

ABSTRACT
Herpes simplex virus 1 (HSV-1) causes recurrent mucocutaneous ulcers and is the leading cause of infectious blindness and sporadic encephalitis in the United States. HSV-1 has been shown to be highly recombinogenic; however, to date, there has been no genome-wide analysis of recombination. To address this, we generated 40 HSV-1 recombinants derived from two parental strains, OD4 and CJ994. The 40 OD4-CJ994 HSV-1 recombinants were sequenced using the Illumina sequencing system, and recombination breakpoints were determined for each of the recombinants using the Bootscan program. Breakpoints occurring in the terminal inverted repeats were excluded from analysis to prevent double counting, resulting in a total of 272 breakpoints in the data set. By placing windows around the 272 breakpoints followed by Monte Carlo analysis comparing actual data to simulated data, we identified a recombination bias toward both high GC content and intergenic regions. A Monte Carlo analysis also suggested that recombination did not appear to be responsible for the generation of the spontaneous nucleotide mutations detected following sequencing. Additionally, kernel density estimation analysis across the genome found that the large, inverted repeats comprise a recombination hot spot.
IMPORTANCE Herpes simplex virus 1 (HSV-1) virus is the leading cause of sporadic encephalitis and blinding keratitis in developed countries. HSV-1 has been shown to be highly recombinogenic, and recombination itself appears to be a significant component of genome replication. To date, there has been no genome-wide analysis of recombination. Here we present the findings of the first genome-wide study of recombination performed by generating and sequencing 40 HSV-1 recombinants derived from the OD4 and CJ994 parental strains, followed by bioinformatics analysis. Recombination breakpoints were determined, yielding 272 breakpoints in the full data set. Kernel density analysis determined that the large inverted repeats constitute a recombination hot spot. Additionally, Monte Carlo analyses found biases toward high GC content and intergenic and repetitive regions.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a double-stranded DNA (dsDNA) virus in the Alphaherpesvirinae subfamily which causes recurrent, mucocutaneous lesions. HSV-1 is the leading cause of both sporadic encephalitis and infectious keratitis in the United States (1, 2). Animal studies have shown that disease severity is dependent on three factors: innate host resistance, the host immune response, and the viral strain (1, 3–16). Previous work of ours examining the role of the viral strain in virulence involved generating recombinant HSV-1 strains through mixed infections with two strains, OD4 and CJ994 (17). The advent of next-generation sequencing technologies has allowed multiple HSV-1 genomes to be sequenced (18, 19), thus allowing the opportunity to sequence previously generated recombinants and examine genome-level recombination phenomena.
The HSV-1 genome is approximately 152 kb in length and is arranged with inverted repeats flanking two unique sequences, the unique long (UL) and unique short (US) regions. The genomic segments invert with four possible, equivalent arrangements (20); however, a more recent report suggests that the effects of both viral strain and cell type may result in an imbalanced isomeric ratio (21). Replication is initiated from the origins of replication: OriL in the UL segment and two copies of OriS in the inverted repeats flanking the US region (22, 23). In HSV-1, replication and homologous recombination appear to be closely linked. While viral DNA was originally demonstrated to be replicated by rolling circle, leading to the formation of head-to-tail genome concatemers (24), subsequent work presented a more complicated mechanism that may include theta replication and rolling circle, leading to the formation of highly branched DNA structures. It is likely that homologous recombination resolves double-strand breaks and assists in the formation of Y-junction origins of replication. Thus, homologous recombination both leads to and resolves the observed DNA branching (25). The 250- to 500-bp a packaging sequence, located at the termini of the repeats, has been identified to be highly recombinogenic; however, it is dispensable for genomic segment isomerization (26–29). It is possible that the a sequence represents a recombination hot spot; however, no comprehensive genomic analysis identifying it as such has been performed. Restriction fragment length polymorphism (RFLP) studies have estimated the genomic recombination frequency to be between 0.26 and 0.7 recombinations per kilobase (30–32). To date, no genomic sequence-based recombination mapping studies have been performed to complement these early estimates.
In this study, we sequenced the genomes of 17 previously described in vivo, ocularly derived HSV-1 recombinants (17), as well as 23 new, in vitro, tissue culture-derived HSV-1 recombinants. The recombination breakpoints were determined for each of the 40 recombinants.
Bioinformatic recombination breakpoint analysis detected a bias toward high GC content, a bias toward intergenic and repetitive regions, and a recombination hot spot in the large inverted repeats. Additionally, recombination does not appear to drive the generation of spontaneous nucleotide mutations.
MATERIALS AND METHODS
Cells.
To generate viral DNA stocks, green monkey kidney (Vero) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 5% serum and antibiotics as described previously (33). For DNA isolation, the infections were performed with 2% serum and antibiotics.
Viruses.
