epithelial cells in the macaque upper respiratory tract is mediated by subepithelial immune cells. J Virol 87:4033–4042. doi: 10.1128/JVI.03258-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mühlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB Jr, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leonard VH, Sinn PL, Hodge G, Miest T, Devaux P, Oezguen N, Braun W, McCray PB Jr, McChesney MB, Cattaneo R. 2008. Measles virus blind to its epithelial cell receptor remains virulent in rhesus monkeys but cannot cross the airway epithelium and is not shed. J Clin Invest 118:2448–2458. doi: 10.1172/JCI35454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeda M, Takeuchi K, Miyajima N, Kobune F, Ami Y, Nagata N, Suzaki Y, Nagai Y, Tashiro M. 2000. Recovery of pathogenic measles virus from cloned cDNA. J Virol 74:6643–6647. doi: 10.1128/JVI.74.14.6643-6647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karp PH, Moninger TO, Weber SP, Nesselhauf TS, Launspach JL, Zabner J, Welsh MJ. 2002. An in vitro model of differentiated human airway epithelia. Methods Mol Med 188:115–137. doi: 10.1385/1-59259-185-X:115. [DOI] [PubMed] [Google Scholar]
- 14.Pezzulo AA, Starner TD, Scheetz TE, Traver GL, Tilley AE, Harvey BG, Crystal RG, McCray PB Jr, Zabner J. 2011. The air-liquid interface and use of primary cell cultures are important to recapitulate the transcriptional profile of in vivo airway epithelia. Am J Physiol Lung Cell Mol Physiol 300:L25–L31. doi: 10.1152/ajplung.00256.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. 2002. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol 76:5654–5666. doi: 10.1128/JVI.76.11.5654-5666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Collins PL, Lamb RA, Pickles RJ. 2011. Comparison of differing cytopathic effects in human airway epithelium of parainfluenza virus 5 (W3A), parainfluenza virus type 3, and respiratory syncytial virus. Virology 421:67–77. doi: 10.1016/j.virol.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villenave R, Touzelet O, Thavagnanam S, Sarlang S, Parker J, Skibinski G, Heaney LG, McKaigue JP, Coyle PV, Shields MD, Power UF. 2010. Cytopathogenesis of Sendai virus in well-differentiated primary pediatric bronchial epithelial cells. J Virol 84:11718–11728. doi: 10.1128/JVI.00798-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mateo M, Navaratnarajah CK, Syed S, Cattaneo R. 2013. The measles virus hemagglutinin β-propeller head β4-β5 hydrophobic groove governs functional interactions with nectin-4 and CD46 but not those with the signaling lymphocytic activation molecule. J Virol 87:9208–9216. doi: 10.1128/JVI.01210-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mateo M, Navaratnarajah CK, Willenbring RC, Maroun JW, Iankov I, Lopez M, Sinn PL, Cattaneo R. 2014. Different roles of the three loops forming the adhesive interface of nectin-4 in measles virus binding and cell entry, nectin-4 homodimerization, and heterodimerization with nectin-1. J Virol 88:14161–14171. doi: 10.1128/JVI.02379-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takai Y, Nakanishi H. 2003. Nectin and afadin: novel organizers of intercellular junctions. J Cell Sci 116:17–27. doi: 10.1242/jcs.00167. [DOI] [PubMed] [Google Scholar]
- 21.Miyoshi J, Takai Y. 2007. Nectin and nectin-like molecules: biology and pathology. Am J Nephrol 27:590–604. doi: 10.1159/000108103. [DOI] [PubMed] [Google Scholar]
- 22.Reymond N, Fabre S, Lecocq E, Adelaide J, Dubreuil P, Lopez M. 2001. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with nectin1/PRR1 through V domain interaction. J Biol Chem 276:43205–43215. doi: 10.1074/jbc.M103810200. [DOI] [PubMed] [Google Scholar]
- 23.Takai Y, Irie K, Shimizu K, Sakisaka T, Ikeda W. 2003. Nectins and nectin-like molecules: roles in cell adhesion, migration, and polarization. Cancer Sci 94:655–667. doi: 10.1111/j.1349-7006.2003.tb01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duprex WP, McQuaid S, Hangartner L, Billeter MA, Rima BK. 1999. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J Virol 73:9568–9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He B, Paterson RG, Ward CD, Lamb RA. 1997. Recovery of infectious SV5 from cloned DNA and expression of a foreign gene. Virology 237:249–260. doi: 10.1006/viro.1997.8801. [DOI] [PubMed] [Google Scholar]
- 26.Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ. 2005. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol 23:222–226. doi: 10.1038/nbt1051. [DOI] [PubMed] [Google Scholar]
