

ABSTRACT
We have previously shown that the Epstein-Barr virus (EBV) likely encodes hundreds of viral long noncoding RNAs (vlncRNAs) that are expressed during reactivation. Here we show that the EBV latency origin of replication (oriP) is transcribed bi-directionally during reactivation and that both leftward (oriPtLs) and rightward (oriPtRs) transcripts are largely localized in the nucleus. While the oriPtLs are most likely noncoding, at least some of the oriPtRs contain the BCRF1/vIL10 open reading frame. Nonetheless, oriPtR transcripts with long 5′ untranslated regions may partially serve noncoding functions. Both oriPtL and oriPtR transcripts are expressed with late kinetics, and their expression is inhibited by phosphonoacetic acid. RNA sequencing (RNA-seq) analysis showed that oriPtLs and oriPtRs exhibited extensive “hyperediting” at their Family of Repeat (FR) regions. RNA secondary structure prediction revealed that the FR region of both oriPtLs and oriPtRs may form large evolutionarily conserved and thermodynamically stable hairpins. The double-stranded RNA-binding protein and RNA-editing enzyme ADAR was found to bind to oriPtLs, likely facilitating editing of the FR hairpin. Further, the multifunctional paraspeckle protein, NONO, was found to bind to oriPt transcripts, suggesting that oriPts interact with the paraspeckle-based innate antiviral immune pathway. Knockdown and ectopic expression of oriPtLs showed that it contributes to global viral lytic gene expression and viral DNA replication. Together, these results show that these new vlncRNAs interact with cellular innate immune pathways and that they help facilitate progression of the viral lytic cascade.
IMPORTANCE Recent studies have revealed that the complexity of lytic herpesviral transcriptomes is significantly greater than previously appreciated with hundreds of viral long noncoding RNAs (vlncRNAs) being recently discovered. Work on cellular lncRNAs over the past several years has just begun to give us an initial appreciation for the array of functions they play in complex formation and regulatory processes in the cell. The newly identified herpesvirus lncRNAs are similarly likely to play a variety of different functions, although these functions are likely tailored to specific needs of the viral infection cycles. Here we describe novel transcripts derived from the EBV latency origin of replication. We show that they are hyperedited, that they interact with a relatively newly appreciated antiviral pathway, and that they play a role in facilitating viral lytic gene expression. These investigations are a starting point to unraveling the complex arena of vlncRNA function in herpesvirus lytic replication.
INTRODUCTION
The Epstein-Barr virus (EBV) is an oncogenic human herpesvirus that infects 95% of the world's population (1). Although infection is typically asymptomatic, the virus will persist in the host for the duration of its life. In most immunocompetent individuals, the virus poses little serious health risk and its presence is unremarkable. Nevertheless, in some individuals, additional events such as genetic alterations and/or immunosuppression lead to life-threatening epithelial and immune cell cancers, including lymphomas, carcinomas, and lymphoproliferative disorders (1).
A defining feature of herpesviruses is their use of two functionally distinct programs that together facilitate their long-lasting tenure in the host, the replicative phase where infectious virus is produced and a “latent” phase where other aspects of the virus infection cycle are carried out. While latency is critical for long-term survival in the host, spread from cell to cell and from host to host requires the lytic phase during which virus is produced.
The orchestration of viral gene expression and virus assembly during reactivation is probably the most complex task that the virus performs during its infection cascade. In general, this process is thought to be regulated primarily through protein coding viral genes. Nevertheless, diverse roles for small noncoding RNAs (ncRNAs) are known to play important roles in EBV infection and have implications to EBV-mediated oncogenesis (2, 3). Recent studies have found evidence for previously unknown viral long noncoding RNAs (vlncRNAs) in EBV, Kaposi's sarcoma-associated herpesvirus, MHV68, and herpes simplex virus that are expressed during the lytic cycle (4–9). For example, evidence from our lab (9, 10) and from Dresang et al. (4) indicates that EBV likely expresses more than a hundred novel lytic noncoding RNAs. Further, extensive alternative splicing is observed (9, 10), illustrating the existence of even greater diversity in the repertoire of expressed coding and noncoding viral transcripts during this process. These lytic vlncRNAs likely serve a diverse set of functions in facilitating and coordinating the lytic cascade, from the regulation of virus gene expression and DNA replication to virion packaging, processing, and transport. Some lytic vlncRNAs may also serve extended functions. For example, lytic lncRNAs that are potentially packaged in virions may convey key signaling events or processes necessary for initial infection of naive B cells or epithelial cells.
Despite these possibilities, we have yet to learn the roles that these vlncRNAs play in reactivation. We report here initial investigations into novel transcripts derived from the EBV latency origin of replication and show that they help facilitate progression of the viral lytic cascade.
MATERIALS AND METHODS
Cell culture.
EBV-positive Akata and Mutu cells were grown in RPMI 1640 medium (Thermo Scientific, catalog no. SH30027) supplemented with 10% fetal bovine serum (FBS; Invitrogen-Gibco, catalog no. 16000-069) and 0.5% penicillin-streptomycin (pen-strep; Invitrogen-Gibco, catalog no. 15070-063). 293T cells were grown in Dulbecco modified Eagle medium (DMEM)/High-Glucose medium (Thermo Scientific, catalog no. SH30243) supplemented with 10% FBS and 0.5% pen-strep. All of the cells were cultured at 37°C in a humidified, 5% CO2 incubator.
Plasmid constructs.
oriPtL sequence spanning nucleotides (nt) 7148 to 9452 of the Akata genome (11) was amplified by PCR and inserted into the pMSCV-puro expression vector. ADAR1 (catalog no. SC119438) and HDLBP (catalog no. SC116829) cDNA clones were purchased from OriGene and their ORFs were inserted in-frame into a 3×FLAG-containing pMSCV-puro expression vector.
Calcium phosphate transfection.
Transfection of 293T cells for retrovirus production and for oriPtLs/ADAR interaction experiment was performed using the calcium phosphate method. Briefly, 293T cells were seeded on 10-cm plates in 10 ml of DMEM/High-Glucose medium (supplemented with 10% FBS and 0.5% pen-strep). The next day, the medium was replaced with 8 ml of fresh supplemented DMEM. For each transfection, 500 μl of HBS (0.5% HEPES, 0.8% NaCl, 0.1% dextrose, 0.01% anhydrous Na2HPO4, and 0.37% KCl at pH 7.2) was mixed with a total of 30 μg of plasmid DNA. For oriPtL/ADAR experiment, 5 μg of each expression plasmid plus 20 μg of carrier pUHD10 plasmid were used. For retrovirus production, 10 μg of each packaging plasmid (pVPACK-Gp-dI and pCI-VSV-G) and 10 μg of the oriPtL or empty vector control were used. A 30-μl portion of 2.5 M CaCl2 was added to each DNA-HBS mixture, and the tubes were vortexed on a low setting. DNA-HBS-CaCl2 mixtures were incubated for 20 min at room temperature and added dropwise to 293T cell cultures. The plates were gently rocked back and forth and then transferred to a 37°C 5% CO2 incubator. The following day, the medium was aspirated and replaced with 10 ml of fresh medium. Cells or supernatant (for retrovirus production) were harvested 48 h later.
Induction of EBV lytic cycle.
Akata and Mutu I cells were spun down and resuspended at a concentration of 106 cells/ml in fresh RPMI 1640 medium (10% FBS, 0.5% pen-strep). Anti-human IgG (Sigma-Aldrich, catalog no. I2136) or anti-human IgM (Sigma-Aldrich, catalog no. I0759) was added to the Akata and Mutu I cell suspensions, respectively, to a final concentration of 10 μg/ml. Treated and untreated cells were harvested 24 h later for RNA and/or protein isolation.
RNA preparation.
Total RNA was isolated using TRIzol reagent (Life Technologies, catalog no. 15596-018) or a miRNeasy minikit (Qiagen, catalog no. 217004) according to the respective vendor's protocols. Nuclear and cytoplasmic RNA was isolated using a cytoplasmic and nuclear RNA purification kit (Norgen Biotek Corp., catalog no. 21000) according to the vendor's protocol. All RNA preparations were subjected to DNase treatment using a DNA-free kit (Life Technologies, catalog no. AM1906).
Standard quantitative RT-PCR (qRT-PCR) and strand-specific qRT-PCR.
Standard reverse transcription (RT) was performed using an iScript cDNA synthesis kit (Bio-Rad, catalog no. 170-8891) according to the manufacturer's protocol. Strand-specific RT was carried out using modified gene-specific primers as described previously (12). cDNA was synthesized at 65°C for 50 min using ThermoScript reverse transcriptase (Life Technologies, catalog no. 12236-022) and treated with exonuclease I (NEB, catalog no. M0293L) to remove the excess gene-specific primers. qPCR analysis was performed using iQ SYBR Green Supermix (Bio-Rad, catalog no. 170-8882) on a Bio-Rad CFX96 instrument as follows. cDNA and 1-μl portions of 10 μM concentrations of the primers were mixed with 10 μl of SYBR Green Supermix and nuclease-free H2O to a 20-μl reaction volume. Polymerase was activated, and cDNA was denatured at 95°C for 5 min. cDNA was then amplified for 40 cycles with 15 s of denaturation at 95°C, 60 s of annealing/extension, and plate reading at 60°C. Melting curve analysis was performed at temperatures from 60 to 90°C, with 0.5°C increments per 5 s. Expression fold changes were calculated using the comparative threshold cycle method (2−ΔΔCT).