The parental HSV-1 strains used to generate the recombinants described in this study were OD4 and CJ994. OD4 and CJ994 are avirulent, plaque-purified, clinical strains originally isolated in Seattle, WA. The ocular virulence characteristics of OD4 and CJ994 have been tested in mice and described previously (33). The OD4-CJ994 recombinants were generated using two methods: in vivo and in vitro methods. Seventeen of the recombinants were generated using an in vivo method, and their disease phenotypes have been described previously (17). All animal experiments followed the Association for Research in Vision and Ophthalomogy and NIH animal welfare guidelines and were approved by the University of Wisconsin—Madison IACUC. Briefly, 3- to 4-week-old BALB/c female mice were infected by corneal scarification with 1 × 105 PFU of strain OD4 and CJ994 (1:1 ratio). The corneas and trigeminal ganglia were removed at 1, 2, 3, 5, 7, and 10 days postinfection. The tissues were homogenized, freeze-thawed, and titrated on a plate of confluent Vero cells. Preparations containing virus were then subjected to three rounds of plaque purification. The purified plaques were picked, and high-titer stocks were prepared by infecting one TC100 plate of Vero cells with a purified plaque and DMEM with 2% serum. Once the cells reached a 100% cytopathic effect (CPE), the cells were harvested and subjected to three freeze-thaw cycles. The resulting lysate was divided to infect confluent Vero cells on five TC100 plates. When the cells reached a 100% CPE, they were harvested and centrifuged at 600 × g for 10 min. The supernatant was removed, and then 5 ml of supernatant was placed back into the tube containing the pellet and vortexed. The vortexed pellets were subjected to three freeze-thaw cycles. The pellet mixture was centrifuged at 600 × g for 10 min. The supernatants were combined and centrifuged at 600 × g for 20 min. The supernatant was layered onto a 36% sucrose cushion in phosphate-buffered saline (PBS) and centrifuged for 80 min at 24,000 × g. Following centrifugation, the supernatant was aspirated, and then the viral pellets were resuspended with 1 ml of DMEM with 2% serum. The samples were then aliquoted and stored at −80°C.
Twenty-three of the recombinants were generated in vitro through tissue culture. First, a plate of confluent Vero cells was infected with 2 × 108 PFU (multiplicity of infection, 10) of OD4 and CJ994 (1:1 ratio) viruses. When the cells reached a 100% CPE, they were harvested and subjected to three freeze-thaw cycles. The samples were then titrated onto Vero cells in six-well plates. The resulting plaques were isolated and further plaque purified an additional two times. High-titer stocks were prepared as described above.
Viral DNA preparation and RFLP screening.
The in vitro plaque-purified viruses were then screened by RFLP to detect which of the samples were likely recombinants. First, a viral DNA preparation was made for each of the isolated plaque preparations using a modification of our previously described protocol (34). One hundred microliters of viral stock was used to infect Vero cells on one TC100 plate with DMEM with 2% serum. Once the cells reached a 100% CPE, the cells were harvested and subjected to three freeze-thaw cycles. The resulting lysate was divided to infect confluent Vero cells on five TC100 plates. When the cells reached a 100% CPE, they were harvested and centrifuged at 600 × g for 10 min. The supernatant was removed, and then 5 ml of supernatant was placed back into the tube containing the pellet and vortexed. The vortexed pellets were subjected to three freeze-thaw cycles. The pellet mixture was centrifuged at 600 × g for 10 min. The supernatants were combined and centrifuged at 600 × g for 20 min. The supernatant was layered onto a 36% sucrose cushion in PBS and centrifuged for 80 min at 24,000 × g. The resulting pellet was resuspended in 5 ml of PBS, layered onto another 36% sucrose cushion in PBS, and centrifuged for 80 min at 26,200 × g. The viral pellet was then resuspended in 3 ml of TE buffer (10 mM Tris [pH 7.4], 1 mM EDTA) with 0.15 M sodium acetate and 50 μg/ml RNase A and then incubated 30 min at 37°C. Proteinase K and SDS (50 μg/ml and 0.1%, respectively) were added, and the solution was incubated for 30 min at 37°C. The viral DNA was purified by phenol-chloroform extraction and ethanol precipitation, resuspended in deionized water, and stored at −20°C.
For the RFLP analysis, 3 μg of DNA from each plaque isolate and parental viruses (OD4 and CJ994) was digested with BamHI (Promega, Madison, WI) overnight at 37°C. The restriction digests were electrophoresed on a 0.8% agarose gel in TBE (Tris-borate-EDTA) and stained with ethidium bromide. The DNA digest patterns were then visually inspected to determine if they had genomic fragments from both of the OD4 and CJ994 parental strains. Plaque isolates containing restriction fragments from both the parental strains were chosen for Illumina sequencing.
Construction and sequencing of Illumina libraries.
The potential recombinants were sequenced using two methods. Twelve of the in vitro-derived recombinants were sequenced using an Illumina HiSeq 2000 sequencing system. One microgram of high-quality genomic DNA was submitted to the University of Wisconsin—Madison DNA Sequencing Facility for paired-end library preparation. Each library was generated using an Illumina TruSeq LT sample preparation kit (Illumina Inc., San Diego, CA, USA) per the manufacturer's specifications, with 300-bp fragments being targeted. The quality and quantity of the DNA were assessed using an Agilent DNA high-sensitivity series chip assay (Agilent Technologies, Santa Clara, CA, USA) and a Qubit dsDNA kit (Life Technologies, Grand Island, NY, USA), respectively, and the concentrations of the libraries were standardized to 2 nM. Paired-end, 100-bp sequencing was performed in a single lane on the Illumina HiSeq 2000 sequencing system using SBS (version 3) kits, and an average of 5 million unique reads (1 Gb) was returned per library. FASTQ reports were created using the CASAVA (version 1.8.2) program.
