

ABSTRACT
Whereas most viruses require only a single protein to bind to and fuse with cells, herpesviruses use multiple glycoproteins to mediate virus entry, and thus communication among these proteins is required. For most alphaherpesviruses, the minimal set of viral proteins required for fusion with the host cell includes glycoproteins gD, gB, and a gH/gL heterodimer. In the current model of entry, gD binds to a cellular receptor and transmits a signal to gH/gL. This signal then triggers gB, the conserved fusion protein, to insert into the target membrane and refold to merge the viral and cellular membranes. We previously demonstrated that gB homologs from two alphaherpesviruses, herpes simplex virus 1 (HSV-1) and saimiriine herpesvirus 1 (SaHV-1), were interchangeable. In contrast, neither gD nor gH/gL functioned with heterotypic entry glycoproteins, indicating that gD and gH/gL exhibit an essential type-specific functional interaction. To map this homotypic interaction site on gH/gL, we generated HSV-1/SaHV-1 gH and gL chimeras. The functional interaction with HSV-1 gD mapped to the N-terminal domains I and II of the HSV-1 gH ectodomain. The core of HSV-1 gL that interacts with gH also was required for functional homotypic interaction. The N-terminal gH/gL domains I and II are the least conserved and may have evolved to support species-specific glycoprotein interactions.
IMPORTANCE The first step of the herpesvirus life cycle is entry into a host cell. A coordinated interaction among multiple viral glycoproteins is required to mediate fusion of the viral envelope with the cell membrane. The details of how these glycoproteins interact to trigger fusion are unclear. By swapping the entry glycoproteins of two alphaherpesviruses (HSV-1 and SaHV-1), we previously demonstrated a functional homotypic interaction between gD and gH/gL. To define the gH and gL requirements for homotypic interaction, we evaluated the function of a panel of HSV-1/SaHV-1 gH and gL chimeras. We demonstrate that domains I and II of HSV-1 gH are sufficient to promote a functional, albeit reduced, interaction with HSV-1 gD. These findings contribute to our model of how the entry glycoproteins cooperate to mediate herpesvirus entry into the cell.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) infects humans and causes recurrent mucocutaneous lesions on the mouth, face, or genitalia. In rare instances, the infection can lead to meningitis or encephalitis. HSV-1 entry into cells requires four glycoproteins: gD, gH, gL, and gB (1–4). Fusion of viral envelope with the cell membrane requires interactions of these glycoproteins with each other and cellular receptors. In the current model of virus entry, gD binding to a cellular receptor activates a gH/gL heterodimer, and this step subsequently triggers gB, the conserved herpesvirus fusion protein, to mediate virus-cell or cell-cell membrane fusion (5).
Crystal structures have been solved for gD, gB, and gH/gL. A comparison of the crystal structures of gD alone (6) or in complex with receptor (7, 8) reveals a conformational change in the C-terminal region of the gD ectodomain that may serve as the trigger for fusion. Structures of gB homologs show that gB is a class III fusion protein (9–11). In contrast, the gH/gL structures do not resemble fusion proteins (12–14). gH/gL is believed to function as a regulator of fusion, possibly transmitting a signal from the gD-receptor complex to the gB fusion protein (5).
Despite multiple studies on the interaction of these four entry glycoproteins, details of the interactions among these glycoproteins are still under investigation, most likely because the interactions are low affinity and/or transient. Purified forms of gH/gL and gB have been shown to associate physically at low pH using a coflotation liposome binding assay (15). Coprecipitation experiments suggest that gD can interact physically with either gH/gL or gB independently (16). Physical interactions of all of the glycoprotein combinations (gD with gH/gL, gD with gB, and gH/gL with gB) have been reported using bimolecular fluorescence complementation (BiFC), but reports disagree over whether the gD interaction with gH/gL or gB requires the presence of a gD receptor (17–19). Disruption of the BiFC with monoclonal antibodies (MAbs) can map physical interaction sites on the glycoproteins (13, 20), but a physical interaction does not indicate necessarily a functional interaction. For example, although BiFC detects a physical interaction between gD-gB, this direct interaction may be dispensable for fusion (5).
gH and gL form a functional heterodimer (gH/gL) (1, 21, 22). The HSV-2 gH/gL structure is boot shaped and composed of three domains (13). The N-terminal gH domain, termed H1, interfaces extensively with gL, with subdomains H1A and H1B flanking either side of gL. H2 is a central helical domain, and H3 comprises a C-terminal β-sandwich. Conservation across the gH domains is uneven. The N-terminal domain H1 is the most divergent, and the membrane-proximal domains H2 and H3 are more conserved.
The Epstein-Barr virus (EBV) gH/gL and pseudorabies virus (PrV) gH structures have a more linear arrangement than HSV-2 gH/gL, with different interdomain packing and modified domain designations (12, 14). When the EBV gH/gL domain designations are applied to HSV gH/gL, the border between H1B and H2 is shifted C-terminally to HSV-1 gH residue 439. As a result, four helices from H2 that run parallel to a β-sheet in H1B are included in the same domain as H1B. This shift transforms an interdomain disulfide bond in HSV-1 gH (between residues 258 and 429) into an intradomain disulfide bond.
Four conserved cysteines in gL are essential for gH/gL association and function (23). The first 147 residues of gL are sufficient for gH association, but the first 161 residues are necessary for cotransport of gH/gL to the cell surface and for gL activity in viral entry and cell-cell fusion (24, 25). gL linker-insertion mutants demonstrate that gL plays a greater role in fusion than simply promoting gH surface expression (26).
To explore functional interactions among alphaherpesvirus entry glycoproteins, we recently attempted to replace HSV-1 entry glycoproteins with heterotypic homologs from several related nonhuman primate alphaherpesviruses. We determined that the gB homologs from HSV-1 and herpesvirus saimiri 1 (SaHV-1), which share 65% sequence identity, are functionally interchangeable (27). SaHV-1 infects squirrel monkeys as its natural host (Saimiri spp.) (28, 29). These results indicate that HSV-1 and SaHV-1 gB homologs retain the ability to interact with gD and/or gH/gL from the heterotypic virus. In contrast, the gD and gH/gL homologs from HSV-1 and SaHV-1 failed to mediate fusion in a heterotypic context. These results led to the conclusion that gD and gH/gL exhibit a homotypic-specific functional interaction. Using HSV-1/SaHV-1 gD chimeras, we mapped the homotypic gH/gL functional interaction site to the ectodomain of gD (27). A functional interaction with the ectodomain of gD is consistent with previous findings that a glycosylphosphatidylinositol (GPI)-anchored form of the gD mutant is functional (30) and that a purified form of the gD ectodomain can mediate fusion (31).
In the current work, we used chimeric constructs to define the homotypic site on gH/gL that interacts functionally with gD. The recent completion of the SaHV-1 genome sequence (32) allowed the alignment of glycoprotein sequences (Fig. 1). The crystal structures of HSV-2 gH/gL, EBV gH/gL, and PrV gH provided structural information for the design of chimeras (12–14). gH is an attractive candidate for domain swapping because each gH domain appears to fold autonomously. A lack of domain insertions and long interdomain extensions simplifies the design of chimeras. In addition, previous mutagenesis studies have shown that gH is relatively tolerant of insertions (33, 34).
FIG 1.