The modified strand-specific primers used for strand-specific RT were as follows: PP4_GSP_L(oriPtL), GTGTTAGAGACAACCAGTGGAGTtataTaaTGTCGGCGT; PP4_GSP_R(oriPtR), GACCAAGACAGGTGAACCATGccGccgCgCTCTATTTGT; PP5_GSP_L(oriPtLs), ATAGCACAATGCCACCACTGAACCaattaTTCAAATTTTA; PP5_GSP_R(oriPtRs), GCCTGCTTCTTCATTCTCCTTCGTggcGCggcTAGAATAACTG; BCRF1_GSP_R, ACTCTTGTTCTCACACGGCAGGAAtaTaTaaCACCTGCGCAGG; and ACTB, GTACAGGTCTTTGCGGATGTttAtaTaACACTTCATG where lowercase letters refer to sequence-modified bases. The primers used for qPCR were as follows: PP1_F, CCTGGACACACAGTCTTAGTT; PP1_R, CCTGCATGCCGTTTAATGATAG; PP2_F, CTCAGCGACCTCGTGAATATG; PP2_R, CGATCTGGAGGACAAGTTACAC; PP3_F, GGGTTAGCGAGGTTACAATCA; PP3_R, GGCCCATAAACAAAGACAAGTG; PP4_F, GTGTTAGAGACAACCAGTGGAG; PP4_R, GACCAAGACAGGTGAACCAT; PP5_F, ATAGCACAATGCCACCACTGAACC; PP5_R, GCCTGCTTCTTCATTCTCCTTCGT; ACTB_F, CACTCTTCCAGCCTTCCTTC; ACTB_R, GTACAGGTCTTTGCGGATGT; ACTB_142_F, CACACTGTGCCCATCTACGA; ACTB_142_R, CCATCTCTTGCTCGAAGTCC; Zta_F, TGGGCACATCTGCTTCAA; Zta_R, AATGCCGGGCCAAGTTTA; Zta_F (for time course), ACATTGGTGTTCCACAGCCT; Zta_R (for time course), GGGGGATAATGGAGTCAACA; Rta_F, CCATACAGGACACAACACCTCA; Rta_R, ACTCCCGGCTGTAAATTCCT; BCRF1_F, ACCCTGAAGCCAAAGACCATGTAA; BCRF1_R, ACTCTTGTTCTCACACGGCAGGAA; KCNQ1OT1_F, TACCGGATCCAGGTTTGCAGTACA; KCNQ1OT1_R, GCTGATAAAGGCACCGGAAGGAAA; ANRIL_F, CCCTCGTCGAAAGTCTTCCATTCT; and ANRIL_R, ATTCAGCCTCCTGATTGGCGGATA.
oriPtL GapmeR knockdown/RNA sequencing.
Customized GapmeRs targeting oriPtL were purchased from Exiqon (sequences: OriPtL Gap 1, GTAAAGCTGTGGAACA; OriPtL Gap 2, GCATTGGTGTAAGAGC). oriPtLs and negative control GapmeRs (control A; Exiqon, catalog no. 300613-04) were transfected into Akata cells by Amaxa electroporation using Nucleofector Solution R (Lonza, catalog no. VCA-1001) and program G-16 on a Nucleofector II device. Briefly, Akata cells were placed in antibiotic-free RPMI medium 2 days before electroporation. For each transfection, 2 × 106 cells in 100 μl of solution R were electroporated with 3 μl of negative control or oriPtLs targeting GapmeRs (200 pmol/μl) and transferred to a six-well plate with 1.5 ml of medium per well. Transfections were performed in triplicate for the negative control and for each of the two oriPtL targeting GapmeRs. After 24 h, 1.5 ml of fresh RPMI medium and anti-human IgG (to a final concentration of 17 μg/ml) was added to each well to induce EBV lytic reactivation. Cells were harvested for RNA isolation 24 h postinduction. cDNA libraries were prepared from ribodepleted RNA (Ribo-Zero; Epicentre, catalog no. MRZH11124) using the strand-specific Illumina TrueSeq protocol (Illumina, catalog no. RS-930-2001) and sequenced on an Illumina HiSeq 2000 instrument. Triplicate samples were multiplexed per sequencing lane.
Constitutive oriPtL expression/RNA sequencing.
Stable oriPtL expressing Mutu I cell lines were generated through retroviral infection and selection of infected pools. Briefly, 293T cells were seeded on 10-cm plates in 10 ml of DMEM/High-Glucose medium (supplemented with 10% FBS and 0.5% pen-strep) 1 day before transfection. Then, 10 μg of each packaging plasmid (pVPACK-Gp-dI and pCI-VSV-G) and 10 μg of the oriPtL or empty vector control (for a total of 30 μg of DNA) were mixed and ethanol precipitated at −20°C overnight to sterilize DNAs. On the day of transfection, the medium on the 293T plates was replaced with 8 ml of fresh supplemented DMEM. Plasmids were spun down, washed with 70% ethanol, and resuspended with 30 μl of nuclease-free H2O. Plasmids were added to 500 μl of HBS (0.5% HEPES, 0.8% NaCl, 0.1% dextrose, 0.01% anhydrous Na2HPO4, and 0.37% KCl at pH 7.2) for each transfection. Thirty microliters of 2.5 M CaCl2 was added to each tube and mixed by vortexing on a low setting.
DNA-HBS-CaCl2 mixtures were incubated for 20 min at room temperature and added dropwise to 293T cell cultures; the plates were gently rocked back and forth and then transferred to a 37°C 5% CO2 incubator. The following day, the medium was aspirated and replaced with 10 ml of fresh medium. Virus-containing supernatant was collected 3 days posttransfection and filtered through a 0.45-μm-pore-size SFCA filter to eliminate residual cells. Mutu I cells were prepared for infection by spinning them down and resuspending them at 2 × 106 to 4 × 106 cells/ml in RPMI (with 10% FBS and 0.5% pen-strep) plus 24 μg of Polybrene/ml. One milliliter of Mutu I cell suspension was added to single wells of a six-well plate. One milliliter of control or oriPtL virus preparations was added to duplicate wells for each, and the plates were spun at 1,000 × g for 1 h, followed by incubation in a 37°C 5% CO2 incubator for 4 h. Cell suspensions were transferred to 15-ml tubes, spun down, and resuspended in 4 ml of fresh RPMI (with 10% FBS and 0.5% pen-strep). The cells were transferred to T-25 flasks and cultured in a 37°C 5% CO2 incubator. Two days later, 4 ml of fresh RPMI (with 10% FBS and 0.5% pen-strep) was added, and 1 μg/ml puromycin was added to a final concentration of 1 μg/ml to start selection. Cells were maintained in RPMI (with 10% FBS, 0.5% pen-strep, and puromycin) for approximately 2 weeks to select for infected pools. RNA was isolated from duplicate infected pools using TRIzol reagent (Life Technologies, catalog no. 15596-018). cDNA libraries were prepared from ribodepleted RNA (Illumina, catalog no. RS-930-2001) using the strand-specific Illumina TrueSeq protocol (Ribo-Zero; Epicentre, catalog no. MRZH11124) and sequenced on an Illumina HiSeq 2000 instrument. Three samples were multiplexed per sequencing lane.
RNA immunoprecipitation.
For each immunoprecipitation reaction, 2 × 107 Akata cells were treated with anti-human IgG for 24 h and then harvested and washed once with phosphate-buffered saline (PBS). Cell pellets were resuspended in 20 ml of 1% formaldehyde-PBS solution and incubated for 10 min at room temperature on an end-to-end rotator. Cross-linking reactions were quenched with 1/20 volume of 2.5 M glycine for 5 min. Cells were washed once with 20 ml of cold PBS, resuspended in 1 ml of cold PBS, and transferred to 1.5-ml tubes. The cells were spun down and lysed in 1.2 ml of radioimmunoprecipitation assay (RIPA) buffer (Santa Cruz, catalog no. SC-24948) with supplements (12 μl of phenylmethylsulfonyl fluoride solution, 12 μl of sodium orthovanadate solution, 12 μl of protease inhibitor cocktail, and 6 μl of SUPERase·In RNase inhibitor [Life Technologies, catalog no. AM2696]). Cell suspensions were split equally into two 1.5-ml tubes and sonicated for 3 × 10 s (with 0.1-s on, 0.1-s off pulses) with 30-s intervals at 30% amplitude using a Branson Digital sonifier 250. Sonicated samples were centrifuged at 16,100 relative centrifugal force for 10 min at 4°C, and the pellets were discarded. Each vial of cell lysate was precleared with 50 μl of Dynabeads Protein G (Life Technologies, catalog no. 10004D) overnight at 4°C on a rotator. Precleared lysate pairs for each sample were pooled and divided into aliquots in 1.5-ml tubes with 0.6 ml per tube. Fifty microliters of the lysate was saved as the input sample for qPCR analysis. Five micrograms of antibody-coated beads was then added to 0.6 ml of cell lysate. After 3 h of incubation at 4°C, the beads were precipitated and washed six times with RIPA buffer supplemented with 0.1 U of SUPERase·In/ml. To reverse cross-linking, the beads were resuspended with 200 μl TE buffer (supplemented with 1 μl of 5 M NaCl, 2.5 μl of proteinase K [20 mg/ml], and 1 μl of SUPERase·In). Supplemented TE buffer was also added to the input sample to a final volume of 200 μl. All samples were incubated at 65°C for 2 h. The samples were then chilled on ice and subjected to RNA isolation using TRIzol. All RNA preparations were treated with DNase using the DNA-free kit. qRT-PCR analysis was performed on these RNA samples.