The remaining recombinants were sequenced using the Illumina MiSeq platform, which produces longer reads. Five hundred nanograms of high-quality genomic DNA was submitted to the University of Wisconsin—Madison DNA Sequencing Facility for paired-end library preparation. Each library was generated using an Illumina TruSeq Nano LT sample preparation kit per the manufacturer's specifications, with 550-bp fragments being targeted. The quality and quantity of the DNA were assessed using an Agilent DNA high-sensitivity series chip assay and Qubit dsDNA kit, respectively, and the concentrations of the libraries were standardized to 2 nM. Paired-end, 250-bp sequencing was performed on the Illumina MiSeq platform using version 2 kits, and an average of 250,000 unique reads (125 Mb) was returned per library. FASTQ reports were created using the CASAVA (version 1.8.2) program.
Genomic assembly.
The paired-end sequencing reads from the 40 recombinants and strain CJ994 were generated using a reference assembly. Between sequencing runs, an updated annotation was made available through GenBank; however, for consistency we decided to use the annotation with GenBank accession number NC_001806. The sequencing reads were aligned to the sequence of HSV-1 strain 17 using a local alignment method, with a consensus sequence subsequently being generated and extracted in a manner similar to that described previously (18). Resulting gaps in the reference assembly were filled in with N′s (any nucleotide) without a proxy sequence, as has been done previously (19, 35). The gapped genomic sequence was annotated and submitted to GenBank (Table 1).
TABLE 1.
Accession numbers for reference and parental HSV-1 strains
| HSV-1 strain | Purpose in study | Origin | GenBank accession no. | No. of mapped reads | Average read length (bp) | Average coverage (no. of times) | Sequence length (bp) |
|---|---|---|---|---|---|---|---|
| 17 | Reference strain | Clinical isolate (UK) | NC_001806 | Not sequenced | Not sequenced | Not sequenced | 152,261 |
| OD4 | Parental strain | Clinical isolate (USA) | JN420342 | Not sequenced | Not sequenced | Not sequenced | 152,015 |
| CJ994 | Parental strain | Clinical isolate (USA) | KR011283 | 294,300 | 251 | 485 | 152,233 |
SNP and indel detection and analysis.
To detect novel single nucleotide polymorphisms (SNPs) and indels in the OD4-CJ994 recombinant viruses, the paired-end reads from each strain were aligned to the parental OD4 genome (GenBank accession number JN420342) as a reference assembly using the CLC-Bio Genomic Workbench. The strain assemblies sequenced with the HiSeq 2000 system were scanned using separate SNP and indel detection tools from Genomic Workbench (version 5.0.2). The recombinant virus assemblies sequenced with the MiSeq platform were analyzed with the quality-based variant detection tool (Genomic Workbench, version 6.0.2). For all base variant detection methods, a minimum read coverage of 4 was required for each base position. Additionally, the sequences of 35% of the reads or a minimum of five reads with a variant base had to differ from the reference sequence. Potential variant bases were filtered on the quality scores at each position and the five neighboring nucleotides on each side of the position. Minimum Phred quality scores of 20 for each variant position and 15 for the adjacent 10 bases were also required for a call. The novel single nucleotide polymorphism (SNP) and indels were logged, and each base position was translated into an HSV-1 strain 17 genomic coordinate. The novel single nucleotide polymorphism (SNP) and insertions/deletion (INDEL) positions translated for strain 17 were also plotted onto the corresponding genomic map for each strain.
To map DNA polymorphisms between the two parental strains, OD4 and CJ994, the genome of each strain was aligned using the MAFFT aligner from the SATé (version 2.2.7) software package (36, 37). The DNA polymorphisms were then mapped using the DNAsp package, a sliding window of 100 bp, and a 25-bp step size. Alignment gaps were excluded from the analysis. The data were visualized using SigmaPlot (version 12.0) software.
To determine if there was a statistically significant difference between the number of novel SNPs/indels between the in vivo- and in vitro-derived recombinants, a Mann-Whitney rank-sum test was performed using SigmaPlot (version 12.0) software.
Breakpoint detection.
In order to characterize the various properties of the breakpoint regions, we first defined the genomic coordinates of the breakpoints by aligning the sequence of the genome of each OD4-CJ994 recombinant to the sequences of the OD4 and CJ994 parental genomes. These alignments can be found at sites.ophth.wisc.edu/brandt/. The Bootscan function from the RDP4 software suite (38, 39) was used to scan the genomic alignment for recombination crossover events, using the Kimura 2-parameter (40) nucleotide substitution model, a sliding window of 1500 bp, and a step size of 750 bp (see Fig. S1A in the supplemental material). Each recombination breakpoint detected by the scan was logged and cross-referenced with the SNP detection reports for verification. Each of the recombination breakpoint positions for each of the recombinants was then translated into the HSV-1 strain 17 genomic coordinates (see Table S1 in the supplemental material). The parental origin of each genomic block was also noted. To more finely map possible recombination events in the inverted repeat regions, the sequence from the terminal repeat long (TRL) and inverted repeat short (IRS) regions for each of the recombinants was aligned to the sequences of parental strains OD4 and CJ994. The alignments of the TRL and IRS regions can also be accessed at sites.ophth.wisc.edu/brandt/. As described above, breakpoints were determined by generating a Bootscan plot and logging crossover coordinates. The breakpoint coordinates were cross-referenced with the alignments of the TRL and IRS regions for verification. The RDP4 bootscan plots were generated using the Kimura 2-parameter model, a sliding window of 500 bp, and a step size of 250 bp (see Fig. S1B and C and Table S1 in the supplemental material). The use of smaller sliding windows was investigated; however, these resulted in an extensive amount of noise and were too difficult to interpret. For analysis, we excluded breakpoints occurring in the terminal repeat regions defined by coordinates 1.0.9212 (flanking UL) and 145585.0.152259 (flanking US) to avoid double counting of these breakpoints in our analysis. The resulting data set has 272 breakpoints in total across the 40 recombinants.