Amino acid alignment of HSV-1 and SaHV-1 gH and gL homologs. The alignments were generated using MAFFT, version 7, and then annotated. Identical (asterisk), strongly similar (colon), and weakly similar (period) residues are marked. Amino acid numbers are shown on the right. Cysteines are highlighted in gray, and disulfide bonds are shown as dashed lines. (A) HSV-1 gH (GenBank accession number AFE62849) and SaHV-1 gH (GenBank ADO13806.1) are predicted to be 820 and 842 amino acids after signal peptide cleavage, respectively. Domains H1A, H1B, H2, and H3 are shown, as defined by the HSV-2 gH/gL structure. Domains I and II, as defined by the EBV gH/gL structure, are in boldface. A disulfide bond that bridges H1B and H2 domains is shown. (B) HSV-1 gL (GenBank U53683.1) and SaHV-1 gL (GenBank ADO13827.1) are predicted to be 205 and 253 amino acids after signal peptide cleavage, respectively. The 161 N-terminal amino acids of HSV-1 gL that are sufficient for HSV-1 gH association are in boldface. An insertion after the signal sequence at the N terminus of mature SaHV-1 gL is marked.
MATERIALS AND METHODS
Cells.
Chinese hamster ovary (CHO-K1) cells (ATTC) were grown in Ham's F-12 medium supplemented with 10% fetal bovine serum (FBS).
Plasmids.
pT7EMCLuc plasmid encoding a firefly luciferase gene under the control of the T7 promoter and the pCAGT7 plasmid encoding T7 RNA polymerase were used to assay cell-cell fusion. Plasmids expressing HSV-1 (KOS) gB (pPEP98), gD (pPEP99), gH (pPEP100), and gL (pPEP101) were described previously (35), as well FLAG-tagged HSV-1 gB, gD, gH, and gL (27) and a plasmid expressing human nectin-1, pBG38 (36). Plasmids expressing wild-type SaHV-1 gB (pQF77), gD (pQF78), gH (pQF79), and gL (pQF80) and FLAG-tagged SaHV-1 gB (pQF81), gD (pQF82), gH (pQF83), and gL (pQF84) were described previously (27). To generate FLAG-tagged gH and gL chimeric constructs for this study, sequences were aligned using MAFFT, version 7 (http://mafft.cbrc.jp/alignment/server/) (Fig. 1). H327S (pQF116) and S335H (pQF117) swap the H1 domain of gH, as defined in the structure of HSV-2 gH/gL (13). H439S (pQF118) and S457H (pQF119) swap domains I and II of gH, as defined in the structure of EBV gH/gL (12). H803S (pQF120) and S835H (pQF121) swap the full gH ectodomains. H439S/SH1A (pQF141), H439S/SH1B (pQF142), and H439S/SH1AB (pQF143) add SaHV-1 gH H1A and H1B subdomain combinations (SH1A, SH1B, and SH1AB, respectively) into H439S, as defined in the HSV-2 gH/gL structure (13). H161S (pQF129) and S216H (pQF130) are gL chimeras in which the core of gL is swapped. For the S75H (pQF128) chimera, an N-terminal extension that is present in SaHV-1 but not HSV-1 gL was added to the N terminus of HSV-1 gL. All constructs were generated by subcloning using the pFLAG-myc-CMV-21 expression vector (E5776; Sigma). Constructs made for this study were sequenced by the Northwestern University Genomic Core Facility.
Western blotting.
Western blotting of whole-cell lysates of transfected cells was performed to determine overall expression of gH or gL chimeras from HSV-1 and SaHV-1. CHO-K1 cells seeded in six-well plates were transfected with 1.5 μg of empty vector or a plasmid encoding FLAG-tagged chimeras and/or wild-type gL or gH from HSV-1 and SaHV-1, using 5 μl of Lipofectamine 2000 (Invitrogen) diluted in Opti-MEM (Invitrogen). After 24 h of incubation, cells were detached using Versene (0.2 g of EDTA/liter in phosphate-buffered saline [PBS]), rinsed with PBS, and lysed with 200 μl of lysis buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 10 mM NaF, 1 mM Na3VO3, 1% Nonidet P-40) containing a protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN). Proteins were separated by SDS-PAGE on 4 to 20% gels after samples were boiled for 5 min under reducing conditions. Proteins were transferred to nitrocellulose and probed with rabbit anti-FLAG antibody (F7425; Sigma) at a 1:1,000 dilution for 1 h at room temperature. Anti-rabbit secondary antibody coupled to horseradish peroxidase (HRP) and ECL Western blotting detection reagents (GE Healthcare) were used to visualize bands. Molecular weight predictions were made using the Expasy Compute pI/MW compute tool (http://web.expasy.org/compute_pi/). N-glycosylation sites were predicted based on an N-X-S/T motif, and the likelihood of site usage was predicted using the NetNGlyc, version 1.0, server (http://www.cbs.dtu.dk/services/NetNGlyc/).
CELISA.
To evaluate the cell surface expression of the FLAG-tagged gH and gL from HSV-1 and SaHV-1, a cell-based enzyme-linked immunosorbent assay (CELISA) was performed as previously described (37). CHO-K1 cells seeded in 96-well plates were transfected with 60 ng of empty vector or plasmids encoding the gH or gL chimeras. FLAG-tagged gH chimeras were cotransfected with wild-type HSV-1 gL (pPEP101) or SaHV-1 gL (pQF80). FLAG-tagged gL chimeras were cotransfected with wild-type HSV-1 gH (pPEP100) or SaHV-1 gH (pQF79) using 30 ng of DNA for each component and 0.15 μl of Lipofectamine 2000. Cell surface protein was bound by anti-FLAG MAb (F1804; Sigma). The cells were washed and fixed in paraformaldehyde and glutaraldehyde. Expression was visualized using biotinylated goat anti-mouse IgG (Sigma), followed by streptavidin-HRP (GE Healthcare) and HRP substrate (BioFX). To examine reactivity with 52S and 53S MAbs (38), CHO-K1 cells in 96-well plates were transfected overnight with 55 ng of each of the plasmids encoding a gH chimera and HSV-1 gL using 0.35 μl of Lipofectamine 2000. 52S or 53S ascites diluted 1:5,000 was added to the cells for 1 h at room temperature. Cells were fixed in paraformaldehyde, and MAb binding was detected using goat anti-mouse-HRP (Cell Signaling Technology) followed by HRP substrate (BioFX).
Cell fusion assay.
Fusion activity of the gH and gL constructs was measured using a quantitative luciferase-based cell-cell fusion assay as previously described (35). CHO-K1 cells were seeded in six-well plates overnight. The CHO-K1 cells (effector cells) were transfected with 400 ng of each plasmid expressing T7 RNA polymerase, gB, gD, gH (or gH chimeras), and gL (or gL chimeras) from HSV-1 and SaHV-1 using 5 μl of Lipofectamine 2000. Target CHO-K1 cells were transfected with 400 ng of a plasmid carrying the firefly luciferase gene under the control of the T7 promoter, plus 1.5 μg of empty vector or plasmid expressing human nectin-1 (pBG38), using 5 μl of Lipofectamine 2000. Six hours after transfection, the cells were detached with Versene and resuspended in 1.5 ml of Ham's F-12 medium supplemented with 10% FBS. Effector and target cells were mixed in a 1:1 ratio and replated in 96-well plates. After 18 h at 37°C, luciferase activity was quantified using a luciferase reporter assay system (Promega) and a Wallac-Victor luminometer (PerkinElmer).