The primary antibodies used for RNA immunoprecipitation were RIPAb+ p54nrb/NonO (Millipore, catalog no. 03-113), rabbit anti-ADAR1 antibody (Abcam, catalog no. ab168809), rabbit IgG control antibody (Abcam, catalog no. ab37415), and mouse Anti-FLAG M2 antibody (Sigma, catalog no. F1804).
RNA FISH and immunofluorescence.
Stellaris fluorescence in situ hybridization (FISH) probes with CAL Fluor Red 610 fluorophore were designed and purchased from Biosearch Technologies. oriPtL FISH was performed according to the Biosearch Technologies online protocol for suspension cells. Anti-human IgG-treated Akata cells were fixed, permeabilized, and incubated with 0.4 μl of 25 μM oriPtL probes overnight at 37°C. The cells were washed on the following day and incubated with DAPI (4′,6′-diamidino-2-phenylindole) for 1 h in the dark. Images were taken with a 100/1.35 oil objective lens on a Leica DMRXA2 deconvolution upright microscope using Slidebook software.
RNA editing analysis.
Sequencing data for RNA editing analysis utilized our previously published ribodepleted strand-specific RNA-seq data from ribodepleted RNA from Akata cells induced for 24 h (9). Sequencing data were aligned to an index containing the human reference genome hg19 (Genome Reference Consortium GRCH37) plus the Akata EBV genome (GenBank accession no. KC207813.1) using Novoalign (Novocraft). Aligned EBV reads were extracted from the output files and separated into sense and antisense read files. Pile-ups for each file were generated using SAMtools (command options: “mpileup −f”) (13) and run through VarScan v2.3.5 (14) to call variants at each nucleotide position and to calculate the frequency of variants. Variants with a frequency >0.01 were reported. Adenosine-to-guanine [A-to-G(I)] editing data were extracted from the output files for downstream RNA editing analysis. A-to-I variations within the repeat regions, IR1, IR2, and IR4, were omitted due to a comparatively high general variation rate (relative to A-to-I variations), likely due to genomic variations from one repeat to another.
oriPt secondary structure analysis.
The oriP FR region falls within a region that was previously predicted to form unusually stable and evolutionarily conserved RNA secondary structure (33). One of the key features used in this study was the thermodynamic z-score of overlapping 120 nt windows. The z-score for the entire oriP FR region was recalculated according to the following formula:
where ΔG°native is the predicted native Gibb's free energy of folding, ΔG°random is the average of the shuffled “mutant” sequences, and σ is the standard deviation of the set of fragment sequences. To model the putative structure of this region, RNAalifold (15) was used to compute the minimum energy structure formed by a set of aligned sequences from five different EBV strains: Akata (KC207813.1, nt 7458 to 7927), EBV1 (NC_007605.1, nt 7450 to 7915), EBV2 (NC_009334.1, nt 7545 to 8014), Mutu (KC207814.1, nt 7363 to 8087), and GD1 (AY961628.3, nt 7492 to 8148). The consensus structure was then manually refined to maximize conservation and identify compensating mutations. A covariance model built using INFERNAL (16) and used to scan herpesvirus genomes for additional instances of the oriP FR structure model.
RNA-seq data analysis (GapmeR and oriPtL ectopic expression studies).
FASTQ files were aligned to human reference genome hg19 and Akata EBV genome (11) using the sequence and junction aligner, TopHat (18). Aligned EBV reads were pulled out from the Tophat output. EBV reads and junctions were loaded on Integrative Genomics Viewer (IGV) (19, 20) for visualization. Gene expression was quantified by SAMMate (21).
Database accession number.
The RNA-seq sequence data has been deposited in the GEO database under accession number GSE68122.
RESULTS
Lytic transcription at the EBV latency origin of replication, oriP.
We recently found evidence for potentially hundreds of previously unknown vlncRNAs that are expressed during the EBV lytic cycle (9). Across the EBV latency origin of replication, we first observed high level transcription in reactivated Akata cells using non-strand-specific RNA-seq data (data not shown). Previous studies showed that the BCRF1/vIL10 open reading frame is transcribed not only from a proximal promoter but also from multiple upstream promoters located within and 5′ to oriP (Fig. 1A) (22–24). Analysis of strand-specific RNA-seq data from induced Akata cells showed strong rightward transcription across the BCRF1 reading frame, much of which likely originates from the proximal BCRF1 promoter (Fig. 1A). In addition, upstream rightward coverage is observed that likely reflects the use of one or more of the upstream promoters (Fig. 1A). Nevertheless, this strand-specific sequencing data also revealed leftward transcription extending across the BCRF1 reading frame and through the entire oriP region (Fig. 1A). Strand specificity calculations as described previously (9) showed fewer than 2 background reads per 1,000 reads, indicating that the observed sense and antisense coverage appropriately represents the levels of transcription on each strand. Higher leftward transcription was observed in sequencing data from ribodepleted RNA compared to poly(A)-selected RNA (Fig. 1A), suggesting that many of these leftward transcripts are terminated through a noncanonical termination mechanism. This contrasts with the BCRF1 transcripts where higher coverage was observed in poly(A)-selected RNA libraries (Fig. 1A). The higher poly(A)+ coverage for the BCRF1 transcripts is consistent with the use of a common poly(A) signal located just downstream from the BCRF1 reading frame.
FIG 1.

Novel lytic transcripts expressed from the EBV latency origin of replication, oriP. (A) Evidence for bidirectional transcription at the oriP locus. Strand-specific RNA-seq coverage data for poly(A)-selected RNA (upper panel) and ribodepleted RNA (lower panel). Rightward-pointing arrows in the top coverage window refer to previously published start sites for transcripts containing the BCRF1 reading frame. The two boxed arrows refer to start sites that are upstream from the genomic region depicted in this figure. Genome features are indicated below the coverage data. PCR primers used throughout the present study are indicated as PP1 through PP5. Gap1 and Gap2 refer to the target sequences of the anti-oriPtL GapmeRs used for functional studies (Fig. 6). Short red bars indicate the FISH probes used for the data shown in panel E. (B) qRT-PCR time course analysis of selected immediate-early, early, and late EBV lytic genes and oriP transcripts during reactivation. Strand-specific qRT-PCR was performed for oriPtRs (PP5) and oriPtLs (PP5). (C) oriPtRs and oriPtLs are sensitive to PAA treatment. Strand-specific qRT-PCR was performed using strand-specific PP5 primers. (D) Relative expression levels of oriPtRs and oriPtLs in the nucleus and cytoplasm analyzed by strand-specific qRT-PCR (PP4 primers). ACTB was analyzed as a cytoplasmic transcript control, and KCNQ1OT1 and ANRIL were analyzed as nuclear transcript controls. (E) FISH showing primarily nuclear localization of leftward oriPts.
Despite the substantial length of the 5′ sequences in the upstream initiated BCRF1 transcripts, no reading frames have been previously annotated in these regions (that might be suggestive of bicistronic BCRF1 mRNAs). Likewise, no reading frames have been previously annotated in the leftward orientation throughout the oriP/BCRF1 genomic region (Fig. 1A). Further, using the Coding Potential Calculator (CPC) (25) with the Akata reference genome (11), these regions were predicted to be noncoding (see Fig. S1 in the supplemental material). The leftward transcripts, which we will refer to here as oriPtLs, are therefore likely new vlncRNAs. Further, we propose that the long 5′ untranslated regions (UTRs) in the upstream initiated BCRF1 transcripts may also play noncoding functions in EBV reactivation (see below), and we refer to this group of upstream initiated transcripts as oriPtRs (Fig. 1).
oriP transcripts are viral late genes.
As previously unknown transcripts, we sought to determine some fundamental characteristics of oriP transcripts that might guide insights into their function. Determining the expression timing of virus genes can imply roles in temporally distinct components of the lytic cascade. Using strand-specific PCR (12) with the PP5 oriP primers shown in Fig. 1A, we found that expression of oriPtRs and oriPtLs peaked at 24 h postinduction, coinciding with expression of the EBV late genes, VCA and gp350 (Fig. 1B). In contrast, the immediate-early and early Zta, Rta, and BMLF1 genes were sharply induced as early as 2 to 4 h after induction. The late timing of oriPtRs and oriPtLs expression was corroborated by an RNA-seq time course experiment (see Fig. S2 in the supplemental material). Further, expression of both oriPtLs and oriPtRs was inhibited by the viral DNA replication inhibitor phosphonoacetic acid (PAA) (Fig. 1C). These data indicate that both the upstream initiated oriPtR transcripts and the oriPtL transcripts are viral late genes, suggesting a role in processes downstream from the early activation events of immediate-early and early genes.
oriPtLs are nuclear RNAs.
To help guide conceptualization of possible oriPt functions, we determined their cytoplasmic/nuclear distribution. Strand-specific qRT-PCR was used to quantify the abundance of oriPtR and oriPtL transcripts in nuclear and cytoplasmic RNA preparations from Akata cells induced for 24 h. The cellular transcripts, KCNQ1OT1 and ANRIL, were used as nuclear RNA controls, and ACTB was used as a cytoplasmic RNA control. Using the PP4 oriP primers (Fig. 1A), both oriPtR and oriPtL transcripts showed nuclear enrichment with a greater degree of enrichment being observed with oriPtL (Fig. 1D). The nuclear localization of oriPtLs was further supported by fluorescence in situ hybridization (Fig. 1E). The finding of nuclear enriched rightward oriP transcripts using the PP4 primers raises the possibility that at least some BCRF1 transcripts, specifically those with extended 5′ UTRs, may be localized in the nucleus. Using primers within the BCRF1 reading frame, we detected a lower nuclear/cytoplasmic ratio than was observed with the PP4 primers, but these levels were still higher than that observed for the cytoplasmic ACTB RNA (Fig. 1D). These data are consistent with the idea that the localization of BCRF1 transcripts is mixed, with proximal promoter initiated transcripts being transported to the cytoplasm for translation and upstream initiated BCRF1 transcripts being retained in the nucleus, where they may have discrete noncoding functions.
oriP transcripts are hyperedited.