To determine if there was a statistically significant differences between the number of breakpoint events between the in vivo- and in vitro-derived recombinants, a Mann-Whitney rank-sum test was performed using SigmaPlot (version 12.0) software.
Breakpoint distribution estimation.
To determine if the breakpoint locations were strongly biased toward a particular region of the genome, we used a kernel density estimation approach to model the distribution of breakpoint occurrences as a function of HSV genome coordinates. We used the R KernSmooth package (41) (http://cran.r-project.org/web/packages/KernSmooth/index.html), which implements a binned approximation to ordinary kernel density estimation. In this approach, a kernel function is centered on each value in the sample, and the heights of the kernel functions are summed in discrete bins that partition the range of the genome coordinates. Our analysis used a Gaussian kernel function and the direct plug-in method (42) for choosing the bandwidth of the kernel. The bandwidth parameter determines the smoothness of the density estimate.
GC bias in breakpoint windows.
We defined a set of 272 breakpoint windows, which consist of subsequences of the strain 17 genome centered on the coordinates of each of the breakpoints identified as described above. Most breakpoint windows were defined by centering a fixed-length window on each breakpoint coordinate. We repeated this procedure with initial window lengths of both 100 and 1,000 bases. In some cases, the windows were truncated so that they would not extend beyond the ends of the genomic sequence being analyzed. In 19 cases, the breakpoints were localized to regions in which there was a high degree of parental sequence uncertainty. In these cases, the windows were enlarged to span a fixed length, in addition to the length of the uncertain region.
To determine if the breakpoint windows exhibited a GC content bias, we first calculated the fraction of the breakpoint windows that consisted of guanines and cytosines and then used a Monte Carlo procedure to assess the extent to which this fraction was biased relative to the background frequencies of these bases in the genome. Specifically, we randomly selected 10,000 sets of windows for comparison. Each of these random window sets comprised 272 windows with the same lengths as the actual breakpoint windows against which they were compared. As with the actual breakpoints, our random windows were calculated using initial lengths of both 1,000 and 100 bases. For each of the random window sets, we calculated the GC fraction in the same way that we calculated the actual breakpoint windows.
Functional properties of breakpoint windows.
To assess the nature of the association between breakpoint windows and the functional properties of the genomic sequence, we employed several types of subsequences annotated in the features table of an NCBI reference sequence (GenBank accession number NC_001806.1). Specifically, we determined the probabilities that each breakpoint overlaps a coding region (inside a coding sequence [CDS] feature), an intergenic region (outside all gene features), and/or a repetitive region (inside a repeat_region feature). Note that these categories are not mutually exclusive, as both coding and intergenic sequences can appear in repetitive regions.
To calculate the probability for a given window and feature type, we assumed that the actual coordinate of the given breakpoint is equally likely to be at any position in its surrounding window, and we then calculated the fraction of the window that overlaps the given feature type. To test the association between breakpoints and each type of region, we again used a Monte Carlo procedure. We randomly selected 10,000 sets of windows for comparison. Each of these random window sets comprised 272 windows with the same lengths as the actual breakpoint windows.
Motif search in breakpoint windows.
We also analyzed the breakpoint windows to determine if they contained any overrepresented sequence motifs. We used the FIRE algorithm (43) to search for motifs that occur more frequently in the breakpoint windows than in the background, which was defined as all other sequence regions outside breakpoint windows. This analysis used 272 breakpoint windows that were initially defined to be 500 bases long, before applying the same truncation and spanning procedures described above for the GC bias analysis. The background sequences that were longer than 1,000 bases were segmented into windows with a maximum length of 500 bases in order to ensure that the background windows had a length distribution similar to that of the breakpoint windows. This procedure resulted in 119 background windows. We ran FIRE with the exptype parameter set to discrete, the nodups parameter set to 1, and the other parameters set to their defaults.
To assess whether the motifs that were found represent statistically significant properties of the breakpoint windows, we used a Monte Carlo procedure in which we ran FIRE on 1,000 sets of randomly chosen pseudobreakpoint windows. Again, each of those random window sets had 272 windows with the same lengths as the actual breakpoint windows. As before, the sequence regions outside the pseudobreakpoint windows were used as the background. This analysis measured the number of motifs found for the actual breakpoint windows and the extent to which these motifs discriminate breakpoint windows from background windows, as indicated by F1 score, a measure of classification accuracy.
Novel SNP association with breakpoint windows.
Finally, we tested whether the positions of novel SNPs in the recombinant strains are related to the breakpoint coordinates. We first determined the empirical distribution of distances between the coordinates of novel SNPs and their nearest breakpoint. We then used a Monte Carlo procedure in which we generated 10,000 pseudorecombinants by randomly selecting the number of breakpoints, the number of SNPs, the breakpoint coordinates, and the SNP coordinates from the empirical distributions of these variables observed in the set of actual recombinants. From the set of 10,000 pseudorecombinants, we then determined the distribution of SNP breakpoint distances as a basis for comparison against the distances from the actual data. We used a kernel density estimation procedure, as described above, to obtain smoothed representations of both distributions.
Nucleotide sequence accession numbers.
The GenBank accession numbers of the 40 HSV-1 recombinants derived from parental strains OD4 and CJ994 are presented in Table 2.
TABLE 2.