RESULTS
HSV-1 and SaHV-1 gH and gL exhibit homotypic requirements for function.
Our previous study demonstrated that gB homologs from SaHV-1 and HSV-1 could replace one another functionally, but the gD and gH/gL were nonfunctional. Although SaHV-1 gH/gL complexes cannot substitute functionally for HSV-1 gH/gL complexes and vice versa (27), whether the gH and gL homologs could function with heterotypic entry glycoproteins when swapped individually was unknown. HSV-1 and SaHV-1 gH homologs share 49% sequence identity, and HSV-1 and SaHV-1 gL homologs share 56% sequence identity. To determine if gH or gL homologs could be exchanged independently, we individually replaced gH and gL with their heterotypic counterparts in a cell-cell fusion assay. Our previous study showed that SaHV-1 can use the HSV-1 entry receptor nectin-1 to mediate cell-cell fusion (27), so we used nectin-1 as the receptor in our fusion assays in the current study. Replacing HSV-1 gH/gL with SaHV-1 gH/gL (or vice versa) resulted in a complete loss of fusion (Fig. 2), in agreement with our previous report (27). Similarly, replacing HSV-1 gH with SaHV-1 gH (or vice versa) resulted in a complete loss of fusion (Fig. 2). Replacing HSV-1 gL with SaHV-1 gL (or vice versa) also abrogated fusion (Fig. 2). These results indicate that gH and gL cannot be replaced functionally by heterotypic counterparts in combination or individually, despite high sequence conservation.
FIG 2.
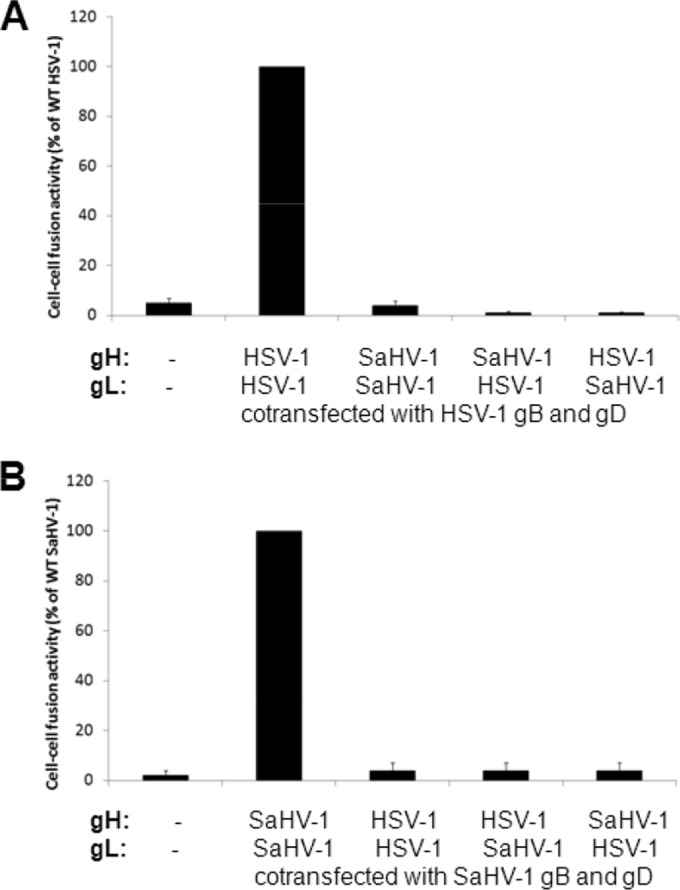
Heterotypic fusion activity of gH and gL from HSV-1 and SaHV-1. Target CHO cells were transfected with a reporter plasmid encoding luciferase under the control of the T7 promoter along with plasmids encoding nectin-1 or empty vector. Effector CHO cells were transfected with a plasmid encoding T7 polymerase and a combination of HSV-1 or SaHV-1 entry glycoproteins. gH and/or gL were swapped for heterotypic counterparts, as indicated. Target and effector cells were coincubated overnight, and luciferase activity was measured as an indication of cell-cell fusion. Data were normalized to the fusion activity measured when a homotypic set of glycoproteins was expressed (either all HSV-1 or all SaHV-1 glycoproteins). Error bars show standard deviation of three independent determinations. (A) Introduction of SaHV-1 glycoproteins into the HSV-1 entry set of glycoproteins. (B) Introduction of HSV-1 glycoproteins into the SaHV-1 entry set of glycoproteins. WT, wild type.
HSV-1 gL acts as a chaperone to promote HSV-1 gH surface expression (21), and HSV-1 gH anchors gL to the cell surface (22). To examine cell surface expression of the mixed dimers (HSV-1 gH/SaHV-1 gL and SaHV-1 gH/HSV-1 gL) by CELISA, cells were transfected with HSV-1 and SaHV-1 gH and gL alone or in various combinations. FLAG-tagged and untagged versions of both gH and gL constructs were cotransfected to allow specific detection of either protein at the surface using an antibody specific for the FLAG epitope tag. As expected, HSV-1 gH was not detected on the surface in the absence of gL (Fig. 3). Surface expression of HSV-1 gH was promoted by HSV-1 gL but not SaHV-1 gL, suggesting a defect in association between HSV-1 gH and SaHV-1 gL. Unexpectedly, SaHV-1 gH did not require gL coexpression for surface expression. gH homologs from other herpesviruses reach the membrane in the absence of gL, including pseudorabies virus (PrV), bovine herpesvirus 4, and murid herpesvirus 4 (39–41). Both HSV-1 and SaHV-1 gL were detected at the cell surface in the absence of gH (Fig. 3). The presence of HSV-1 gL on the cell surface in the absence of gH has been reported previously (26). Since gL lacks a transmembrane domain, its presence on the surface may be due to binding to the cell surface or incomplete cleavage of the signal peptide in the absence of gH.
FIG 3.
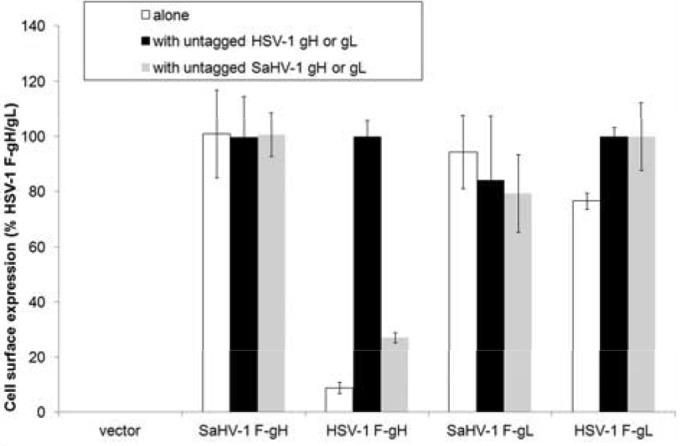
Expression of gH or gL from HSV-1 and SaHV-1. Cell surface expression of FLAG-tagged constructs was measured by CELISA. CHO-K1 cells in a 96-well plate were transfected overnight with empty vector or plasmids encoding FLAG (F)-tagged gH or gL from HSV-1 and SaHV-1. These plasmids were transfected alone (white bars) or with gL or gH from HSV-1 (black bars) or SaHV-1 (gray bars), as indicated. The cells were washed and incubated with an anti-FLAG M2 antibody. After multiple rinses, cells were fixed and incubated with an anti-mouse secondary antibody for detection. Error bars show standard deviations of three independent determinations. Background signal (vector) was abstracted from the values. Data were normalized to the expression level of HSV-1 gH/gL, which was set at 100%.