While visualizing RNA-seq alignment data from induced Akata cells, we noticed a strikingly high density of adenosine to guanine (A-to-G) conversions, indicative of adenosine to inosine (A-to-I) RNA editing, on both strands of the Family of Repeats (FR) region of oriP (Fig. 2; see also Table S1 in the supplemental material). Although substantially less pervasive, we also noted evidence of A-to-I editing at the oriP dyad symmetry (DS) element (Fig. 2). Notably, evidence of A-to-I editing was observed in both the BHRF1 and the BamHI A microRNA clusters (Fig. 2A and C and see Table S2 in the supplemental material), with some of these sites having been previously reported (11, 26, 27). Among all of these regions, the most pervasive RNA-editing was observed within the FR region, where 66 and 73% of adenosines showed evidence of editing in the rightward and leftward transcripts, respectively. Further, the extent of editing at individual residues is as high as 74% in rightward transcripts and as high as 68% in leftward transcripts. This extensive editing of rightward and leftward transcripts is consistent with the classification as “hyperedited” RNAs (28, 29).
FIG 2.

RNA editing of EBV transcripts. (A) RNA-editing (A-to-I) evidence across the EBV genome generated from strand-specific RNA-seq of Akata cells induced for 24 h. The percent conversions at each adenosine residue are depicted for sense (positive values) and antisense (negative values) transcripts. (B) Expanded view of high-density editing at the FR repeats. (C) Expanded genome view of RNA-editing at the BART and BHRF1 microRNA loci.
Evidence that FR containing oriPtL and oriPtR transcripts can form evolutionarily conserved long hairpin structures.
RNA editing classically occurs through the action of the adenosine deaminase acting on RNA (ADAR) family of enzymes that recognize double-stranded RNA regions (30), commonly through long hairpin structures, e.g., those found in the 3′ UTRs of humans (31) and nematodes (32). Interestingly, a previous bioinformatics scan of the EBV genome discovered evidence for thermodynamically stable (likely functional) RNA secondary structure in the FR region (33). Initial secondary structure predictions in this region posited two mid-sized hairpins (33); however, with the addition of our recently sequenced Akata strain genome (11), it was possible to remodel the structure here to form very large (470-nt) hairpin structures in both forward and reverse strands (Fig. 3). Interestingly, in each hairpin the FR repeat sequences are annealed together in stems (see Fig. S3 in the supplemental material), although a functional significance for a double-stranded RNA version of a double-stranded DNA motif is unclear.
FIG 3.
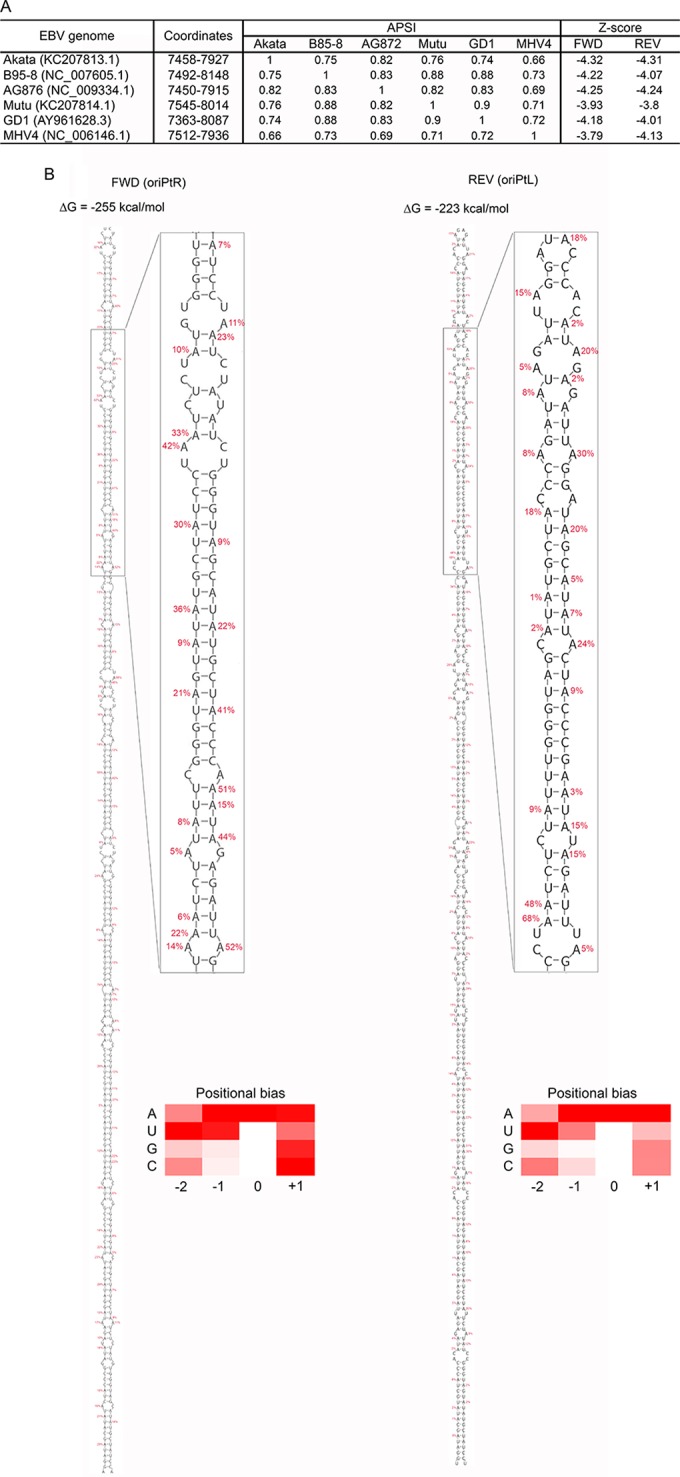
oriPt secondary structures. (A) Consensus hairpin structures were predicted in the FR region of oriP across five EBV strains (Akata, B95-8, G876, Mutu, and GD1) and macacine herpesvirus 4 (MHV4). The average pairwise sequence identity (APSI) was calculated for the regions where the hairpin structures were identified in the six virus strains. The z-scores were calculated for the hairpin structures in sense (Fwd) and antisense (Rev) transcripts in each virus. (B) Predicted hairpin structures for oriPtR (left) and oriPtL (right) with associated folding free energy values. Editing frequencies for each adenosine through each hairpin are shown (an expanded view is depicted in Fig. S3 in the supplemental material for visualization in greater detail). Heat maps show relative average editing for A's, U's, G's, and C's located at positions −2, −1, 0, and +1 relative to the edited adenosine residue (0 position).
The metric used for measuring propensity to form structure, the thermodynamic z-score (34), has equally large magnitude in both strands: −4.3. This number predicts that the native sequence is ∼4 standard deviations more thermodynamically stable than expected for random sequences with equal dinucleotide composition, suggesting that evolution is acting to preserve base pairing here. Indeed, we were able to build consensus structures and alignments for the Akata strain versus four other EBV strains (B95-8, AG876, Mutu, and GD1) and use these models to search for structural homologs in other lymphocryptoviruses (using the INFERNAL package [16, 35]). This search returned highly similar (∼70% conserved) hairpins in a syntenic region of the rhesus lymphocryptovirus (rLCV) genome (also known as Macacine herpesvirus 4): NC_006146.1 (nt 7512 to 7936). Overall conservation of the hairpin structure (in EBV strains and rLCV) is high in both strands (82.4 and 87.6% for the forward and reverse strands, respectively). Furthermore, both hairpin models are supported by compensatory mutations (double point mutations preserving base pairs) and consistent mutations (single point mutations preserving base pairs). The predicted z-score and overall folding energy (ΔG°) in each homologous sequence are of a magnitude similar to that for the Akata strain (in both strands). These results suggest that evolution has “tuned” this region to fold in both strands and that it is acting to preserve these hairpins to perform some important conserved function.
The high degree of secondary structure at the FR regions likely provides a key configuration for recognition by RNA editing enzymes (36). We also assessed the average percent editing for each of A, U, G or C for the two preceding bases and for the following base across both the rightward transcript and the leftward transcript to determine recognition biases. This analysis generally showed consistency with previous studies (28, 29, 37), with A/U enrichment at the −1 position, U enrichment at the −2 position, and A enrichment at the +1 position.
Molecular interaction between oriPtLs and ADAR during reactivation.
There are currently three known members of the ADAR family in humans: ADAR, ADARB1, and ADARB2. Using RNA-seq data generated for the functional studies outlined below, we determined the mRNA levels of each member of this family in the two EBV-positive Burkitt's lymphoma cell lines: Akata and Mutu. Of the three members of the ADAR family, only ADAR transcripts were detected in either cell line (Fig. 4A).
FIG 4.