- 27.Rose SD, Kim DH, Amarzguioui M, Heidel JD, Collingwood MA, Davis ME, Rossi JJ, Behlke MA. 2005. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res 33:4140–4156. doi: 10.1093/nar/gki732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noyce RS, Richardson CD. 2012. Nectin 4 is the epithelial cell receptor for measles virus. Trends Microbiol 20:429–439. doi: 10.1016/j.tim.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 29.Krishnamurthy S, Behlke MA, Ramachandran S, Salem AK, McCray PB Jr, Davidson BL. 2012. Manipulation of cell physiology enables gene silencing in well-differentiated airway epithelia. Mol Ther Nucleic Acids 1:e41. doi: 10.1038/mtna.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramachandran S, Krishnamurthy S, Jacobi AM, Wohlford-Lenane C, Behlke MA, Davidson BL, McCray PB Jr. 2013. Efficient delivery of RNA interference oligonucleotides to polarized airway epithelia in vitro. Am J Physiol Lung Cell Mol Physiol 305:L23–L32. doi: 10.1152/ajplung.00426.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurita S, Ogita H, Takai Y. 2011. Cooperative role of nectin-nectin and nectin-afadin interactions in formation of nectin-based cell-cell adhesion. J Biol Chem 286:36297–36303. doi: 10.1074/jbc.M111.261768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLean PF, Cooley L. 2013. Protein equilibration through somatic ring canals in Drosophila. Science 340:1445–1447. doi: 10.1126/science.1234887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swaminathan R, Hoang CP, Verkman AS. 1997. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys J 72:1900–1907. doi: 10.1016/S0006-3495(97)78835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patterson GH, Lippincott-Schwartz J. 2002. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- 35.Greber UF, Way M. 2006. A superhighway to virus infection. Cell 124:741–754. doi: 10.1016/j.cell.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 36.Jeffree CE, Brown G, Aitken J, Su-Yin DY, Tan BH, Sugrue RJ. 2007. Ultrastructural analysis of the interaction between F-actin and respiratory syncytial virus during virus assembly. Virology 369:309–323. doi: 10.1016/j.virol.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 37.Kallewaard NL, Bowen AL, Crowe JE Jr. 2005. Cooperativity of actin and microtubule elements during replication of respiratory syncytial virus. Virology 331:73–81. doi: 10.1016/j.virol.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Katayama H, Hori M, Sato K, Kajita M, Ozaki H, Karaki H, Ohashi K, Kai C. 2004. Role of actin microfilaments in canine distemper virus replication in Vero cells. J Vet Med Sci 66:409–415. doi: 10.1292/jvms.66.409. [DOI] [PubMed] [Google Scholar]
- 39.Miazza V, Mottet-Osman G, Startchick S, Chaponnier C, Roux L. 2011. Sendai virus induced cytoplasmic actin remodeling correlates with efficient virus particle production. Virology 410:7–16. doi: 10.1016/j.virol.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Bohn W, Rutter G, Hohenberg H, Mannweiler K, Nobis P. 1986. Involvement of actin filaments in budding of measles virus: studies on cytoskeletons of infected cells. Virology 149:91–106. doi: 10.1016/0042-6822(86)90090-5. [DOI] [PubMed] [Google Scholar]
- 41.Koethe S, Avota E, Schneider-Schaulies S. 2012. Measles virus transmission from dendritic cells to T cells: formation of synapse-like interfaces concentrating viral and cellular components. J Virol 86:9773–9781. doi: 10.1128/JVI.00458-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stallcup KC, Raine CS, Fields BN. 1983. Cytochalasin B inhibits the maturation of measles virus. Virology 124:59–74. doi: 10.1016/0042-6822(83)90290-8. [DOI] [PubMed] [Google Scholar]
- 43.Tyrrell DL, Norrby E. 1978. Structural polypeptides of measles virus. J Gen Virol 39:219–229. doi: 10.1099/0022-1317-39-2-219. [DOI] [PubMed] [Google Scholar]
- 44.Udem SA. 1984. Measles virus: conditions for the propagation and purification of infectious virus in high yield. J Virol Methods 8:123–136. doi: 10.1016/0166-0934(84)90046-6. [DOI] [PubMed] [Google Scholar]
- 45.Berghäll H, Wallen C, Hyypia T, Vainionpaa R. 2004. Role of cytoskeleton components in measles virus replication. Arch Virol 149:891–901. doi: 10.1007/s00705-003-0264-9. [DOI] [PubMed] [Google Scholar]
- 46.Moyer SA, Baker SC, Horikami SM. 1990. Host cell proteins required for measles virus reproduction. J Gen Virol 71(Part 4):775–783. doi: 10.1099/0022-1317-71-4-775. [DOI] [PubMed] [Google Scholar]
- 47.Wakimoto H, Shimodo M, Satoh Y, Kitagawa Y, Takeuchi K, Gotoh B, Itoh M. 2013. F-actin modulates measles virus cell-cell fusion and assembly by altering the interaction between the matrix protein and the cytoplasmic tail of hemagglutinin. J Virol 87:1974–1984. doi: 10.1128/JVI.02371-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dietzel E, Kolesnikova L, Maisner A. 2013. Actin filaments disruption and stabilization affect measles virus maturation by different mechanisms. Virol J 10:249. doi: 10.1186/1743-422X-10-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Racaniello V. 2011. An exit strategy for measles virus. Science 334:1650–1651. doi: 10.1126/science.1217378. [DOI] [PubMed] [Google Scholar]
- 50.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakisaka T, Taniguchi T, Nakanishi H, Takahashi K, Miyahara M, Ikeda W, Yokoyama S, Peng YF, Yamanishi K, Takai Y. 2001. Requirement of interaction of nectin-1α/HveC with afadin for efficient cell-cell spread of herpes simplex virus type 1. J Virol 75:4734–4743. doi: 10.1128/JVI.75.10.4734-4743.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bergelson JM. 2009. Intercellular junctional proteins as receptors and barriers to virus infection and spread. Cell Host Microbe 5:517–521. doi: 10.1016/j.chom.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 53.Mateo M, Generous A, Sinn PL, Cattaneo R. 2015. Connections matter—how viruses use cell-cell adhesion components. J Cell Sci 128:431–439. doi: 10.1242/jcs.159400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.