Expression and fusion activity of HSV-1/SaHV-1 gH chimeras.
To map the homotypic requirement on gH/gL, we generated chimeric gH molecules (Fig. 4A) and assessed their expression and fusion function. The chimeras were designed to swap gH structural domains, as defined by the crystal structures of HSV-2 and EBV gH/gL. The crystal structures of HSV-2 and EBV gH/gL adopt similar folds, but the relative positions of the domains differ in the two structures. As a result, domain designations at the N terminus of gH/gL differ somewhat between HSV-2 and EBV (Fig. 4A). EBV domain I roughly corresponds to HSV-2 domain H1A; however, EBV domain II encompasses the HSV-2 domain H1B plus additional downstream sequence. To create HSV-1/SaHV-1 chimeras, the gH N-terminal domains were swapped using both the HSV-2 H2B and EBV domain II definitions. For detection, a FLAG epitope tag was added to the N terminus of each chimera.
FIG 4.

Schematic representation of gH and gL chimeras from HSV-1 and SaHV-1. Black bars represent HSV-1 sequence, whereas gray bars represent SaHV-1 sequence. For all constructs, an exogenous signal sequence and N-terminal FLAG tag replace the native signal sequence. Amino acids present in each construct are indicated, using HSV-1 (bold font) and SaHV-1 (italics) numbering. (A) gH chimeras are shown. Domains of gH were defined by the HSV-2 or EBV gH/gL structures and are identified at the top. H439S includes HSV-1 gH D-I and D-II (as defined by the EBV gH/gL structure) and the SaHV-1 C terminus. (B) gL chimeras are shown. Lines above the wild-type HSV-1 and SaHV-1 bars delineate regions that were swapped. HSV-1 gL residues 1 to 161 are sufficient for gH association. Sequence alignment revealed an N-terminal insertion in SaHV-1 gL (A27-A75) that is absent in HSV-1 gL. The table on the right summarizes the cell surface expression of each construct when it is coexpressed with HSV-1 or SaHV-1 gL (first symbol) and the ability each construct to mediate fusion when it is coexpressed with entry glycoproteins from either HSV-1 or SaHV-1 (second symbol). A negative sign indicates insubstantial expression/fusion, a positive sign indicates reduced but substantial levels, and “wt” indicates nearly wild-type levels.
Cells were transfected with the FLAG-tagged gH chimeras together with HSV-1 gL or SaHV-1 gL. Western blotting of total cell lysates confirmed expression of gH chimeras (Fig. 5A). The chimeras migrated as expected. Molecular mass predictions based on the amino acid composition of the mature forms of the chimeras ranged from 88 to 92 kDa. Glycosylation accounts for the larger apparent size of the chimeras by SDS-PAGE, and differences in migration among the chimeras likely reflect differences in glycosylation. HSV-1 gH has eight potential N-glycosylation sites, whereas SaHV-1 gH has five potential N-glycosylation sites, only three of which are predicted to be used. H439A and H803S migrated more slowly than the other chimeras, and each has eight N-glycosylation sites. The other chimeras migrated faster and contain five to seven N-glycosylation sites, three to six of which are predicted to be used. The gH chimeras migrated similarly when coexpressed with either HSV-1 gL or SaHV-1 gL, with the exception of H803S. H803S migrated faster when coexpressed with heterotypic SaHV-1 gL than with HSV-1 gL, potentially due to incomplete glycosylation from improper cellular processing.
FIG 5.
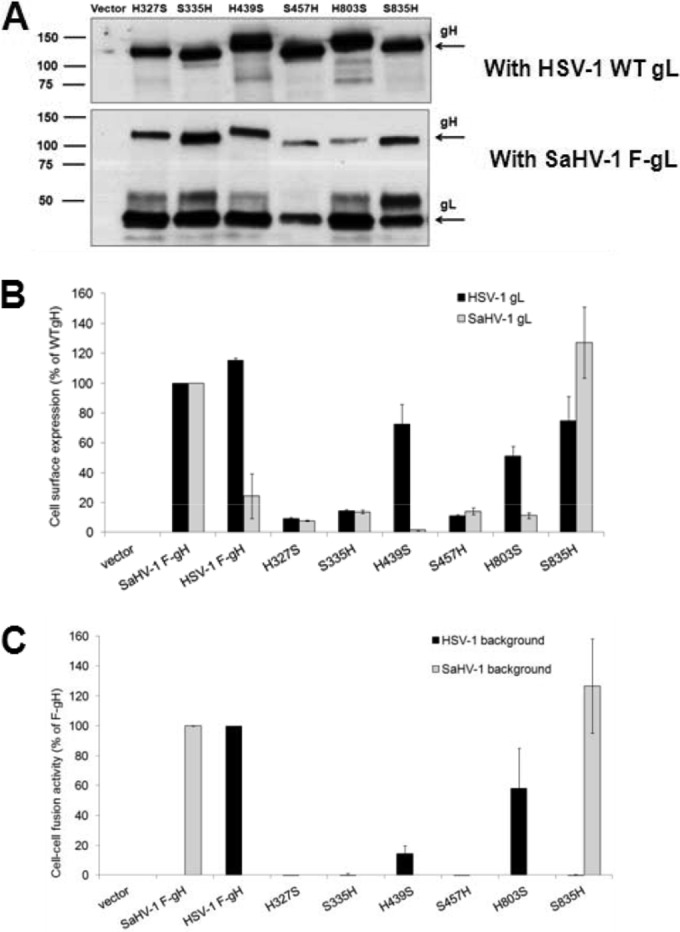
Expression and fusion activity of HSV-1 and SaHV-1 gH chimeras. (A) Total protein expression measured by Western blotting of cell lysates. CHO-K1 cells were transfected with plasmids encoding FLAG-tagged gH chimeras and either wild-type (WT) HSV-1 gL or FLAG (F)-tagged SaHV-1 gL. Cells lysates were resolved by SDS-PAGE and transferred to nitrocellulose. Blots were probed with rabbit anti-FLAG antibody. (B) Cell surface expression measured by CELISA. CHO-K1 cells in a 96-well plate were transfected overnight with plasmids encoding FLAG (F)-tagged gH from HSV-1 or SaHV-1, FLAG-tagged gH chimeras, or empty vector, together with wild-type HSV-1 (black bars) or SaHV-1 (gray bars) gL. The cells were rinsed and incubated with an anti-FLAG M2 MAb. After multiple rinses, cells were fixed and incubated with an anti-mouse secondary antibody for detection. Each bar shows the mean and standard deviation of three independent determinations. Background signals detected after transfection with vector alone were subtracted from the values. Data were normalized to the expression level of SaHV-1 F-gH with either HSV-1 gL or SaHV-1 gL. (C) Fusion activity. Target CHO-K1 cells were transfected with a reporter plasmid encoding luciferase under the control of the T7 promoter and a plasmid encoding nectin-1. Effector CHO-K1 cells were transfected with plasmids encoding FLAG (F)-tagged gH from HSV-1 or SaHV-1, FLAG-tagged gH chimeras, or empty vector, together with plasmids encoding T7 polymerase and gD, gB, and gL from HSV-1 or SaHV-1. The cells were mixed, and, after coincubation overnight, luciferase activity was measured as an indication of cell-cell fusion. The cell fusion results are expressed as percentage of wild-type HSV-1 and wild-type SaHV-1 gH activity, respectively. Background values (vector control) were subtracted, and the means and standard deviations of three independent experiments are shown.
