

ABSTRACT
Persistent infections with certain human papillomaviruses (HPV) such as HPV16 are a necessary risk factor for the development of anogenital and oropharyngeal cancers. HPV16 genomes replicate as low-copy-number plasmids in the nucleus of undifferentiated keratinocytes, which requires the viral E1 and E2 replication proteins. The HPV16 E8^E2C (or E8^E2) protein limits genome replication by repressing both viral transcription and the E1/E2-dependent DNA replication. How E8^E2C expression is regulated is not understood. Previous transcript analyses indicated that the spliced E8^E2C RNA is initiated at a promoter located in the E1 region upstream of the E8 gene. Deletion and mutational analyses of the E8 promoter region identify two conserved elements that are required for basal promoter activity in HPV-negative keratinocytes. In contrast, the transcriptional enhancer in the upstream regulatory region of HPV16 does not modulate basal E8 promoter activity. Cotransfection studies indicate that E8^E2C inhibits, whereas E2 weakly activates, the E8 promoter. Interestingly, the cotransfection of E1 and E2 induces the E8 promoter much more strongly than the major early promoter, and this is partially dependent upon binding of E2 to Brd4. Mutation of E8 promoter elements in the context of HPV16 genomes results in an increased genome copy number and elevated levels of viral early and late transcripts. In summary, the promoter responsible for the expression of E8^E2C is both positively and negatively regulated by viral and cellular factors, and this regulatory circuit may be crucial to maintain a low but constant copy number of HPV16 genomes in undifferentiated cells.
IMPORTANCE HPV16 replicates in differentiating epithelia and can cause cancer. How HPV16 maintains its genome in undifferentiated cells at a low but constant level is not well understood but may be relevant for the immunological escape of HPV16 in the basal layers of the infected epithelium. This study demonstrates that the expression of the viral E8^E2C protein, which is a potent inhibitor of viral replication in undifferentiated cells, is driven by a separate promoter. The E8 promoter is both positively and negatively regulated by viral proteins and thus most likely acts as a sensor and modulator of viral copy number.
INTRODUCTION
Persistent infections with high-risk (HR) human papillomaviruses (HPV) are a necessary risk factor for the development of anogenital and oropharyngeal cancers of the cervix uteri and other epithelial cancers. HPV16 is an exceptional human carcinogen, as it is responsible for more than 50% of cervical cancers and is highly prevalent in other HPV-related cancers (1–4).
HPV have a covalently closed, double-stranded DNA genome of approximately 8 kb. After infection of keratinocytes in the basal layer of the mucosal epithelium, viral genomes are established at 10 to 100 extrachromosomal copies per cell and only viral early genes are transcribed (5). In suprabasal cells, viral genomes are amplified to several thousands of copies and viral late genes are expressed (5). When maintained in an undifferentiated state, the viral copy number remains relatively constant in HR-HPV-positive keratinocytes, which indicates that regulatory circuits are active to control copy number (5). The replication of HR-HPV genomes in undifferentiated cells requires the expression of the viral E1 and E2 proteins, which recognize specifically the viral origin of replication (6). The major HR-HPV, HPV16, -18, and -31, express a spliced mRNA that links the E8 gene to the E2 gene and has the capability to encode an E8^E2C (or E8/E2 or E8^E2) protein (7–10). Genetic analyses have revealed that E8^E2C inhibits genome replication of HR-HPV16, -18, and -31 in undifferentiated cells, and there is also evidence that E8^E2C limits productive replication of HPV16 in differentiated cells (8–11). The HPV16, -18, and -31 E8^E2C proteins bind to the viral origin of replication and are potent repressors of transcription and the E1/E2-dependent replication (7, 9, 11–14). Both activities require the conserved E8 part, which recruits cellular corepressor molecules (14–16). In summary, it is very likely that the expression of E8^E2C is tightly regulated.
The transcriptomes of the major HR-HPV HPV16, -18, and -31 have been extensively analyzed, which revealed that HR-HPV have two main promoters: the major early promoter in front of the E6 gene (P97 for HPV16) and the major late promoter in the E7 gene (P670 for HPV16) (http://pave.niaid.nih.gov/#explore/transcript_maps). The major early promoter is highly active in both undifferentiated and differentiated keratinocytes, whereas the major late promoter becomes highly active only in differentiated keratinocytes (17–19). Primary P97 transcripts include all early viral genes (E1, E2, and E4 to E8) and are processed at the early polyadenylation signal downstream of the E5 gene. Facultative splicing signals within the E6, E1, and E2 genes result in more than 10 polycistronic transcripts with different coding potential. HPV use unconventional translation mechanisms, as E1, E2, and E7 proteins can be efficiently translated from polycistronic mRNAs despite one or more upstream open reading frames (ORFs) (20–22). Transcripts initiated at the major late promoter can be processed at either the early or the late polyadenylation signal downstream of L1 and thus have the capacity to carry all viral genes except E6 and E7. Late transcripts are also processed at facultative splice sites in E1, E2, and L1, giving rise to multiple species with differing coding potential.
Theoretically, E8^E2C could be translated from spliced mRNAs initiated at the major early or late promoter by an unconventional translation mechanism. However, the analysis of polyadenylated transcripts isolated from differentiated, HPV16-positive W12 cells by 5′ rapid amplification of cDNA ends (5′RACE) identified an E8^E2C/E5/L2/L1 transcript that initiated at nucleotide (nt) 1140 within E1 (23). Sequencing of HPV16 transcripts in cervical intraepithelial neoplasia grade 2 (CIN2) and CIN3 lesions also revealed a spliced E8 transcript starting at nt 1127 (24). Consistent with these data, a competitive reverse transcriptase (RT)-PCR approach indicated that the major start site for the HPV16 E8^E2C transcript maps to nt 1126 to 1142 in undifferentiated cells (9). Furthermore, the mapping of HPV18 transcripts during productive viral infection by 5′RACE identified an E8^E2C transcript initiating at nt 1202 (19). Taken together, these data suggest that HPV16 and 18 E8^E2C transcripts initiate 120 to 130 nt upstream of the E8 ATG in E1.