Accession numbers and additional information for the OD4-CJ994 recombinants
| OD4-CJ994 recombinant name | Derivation method | Sequencing method | GenBank accession no. | No. of mapped reads | Average read length (bp) | Average coverage (no. of times) | Gapped sequence length (bp) |
|---|---|---|---|---|---|---|---|
| 2-4-2 | In vivo | Illumina HiSeq 2000 | KR011288 | 944,906 | 88.8 | 546 | 152,245 |
| 2-5-3 | In vivo | Illumina HiSeq 2000 | KR011292 | 1,347,097 | 86.1 | 742 | 152,249 |
| 5-1-1 | In vivo | Illumina HiSeq 2000 | KR011299 | 3,736,106 | 89.7 | 2,188 | 152,242 |
| 5-2-1 | In vivo | Illumina MiSeq | KR011285 | 346,024 | 251 | 554 | 152,197 |
| 5-4-2 | In vivo | Illumina HiSeq 2000 | KR011311 | 2,518,022 | 89.33 | 1,468 | 152,259 |
| 5-5-2 | In vivo | Illumina MiSeq | KR011295 | 345,902 | 251 | 554 | 152,238 |
| 10-1-2 | In vivo | Illumina HiSeq 2000 | KR011302 | 2,443,268 | 89.1 | 1,416 | 152,263 |
| 10-2-2 | In vivo | Illumina HiSeq 2000 | KR011277 | 2,928,588 | 89.5 | 1,708 | 152,240 |
| 10-2-3 | In vivo | Illumina MiSeq | KR011274 | 355,262 | 251 | 569 | 152,236 |
| 10-5-1 | In vivo | Illumina HiSeq 2000 | KR011301 | 4,348,509 | 89.9 | 2,553 | 152,238 |
| 10-6-1 | In vivo | Illumina MiSeq | KR011296 | 318,649 | 251 | 510 | 152,227 |
| 10-6-2 | In vivo | Illumina HiSeq 2000 | KR011306 | 3,125,257 | 89.3 | 1,813 | 152,236 |
| 10-6-3 | In vivo | Illumina MiSeq | KR011284 | 303,892 | 251 | 486 | 152,185 |
| 10-7-1 | In vivo | Illumina HiSeq 2000 | KR011290 | 642,463 | 87.6 | 364 | 152,248 |
| 10-11-2 | In vivo | Illumina HiSeq 2000 | KR011287 | 394,026 | 87.9 | 224 | 152,256 |
| 10-11-3 | In vivo | Illumina HiSeq 2000 | KR011309 | 1,662,613 | 90.3 | 981 | 152,233 |
| 10-14-1 | In vivo | Illumina HiSeq 2000 | KR011291 | 583,725 | 87.3 | 328 | 152,235 |
| 3M | In vitro | Illumina MiSeq | KR011282 | 31,569 | 251 | 51 | 152,219 |
| 4M | In vitro | Illumina MiSeq | KR011278 | 259,115 | 251 | 419 | 152,172 |
| 8S | In vitro | Illumina MiSeq | KR011280 | 115,226 | 251 | 185 | 152,189 |
| 11M | In vitro | Illumina MiSeq | KR011294 | 83,354 | 251 | 134 | 152,140 |
| 16S | In vitro | Illumina MiSeq | KR011303 | 153,529 | 251 | 246 | 152,238 |
| 19Lsyn | In vitro | Illumina MiSeq | KR011293 | 92,653 | 251 | 150 | 152,122 |
| 20L | In vitro | Illumina MiSeq | KR011289 | 135,116 | 251 | 218 | 152,188 |
| 26S | In vitro | Illumina MiSeq | KR011308 | 129,969 | 251 | 208 | 152,207 |
| 27S | In vitro | Illumina MiSeq | KR011297 | 40,002 | 251 | 64 | 152,223 |
| 31XL | In vitro | Illumina MiSeq | KR011304 | 56,103 | 251 | 90 | 152,277 |
| 34L | In vitro | Illumina MiSeq | KR011275 | 40,046 | 251 | 64 | 152,279 |
| 36L | In vitro | Illumina MiSeq | KR011279 | 27,109 | 251 | 42 | 152,272 |
| 47M | In vitro | Illumina MiSeq | KR011305 | 193,211 | 251 | 313 | 152,298 |
| 57M | In vitro | Illumina MiSeq | KR011276 | 77,036 | 251 | 124 | 152,312 |
| 65M | In vitro | Illumina MiSeq | KR011312 | 200,626 | 251 | 324 | 152,178 |
| 66M | In vitro | Illumina MiSeq | KR011281 | 115,879 | 251 | 187 | 152,278 |
| 76M | In vitro | Illumina MiSeq | KR011300 | 174,352 | 251 | 279 | 152,249 |
| 78S | In vitro | Illumina MiSeq | KR052507 | 124,762 | 251 | 199 | 152,234 |
| 81L | In vitro | Illumina MiSeq | KR052508 | 65,176 | 251 | 105 | 152,263 |
| 82S | In vitro | Illumina MiSeq | KR011307 | 374,518 | 251 | 604 | 152,148 |
| 83M | In vitro | Illumina MiSeq | KR011310 | 37,704 | 251 | 61 | 152,232 |
| 12-12-2 | In vitro | Illumina MiSeq | KR011298 | 57,116 | 251 | 91 | 152,288 |
| 12-12-67 | In vitro | Illumina MiSeq | KR011286 | 15,024 | 251 | 24 | 152,276 |
RESULTS AND DISCUSSION
Genomic sequencing and assembly.