Cell surface expression of the chimeras was assayed using CELISA (Fig. 5B). Three of the gH chimeras, H327S, S335H, and S457H, failed to express on the cell surface when cotransfected with either of the gL homologs. These gH chimeras may be misfolded and retained within the cell. Chimeras that included a longer portion of the HSV-1 ectodomain, H439S and H803S, were expressed at reduced but readily detectable levels on the cell surface when cotransfected with HSV-1 gL. This is consistent with the fact that gL associates with the N terminus of gH (13). Both of these chimeras failed to reach the surface when coexpressed with SaHV-1 gL, consistent with the observation that wild-type HSV-1 gH does not reach the surface when coexpressed with SaHV-1 gL (Fig. 3A). S835H was detected on the surface when coexpressed with either gL construct, as seen for the wild-type SaHV-1 gH. These results indicate that all gH chimeras were expressed but that only a subset were transported to the cell surface.
As expected, the three chimeras that failed express on the cell surface (H327S, S335H, and S457H) also failed to mediate cell-cell fusion (Fig. 5C). The two chimeras that were surface expressed only when cotransfected with HSV-1 gL (H439S and H803S) mediated reduced but detectable levels of cell-cell fusion when coexpressed with HSV-1 gL, gD, and gB. Neither of these chimeras functioned when coexpressed with SaHV-1 gL, as expected based on their expression profiles. The one chimera that reached the surface when cotransfected with either gL homolog, S835H, mediated fusion only when coexpressed with SaHV-1 gL, gD, and gB. The failure to mediate fusion with HSV-1 glycoproteins demonstrates that surface expression alone is insufficient for the function of the chimera. The gH/gL structure demonstrates that gH and gL require each other for proper folding (13) and that S835H requires a homotypic gL for function. S835H contains the entire SaHV-1 gH ectodomain and behaves similarly to wild-type SaHV-1 gH, indicating that the homotypic requirement exhibited by SaHV-1 gH maps to its ectodomain and not its transmembrane or cytoplasmic domain. The functionality of H439S maps the homotypic requirement exhibited by HSV-1 gH to domain I of the HSV-1 gH ectodomain.
HSV-1 gH domain I and II contain the homotypic functional interaction site.
To further examine the site within domain I of HSV-1 gH required for a functional homotypic interaction, derivatives of the H439S chimera were created. These chimeras contain shorter fragments of HSV-1 gH in order to refine the region required for a functional interaction with HSV-1 glycoproteins. HSV-1 gH domains H1A (G48-G115), H1B (P137-L327), or both H1A and H1B (G48-L327) were swapped with homologous sequence from SaHV-1 gH to create the chimeras H439S/SH1A, H439S/SH1B, and H439S/SH1AB, respectively (Fig. 4A). Loss of function with HSV-1 glycoproteins would indicate a disruption of the homotypic functional site. A restoration of function with SaHV-1 glycoproteins would map the homotypic functional site on SaHV-1 gH.
Expression and fusion function of these chimeras were assessed during coexpression of either HSV-1 or SaHV-1 entry glycoproteins. Western blot analysis of cell lysates showed that the chimeras migrated as expected (Fig. 6A). All three chimeras were expressed on the cell surface when cotransfected with HSV-1 gL but not with SaHV-1 gL (Fig. 6B). Thus, derivatives of the H439S chimera retained interactions with HSV-1 gL but did not gain interactions with SaHV-1 gL. None of the chimeras mediated fusion when they were coexpressed with SaHV-1 glycoproteins, as expected since SaHV-1 gL failed to promote the surface expression of the chimeras (Fig. 6C). Unfortunately, the chimeras also failed to mediate detectable fusion when coexpressed with HSV-1 glycoproteins, indicating that surface expression and association with HSV-1 gL are insufficient to mediate fusion.
FIG 6.

Expression and fusion activity of H439S and its derivatives. (A) Total protein expression measured by Western blotting of cell lysates. CHO-K1 cells were transfected with plasmids encoding FLAG-tagged gH chimeras and FLAG-tagged HSV-1 gL. Cells lysates were resolved by SDS-PAGE and transferred to nitrocellulose. Blots were probed with rabbit anti-FLAG antibody. (B) Cell surface expression measured by CELISA. CHO-K1 cells in a 96-well plate were transfected overnight with plasmids encoding FLAG-tagged gH chimeras or empty vector, together with wild-type HSV-1 or SaHV-1 gL. The cells were rinsed and incubated with an anti-FLAG M2 MAb. After multiple rinses, cells were fixed and incubated with an anti-mouse secondary antibody for detection. Each bar shows the mean and standard deviation of three independent determinations. Background signals detected after transfection with vector alone were subtracted from the values. Data for each set of glycoproteins were normalized to the expression level of H439S with HSV-1 gL. (C) Fusion activity. Target CHO-K1 cells were transfected with a reporter plasmid encoding luciferase under the control of the T7 promoter and a plasmid encoding nectin-1. Effector CHO-K1 cells were transfected with plasmids encoding FLAG-tagged gH from HSV-1 or SaHV-1, FLAG-tagged gH chimeras, or empty vector, together with plasmids encoding T7 polymerase and gD, gB, and gL from HSV-1 or SaHV-1. The cells were mixed, and, after coincubation overnight, luciferase activity was measured as an indication of cell-cell fusion. The cell fusion results are expressed as a percentage of wild-type HSV-1 gH activity. Background values (vector control) were subtracted, and the means and standard deviations of three independent experiments are shown. (D) Reactivity with conformation-specific MAbs measured by CELISA. CHO-K1 cells were transfected overnight with plasmids encoding HSV-1 gL and FLAG-tagged gH chimeras or empty vector. The cells were incubated with 52S or 53S MAbs, fixed, and incubated with an anti-mouse secondary antibody for detection. Background signals detected after transfection with vector alone were subtracted from the values. The standard deviations of triplicate samples are shown.
To examine the conformation of these gH chimeras, binding by the conformation-dependent neutralizing MAbs 52S and 53S (38) was determined by CELISA (Fig. 6D). The gH chimeras were coexpressed with HSV-1 gL, and the functional chimera H803S was included for comparison. Both H803S and H439S displayed nearly wild-type reactivity with MAb 53S; however, derivatives of the H439S containing H1A and/or H1B sequence from SaHV-1 gH failed to react with 53S. These data suggest that elements of the 53S epitope lie within the H1A and H1B regions (25) or that these derivative chimeras are misfolded. MAb 52S failed to bind to H439S and its derivative chimeras, most likely because the 52S epitope maps to domain III (42), a region comprised of SaHV-1 gH sequence in these chimeras.
These results suggest that the full HSV-1 gH domains I and II (residues 1 to 439) are required for functional interaction with HSV-1 entry glycoproteins, including residues 1 to 48 and/or residues 328 to 439. Since our previous study mapped this homotypic fusion requirement to an interaction between gH/gL and gD, this work suggests that gH domains I and II within HSV-1 gH/gL exhibit a functional interaction with HSV-1 gD.
Expression and fusion activity of HSV-1 and SaHV-1 gL chimeras.