MATERIALS AND METHODS
Recombinant plasmids.
Reporter plasmids pGL16 863/1268, 958/1268, 1038/1268, and 1142/1268 are based upon pGL3-basic (Promega) and were constructed by PCR using the cloned HPV16 114b genome and primer HPV16 E7 649F or HPV16-specific primers adding a 5′ NcoI restriction site and a reverse primer creating a fusion between HPV16 and the luciferase gene (16 E8/luc NarI R, 5′-ATAGAATGGCGCCGGGCCTTTCTTTATGTTTTTGGCGTCTTCCATACCCGCTGTCTTCGCT-3′). Amplicons were cloned between the NcoI and NarI restriction sites of pGL3-basic (Promega). Plasmids pGL16 863/1109 and 863/1199 were constructed by PCR using primer HPV16 E7 649F and HPV16-specific primers adding a 3′ NcoI restriction site. Amplicons were cloned into the NcoI restriction site of pGL3-basic (Promega). Plasmid pGL16 URR (where URR is upstream regulatory region) has been previously described (7). Plasmid pGL16 7154/868 was constructed by PCR, and HPV16 nt 7154 to 7906 and 1 to 868 were inserted between the XhoI and NcoI restriction sites of pGL3-basic. To generate plasmid pGL16 7154/1268, the XhoI/NcoI fragment from pGL16 7154/868 was inserted into XhoI/NcoI-digested pGL16 863/1268. Expression plasmids pSG16 E2, E8^E2C, and E8^E2C KWK mt are based upon pSG5 (Stratagene) and have been previously described (7, 11). Plasmid pSG16 E2 R37A/I73A was constructed by exchanging an in vitro-synthesized EcoRI/MscI fragment of a mutated version of HPV16 E2 (Life Technologies). A human codon optimized version of HPV16 E1 was synthesized by Life Technologies and cloned into the BglII site of pSG5, giving rise to pSG16 E1co. Mutations of promoter elements were generated by overlap extension PCR, and the resulting amplicons were cloned into plasmid pGL16 863/1268, pGL16 7154/1268, or pBS HPV16 114b. The inserts of all newly constructed plasmids were verified by DNA sequencing.
Cell culture.
The HPV-negative human keratinocyte cell line RTS3b (25) and normal human keratinocytes (NHK) derived from foreskin were maintained as previously described (11). HPV16-positive human keratinocytes were established by transfection with ligated HPV16 wild-type or mutant genomes and maintained as described previously (11).
Quantification of viral copy number and viral transcripts in stable cell lines.
Copy numbers in low-molecular-weight DNA were quantified by quantitative real-time PCR (qPCR) using amplicons in the HPV16 E2 and the cellular ACTB genes and copy number standards as described previously (11). Isolation of RNA and cDNA synthesis was performed as described previously (11). The analysis of HPV16 E1, E2, E6, E7, and L1 transcripts was done by qPCR using primer pairs previously described (11). For the analysis of spliced E8^E2C, E8^E1/E2, E8/L1, and E8^E2H^L2/L1 transcripts, primer pairs 1079F (5′-CTGCACAGGAAGCAAAACAA-3′)/3403R (5′-GGCCAAGTGCTGCCTAATAA-3′), 1281F (26)/3719R (5′-TCCAATGCCATGTAGACGAC-3′), 1259F (11)/2857R (5′-CACATTCTAGGCGCATGTGT-3′), 16E8L1 F (5′-TGAAGTGGAAACTCAGCAGATG-3′)/5764R (5′-TAGGGATGTCCAACTGCAAG-3′), and 1281F/5690R (5′-CTGGGACAGGAGGCAAGTAG-3′) were selected with the Primer3plus algorithm (27).
Qualitative RT-PCR.
RNA (1 μg) isolated from undifferentiated HPV16 wild-type (wt)-positive keratinocytes was reverse transcribed and amplified with primer pair 1079F/3403R, 1259F/2857R, or 1259F/5674R using the One-Step RT-PCR kit (Qiagen). Amplicons were isolated from agarose gels and cloned into pJET1.2 (Thermo Scientific), and single clones were sequenced. The corresponding plasmids were labeled pJET 1079-1301/3358-3403, pJET HPV16 E8^E1, E2, pJET HPV16 E8^E2H^L2, L1, and pJET HPV16 E8^X, L1.
Reporter gene assays.
A total of 3 × 104 RTS3b or 5 × 104 NHK cells were seeded into 24-well dishes 1 day before transfection. Cells were transfected with reporter plasmids alone or together with pSG 16 E1 co, pSG 16 E2 (or pSG16 E2 R37A/I73A), or pSG 16 E8^E2C (or 16 E8^E2C KWK mt) expression constructs or the empty vector pSG5 as indicated in the figure legends using Fugene HD (Promega) and Opti-MEM (Life Technologies). In addition, the pCMV-Gluc plasmid (New England BioLabs) was cotransfected as an internal control. Gaussia and firefly luciferase assays were carried out 48 h after transfection.
Quantification of transiently replicating reporter plasmids.
The remaining whole-cell extracts used to determine luciferase activity were transferred into 1.5-ml tubes. Total DNA was then extracted with the EZ1 DNA tissue kit and the EZ1 instrument according to the manufacturer′s instructions (Qiagen). Ten-microliter aliquots were either digested with DpnI (20 U) or left untreated for 2 h, and then aliquots thereof were subjected to qPCR using primers that detect an amplicon in the luciferase gene that includes two DpnI restriction sites (luc qPCR F, 5′-CCAGGGATTTCAGTCGATGT-3′, and luc qPCR R, 5′-AATCTCACGCAGGCAGTTCT-3′). Copy number standards were run in parallel, and values are given as the plasmid fraction resistant to DpnI digestion.
Southern blot hybridization.