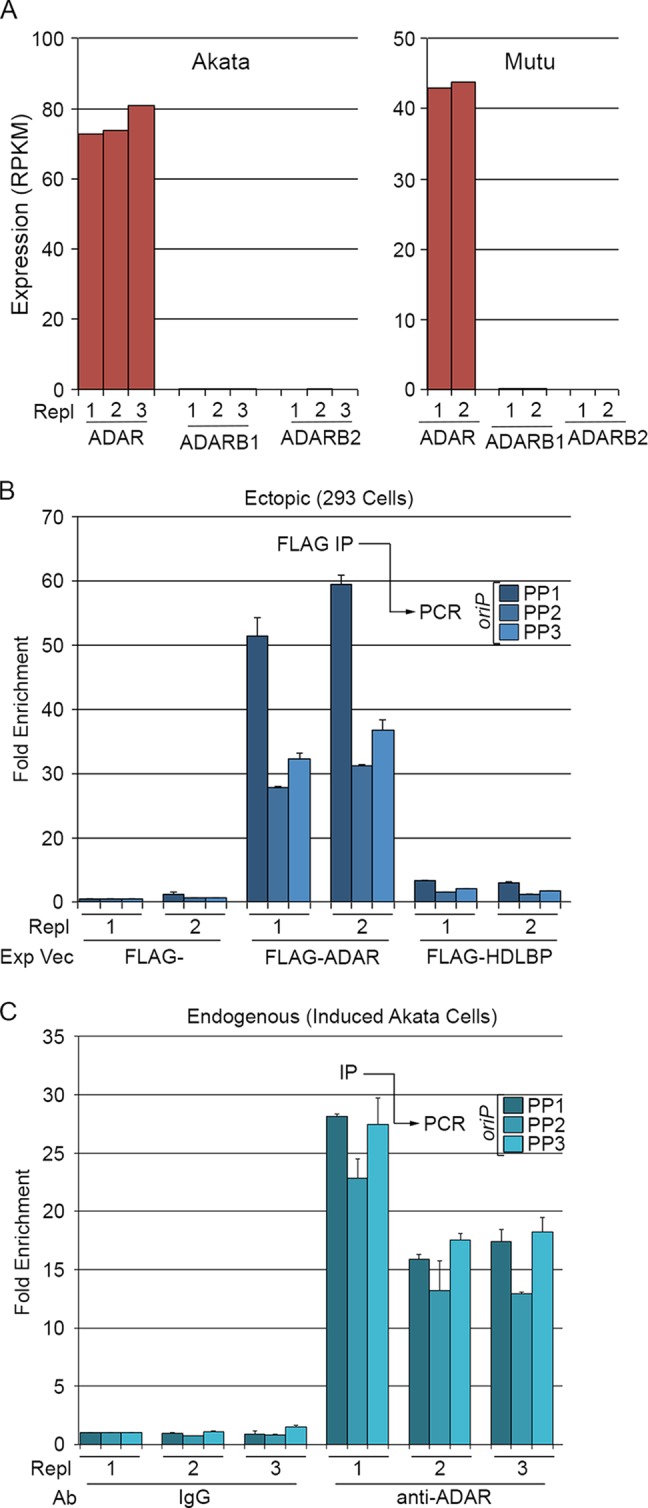
oriPts associate with the adenosine deaminase acting on RNA, ADAR. (A) The adenosine deaminase family member ADAR is expressed in Akata and Mutu cells. Expression values of the three known RNA adenosine deaminases in Akata and Mutu cells were determined by RNA-seq analysis of triplicate or duplicate biological replicates. (B) oriPtL binds FLAG-tagged ADAR in 293 cells. Duplicate FLAG, FLAG-ADAR, and FLAG-HDLBP cotransfections with oriPtL were analyzed using three different sets of oriPt primer pairs (shown in Fig. 1A). (C) Endogenous oriPts coimmunoprecipitate with endogenous ADAR during reactivation in Akata cells. Triplicate induced Akata cells inductions were immunoprecipitated with control or anti-ADAR antibodies and analyzed for oriPts by qRT-PCR using the indicated three oriPt primer pairs (shown in Fig. 1A).
We next tested whether ADAR can interact with oriPtL in 293T cells cotransfected with a FLAG-tagged ADAR expression vector and an oriPtL retroviral expression vector (depicted in Fig. 1A). Cell extracts were prepared 3 days posttransfection and subjected to immunoprecipitation with an anti-FLAG antibody. RNA was prepared from the immunoprecipitated complexes, and qRT-PCR was used to assess enrichment of oriPtL transcripts. Using PCR primers for three different regions of oriP, oriPtL transcripts were found to be enriched in FLAG-ADAR transfected cells but not in cells transfected with an empty vector or cells transfected with a vector expressing a FLAG-tagged HDLBP gene (Fig. 4B).
To determine whether endogenous ADAR binds oriPts under reactivation conditions, Akata cells were induced for 24 h, and ADAR-containing complexes were immunoprecipitated with an anti-ADAR antibody. oriPts were enriched in each biological replicate of anti-ADAR immunoprecipitations (Fig. 4C), indicating that oriPt RNAs interact with ADAR during reactivation, likely explaining the observed hyperediting of these transcripts.
oriPtL interacts with the paraspeckle complex assembly factor, NONO.
Previous studies have shown that some hyperedited nuclear RNAs are sequestered to subnuclear compartments called paraspeckles through specific interactions between the key paraspeckle assembly factor, NONO, and poly(I)-containing transcripts (36). The presence of paraspeckles in cells is highly tissue specific and depends exquisitely on tissue specific expression of the 23-kb nuclear paraspeckle scaffolding RNA, NEAT1_2 (38, 39). A short form of NEAT1, referred to as NEAT1_1 (Fig. 5A) likely also plays a role in assembly but appears to be less critical than NEAT1_2 (38, 39). Interestingly, NEAT1/paraspeckles appear to have innate immunity functions in viral infection (40–42) through infection-mediated induction of NEAT1 and the resulting paraspeckle formation (40). As is commonly the case for successful viruses, at least HIV is known to have adapted to utilize this pathway to regulate its own infection cascade (42).
FIG 5.
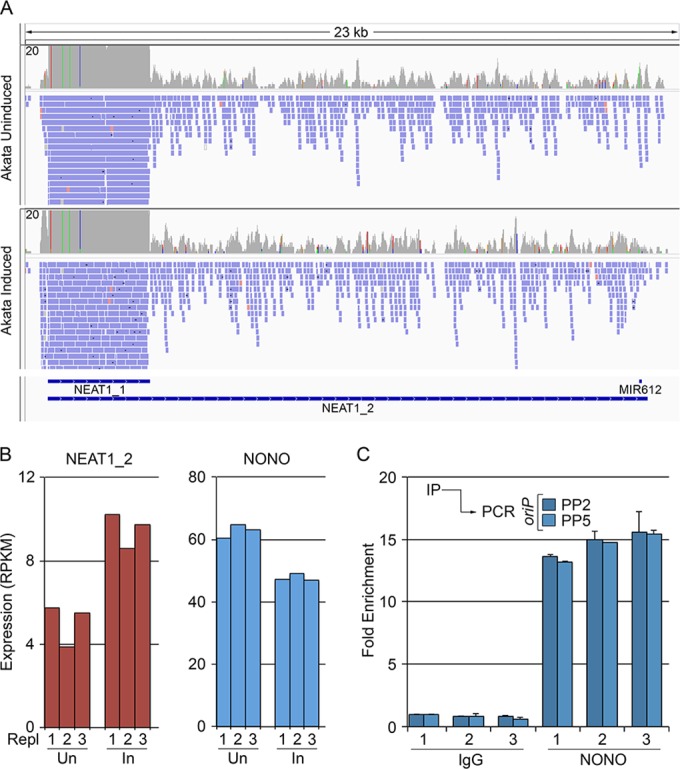
oriPts bind the multifunctional paraspeckle component, NONO. (A) Coverage of short (NEAT1_1) and long (NEAT1_2) forms of NEAT1. (B) Expression values of NEAT1_2 and NONO in uninduced and induced Akata cells as determined by biological triplicate RNA-seq data sets. (C) Endogenously expressed oriPts bind NONO during reactivation. Triplicate inductions were immunoprecipitated with control or anti-NONO antibody, and oriPt enrichment was assessed by qRT-PCR using the indicated primer pairs.
Although we did not observe substantial differences in NEAT1 expression between EBV positive and EBV negative Akata cells (data not shown), both NEAT1_1 and NEAT1_2 are expressed in Akata cells and in Mutu cells (Fig. 5A and B and see Table S3 in the supplemental material). Further, key protein factors that are required for paraspeckle assembly, including NONO, are also expressed in these cell lines (Fig. 5B and see Table S3 in the supplemental material). As an initial investigation into a possible interaction between oriPts and this antiviral/stress response pathway, we assessed whether oriPt associates with NONO during reactivation. As shown in Fig. 5C, endogenously expressed oriPt transcripts were found to be enriched in NONO containing complexes during reactivation. These results show a physical interaction between oriPt and paraspeckle factors, raising the possibility that oriPts interact with this antiviral/stress response pathway in some way.
oriPtL transcripts support viral lytic gene expression and DNA replication.
The substantial expression of oriPtL during reactivation and its association with specific nuclear factors support the contention that these transcripts play a role in remodeling the nuclear signaling environment, presumably supporting some aspect of the viral lytic replication phase. A contrasting model is that these are spuriously generated transcripts that play no role in the lytic cascade but whose ADAR modification and possibly paraspeckle sequestration is part of an antiviral response. To distinguish between these two opposing models, we investigated whether oriPtL plays a role in viral gene expression and/or viral DNA replication.
Since the small interfering RNA (siRNA) machinery is primarily localized in the cytoplasm, there is some question as to the ability of siRNAs to degrade nuclear transcripts. We therefore explored the application of antisense LNA-DNA-LNA GapmeR oligonucleotides (43), which degrade their target RNAs through nuclear localized RNase H activity. We tested five oriPtL GapmeRs and, while most of them showed good inhibition of oriPtL expression, two of the better-performing GapmeRs (Gap1 and Gap2, shown in Fig. 1A) were selected for functional experiments.