The 40 HSV-1 recombinants were sequenced using two subplatforms of the Illumina next-generation system: the HiSeq 2000 and the MiSeq platforms (Table 2). The genomes were then assembled using the sequence of HSV-1 strain 17 as a reference. The number of mapped reads obtained using the HiSeq 2000 platform ranged from 394,026 in strain 10-11-2 to 4,348,509 in strain 10-5-1 (Table 2). The average read length for the samples sequenced using the HiSeq 2000 platform ranged from a low of 86.1 in strain 2-5-3 to 90.3 in strain 10-11-3, which resulted in an average coverage of from 224 times in strain 10-11-2 to 2,553 times in strain 10-5-1 (Table 2). The average number of mapped reads obtained using the MiSeq platform ranged from 15,024 in strain 12-12-67 to 374,518 in strain 82S (Table 2). The average read length for all of the samples sequenced using the MiSeq platform was approximately 251, which resulted in an average coverage range of from 24 times in strain 12-12-67 to 604 times in strain 82S.
Breakpoint detection.
Following genomic sequencing and assembly, recombination breakpoints were determined and mapped using the Bootscan algorithm in conjunction with SNP/INDEL analysis (Fig. 1). To avoid double counting of the number of breakpoints in the inverted repeats, the TRL and TRS repeats were excluded from analysis, yielding a total of 272 recombination breakpoints in the data set. An examination of the breakpoint map shows a greater number of breakpoints in the strains derived from the cornea in vivo than in the in vitro, tissue culture-derived strains. The in vivo-derived strains had an average of 13.8 recombination events per genome, while the in vitro-derived recombinants had an average of 6.4 recombination events per genome. The median number of breakpoints from the in vivo- and in vitro-derived samples was analyzed using the Mann-Whitney rank-sum test, and a statistically significant difference between the two groups (P < 0.001) was found. It is unclear what is responsible for this observation; however, it may be that the in vivo-derived viruses were able to undergo an increased number of replication cycles, and therefore, this may have increased the chances of recombination events in the in vivo-derived viruses compared to those in the in vitro, tissue culture-derived recombinants. An additional possibility is that either the cell type or the environment in the cornea selected the most evolutionarily fit viruses, which were in turn highly mosaicized recombinants that possessed advantageous phenotypic attributes of both parents. An examination of Fig. 1 shows what appears to be a greater prevalence of CJ994 genomic segments in the in vivo-derived viruses compared to the amount in the in vitro-derived recombinants, and this may suggest a selection bias for replication in the cornea.
FIG 1.

Recombination breakpoint map of OD4-CJ994 recombinant HSV-1 strains. The strains are divided into in vivo- and in vitro-derived strains. A key to the map has been placed at the top of each column of viruses. Briefly, red blocks denote genomic blocks of CJ994 parental origin, white blocks denote genomic blocks of OD4 parental origin, and purple blocks are of uncertain origin. Additionally, the locations of novel coding and noncoding SNPs and indels have been placed on the map and are defined in the key.
Breakpoint distribution estimation.
To determine if the breakpoint locations were strongly biased toward a particular region of the genome, we used a kernel density estimation approach to model the distribution of breakpoint occurrences as a function of HSV genome coordinates, with the terminal inverted repeats being excluded from the analysis to prevent double counting of recombination crossovers in the repeat regions (Fig. 2). Figure 2A shows that the distribution of breakpoint positions is uniform, with the exception of a notable peak in the internal repetitive regions. It is unlikely that a large number of recombination events in the coding regions were missed due to crossovers between SNPs, as a DNA polymorphism analysis between the two parental strains, OD4 and CJ994 (Fig. 2B), showed that the DNA polymorphism differences between the two strains were generally evenly distributed along the genome. The signal strength observed in the inverted repeats is likely an underestimate, as several recombinants had lower sequencing coverage in those areas, and therefore, it is likely that several recombination breakpoints were uncounted. The identification of the inverted repeats as recombination hot spots was not unexpected, as previous studies detected a large amount of recombination in the a packaging sequence, as well as recombination in OriS (27–29, 44). Future studies incorporating more recombinants may result in the detection of additional recombination hot spots in the genome.
FIG 2.

Estimated probability density function for breakpoint occurrence across the genome. (A) Estimated probability density function of breakpoint occurrence in terms of HSV genome coordinates. An HSV-1 genomic map appears above the plot. This plot was calculated using a kernel density estimation approach, which centers a smooth Gaussian kernel function at each observed breakpoint and sums the influence from each Gaussian kernel function to determine an overall density estimate. (B) DNA polymorphisms between parental strains OD4 and CJ994 across the genome (terminal repeats were excluded). (C) Repetitive regions, protein-coding regions, and intergenic regions in the genome. For both plots, terminal repetitive regions were excluded from consideration to avoid overcounting of breakpoints in internal repetitive regions. Repetitive regions are hot spots for breakpoints. Red dashed lines, peak of the density function.
Novel SNPs/INDELs.
Following SNP and indel detection, novel or spontaneously occurring SNPs/indels in each of the 40 recombinants were cataloged. This was performed by cataloging the SNPs/indels that did not match either the OD4 or the CJ994 parental sequence. Recombinant strains 5-5-2, 16S, 26S, 27S, 31XL, 76M, 78S, and 83M did not have any detected novel mutations (Fig. 1; see also Table S5 in the supplemental material). In contrast, recombinant strains 10-11-2 and 10-2-2 had 13 and 14 spontaneously occurring mutations, respectively (Fig. 1; see also Table S2 in the supplemental material). The in vivo-derived strains had a higher average number of SNPs/indels of 4.35 per genome than the in vitro-derived strains, which had 1.95 per genome. The median number of novel SNPs/indels from the in vivo- and in vitro-derived samples was analyzed using the Mann-Whitney rank-sum test, and a statistically significant difference between the two groups was found (P = 0.006).