Low-molecular-weight DNA from HPV16-positive cell lines was isolated as described previously (10). DNA was digested with XbaI, a noncutter for HPV16, and run in a 0.8% agarose gel. Blotting and hybridization to a 32P-labeled HPV16 probe were carried out as previously described (10). After exposure of the membrane to PhosphorImager screens, HPV16 genomes were visualized using the AIDA software package (Raytest).
EMSAs.
For electrophoretic mobility shift assays (EMSAs), nuclear extracts from RTS3b cells were incubated with unlabeled double-stranded oligonucleotides (4 pmol) in the presence of 20 mM HEPES (pH 7.9), 50 mM KCl, 0.5 mM EDTA, 1 mM dithiothreitol (DTT), 5% glycerol (vol/vol), and 50 μg/ml poly(dI·dC) for 5 min at room temperature. Then, double-stranded 5′-DY681-labeled oligonucleotides (40 fmol) were added and incubated for an additional 15 min and then separated in 5% polyacrylamide (PAA)–0.5× Tris-borate-EDTA (TBE) gels at 100 V. Fluorescent signals were recorded with an OdysseyFC infrared imaging system (LI-COR Biosciences).
Statistical analyses.
Statistical analyses were performed with GraphPad Prism 5.02 software. Some data (see Fig. 5 and 6B) were analyzed with a one-way analysis of variance (ANOVA) and Tukey's multiple-comparison test; some (see Fig. 7B) were analyzed with a two-tailed paired t test.
FIG 5.
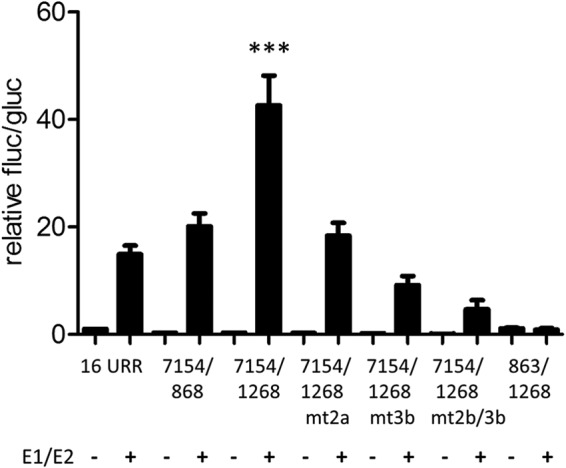
Regulation of the E8 promoter by E1 and E2. RTS3b cells were cotransfected with 0.5 ng of pCMV-Gluc, the indicated reporter plasmids (50 ng), and the empty vector (−) or a combination of 100 ng pSG16 E1co and 10 ng pSG16 E2 (+).Values are presented as the ratios of fluc to gluc activities relative to 16URR (set as 1). Error bars indicate the SEM. Statistical significance was calculated with a one-way ANOVA and Tukey's multiple-comparison test (***, P < 0.001).
FIG 6.
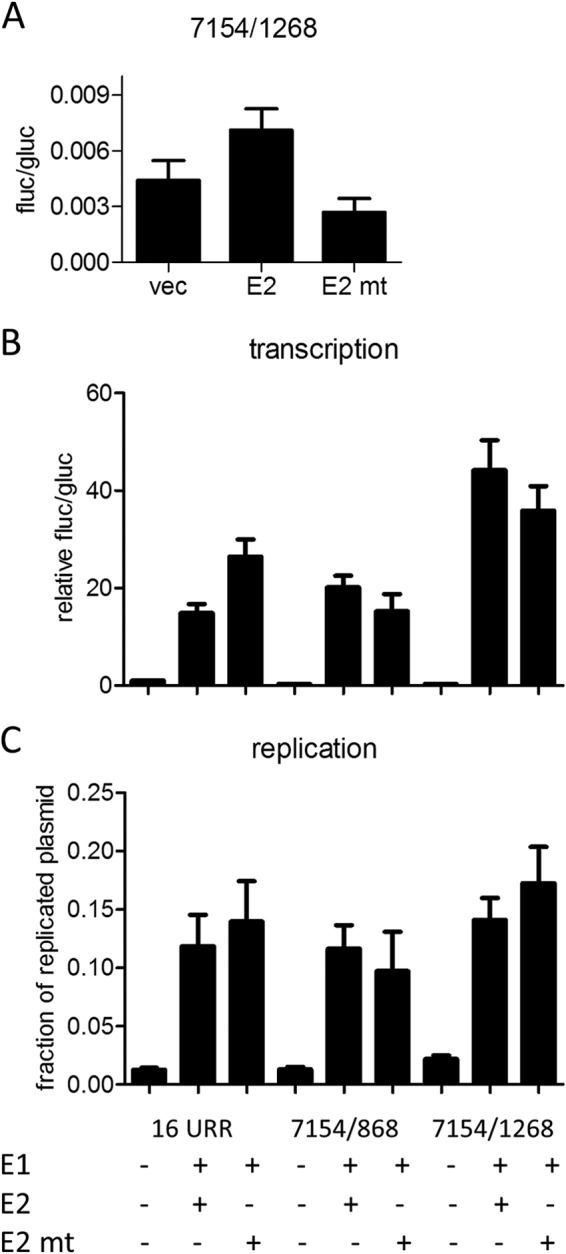
Influence of a BRD4-binding-deficient E2 mutant on E8 promoter activity. (A) RTS3b cells were cotransfected with 0.5 ng of pCMV-Gluc, 50 ng reporter, and 30 ng of the empty vector (vec), pSG16 E2 (E2), or pSG16 E2 R37A/I73A (E2 mt). Values are presented as the ratios of fluc to gluc activities. (B) RTS3b cells were cotransfected with 0.5 ng of pCMV-Gluc, the indicated reporter plasmids (50 ng), and the empty vector (−) or combinations of 100 ng pSG16 E1co and 10 ng pSG16 E2 or pSG 16 E2 R37A/I73A (E2 mt). Values are presented as the ratios of fluc to gluc activities relative to that of 16 URR (set as 1). (C) The amount of DNA replication was determined by qPCR and is presented as the fraction of plasmid DNA resistant to DpnI digest. Error bars indicate the SEM.