To investigate the influence of oriPtL on viral gene expression, Akata cells were nucleofected in triplicate with no GapmeR, a control GapmeR, Gap1, and Gap2. After 24 h, the control GapmeR, Gap1 and Gap2 transfected cells were treated with anti-IgG to induce reactivation. After another 24 h, the cells were harvested, and RNA was prepared. qRT-PCR showed that Gap1 and Gap2 inhibited oriPtL induction by ∼4-fold (Fig. 6A). These RNAs were then subjected to strand-specific TruSeq 100 base single-end Illumina sequencing. Sequencing reads were aligned to a human plus virus genome index, and strand-specific gene expression was quantified using SAMMate (21) (see Table S4 in the supplemental material for viral gene expression values). The average fold inductions were calculated for induced cells transfected with control, Gap1, and Gap2 GapmeRs (Fig. 6B). The statistical R package, EBseq (44), was used to test for differentially expressed genes across the four conditions: uninduced, induced cntl-GapmeR, induced Gap1, and induced Gap2. Of the 15 possible differential gene expression patterns, we specifically investigated genes called as statistically significant in the pattern showing change in induced cells and an intermediate change in both GapmeR transfected conditions (pattern 14, see Table S5 in the supplemental material). Twenty viral genes were identified as statistically significantly associated with this pattern (see Table S5 in the supplemental material) and, in these cases, the expression was higher in induced cells, with an intermediate expression in induced cells treated with Gap1 or Gap2 (Fig. 6B). In addition, although not determined to be statistically significant in this analysis, most EBV genes found to be induced by reactivation conditions were found to be induced to a lower level in cells treated with Gap1 and Gap2 (Fig. 6B). Although the observed changes in this group of EBV genes further support a role for oriPtL in facilitating viral transcriptional increases, statistical significance was likely not achieved because of variance in biological replicates combined with the moderate decreases in gene expression in the presence of the oriPtL GapmeRs. It is notable that the moderate nature of the decreases likely result from the combination of incomplete abrogation of oriPtL transcripts (4-fold decreases) and potential compensatory functioning of the oriPtR transcripts. Nevertheless, the findings that the expression of most induced viral genes were decreased in the presence of both oriPtL GapmeRs provide strong support that oriPtL plays a role in supporting EBV lytic gene expression.
FIG 6.

Involvement of oriPtL in reactivation. (A) Changes in oriPtL expression in GapmeR-transfected Akata cells (left panel) and in retrovirally transduced Mutu cells (right panel). (B) oriPtL regulates lytic viral gene expression. The left heat map shows results from oriPtL GapmeR knockdown in Akata cells. Triplicate uninduced cells and triplicate induced cells transfected with a control or two different anti-oriPtL GapmeRs were subjected to RNA-seq, and the average fold inductions for control, GapmeR1 (Gap1), and GapmeR2 (Gap2) transfected cells are displayed (raw values for each are shown in Table S4 in the supplemental materials). The heat map on the right displays the average gene expression in duplicate transduced control or oriPtL retroviral expression vector (raw values for each are shown in Table S4 in the supplemental material). (C) oriPtL knockdown reduces viral lytic DNA replication.
To further assess a role for oriPtL in supporting the expression of viral lytic genes, we transduced Mutu cells with an empty control retrovirus or a retrovirus containing the oriPtL sequences and selected cells for stable integration for ∼2 weeks. oriPtL-transduced cells expressed >150 times the levels of oriPtL in uninduced cells (Fig. 6A), and RNA-seq analysis showed that the expression of most viral RNAs was higher in duplicate oriPtL transductants (Fig. 6B and see Table S5 in the supplemental material). Lastly, we found that Gap2 inhibited the increase in endogenous viral genomes in induced Akata cells 96 h after induction (Fig. 6C), suggesting that oriPtL, in addition to playing a role in viral lytic gene expression, contributes to the production of progeny viral genomes.
DISCUSSION
Pervasiveness and functional significance of lytic vlncRNAs.
Tiling array experiments by Dresang et al. (4) suggested the presence of additional previously unknown antisense transcripts expressed during EBV reactivation. In recent studies from our lab (9, 10, 45), we estimated that there are hundreds of previously unknown viral long noncoding transcripts expressed during the EBV lytic cycle. One possible viewpoint is that these are spurious transcripts resulting from the open chromatin environment of highly transcribed viral genomes during reactivation. Nevertheless, here we show that leftward oriP derived vlncRNAs help facilitate lytic viral gene expression. Our investigations of other newly identified vlncRNAs have shown that they also seem to play roles in modulating the replicative environment; through similarly regulating viral gene expression (M. Concha et al., unpublished data) and through sequestration of microRNAs (M. J. Strong et al., unpublished data). We therefore support the contention that most of these new vlncRNAs likely function in processes facilitating the viral replication cascade.
Although lncRNAs are well known as chromatin regulators, their protein scaffolding properties make them suitable candidates in other cellular processes that require protein bridging and/or the assembly of various types of regulatory factories. Nevertheless, the breadth of their repertoire of activities is likely poorly appreciated at this time. During viral replication, a vast number of diverse tasks are carried out, raising questions about possible new vlncRNA functions in processes, such as pathway signaling and virus assembly, trafficking, and secretion. In line with there being varied possibilities, we found that some vlncRNAs are localized primarily in the nucleus while others are predominantly localized in the cytoplasm (data not shown). Many of the vlncRNAs that we have identified to date appear to be expressed with late kinetics, supporting possible roles in downstream assembly events. Further, we hypothesize that some vlncRNAs are likely packaged in virions where they may play structural roles in maintaining virion integrity and/or where they may provide rapid signaling responses following de novo infection of naive cells.
Are upstream initiated BCRF1 transcripts lncRNAs?
Although not expressed as abundantly as the leftward oriP transcripts, the previously described upstream initiated BCRF1 transcripts (22–24) are similarly likely to perform functions in the reactivation cascade, and we propose that these roles are distinct from making BCRF1 protein. Since long 5′ UTRs tend to be inefficient substrates for translation, it is more likely that the 5′ extended BCRF1 transcripts have noncoding functions. PCR analysis using primers that recognize the upstream initiated BCRF1 transcripts show significant nuclear localization, suggesting that these transcripts likely perform nuclear noncoding functions. Primers hybridizing to the BCRF1 reading frame show a lesser degree of nuclear localization likely reflecting the detection of both upstream initiated nuclear localized BCRF1 transcripts and cytoplasmic localized downstream initiated transcripts (which are presumably recruited to ribosomes for translation). So why do the upstream initiated transcripts even include the BCRF1 reading frame? Poly(A) site sharing seems to be relatively common in viruses. For example, the BRLF1 gene uses the poly(A) signal from the BZLF1 gene (46). We have also observed additional examples of extended 5′ UTRs for other EBV genes (C. O'Grady et al., unpublished data). It is therefore likely that the BCRF1 locus is just one of a repertoire of many loci where upstream initiated transcripts play primarily noncoding functions despite the presence of downstream reading frames. This does not exclude the possibility that some of these may have cryptic internal ribosomal entry sites, but this may be the exception rather than the rule.
Regulation of lytic viral gene expression by oriPtL.
Remarkably, most EBV genes that were induced during reactivation in Akata cells were inhibited by the oriPtL GapmeRs (Fig. 6B and see Table S4 in the supplemental material). Notably, although oriPts are expressed with late kinetics, the expression of immediate-early and early genes were also found to be supported by oriPtL. Although seemingly contradictory, this may indicate that oriPtL plays a role in sustaining the expression of this earlier classes of genes and/or that oriPtLs can also support initial reactivation through export mechanisms involving packaging of the transcripts in either virions or microvesicles. In contrast to the influence on EBV lytic genes, oriPtL GapmeRs did not inhibit the expression of most cellular genes, and they did not inhibit the expression of the few EBV genes that are not induced during reactivation; for example, the polymerase III-transcribed genes, EBER1 and EBER2. Although these observations show specificity (to primarily viral lytic genes), this raises the possibility that oriPtLs operate through modulating a central upstream stimuli and/or that it operates broadly on genes through a common reactivation inducible regulatory context.
The localization of oriPtLs to the nucleus is consistent with a potential regulatory role in gene expression. Our attempts to identify associations between oriPtL and viral chromatin using a ChIRP assay (47), however, failed to show high-confidence binding (data not shown). This raises the possibility that oriPtL operates through less direct mechanisms.
Imamura et al. (40) showed that herpes simplex virus 1 (HSV-1) and influenza virus infections induced NEAT1 expression, resulting in the formation of paraspeckles. In their study, paraspeckle assembly caused a redistribution of the otherwise diffuse and/or scattered localization of the immune gene transcriptional repressor, SFPQ, to paraspeckles. Sequestration of the SFPQ repressor led to the derepression of a panel of immunoregulatory genes, including antiviral factors such as interleukin-8 (IL-8), CCL5, RIG-I, and MDA5. These findings reinforce the contention that the NEAT1/paraspeckle pathway is a cellular antiviral pathway. Notably, SFPQ functions to a large extent as a heterodimer with NONO (48). Since viruses often hijack antiviral pathways and remodel them to accommodate their own needs, we considered the possibility that oriPtL might alter the function and/or localization of SFPQ/NONO complexes in a way that causes derepression of lytic gene expression. We searched the EBV genome for the SFPQ binding sequence identified in the IL-8 gene (40) and found 17 sites scattered throughout the genome (see Fig. S4 in the supplemental material), with one of these located just upstream from oriP (see Fig. S4 in the supplemental material). It remains to be determined, however, whether SFPQ function is altered in the presence of oriPtL in a way that leads to derepression at these SFPQ and/or other cryptic SFPQ binding sites throughout the EBV genome.