To examine the contribution of gL to the functional interaction with gD, FLAG-tagged gL chimeras containing HSV-1 and SaHV-1 sequence were created (Fig. 4B). The first 161 residues of HSV-1 gL are sufficient for gH association (25) and were swapped with homologous SaHV-1 sequence to create chimeras H161S and S216H. SaHV-1 gL has an extended N terminus compared to that of HSV-1 gL, and this SaHV-1 gL N-terminal region was added to HSV-1 gL to create S75H.
Cell surface expression was evaluated after cotransfection of plasmids encoding either wild-type HSV-1 gH or SaHV-1 gH. gL lacks a transmembrane domain, and gH serves to anchor gL to the cell surface. As shown in Fig. 3, both HSV-1 and SaHV-1 gL are retained on the cell surface also in the absence of gH. Western blotting of total cell lysate from cells transfected with the chimeric constructs confirmed expression of the gL chimeras (Fig. 7A). Based on amino acid composition, HSV-1 gL and SaHV-1 gL are predicted to be 23 kDa and 28 kDa, respectively. Each wild-type gL homolog and gL chimera contains a single N-glycosylation site. The slower-migrating bands likely represent aggregation of the gL chimeras. The presence of the N-terminal 75 residues of SaHV-1 gL appears to enhance the aggregation. All of the gL chimeras had reduced but detectable levels of surface expression when they were coexpressed with either wild-type gH homolog (Fig. 7B).
FIG 7.
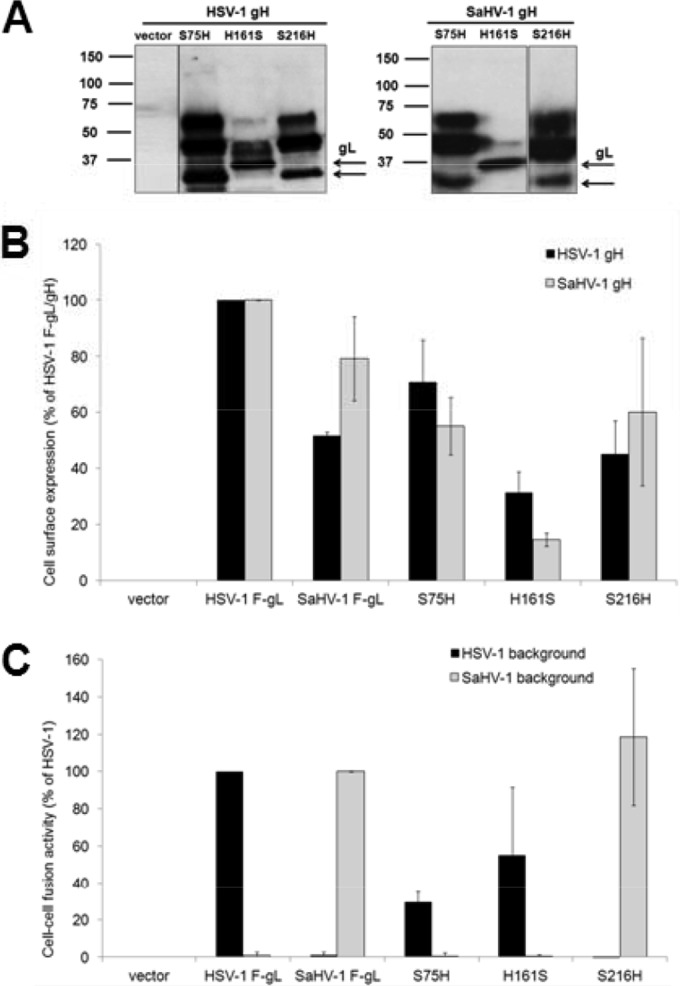
Expression and fusion activity of HSV-1 and SaHV-1 gL chimeras. (A) Total protein expression measured by Western blotting of cell lysates. CHO-K1 cells were transfected with plasmids encoding FLAG-tagged gL chimeras and wild-type HSV-1 or SaHV-1 gH. Cell lysates were resolved by SDS-PAGE and transferred to nitrocellulose. Blots were probed with rabbit anti-FLAG antibody. Arrows mark the bands that correspond to HSV-1 gL and SaHV-1 gL. (B) Cell surface expression measured by CELISA. CHO-K1 cells in a 96-well plate were transfected overnight with plasmids encoding FLAG (F)-tagged gL from HSV-1 or SaHV-1, FLAG-tagged gL chimeras, or empty vector, together with wild-type HSV-1 or SaHV-1 gH. The cells were rinsed and incubated with an anti-FLAG M2 MAb. After multiple rinses, cells were fixed and incubated with an anti-mouse secondary antibody for detection. Each bar shows the mean and standard deviation of three independent determinations. Background signals detected after transfection with vector alone were subtracted from the values. Data were normalized to the expression level of HSV-1 FLAG-tagged gL with either HSV-1 gH or SaHV-1 gH. (C) Fusion activity. Target CHO-K1 cells were transfected with a reporter plasmid encoding luciferase under the control of the T7 promoter and a plasmid encoding nectin-1. Effector CHO-K1 cells were transfected with plasmids encoding FLAG-tagged gL from HSV-1 or SaHV-1, FLAG-tagged gL chimeras, or empty vector, together with plasmids encoding T7 polymerase and gD, gB, and gH from HSV-1 or SaHV-1. The cells were mixed, and, after coincubation overnight, luciferase activity was measured as an indication of cell-cell fusion. The cell fusion results are expressed as a percentage of wild-type HSV-1 and wild-type SaHV-1 gL activity, respectively. Background values (vector control) were subtracted, and the means and standard deviations of three independent experiments are shown.
Fusion function of the gL chimeras corresponded to the N-terminal gH binding region of the chimera. The gL chimera with N-terminal sequence from HSV-1, H161S, retained a fusion function when cotransfected with HSV-1 glycoproteins but not SaHV-1 glycoproteins (Fig. 7C). Similarly, the gL chimeras with N-terminal sequence from SaHV-1 gL, S216H, retained a fusion function when they were cotransfected with SaHV-1 glycoproteins but not HSV-1 glycoproteins. For the gL chimera S75H, the addition of the extended N-terminal SaHV-1 gL sequence to HSV-1 gL did not impart a capacity for functional interaction with SaHV-1 glycoproteins and did not abolish functional interaction with HSV-1 glycoproteins. The results suggest that the C terminus of gL is not critical for a homotypic interaction with gD.
DISCUSSION
We recently reported that SaHV-1 gH/gL cannot substitute for HSV-1 gH/gL in fusion (27), despite high sequence conservation between the viruses. In this study, we extended our analysis of this homotypic requirement for function. We demonstrate that gH and gL are not individually interchangeable (Fig. 2). SaHV-1 gH does not function with HSV-1 gL, gD, and gB, and HSV-1 gH does not function with SaHV-1 gL, gD, and gB. Similarly, both gL homologs fail to function in a heterotypic context. SaHV-1 gH and both gL homologs reached the cell surface when expressed alone (Fig. 3), so surface expression is insufficient for function. We found that a homotypic gH/gL complex was required for fusion since mixed complexes did not function in fusion. Combined with our previous study (27), these results indicate that gH/gL interacts with gD as a functional unit.