FIG 7.

Analysis of HPV16 E8 promoter mutant genomes. (A) Southern blot analysis of low-molecular-weight DNA from two different sets of HPV16 wt (wt) and E8P mt2b/3b (mt) NHK cell lines. The linearized HPV16 genome (100 pg) was used as the molecular size marker (M). Viral copy number in the low-molecular-weight fraction was determined by qPCR and indicates the number of HPV16 E2 copies/2 beta-actin copies. (B) qPCR analysis of different viral transcripts. Values represent the number of viral transcript copies per cellular PGK1 copies.
RESULTS
E8^E2C transcripts initiate within E1 and are the dominant species among HPV16 RNAs processed at SD1302.
We first aimed to characterize in detail the structures of E8 exon-containing transcripts and performed qualitative RT-PCR experiments using primer pairs 1079F/3403R, 1259F/2857R, and 1259F/5764R and RNA from keratinocytes harboring episomal HPV16 genomes (11). Cloning and sequencing of RT-PCR products revealed that E8^E2C transcripts might also initiate upstream of nt 1126, that SD1302 can also be linked to SA2709, and that the corresponding mRNA could be translated into an E8^E1 (nt 616 to 649) fusion protein and encode E2 (Fig. 1A). SD1302 can also be directly linked to SA5639, giving potentially rise to an E8 fusion protein with an undesignated ORF (X) and also L1. Alternatively, SD1302 can be first linked to an exon between SA3358 and SD3632 and then linked to SA5639. This transcript may encode an E8^E2H^L2 fusion protein consisting of E8, the E2 hinge region, and the final 6 residues of L2 and also L1. This indicated that the E8 exon is part of additional transcripts that may express novel E8 fusion proteins as well as full-length E2 and L1 proteins (Fig. 1A).
FIG 1.

E8^E2C transcripts are the dominant species among HPV16 RNAs processed at SD 1302. (A) Schematic representation of a partial HPV16 genome from the E1 to the L1 gene. Splice donor (SD) and acceptor (SA) sites as well as the early (pAE) and late (pAL) polyadenylation sites are indicated. The structures of RNAs and their coding potential present in HPV16 wt cells processed at SD1302 are shown below the genome representations. Primers used for the detection by qPCR are indicated by arrows above the different RNA species. (B) Quantification of the different RNA species in undifferentiated and differentiated HPV16 wt cells by qPCR. Error bars indicate the standard errors of the means (SEM).
To quantify the amounts of the different transcripts, real-time PCR experiments were carried out using RNA from different HPV16wt cell lines maintained in an undifferentiated state or grown in organotypic raft cultures (11). E8^E1, E2 transcripts were detected with primers 1259F and 2857R. E8^E2C transcripts were detected with primer 1281F, which binds to the SD1302/SA3358 fusion sequence and primer 3719R downstream of SA3632. The E8^E2H^L2, L1 transcript was detected with primers 1281F and 5690R, and the E8^X, L1 transcript was detected with primer E8/L1 F binding to the SD1302/SA5639 fusion sequence and 5764R. E8^E2C transcripts that are initiated upstream of nt 1126 were detected with the primer pair 1079F/3403R. These analyses revealed that the E8^E2C encoding transcript is the most abundant transcript processed at SD1302 in undifferentiated and differentiated cells (Fig. 1B). E8^E1,E2 transcripts are 5- and 10-fold less abundant in undifferentiated and differentiated cells, respectively. E8^E2H^L2,L1 and E8^X,L1 transcripts are 5- and 13-fold less abundant in both undifferentiated and differentiated cells (Fig. 1B). Furthermore, E8^E2C transcripts detected with the primer pair 1079F/3403R are 17-fold less abundant than transcripts detected with the primer pair 1281F/3719R in both undifferentiated and differentiated cells (Fig. 1B). In organotypic cultures, the amounts of all transcripts processed at SD1302 were increased 2- to 5-fold compared to undifferentiated cultures, which is consistent with previous findings (11). This indicates that E8^E2C transcripts are present throughout the life cycle of HPV16 and that these transcripts are initiated mainly downstream of nt 1098, confirming a separate promoter for E8^E2C transcripts (9).
Two conserved elements are required for HPV16 E8 promoter activity.
To characterize the E8 promoter in more detail, HPV16 nt 863 to 1268 were inserted in the promoterless firefly luciferase reporter pGL3-basic in such a way that the E8 ATG (nt 1265 to 1268) was used for the translation of luciferase. This construct is devoid of any regulatory elements that have been implicated in the control of the major late promoter P670 (28, 29). In addition, 5′- and 3′-deletion constructs were generated. The different constructs were transfected into HPV-negative RTS3b or NHK cells, and luciferase activity was determined. In both cell lines, activity remained constant when nt 863 to 1038 were deleted (Fig. 2). In contrast, deletion of nt 1038 to 1094 dramatically decreased activity. The removal of nt 1199 to 1268 did not change activity in NHK and increased transcription in RTS3b cells. The deletion of nt 1109 to 1199 resulted in greatly decreased activities (Fig. 2). This strongly suggested that promoter activity in the 5′ region of E1 is dependent upon two separate elements. A sequence alignment of the E1 coding region of HPV16, 11, 18, 31, and 33, which all transcribe E8^E2C messages, identified two conserved elements in the region between nt 1038 to 1094 and one in the region between nt 1109 and 1199 (Fig. 3A). Mutational analysis of CE1, CE2, and CE3 in the context of the 863/1268 reporter plasmid demonstrated that CE1 does not contribute to activity in NHK and contributes only moderately in RTS3b cells (Fig. 3B). In contrast, mutation of CE2 (mt2a) or CE3 (mt3a) dramatically reduced activity. Additional mutations in CE2 (mt2b and mt2d) revealed that single nucleotide exchanges in the central GG nucleotides were sufficient to diminish activity significantly. Also, the mutation of five conserved nucleotides in CE3 greatly reduced promoter activity. EMSAs revealed that CE2 and CE3 interacted specifically with nuclear proteins, as only wt but not mutant sequences could compete for binding (Fig. 3C).