Like SFPQ, NONO is a multifunctional DNA-binding protein that plays a role in transcriptional regulation (49–53). Although NONO is a known transcriptional activator (49, 50, 54), it, like SFPQ, can play a repressive role (51). A study by Liu et al. (51) showed that dephosphorylation of NONO by the phosphatase, PP1, can disrupt its interaction with Sin3A and HDAC1, changing it from a transcriptional inhibitor to a transcriptional activator. We were able to identify 17 unique NONO binding sites in the EBV genome (see Fig. S4 in the supplemental material), three of which are in or near the oriPtL and oriPtR transcript regions (see Fig. S4 in the supplemental material). It is interesting to consider the possibility that the binding of oriPtL to NONO either alters its structure directly, inhibits the ability of kinases to phosphorylate NONO or alters its interaction with PP1 or other factors in a way that transforms NONO from a transcriptional repressor into a transcriptional activator. In such a scenario, the candidate binding sites shown in Fig. S4 in the supplemental material and/or other cryptic NONO binding sites may be transitioned from repressor to activator cis response elements, thereby inducing local viral lytic gene expression.
Irrespective of the exact mechanism of action of oriPtL in modulating lytic gene expression, it is notable that ChIRP pulldowns of oriPtL transcripts did not show enrichment of NEAT1 transcripts (data not shown). This is probably significant because it suggests that oriPtL is not simply sequestered to paraspeckles as part of a cellular antiviral response. Instead, oriPtL likely interacts with this pathway in a way that ultimately leads to alterations in cellular signaling that helps facilitate virus production.
In addition to its roles in facilitating paraspeckle structures and in transcriptional regulation, NONO also regulates splicing through its association with the carboxyl-terminal domain of RNA polymerase II during the elongation process (52). NONO is also involved in polyadenylation (55, 56), and it plays a role in DNA damage response (57, 58). We previously reported extensive alternative splicing at the EBV LMP2 gene locus under reactivation conditions that was not observed in type III latency (10). The mechanisms driving LMP2 alternative splicing during reactivation are unknown at this time. Considering the hypothesis that a lytic gene facilitates LMP2 alternative splicing, we tested whether the ratio of noncanonical to canonical LMP2 splicing changed in the presence or absence of oriPtL GapmeRs. This analysis, however, showed no notable differences under these conditions (data not shown). Whether this experimental design is adequate to appropriately address a possible role for oriPtL and/or NONO in LMP2 alternative splicing or whether alternative splicing is controlled through some other viral or cellular factors is unclear at this time.
Does the interaction between oriPtL and ADAR have additional consequences besides editing of oriPtL?
The extensive A-to-I editing that we observed in these transcripts (Fig. 2 and 3) and their structural similarity to other massive hairpin substrates of ADAR (30–32) suggests recruitment of ADAR as a possible function of the oriP hairpins. ADAR normally undergoes nucleocytoplasmic shuttling (59, 60), but an abundant nuclear RNA such as the oriPts could play a role in sequestering ADAR during lytic reactivation, thereby leading to aberrant partitioning of this important host protein. Interestingly, the nuclear localization signal of human ADAR partially overlaps one of its double-stranded RNA-binding domains and interactions with RNA substrates were shown to affect its nucleocytoplasmic distribution (61). If the oriP hairpins are affecting ADAR shuttling in lytic infection, there could be wide-ranging effects on the host cell. For example, ADAR editing is estimated to occur in ∼10% of human transcripts (62): editing occurs in 3′ UTRs, where it may affect localization and translation (36); in coding regions, where it can alter amino acid codons (63); in pri-miRNA hairpins, where it affects miRNA processing (64) and target specificity (65, 66); and in introns, where it can affect splicing (67), for example.
Final thoughts.
We have identified novel oriP-derived leftward vlncRNAs that are involved in facilitating viral lytic gene expression. These transcripts seem to integrate into the nuclear antiviral NEAT1/paraspeckle pathway, possibly in a way that supports the lytic cascade. However, we have only scratched the surface regarding the possible functional significance of this interaction, or more specifically the interaction with ADAR and/or NONO. With NONO in particular having so many known functions, it will be interesting to explore whether the binding of oriPts to NONO influences one or more of these activities. For example, is there any role in altering polyadenylation of viral versus cellular genes? Lytic replication in many ways establishes a DNA damage-like setting (for example, with the presence of de novo genome ends derived from replicating DNA). Does the interaction between oriPtL and NONO alter how the cell responds in these settings, and/or does NONO or an oriPtL complex play any role in cleavage or processing of concatenated genomes. It will be interesting to explore the many possible functions of NONO and/or oriPtL in the viral replication cascade in the coming years.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health grants R01CA138268, R01AI101046, and R01AI106676 to E.K.F., F30CA177267 to M.J.S., and F31CA180449 to T.O. (and P20GM103518 to Prescott Deininger, Tulane Cancer Center), a Louisiana Clinical and Translational Science Center Pilot grant to Z.L., and American Cancer Society grant 126117-PF-14-139-01-MPC to W.N.M.
Next-generation sequencing was performed at the University of Wisconsin Biotechnology Center. Data analysis was carried out in the Tulane Cancer Center Next Generation Sequence Analysis Core using core computational resources.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00608-15.
REFERENCES
- 1.Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat Rev Cancer 4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 2.Moss WN, Lee N, Pimienta G, Steitz JA. 2014. RNA families in Epstein-Barr virus. RNA Biol 11:10–17. doi: 10.4161/rna.27488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee N, Moss WN, Yario TA, Steitz JA. 2015. EBV noncoding RNA binds nascent RNA to drive host PAX5 to viral DNA. Cell 160:607–618. doi: 10.1016/j.cell.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dresang LR, Teuton JR, Feng H, Jacobs JM, Camp DG II, Purvine SO, Gritsenko MA, Li Z, Smith RD, Sugden B, Moore PS, Chang Y. 2011. Coupled transcriptome and proteome analysis of human lymphotropic tumor viruses: insights on the detection and discovery of viral genes. BMC Genomics 12:625. doi: 10.1186/1471-2164-12-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandriani S, Ganem D. 2010. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi's sarcoma-associated herpesvirus. J Virol 84:5565–5573. doi: 10.1128/JVI.02723-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson LS, Willert EK, Virgin HW. 2010. Redefining the genetics of murine gammaherpesvirus 68 via transcriptome-based annotation. Cell Host Microbe 7:516–526. doi: 10.1016/j.chom.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fakhari FD, Dittmer DP. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J Virol 76:6213–6223. doi: 10.1128/JVI.76.12.6213-6223.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toth Z, Brulois KF, Wong LY, Lee HR, Chung B, Jung JU. 2012. Negative elongation factor-mediated suppression of RNA polymerase II elongation of Kaposi's sarcoma-associated herpesvirus lytic gene expression. J Virol 86:9696–9707. doi: 10.1128/JVI.01012-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Grady T, Cao S, Strong MJ, Concha M, Wang X, Splinter Bondurant S, Adams M, Baddoo M, Srivastav SK, Lin Z, Fewell C, Yin Q, Flemington EK. 2014. Global bidirectional transcription of the Epstein-Barr virus genome during reactivation. J Virol 88:1604–1616. doi: 10.1128/JVI.02989-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Concha M, Wang X, Cao S, Baddoo M, Fewell C, Lin Z, Hulme W, Hedges D, McBride J, Flemington EK. 2012. Identification of new viral genes and transcript isoforms during Epstein-Barr virus reactivation using RNA-Seq. J Virol 86:1458–1467. doi: 10.1128/JVI.06537-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin Z, Wang X, Strong MJ, Concha M, Baddoo M, Xu G, Baribault C, Fewell C, Hulme W, Hedges D, Taylor CM, Flemington EK. 2013. Whole-genome sequencing of the Akata and Mutu Epstein-Barr virus strains. J Virol 87:1172–1182. doi: 10.1128/JVI.02517-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng L, Lintula S, Ho TH, Anastasina M, Paju A, Haglund C, Stenman UH, Hotakainen K, Orpana A, Kainov D, Stenman J. 2012. Technique for strand-specific gene-expression analysis and monitoring of primer-independent cDNA synthesis in reverse transcription. Biotechniques 52:263–270. doi: 10.2144/0000113842. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. 2012. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. 2008. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics 9:474. doi: 10.1186/1471-2105-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nawrocki EP, Eddy SR. 2013. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edgar R, Domrachev M, Lash AE. 2002. Gene Expr. Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu G, Deng N, Zhao Z, Judeh T, Flemington E, Zhu D. 2011. SAMMate: a GUI tool for processing short read alignments in SAM/BAM format. Source Code Biol Med 6:2. doi: 10.1186/1751-0473-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Touitou R, Cochet C, Joab I. 1996. Transcriptional analysis of the Epstein-Barr virus interleukin-10 homologue during the lytic cycle. J Gen Virol 77(Pt 6):1163–1168. [DOI] [PubMed] [Google Scholar]
- 23.Lau R, Middeldorp J, Farrell PJ. 1993. Epstein-Barr virus gene expression in oral hairy leukoplakia. Virology 195:463–474. doi: 10.1006/viro.1993.1397. [DOI] [PubMed] [Google Scholar]