We hypothesized that recombination may be causing the generation of spontaneous mutations, because of the higher number of both recombination events and novel SNPs/INDELs in the in vivo-derived strains, and that recombination involves both strand exchange and DNA repair mechanisms. To test this, we first determined empirically the distribution of the distances between the coordinate of each novel SNP and its nearest breakpoint. We then compared the observed distribution to a simulated data set that preserved the distribution of SNPs and breakpoint positions while selecting them independently. This procedure was designed to test whether SNP positions are dependent on breakpoint positions. The results, shown in Fig. 3, suggest that there is no correlation between recombination breakpoints and spontaneous mutations; thus, the hypothesis was false. It is unclear what influences the number of novel SNPs/INDELs in a given strain. The number of novel mutations that we detected is higher than that which may be expected, given that HSV-1 polymerase has a lower error rate than eukaryotic polymerases (45). Recombination studies in vaccinia virus found a small number of rare mutations, with a low estimated error rate of 1 × 10−8 mutation per nucleotide copied per cycle of replication (46). In HSV, additional factors affect the mutation rate; for instance, when the more error-prone HSV-2 polymerase is placed into HSV-1, the error rate is similar to that in wild-type HSV-1 in thymidine kinase assays, indicating that multiple factors are involved (47). Calculation of an error rate for the current study is problematic, given the number of assumptions required, that the amount of viral DNA produced during infection is higher than the amount packaged, and that viruses with lethal mutations would not be sequenced. Further, in a previous study comparing the genomes of 31 global HSV-1 strains, most of which were sequenced using the Illumina system, we estimated a substitution rate of 1.38 × 10−7, which was a log unit higher than previous estimates (48). In the present study, the sequence coverage of the SNPs/indels detected in the strains sequenced on the HiSeq 2000 platform was an average of 1,401 times, and the sequence coverage of the SNPs/indels detected in the strains sequenced on the MiSeq platform averaged 171 times; thus, it is unlikely that the SNPs/indels are a result of sequencing errors, unless the high GC content of the HSV-1 genome results in a higher error rate for the Illumina system. An examination of the novel SNPs determined that 89% are transversions, which is consistent with most mutations, increasing the likelihood that they do not represent sequencing errors. It is possible that the viral strains containing a high number of novel mutations have an altered ability to manipulate the host cell DNA repair mechanisms compared to that of the recombinant strains that have a low number of novel mutations. All of the detected novel mutations were in the UL- and US-coding regions due to the lower sequence coverage and read quality in the inverted repeat regions. It is almost certain that additional novel SNPs and indels exist in the inverted repeats as well; however, current sequencing and detection technology precludes their identification in the context of large, multisample sequencing studies.
FIG 3.
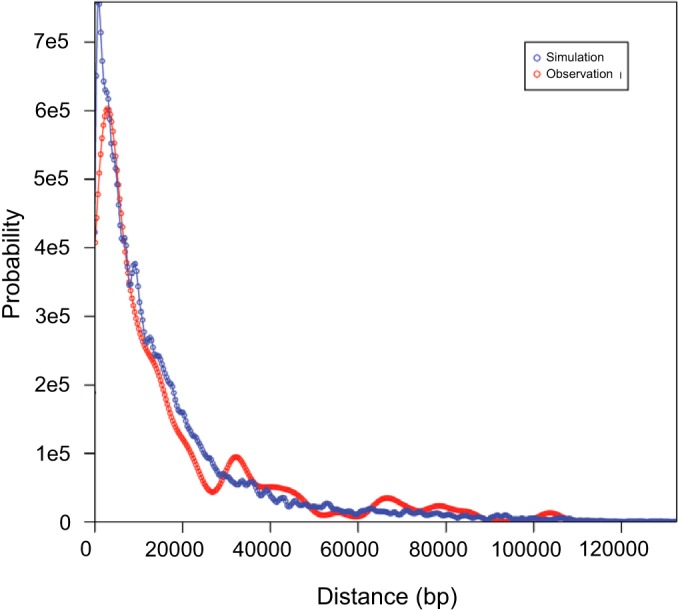
Distribution of lengths between novel SNPs and the nearest breakpoints in actual and simulated data. Red curve, estimated probability density function representing the distance between novel SNPs and the nearest breakpoint in the actual recombinants; blue curve, distribution of simulated data from a Monte Carlo process in which simulated SNP coordinates and breakpoint coordinates were sampled independently from the observed distributions of these two variables. The curves were calculated using a kernel density estimation approach to obtain smoothed representations of both the actual distances and the simulated distances.
Functional properties of breakpoint windows.