FIG 2.

Two regions are required for E8 promoter activity in keratinocytes. RTS3b or NHK cells were transfected with 0.5 ng of pCMV-Gluc and 50 or 300 ng of the indicated reporter plasmids, respectively. (Upper panels) Values are presented as the ratios of firefly luciferase (fluc) to gaussia luciferase (gluc) activities, and error bars indicate the SEM. (Lower panel) Schematic representation of reporter constructs aligned to the HPV genome.
FIG 3.

Two conserved elements contribute to E8 promoter activity. (A) Sequence alignments identified three conserved elements (CE1 to CE3) in alpha-PV genomes reported to transcribe E8^E2C mRNA. Mutated bases are shown below the alignments. (B) RTS3b or NHK cells were transfected as described in the legend to Fig. 2. Values are presented as the ratios of firefly luciferase (fluc) to gaussia luciferase (gluc) activities, and error bars indicate the SEM. (C) EMSA analysis of nuclear proteins binding to conserved element 2 (CE2) or CE3. Nuclear extracts (NE) were incubated with the respective wt DY681-labeled oligonucleotides and unlabeled competitor oligonucleotides as indicated. Major shifted bands that can be competed only by wt but not mt sequences are indicated by arrows.
The E8 promoter is repressed by E8^E2C and activated by E1 and E2.
The major early and late promoters are controlled by sequences in the viral upstream regulatory region (URR) (30). To test whether the E8 promoter is also influenced by the URR, HPV16 nt 7154 to 1268 were inserted into pGL3-basic, adding the URR, P97, E6, E7, and P670 to the E8 promoter. Interestingly, activities from pGL16 7154/1268 were similar to those of pGL16 863/1268, indicating that neither the regulatory elements in the URR nor the major early and late promoters contribute to E8 expression in RTS3b and NHK cells (Fig. 4A). Mutation of CE2 (mt2a) or CE3 (mt3b) alone or in combination (mt2b/3b) decreased basal activity, strongly indicating that luciferase expression of the 7154/1268 construct is driven mainly by the E8 promoter (Fig. 4A). In order to understand how E8 promoter activity is influenced by viral regulator proteins, cotransfection experiments with an expression vector for HPV16 E8^E2C or the mutant E8^E2C KWK mt protein, which no longer inhibits genome replication and E1/E2-dependent ori replication, were carried out (11). In both cell lines, the wild-type repressor inhibited expression from the 7154/1268 construct 10- and 7-fold, respectively, whereas the mutant had no effect (Fig. 4B). This is consistent with observations for HPV31 indicating that E8^E2C proteins have long-distance repression activity (12). We also determined E8^E2 transcript levels in previously described cell lines harboring HPV16 wt or E8− genomes (11). This revealed that the loss of E8^E2C increases E8^E2C transcription ∼12-fold (Fig. 4C), which supports the idea that E8^E2C autoregulates its own expression.
FIG 4.

Regulation of the E8 promoter by the URR and E8^E2C. (A, B) RTS3b or NHK cells were transfected with 0.5 ng of pCMV-Gluc and 50 or 300 ng of the indicated reporter plasmids, respectively. Values are presented as the ratios of fluc to gluc activities, and error bars indicate the SEM. (A) Basal promoter activities. (B) Cells were cotransfected with the pGL16 7154/1268 construct and 10 ng (NHK) or 30 ng (RTS3b) of either the empty vector (vec) or HPV16 E8^E2C or HPV16 E8^E2C KWK mt expression vectors. (C) The amount of E8^E2C RNA is greatly enhanced by a loss of E8^E2C. RNA from NHK-HPV16 wt or HPV16 E8− cell lines (11) was analyzed by qPCR with E8^E2C and PGK1 specific primers. Error bars indicate the SEM.
Coexpression of the HPV16 URR construct (16 URR) with HPV16 E1 and E2 strongly activates reporter gene expression, which is due mainly to the induction of plasmid replication and therefore an increase in copy number (11). We therefore cotransfected the 7154/1268 E8 promoter construct with expression vectors for E1 and E2 into RTS3b cells. Interestingly, cotransfection of E1 and E2 induced a statistically significant, ∼3-fold-higher activity from 7154/1268 than that obtained with the HPV16 URR (Fig. 5). Calculating the extent of induction related to the corresponding basal activities revealed that the URR construct is activated 15-fold by E1 and E2 whereas the 7154/1268 construct is induced 139-fold. The E1/E2-induced activity of 7154/1268 was strongly dependent on the activity of the E8 promoter, as single or combined mutations of CE2 and CE3 (mt2a, mt3b, and mt2b/3b [Fig. 3A]) were greatly impaired in their response to E1/E2 (Fig. 5). The E1/E2-induced activity of the 7154/1268 construct was also highly dependent on URR sequences, as the 863/1268 construct did not respond to E1 and E2 (Fig. 5). Furthermore, a plasmid that extends the URR construct to the ATG of E1 (pGL16 7154/868) and thus includes the late promoter P670 was activated by E1/E2 to levels similar to those of 16 URR. The HPV E2 protein has been described to repress the major early promoter but has also the ability to activate promoters when E2 binding sites are placed in an enhancer configuration (31). Both activities have been linked to the interaction of E2 with the cellular Brd4 protein (32). Thus, the different responses of the major early promoter in the 16 URR construct and the E8 promoter in the 7154/1268 construct to E1/E2 could be due to transcriptional modulation by E2. To test this, a mutant of HPV16 E2 (R37A/I73A) that no longer binds to the cellular Brd4 protein and therefore cannot activate transcription was used (33–35). Consistent with published data, HPV16 E2 R37A/I73A almost completely lost the ability to activate the E2-dependent promoter