HSV-1/SaHV-1 gH chimeras H439S, H803S, and S835H demonstrate that the homotypic requirement for function maps to the gH ectodomain (Fig. 5C). This finding is consistent with reports that a purified form of the HSV gH/gL ectodomains can mediate fusion in the presence of gD and gB (5). Other reports demonstrate that the gH transmembrane domain and short cytoplasmic tail affect fusion. Deletion of the final six residues of the gH cytoplasmic tail has no effect on fusion, but substitutions or deletions within the proximal region of the tail inhibit entry and syncytium formation (43–45). Similarly, an insertion at the border of the gH transmembrane and cytoplasmic tail abrogates the fusion function without affecting surface expression (33). Substitution of the gH transmembrane and/or cytoplasmic domains with heterologous sequence from CD8 or gD similarly abrogated the fusion function (43). Although this heterologous substitution was not permitted, our chimeras H803S and S835H demonstrate that the gH transmembrane and cytoplasmic domains can be replaced by homologous gH sequence from a similar virus. Our results suggest that the functional defects in the previously described gH transmembrane and cytoplasmic tail mutants are not due to a loss of the ability to interact with gD. The functional homotypic gD-gH/gL interaction maps to their ectodomains (27).
Results from the chimera H439S mapped the homotypic functional interaction site to domains I and II of HSV-1 gH (Fig. 6C). In combination with our previous study (27), we conclude that HSV-1 gH domains I and II functionally interact with gD. The N-terminal gH domains I and II are less conserved than the C-terminal domains III and IV and may have evolved to promote type-specific interactions with entry glycoproteins. The EBV receptor-binding protein gp42 was shown recently to interact with gH at the domain II-III interface using electron microscopy of purified protein complexes (46).
The HSV-2 gH/gL structure was solved using a purified soluble form of gH/gL that is called gHΔ48/gL and lacks the N-terminal 28 gH residues after the signal sequence (13). Recently, gHΔ48/gL was reported to mediate low levels of fusion in the absence of gD and a gD receptor (47). gHΔ48/gL may represent a partially triggered form of gH/gL. This implies that the gD interaction with gH/gL may alter the conformation of the N-terminal domain I in wild-type gH/gL, a scenario that is consistent with gD exhibiting a functional interaction with gH/gL domains I and II.
The chimera H439S demonstrated that the HSV-1 gH domains I and II could be swapped as a functional unit, whereas the chimera H327S showed that the HSV-1 gH H1 domain was unable to be transferred as a functional unit (Fig. 5). Similarly, chimeras H439S/SH1A and H439S/SH1B demonstrated that the subdomains H1A and H1B failed to retain function when swapped individually (Fig. 6). These results support the functional relevance of the domain designations proposed in the EBV gH/gL and PrV gH structures (12, 14). A disulfide bond between HSV-1 gH cysteines 258 and 429 bridges the domains designated H1B and H2 (13). Substitution of the H1 domain alone may fail to reconstitute that disulfide bond properly. The C258-C429 disulfide bond is not conserved in SaHV-1 gH. In SaHV-1 gH, the residue that corresponds to HSV-1 C285 is a serine. Although a disulfide bond between cysteines 258 and 429 is not essential for HSV-1 gH function (23), the presence of two free cysteines in the H327S chimera may contribute to its failure to reach the cell surface (Fig. 5B).
Homotypic gL was required to support the functional interaction between gH/gL and gD. Residues downstream from HSV-1 gL residue 161 did not contribute to the homotypic functional interaction with gD (Fig. 7), nor are they required for gH/gL function (24). The extended N terminus of SaHV-1 gL also failed to contribute to the homotypic specificity of the gH/gL-gD interaction.
gH/gL likely has multiple interaction sites that may change in conformation as fusion is triggered. gH/gL has been reported to interact with both gD and gB (16, 17, 19), and neutralizing MAbs map to opposite faces of HSV gH/gL (13). The neutralizing MAb LP11 that maps to domain II was reported to block the gH/gL-gB physical interaction, based on bimolecular fluorescence complementation (13) (Fig. 8). The neutralizing MAb 52S maps to domain III (Fig. 8), on the opposite face of gH/gL from LP11 (38, 42).
FIG 8.

HSV-2 gH/gL structure. (A) Linear representations of HSV-1 gH and gL including known disulfide bonds in gH. gH domain designations based on the HSV-2 gH/gL (13) and EBV gH/gL (12) structures are labeled above and below gH, respectively. (B) The HSV-2 gH/gL ectodomain (Protein Data Bank [PDB] accession number 3M1C) is shown from the front and back. Coloring is identical in both images. Domain designations from HSV-2 (H1A, H1B, H2, and H3) and EBV (D-I, D-II, D-III, and D-IV) are labeled on the left and right, respectively. The yellow helices are assigned to different domains in the two naming schemes. gH domain H1 flanks gL (blue) and is divided into H1A (purple) and H1B (green). Domain H2 (yellow and orange) and domain H3 (red) are shown. Using EBV gH/gL designations, domain I comprises gL (blue) and the N-terminal region of gH (purple). Domain II (green and yellow) correlates to H1B and part of H2. Domains III (orange) and IV (red) are shown. A disulfide bond forms between domains H1B and H2 (yellow and green spheres). Mutations that confer resistance to neutralization by MAbs LP11 (magenta spheres) or 52S (cyan spheres) are shown. LP11 binding is proposed to overlap a gB interaction site. Chimera H439S was functional when coexpressed with HSV-1 gL, gB, and gD and includes domains I and II (purple, blue, green, and yellow) from HSV-1, ending at gH residue S439 (red spheres). TM, transmembrane domain.
As expected from previous reports (21), HSV-1 gH did not reach the cell surface without gL. Unexpectedly, SaHV-1 gH was expressed on the cell surface in the absence of gL; however, SaHV-1 gH was nonfunctional in the absence of SaHV-1 gL. The core of HSV-1 or SaHV-1 gL was required for homotypic gH/gL function. This gL core likely supports the proper conformation of gH. In addition, gL may directly participate in triggering fusion mediated by gB. A functional interaction between gL and gB was mapped previously to two gL residues using gL chimeras from EBV and a related gammaherpesvirus, rhesus lymphocryptovirus (48).
ACKNOWLEDGMENTS
We thank N. Susmarski for timely and excellent technical assistance and members of the Longnecker laboratory for their help in these studies. Sequencing services were performed at the Northwestern University Genomics Core Facility.
R.L. is the Dan and Bertha Spear Research Professor in Microbiology-Immunology. This work was supported by NIH grants CA021776 and AI067048 to R.L.