- 24.Hudson GS, Bankier AT, Satchwell SC, Barrell BG. 1985. The short unique region of the B95-8 Epstein-Barr virus genome. Virology 147:81–98. doi: 10.1016/0042-6822(85)90229-6. [DOI] [PubMed] [Google Scholar]
- 25.Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. 2007. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35:W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iizasa H, Wulff BE, Alla NR, Maragkakis M, Megraw M, Hatzigeorgiou A, Iwakiri D, Takada K, Wiedmer A, Showe L, Lieberman P, Nishikura K. 2010. Editing of Epstein-Barr virus-encoded BART6 microRNAs controls their dicer targeting and consequently affects viral latency. J Biol Chem 285:33358–33370. doi: 10.1074/jbc.M110.138362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lei T, Yuen KS, Tsao SW, Chen H, Kok KH, Jin DY. 2013. Perturbation of biogenesis and targeting of Epstein-Barr virus-encoded miR-BART3 microRNA by adenosine-to-inosine editing. J Gen Virol 94:2739–2744. doi: 10.1099/vir.0.056226-0. [DOI] [PubMed] [Google Scholar]
- 28.Polson AG, Bass BL. 1994. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J 13:5701–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishikura K, Yoo C, Kim U, Murray JM, Estes PA, Cash FE, Liebhaber SA. 1991. Substrate specificity of the dsRNA unwinding/modifying activity. EMBO J 10:3523–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eggington JM, Greene T, Bass BL. 2011. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2:319. doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morse DP, Aruscavage PJ, Bass BL. 2002. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc Natl Acad Sci U S A 99:7906–7911. doi: 10.1073/pnas.112704299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morse DP, Bass BL. 1999. Long RNA hairpins that contain inosine are present in Caenorhabditis elegans poly(A)+ RNA. Proc Natl Acad Sci U S A 96:6048–6053. doi: 10.1073/pnas.96.11.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moss WN, Steitz JA. 2013. Genome-wide analyses of Epstein-Barr virus reveal conserved RNA structures and a novel stable intronic sequence RNA. BMC Genomics 14:543. doi: 10.1186/1471-2164-14-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clote P, Ferre F, Kranakis E, Krizanc D. 2005. Structural RNA has lower folding energy than random RNA of the same dinucleotide frequency. RNA 11:578–591. doi: 10.1261/rna.7220505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nawrocki EP, Kolbe DL, Eddy SR. 2009. Infernal 1.0: inference of RNA alignments. Bioinformatics 25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hundley HA, Bass BL. 2010. ADAR editing in double-stranded UTRs and other noncoding RNA sequences. Trends Biochem Sci 35:377–383. doi: 10.1016/j.tibs.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehmann KA, Bass BL. 2000. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 39:12875–12884. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 38.Naganuma T, Hirose T. 2013. Paraspeckle formation during the biogenesis of long non-coding RNAs. RNA Biol 10:456–461. doi: 10.4161/rna.23547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakagawa S, Hirose T. 2012. Paraspeckle nuclear bodies–useful uselessness? Cell Mol Life Sci 69:3027–3036. doi: 10.1007/s00018-012-0973-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imamura K, Imamachi N, Akizuki G, Kumakura M, Kawaguchi A, Nagata K, Kato A, Kawaguchi Y, Sato H, Yoneda M, Kai C, Yada T, Suzuki Y, Yamada T, Ozawa T, Kaneki K, Inoue T, Kobayashi M, Kodama T, Wada Y, Sekimizu K, Akimitsu N. 2014. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL-8 expression upon immune stimuli. Mol Cell 53:393–406. doi: 10.1016/j.molcel.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 41.Saha S, Murthy S, Rangarajan PN. 2006. Identification and characterization of a virus-inducible noncoding RNA in mouse brain. J Gen Virol 87:1991–1995. doi: 10.1099/vir.0.81768-0. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Q, Chen CY, Yedavalli VS, Jeang KT. 2013. NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. mBio 4:e00596–12. doi: 10.1128/mBio.00596-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grunweller A, Wyszko E, Bieber B, Jahnel R, Erdmann VA, Kurreck J. 2003. Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates, and small interfering RNA. Nucleic Acids Res 31:3185–3193. doi: 10.1093/nar/gkg409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BM, Haag JD, Gould MN, Stewart RM, Kendziorski C. 2013. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29:1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao S, Strong MJ, Wang X, Moss WN, Concha M, Lin Z, O'Grady T, Baddoo M, Fewell C, Renne R, Flemington EK. 2015. High-throughput RNA sequencing-based virome analysis of 50 lymphoma cell lines from the Cancer Cell Line Encyclopedia Project. J Virol 89:713–729. doi: 10.1128/JVI.02570-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manet E, Gruffat H, Trescol-Biemont MC, Moreno N, Chambard P, Giot JF, Sergeant A. 1989. Epstein-Barr virus bicistronic mRNAs generated by facultative splicing code for two transcriptional trans-activators. EMBO J 8:1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. 2011. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 44:667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shav-Tal Y, Zipori D. 2002. PSF and p54(nrb)/NonO–multi-functional nuclear proteins. FEBS Lett 531:109–114. doi: 10.1016/S0014-5793(02)03447-6. [DOI] [PubMed] [Google Scholar]
- 49.Basu A, Dong B, Krainer AR, Howe CC. 1997. The intracisternal A-particle proximal enhancer-binding protein activates transcription and is identical to the RNA- and DNA-binding protein p54nrb/NonO. Mol Cell Biol 17:677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yadav SP, Hao H, Yang HJ, Kautzmann MA, Brooks M, Nellissery J, Klocke B, Seifert M, Swaroop A. 2014. The transcription-splicing protein NonO/p54nrb and three NonO-interacting proteins bind to distal enhancer region and augment rhodopsin expression. Hum Mol Genet 23:2132–2144. doi: 10.1093/hmg/ddt609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu L, Xie N, Rennie P, Challis JR, Gleave M, Lye SJ, Dong X. 2011. Consensus PP1 binding motifs regulate transcriptional corepression and alternative RNA splicing activities of the steroid receptor coregulators, p54nrb and PSF. Mol Endocrinol 25:1197–1210. doi: 10.1210/me.2010-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emili A, Shales M, McCracken S, Xie W, Tucker PW, Kobayashi R, Blencowe BJ, Ingles CJ. 2002. Splicing and transcription-associated proteins PSF and p54nrb/nonO bind to the RNA polymerase II CTD. RNA 8:1102–1111. doi: 10.1017/S1355838202025037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dong X, Sweet J, Challis JR, Brown T, Lye SJ. 2007. Transcriptional activity of androgen receptor is modulated by two RNA splicing factors, PSF and p54nrb. Mol Cell Biol 27:4863–4875. doi: 10.1128/MCB.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang YS, Hanke JH, Carayannopoulos L, Craft CM, Capra JD, Tucker PW. 1993. NonO, a non-POU-domain-containing, octamer-binding protein, is the mammalian homolog of Drosophila nonAdiss. Mol Cell Biol 13:5593–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosonina E, Ip JY, Calarco JA, Bakowski MA, Emili A, McCracken S, Tucker P, Ingles CJ, Blencowe BJ. 2005. Role for PSF in mediating transcriptional activator-dependent stimulation of pre-mRNA processing in vivo. Mol Cell Biol 25:6734–6746. doi: 10.1128/MCB.25.15.6734-6746.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang S, Lutz CS. 2006. p54nrb is a component of the snRNP-free U1A (SF-A) complex that promotes pre-mRNA cleavage during polyadenylation. RNA 12:111–121. doi: 10.1261/rna.2213506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Straub T, Grue P, Uhse A, Lisby M, Knudsen BR, Tange TO, Westergaard O, Boege F. 1998. The RNA-splicing factor PSF/p54 controls DNA-topoisomerase I activity by a direct interaction. J Biol Chem 273:26261–26264. doi: 10.1074/jbc.273.41.26261. [DOI] [PubMed] [Google Scholar]
- 58.Straub T, Knudsen BR, Boege F. 2000. PSF/p54(nrb) stimulates “jumping” of DNA topoisomerase I between separate DNA helices. Biochemistry 39:7552–7558. doi: 10.1021/bi992898e. [DOI] [PubMed] [Google Scholar]
- 59.Poulsen H, Nilsson J, Damgaard CK, Egebjerg J, Kjems J. 2001. CRM1 mediates the export of ADAR1 through a nuclear export signal within the Z-DNA binding domain. Mol Cell Biol 21:7862–7871. doi: 10.1128/MCB.21.22.7862-7871.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fritz J, Strehblow A, Taschner A, Schopoff S, Pasierbek P, Jantsch MF. 2009. RNA-regulated interaction of transportin-1 and exportin-5 with the double-stranded RNA-binding domain regulates nucleocytoplasmic shuttling of ADAR1. Mol Cell Biol 29:1487–1497. doi: 10.1128/MCB.01519-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strehblow A, Hallegger M, Jantsch MF. 2002. Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol Biol Cell 13:3822–3835. doi: 10.1091/mbc.E02-03-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, Olshansky M, Rechavi G, Jantsch MF. 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 63.Keegan LP, Gallo A, O'Connell MA. 2001. The many roles of an RNA editor. Nat Rev Genet 2:869–878. doi: 10.1038/35098584. [DOI] [PubMed] [Google Scholar]
- 64.Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. 2006. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol 13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. 2007. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kume H, Hino K, Galipon J, Ui-Tei K. 2014. A-to-I editing in the miRNA seed region regulates target mRNA selection and silencing efficiency. Nucleic Acids Res 42:10050–10060. doi: 10.1093/nar/gku662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laurencikiene J, Kallman AM, Fong N, Bentley DL, Ohman M. 2006. RNA editing and alternative splicing: the importance of cotranscriptional coordination. EMBO Rep 7:303–307. doi: 10.1038/sj.embor.7400621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.