plasmid pC18-Sp1-luc (data not shown). Wild-type E2, in contrast to E2 R37A/I73A, weakly activated the 7154/1268 construct (Fig. 6A). This suggested that E2 alone is a weak activator of the E8 promoter. In combination with E1, HPV16 E2 R37A/I73A induced slightly lower luciferase levels from the 7154/1268 and the 7154/868 construct than did wt E2 (Fig. 6B). In contrast, E1 and E2 R37A/I73A induced a higher luciferase activity from the 16 URR construct. The differences between 16 URR + E1/E2 and 7154/1268 + E1/E2 or 7154/1268 + E1/E2mt were highly statistically significant, whereas the differences between 16 URR + E1/E2 mt and 7154/1268 + E1/E2 or 7154/1268 + E1/E2mt were not, which suggests that the recruitment of Brd4 by E2 contributes to the differences observed with wt E2. Furthermore, DNA replication levels of the different promoter plasmids were determined by qPCR. This revealed that all plasmids replicated to similar levels in the presence of wt E2 and the R37A/I73A mutant (Fig. 6C). The replication competence of the R37A/I73A mutant in RTS3b cells seen here differs from observations in C33a cells reported by Wang et al. (36). However, a recent publication demonstrated that the failure of the HPV16 E2 R37A mutant to replicate the viral origin together with E1 in C33a cells can be overcome by greatly increasing the amount of expression vector (37). Thus, the replication phenotype of R37A/I73A may depend on E2 protein levels. In summary, our data suggest that the differences in transcription levels are not due to differences in replication but are at least partially caused by the interaction of E2 with Brd4, which weakly represses the major early promoter on one hand and weakly activates the E8 promoter on the other hand.
Mutations of CE2 and CE3 in HPV16 genomes enhance viral replication and transcription in human keratinocytes.
To evaluate the contribution of E8 promoter elements to viral replication, mutations in CE2 (mt2b) and CE3 (mt3b) that disrupt basal and E1/E2-induced promoter activity but are silent in the overlapping E1 gene were introduced into the HPV16 genome (HPV16-E8P mt2b/3b). Two independent sets of stable NHK cells obtained from two different donors were established by transfection with HPV16 wild-type or E8P mt2b/3b mutant genomes and drug selection. Following selection, colonies were pooled and expanded. Low-molecular-weight DNA was isolated from low-passage-number cells and subjected to qPCR and Southern blot analysis. This revealed that mutant genomes were maintained episomally with a 1.9-fold- and 2.6-fold-increased copy number compared to that of the wild type (Fig. 7A). In addition, viral early and late transcription in wt and E8P-mt2b/3b cell lines was quantified by qPCR. This revealed that E6 transcripts were significantly increased 1.9-fold, E7 transcripts 1.7-fold, E1 transcripts 3.6-fold, E2 transcripts 2.3-fold, and L1 transcripts 3.3-fold compared to the corresponding HPV16 wild-type cells (Fig. 7B). In contrast, the levels of E8^E2C transcripts were slightly but not significantly reduced in mutant cell lines (Fig. 7B). In summary, these data are consistent with the idea that the CE2 and CE3 are E8 promoter elements that regulate E8^E2C expression from the HPV16 genome, as their inactivation increases both genome copy number and viral transcription.
DISCUSSION
Several papillomaviruses have been shown to maintain their genomes at 10 to 100 copies per cell in undifferentiated cells over many passages. The underlying mechanisms have not been fully elucidated, but studies using HR-HPV genomes that no longer express the E8^E2C protein have suggested that E8^E2C plays an important role in limiting copy number (8–11). How E8^E2C expression is regulated is currently unknown. Recent studies have indicated that HPV16 and 18 E8^E2C mRNAs initiate within the E1 gene 120 to 130 nt upstream of the E8 ATG start codon and also suggested the existence of additional transcripts that use SD1302 but do not encode E8^E2C (9, 19, 23, 24). Qualitative RT-PCR experiments confirmed that SD1302 is used in at least three additional viral transcripts that do not encode E8^E2C (Fig. 1A). However, qPCR analyses strongly suggest that E8^E2C-encoding transcripts account for approximately 70% of the transcripts processed at SD1302 in both undifferentiated and differentiated cells. Furthermore, these experiments also confirmed that E8^E2C transcripts are derived from a separate promoter close to the E8 ATG as suggested by Lace and coworkers and not from the viral major early or late promoters (9). Consistent with this, we found that an HPV16 fragment from the E1 ATG to the E8 ATG (nt 863 to 1268) displayed promoter activity in normal and immortalized human keratinocytes (Fig. 2). Deletion and mutational analyses identified two conserved elements as being important for promoter activity: CE2 (nt 1084 to 1090) and CE3 (nt 1121 to 1134). CE2 interacted specifically with nuclear proteins, and based on the importance of the central GG motif for binding and promoter activity, it is possible that CE2 interacts with members of the ets protein family that recognize GGAA/T sequences (38). Humans carry 28 different ets genes, which have very similar DNA recognition sites, and 14 to 25 family members can be coexpressed in different cell types (38). Thus, it is possible that different ets members interact with CE2, which may explain that several specific shifted bands were observed in EMSAs (Fig. 3C). CE3 (nt 1121 to 1134) also specifically interacted with nuclear proteins (Fig. 3C). CE3 overlaps with one of the reported start sites and is very close to the other one (23, 24) and thus might have initiator activity.