REFERENCES
- 1.Roop C, Hutchinson L, Johnson DC. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J Virol 67:2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner A, Bruun B, Minson T, Browne H. 1998. Glycoproteins gB, gD and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J Virol 72:873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. 2005. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J 24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell 8:169–179. doi: 10.1016/S1097-2765(01)00298-2. [DOI] [PubMed] [Google Scholar]
- 8.Di Giovine P, Settembre EC, Bhargava AK, Luftig MA, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C, Carfi A. 2011. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog 7:e1002277. doi: 10.1371/journal.ppat.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc Natl Acad Sci U S A 106:2880–2885. doi: 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Backovic M, Jardetzky TS. 2011. Class III viral membrane fusion proteins. Adv Exp Med Biol 714:91–101. doi: 10.1007/978-94-007-0782-5_3. [DOI] [PubMed] [Google Scholar]
- 11.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 12.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc Natl Acad Sci U S A 107:22641–22646. doi: 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney MC, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc Natl Acad Sci U S A 107:22635–22640. doi: 10.1073/pnas.1011507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cairns TM, Whitbeck JC, Lou H, Heldwein EE, Chowdary TK, Eisenberg RJ, Cohen GH. 2011. Capturing the herpes simplex virus core fusion complex (gB-gH/gL) in an acidic environment. J Virol 85:6175–6184. doi: 10.1128/JVI.00119-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL through the C-terminal profusion domain. J Biol Chem 284:17370–17382. doi: 10.1074/jbc.M109.005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avitabile E, Forghieri C, Campadelli-Fiume G. 2007. Complexes between herpes simplex virus glycoproteins gD, gB gH detected in cells by complementation of split enhanced green fluorescence protein. J Virol 81:11532–11537. doi: 10.1128/JVI.01343-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Avitabile E, Forghieri C, Campadelli-Fiume G. 2009. Cross talk among the glycoproteins involved in herpes simplex virus entry and fusion: the interaction between gB and gH/gL does not necessarily require gD. J Virol 83:10752–10760. doi: 10.1128/JVI.01287-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci U S A 104:18718–18723. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of Herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J Virol 84:3825–3834. doi: 10.1128/JVI.02687-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J Virol 66:2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dubin G, Jiang H. 1995. Expression of herpes simplex virus type 1 glycoprotein L (gL) in transfected mammalian cells: evidence that gL is not independently anchored to cell membranes. J Virol 69:4564–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cairns TM, Landsburg DJ, Charles Whitbeck J, Eisenberg RJ, Cohen GH. 2005. Contribution of cysteine residues to the structure and function of herpes simplex virus gH/gL. Virology 332:550–562. doi: 10.1016/j.virol.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Klyachkin YM, Stoops KD, Geraghty RJ. 2006. Herpes simplex virus type 1 glycoprotein L mutants that fail to promote trafficking of glycoprotein H and fail to function in fusion can induce binding of glycoprotein L-dependent anti-glycoprotein H antibodies. J Gen Virol 87:759–767. doi: 10.1099/vir.0.81563-0. [DOI] [PubMed] [Google Scholar]
- 25.Peng T, Ponce de Leon M, Novotny MJ, Jiang H, Lambris JD, Dubin G, Spear PG, Cohen GH, Eisenberg RJ. 1998. Structural and antigenic analysis of a truncated form of the herpes simplex virus glycoprotein gH-gL complex. J Virol 72:6092–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan Q, Lin E, Spear PG. 2009. Insertional mutations in herpes simplex virus type 1 gL identify functional domains for association with gH and for membrane fusion. J Virol 83:11607–11615. doi: 10.1128/JVI.01369-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan Q, Longnecker R, Connolly SA. 2014. Substitution of herpes simplex virus 1 entry glycoproteins with those of saimiriine herpesvirus 1 reveals a gD-gH/gL functional interaction and a region within the gD profusion domain that is critical for fusion. J Virol 88:6470–6482. doi: 10.1128/JVI.00465-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daniel MD, Karpas A, Melendez LV, King NW, Hunt RD. 1967. Isolation of herpes-T virus from a spontaneous disease in squirrel monkeys (Saimiri sciureus). Arch Gesamte Virusforsch 22:324–331. doi: 10.1007/BF01242953. [DOI] [PubMed] [Google Scholar]
- 29.King NW, Hunt RD, Daniel MD, Melendez LV. 1967. Overt herpes-T infection in squirrel monkeys (Saimiri sciureus). Lab Anim Care 17:413–423. [PubMed] [Google Scholar]
- 30.Jones NA, Geraghty RJ. 2004. Fusion activity of lipid-anchored envelope glycoproteins of herpe simplex virus type 1. Virology 324:213–228. doi: 10.1016/j.virol.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 31.Cocchi F, Fusco D, Menotti L, Gianni T, Eisenberg RJ, Cohen GH, Campadelli-Fiume G. 2004. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc Natl Acad Sci U S A 101:7445–7450. doi: 10.1073/pnas.0401883101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tyler S, Severini A, Black D, Walker M, Eberle R. 2011. Structure and sequence of the saimiriine herpesvirus 1 genome. Virology 410:181–191. doi: 10.1016/j.virol.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson JO, Lin E, Spear PG, Longnecker R. 2010. Insertion mutations in herpes simplex virus 1 glycoprotein H reduce cell surface expression, slow the rate of cell fusion, or abrogate functions in cell fusion and viral entry. J Virol 84:2038–2046. doi: 10.1128/JVI.02215-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galdiero M, Whiteley A, Bruun B, Bell S, Minson T, Browne H. 1997. Site-directed and linker insertion mutagenesis of herpes simplex virus type 1 glycoprotein H. J Virol 71:2163–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pertel P, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324. doi: 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 36.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 37.Lin E, Spear PG. 2007. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc Natl Acad Sci U S A 104:13140–13145. doi: 10.1073/pnas.0705926104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Showalter SD, Zweig M, Hampar B. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect Immun 34:684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klupp BG, Fuchs W, Weiland E, Mettenleiter TC. 1997. Pseudorabies virus glycoprotein L is necessary for virus infectivity but dispensable for virion localization of glycoprotein H. J Virol 71:7687–7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lete C, Machiels B, Stevenson PG, Vanderplasschen A, Gillet L. 2012. Bovine herpesvirus type 4 glycoprotein L is nonessential for infectivity but triggers virion endocytosis during entry. J Virol 86:2653–2664. doi: 10.1128/JVI.06238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gillet L, May JS, Colaco S, Stevenson PG. 2007. Glycoprotein L disruption reveals two functional forms of the murine gammaherpesvirus 68 glycoprotein H. J Virol 81:280–291. doi: 10.1128/JVI.01616-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gompels UA, Carss AL, Saxby C, Hancock DC, Forrester A, Minson AC. 1991. Characterization and sequence analyses of antibody-selected antigenic variants of herpes simplex virus show a conformationally complex epitope on glycoprotein H. J Virol 65:2393–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harman A, Browne H, Minson T. 2002. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J Virol 76:10708–10716. doi: 10.1128/JVI.76.21.10708-10716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson DW, Davis-Poynter N, Minson AC. 1994. Mutations in the cytoplasmic tail of herpes simplex virus glycoprotein H suppress cell fusion by a syncytial strain. J Virol 68:6985–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Browne HM, Bruun BC, Minson AC. 1996. Characterization of herpes simplex virus type 1 recombinants with mutations in the cytoplasmic tail of glycoprotein H. J Gen Virol 77:2569–2573. doi: 10.1099/0022-1317-77-10-2569. [DOI] [PubMed] [Google Scholar]
- 46.Sathiyamoorthy K, Jiang J, Hu YX, Rowe CL, Mohl BS, Chen J, Jiang W, Mellins ED, Longnecker R, Zhou ZH, Jardetzky TS. 2014. Assembly and architecture of the EBV B cell entry triggering complex. PLoS Pathog 10:e1004309. doi: 10.1371/journal.ppat.1004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Atanasiu D, Cairns TM, Whitbeck JC, Saw WT, Rao S, Eisenberg RJ, Cohen GH. 2013. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. mBio 4(2):e00046–13. doi: 10.1128/mBio.00046-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plate AE, Smajlovic J, Jardetzky TS, Longnecker R. 2009. Functional analysis of glycoprotein L (gL) from rhesus lymphocryptovirus in Epstein-Barr virus-mediated cell fusion indicates a direct role of gL in gB-induced membrane fusion. J Virol 83:7678–7689. doi: 10.1128/JVI.00457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]