In addition to being activated by cellular factors, the E8 promoter can be inhibited by E8^E2C in an E8 domain dependent-manner, suggesting that E8^E2C transcription is autoregulated. Consistent with this, E8^E2C transcripts are strongly upregulated when E8^E2C cannot be expressed from HPV16 genomes (Fig. 4C). On the other hand, the E8 promoter responds to E1 and E2 more strongly than do P97 or P670 promoter constructs. A part of this stimulation appears to be due to the recruitment of Brd4 by E2 (Fig. 6). However, in the presence of E1, the Brd4-binding-deficient E2 mutant showed only a slightly decreased activation of the E8 promoter construct, whereas an increased activation of the P97 (16 URR) construct was observed. Thus, the differential responses of the P97 and the E8 promoter to E1 and wild-type E2 might be due both to a repression of P97 and to an activation of the E8 promoter via E2-Brd4. It is also possible that E1 contributes to the transcriptional activation of the E8 promoter, as it has been described that HPV E1 proteins display transcriptional activation properties in yeast and mammalian cells (39–41). A recent report suggested that this is due to the interaction of E1 with the p62 subunit of TFIIH (42). Unfortunately, an E1 mutant that no longer stimulates transcription also displays a reduced DNA replication activity, making it difficult to dissect the transactivation from replication activities of E1 (42). It is tempting to speculate that the E1/E2 complex not only induces DNA replication but also recruits BRD4 and p62 (and possibly other proteins), which preferentially activate expression from the E8 promoter, as this would link copy number to E8^E2C expression.
The disruption of CE2 and CE3 in the context of the HPV16 genome increased copy number and viral early and late transcription with the exception of E8^E2C transcripts (Fig. 7). The weak effects compared to those of E8− genomes (9, 11) may be due to the finding that the inactivation of CE2 and CE3 does not completely abolish basal or E1/E2-induced activity (Fig. 4A and 5) and support the idea that the E8 promoter responds positively to the levels of E1 and E2 and negatively to the levels of E8^E2C. A reduced expression of E8^E2C should result in higher transcription of E1 and E2 and thus enhanced DNA replication, which would in turn activate expression of E8^E2C, which then again would limit the expression of E8^E2C. In summary, the E8 promoter that is responsible for the expression of E8^E2C is tightly regulated by both cellular and viral proteins in order to allow limiting replication but preventing overreplication in undifferentiated cells.
ACKNOWLEDGMENT
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (Stu 218/4-1) to F.S.
REFERENCES
- 1.Cogliano V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F. 2005. Carcinogenicity of human papillomaviruses. Lancet Oncol 6:204. doi: 10.1016/S1470-2045(05)70086-3. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F. 2006. Chapter 2: the burden of HPV-related cancers. Vaccine 24(Suppl 3):S3/11–25. doi: 10.1016/j.vaccine.2005.01.101. [DOI] [PubMed] [Google Scholar]
- 3.Schiffman M, Clifford G, Buonaguro FM. 2009. Classification of weakly carcinogenic human papillomavirus types: addressing the limits of epidemiology at the borderline. Infect Agents Cancer 4:8. doi: 10.1186/1750-9378-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.zur Hausen H. 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 5.Stubenrauch F, Laimins LA. 1999. Human papillomavirus life cycle: active and latent phases. Semin Cancer Biol 9:379–386. doi: 10.1006/scbi.1999.0141. [DOI] [PubMed] [Google Scholar]
- 6.Stenlund A. 2003. Initiation of DNA replication: lessons from viral initiator proteins. Nat Rev Mol Cell Biol 4:777–785. doi: 10.1038/nrm1226. [DOI] [PubMed] [Google Scholar]
- 7.Fertey J, Hurst J, Straub E, Schenker A, Iftner T, Stubenrauch F. 2011. Growth inhibition of HeLa cells is a conserved feature of high-risk human papillomavirus E8^E2C proteins and can also be achieved by an artificial repressor protein. J Virol 85:2918–2926. doi: 10.1128/JVI.01647-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurg R, Uusen P, Vosa L, Ustav M. 2010. Human papillomavirus E2 protein with single activation domain initiates HPV18 genome replication, but is not sufficient for long-term maintenance of virus genome. Virology 408:159–166. doi: 10.1016/j.virol.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 9.Lace MJ, Anson JR, Thomas GS, Turek LP, Haugen TH. 2008. The E8–E2 gene product of human papillomavirus type 16 represses early transcription and replication but is dispensable for viral plasmid persistence in keratinocytes. J Virol 82:10841–10853. doi: 10.1128/JVI.01481-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stubenrauch F, Hummel M, Iftner T, Laimins LA. 2000. The E8^E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J Virol 74:1178–1186. doi: 10.1128/JVI.74.3.1178-1186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Straub E, Dreer M, Fertey J, Iftner T, Stubenrauch F. 2014. The viral E8^E2C repressor limits productive replication of human papillomavirus 16. J Virol 88:937–947. doi: 10.1128/JVI.02296-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stubenrauch F, Zobel T, Iftner T. 2001. The E8 domain confers a novel long-distance transcriptional repression activity on the E8^E2C protein of high-risk human papillomavirus type 31. J Virol 75:4139–4149. doi: 10.1128/JVI.75.9.4139-4149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toots M, Mannik A, Kivi G, Ustav M Jr, Ustav E, Ustav M. 2014. The transcription map of human papillomavirus type 18 during genome replication in U2OS cells. PLoS One 9:e116151. doi: 10.1371/journal.pone.0116151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zobel T, Iftner T, Stubenrauch F. 2003. The papillomavirus E8-E2C protein represses DNA replication from extrachromosomal origins. Mol Cell Biol 23:8352–8362. doi: 10.1128/MCB.23.22.8352-8362.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ammermann I, Bruckner M, Matthes F, Iftner T, Stubenrauch F. 2008. Inhibition of transcription and DNA replication by the papillomavirus E8-E2C protein is mediated by interaction with corepressor molecules. J Virol 82:5127–5136. doi: 10.1128/JVI.02647-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Powell ML, Smith JA, Sowa ME, Harper JW, Iftner T, Stubenrauch F, Howley PM. 2010. NCoR1 mediates papillomavirus E8;E2C transcriptional repression. J Virol 84:4451–4460. doi: 10.1128/JVI.02390-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grassmann K, Rapp B, Maschek H, Petry KU, Iftner T. 1996. Identification of a differentiation-inducible promoter in the E7 open reading frame of human papillomavirus type 16 (HPV-16) in raft cultures of a new cell line containing high copy numbers of episomal HPV-16 DNA. J Virol 70:2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hummel M, Hudson JB, Laimins LA. 1992. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J Virol 66:6070–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Meyers C, Wang HK, Chow LT, Zheng ZM. 2011. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J Virol 85:8080–8092. doi: 10.1128/JVI.00670-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alloul N, Sherman L. 1999. The E2 protein of human papillomavirus type 16 is translated from a variety of differentially spliced polycistronic mRNAs. J Gen Virol 80(Part 1):29–37. [DOI] [PubMed] [Google Scholar]
- 21.Remm M, Remm A, Ustav M. 1999. Human papillomavirus type 18 E1 protein is translated from polycistronic mRNA by a discontinuous scanning mechanism. J Virol 73:3062–3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan TM, Gloss B, Bernard HU, Ting RC. 1994. Mechanism of translation of the bicistronic mRNA encoding human papillomavirus type 16 E6-E7 genes. J Gen Virol 75(Part 10):2663–2670. doi: 10.1099/0022-1317-75-10-2663. [DOI] [PubMed] [Google Scholar]
- 23.Milligan SG, Veerapraditsin T, Ahamet B, Mole S, Graham SV. 2007. Analysis of novel human papillomavirus type 16 late mRNAs in differentiated W12 cervical epithelial cells. Virology 360:172–181. doi: 10.1016/j.virol.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Xue Y, Poidinger M, Lim T, Chew SH, Pang CL, Abastado JP, Thierry F. 2014. Mapping of HPV transcripts in four human cervical lesions using RNAseq suggests quantitative rearrangements during carcinogenic progression. Virology 462-463:14–24. doi: 10.1016/j.virol.2014.05.026. [DOI] [PubMed] [Google Scholar]
- 25.Purdie KJ, Sexton CJ, Proby CM, Glover MT, Williams AT, Stables JN, Leigh IM. 1993. Malignant transformation of cutaneous lesions in renal allograft patients: a role for human papillomavirus. Cancer Res 53:5328–5333. [PubMed] [Google Scholar]
- 26.Soeda E, Ferran MC, Baker CC, McBride AA. 2006. Repression of HPV16 early region transcription by the E2 protein. Virology 351:29–41. doi: 10.1016/j.virol.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 27.Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. 2007. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35:W71–W74. doi: 10.1093/nar/gkm306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bodily JM, Hennigan C, Wrobel GA, Rodriguez CM. 2013. Regulation of the human papillomavirus type 16 late promoter by E7 and the cell cycle. Virology 443:11–19. doi: 10.1016/j.virol.2013.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kukimoto I, Kanda T. 2001. Displacement of YY1 by differentiation-specific transcription factor hSkn-1a activates the P(670) promoter of human papillomavirus type 16. J Virol 75:9302–9311. doi: 10.1128/JVI.75.19.9302-9311.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bodily JM, Meyers C. 2005. Genetic analysis of the human papillomavirus type 31 differentiation-dependent late promoter. J Virol 79:3309–3321. doi: 10.1128/JVI.79.6.3309-3321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McBride AA. 2013. The papillomavirus E2 proteins. Virology 445:57–79. doi: 10.1016/j.virol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McBride AA, Jang MK. 2013. Current understanding of the role of the Brd4 protein in the papillomavirus lifecycle. Viruses 5:1374–1394. doi: 10.3390/v5061374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. 2006. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J Virol 80:9530–9543. doi: 10.1128/JVI.01105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakai H, Yasugi T, Benson JD, Dowhanick JJ, Howley PM. 1996. Targeted mutagenesis of the human papillomavirus type 16 E2 transactivation domain reveals separable transcriptional activation and DNA replication functions. J Virol 70:1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schweiger MR, You J, Howley PM. 2006. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J Virol 80:4276–4285. doi: 10.1128/JVI.80.9.4276-4285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Helfer CM, Pancholi N, Bradner JE, You J. 2013. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J Virol 87:3871–3884. doi: 10.1128/JVI.03068-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gauson EJ, Donaldson MM, Dornan ES, Wang X, Bristol M, Bodily JM, Morgan IM. 2015. Evidence supporting a role for TopBP1 and Brd4 in the initiation but not continuation of human papillomavirus 16 E1/E2-mediated DNA replication. J Virol 89:4980–4991. doi: 10.1128/JVI.00335-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hollenhorst PC, McIntosh LP, Graves BJ. 2011. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu Rev Biochem 80:437–471. doi: 10.1146/annurev.biochem.79.081507.103945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Demeret C, Goyat S, Yaniv M, Thierry F. 1998. The human papillomavirus type 18 (HPV18) replication protein E1 is a transcriptional activator when interacting with HPV18 E2. Virology 242:378–386. doi: 10.1006/viro.1997.9023. [DOI] [PubMed] [Google Scholar]
- 40.Titolo S, Pelletier A, Sauve F, Brault K, Wardrop E, White PW, Amin A, Cordingley MG, Archambault J. 1999. Role of the ATP-binding domain of the human papillomavirus type 11 E1 helicase in E2-dependent binding to the origin. J Virol 73:5282–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yasugi T, Benson JD, Sakai H, Vidal M, Howley PM. 1997. Mapping and characterization of the interaction domains of human papillomavirus type 16 E1 and E2 proteins. J Virol 71:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morin G, Fradet-Turcotte A, Di Lello P, Bergeron-Labrecque F, Omichinski JG, Archambault J. 2011. A conserved amphipathic helix in the N-terminal regulatory region of the papillomavirus E1 helicase is required for efficient viral DNA replication. J Virol 85:5287–5300. doi: 10.1128/JVI.01829-10. [DOI] [PMC free article] [PubMed] [Google Scholar]