We also assessed the strength of the association between breakpoint windows and several functional classes of the genomic sequence: coding regions (inside a CDS feature in the NCBI reference sequence), intergenic regions (outside all gene features), and repetitive regions (inside a repeat_region feature). Note that these categories are not mutually exclusive, as both coding and intergenic sequences can appear in repetitive regions. Figure 4 shows the estimated fraction of breakpoints that overlap each of these sequence categories compared to the fractions that we observed when randomly simulating breakpoint positions. These results indicate that there is a very strong bias toward the occurrence of breakpoints in repetitive regions and somewhat less pronounced but still strong biases in favor of intergenic regions and against coding regions. This finding is consistent with the estimated breakpoint distribution shown in Fig. 2. The mechanism driving the observed recombination bias in favor of intergenic regions is unclear; however, high intergenic recombination rates have been observed in norovirus (49, 50). Additionally, in lytic infection, the majority of viral DNA is not nucleosome associated (51), so chromatin structure is unlikely to play a major role in influencing recombination location bias. The intergenic recombination bias in norovirus is thought to result in new combinations of genes, which may increase fitness and may account for the intergenic recombination bias in HSV-1 as well.
FIG 4.

Occurrence of actual breakpoints and simulated breakpoints in specific genomic region types. Red circles, the fractions of breakpoints occurring in the three types of sequence regions. The box plots show the distribution of simulated breakpoints generated by the Monte Carlo process in each of the three region types. The blue box in a given box plot represents the range from the first quartile to the third quartile and contains a black thick bar indicating the median. The two horizontal dashed lines, called whiskers, extending from the blue box represent the range from the smallest nonoutlier to the largest nonoutlier. Black circles represent outliers, which are defined as those points occurring more than one and a half times the length of the blue box from either end. The results indicate that actual breakpoints occur in repetitive regions much more frequently than the frequency expected by chance. Actual breakpoints also occur in intergenic regions somewhat more frequently than expected by chance and in coding regions less frequently than expected by chance.
GC bias in breakpoint windows.
Based on previous reports of recombination bias toward high GC content regions in Saccharomyces, pigs, humans, and poliovirus (52–55), we tested the hypothesis that breakpoints are more likely to occur in GC-rich regions of the genome. The box plots shown in Fig. 5 illustrate the GC composition of windows surrounding breakpoints compared to the GC composition of randomly selected windows. In each plot, the red circle indicates the fraction of GC bases for the actual breakpoint windows, whereas the other components of the box plot illustrate the distribution of the GC composition for the random windows. These results indicate that the breakpoint positions are strongly biased toward GC-rich regions, and the bias is even more apparent when we consider shorter windows. The observed GC content was more extreme than the GC composition recorded in any of the 10,000 simulated breakpoints. The observed bias toward both GC content and repetitive sequences appears to be entwined, with the areas of high GC content inevitably giving rise to repetitive sequences. The kernel density estimation data as well as the recombination bias toward GC content and repetitive regions identified the inverted repeats to be recombination hot spots. The GC content of the inverted repeats is approximately 74.6%, that of the UL-coding region is 66.8%, and that of the US-coding region is 64.3%; thus, the increased recombination frequency in the repeats may be a function of higher GC content. A heightened recombination frequency in the inverted repeats is likely critical for the formation of identical inverted repeat sequences as well as genome isomerization.
FIG 5.

GC content of windows around breakpoints. Red circles, GC content of windows around actual breakpoints. The top plot represents windows of 1,000 bases, and the bottom plot represents windows of 100 bases. The box plots show the GC content for windows around simulated breakpoints generated by the Monte Carlo process. The GC content for the observed breakpoint windows is located outside the whiskers' range, suggesting that there is a strong bias in GC content near breakpoints.
Motif search in breakpoint windows.
We also analyzed the breakpoint windows to determine if they contain any overrepresented sequence motifs. We used the FIRE algorithm (43) to search for motifs that occur more frequently in the breakpoint windows than in the background (all other sequence regions outside breakpoint windows). To assess whether the motifs that were found represent statistically significant properties of the breakpoint windows, we used a Monte Carlo procedure in which we ran FIRE on 1,000 sets of randomly chosen pseudobreakpoint windows. This analysis showed that the number of motifs found for the actual breakpoint windows and the extent to which these motifs discriminate breakpoint windows from background windows fell into the middle of the distributions of these values from the Monte Carlo runs. From this result, we conclude that the motifs found by FIRE for the actual breakpoint windows do not represent statistically significant properties of the neighboring breakpoints of the sequences.
Summary.
In conclusion, the genomes of 40 OD4-CJ994 viral recombinants were sequenced, and then the recombination breakpoints were determined, yielding 272 breakpoints in the final data set. Kernel density estimation analysis identified the large inverted repeats to be a recombination hot spot. Monte Carlo simulation of the breakpoint data determined recombination bias toward both high GC content and repetitive sequences and that recombination does not appear to be responsible for spontaneous mutations. Motif analysis of the recombination breakpoint windows did not find any sequence motifs that were statistically significantly overrepresented. The kernel density estimation results in conjunction with the identified recombination bias toward high GC content and repetitive sequences appear to be linked and suggest that the heightened recombination frequency in the inverted repeats is likely critical for the formation of identical inverted repeat sequences as well as genome isomerization.
Supplementary Material
ACKNOWLEDGMENTS
We express appreciation to the University of Wisconsin—Madison Next-Gen Sequencing Core and Kevin Hunt for their assistance.
This study was supported by a grant from the NIH (R01EY023292), a Core Grant for Vision Research (P30EY016665), NIH training grant LMT15007359, and an unrestricted grant to the Department of Ophthalmology and Visual Sciences from Research to Prevent Blindness, Inc.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00880-15.
REFERENCES
- 1.Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Whitley RJ. 1996. Herpes simplex viruses, p 2297–2342. In Fields BN, Knipe DM, Howley PM (ed), Fields virology, 3rd ed, vol 2 Lippincott-Raven, Philadelphia, PA. [Google Scholar]
- 3.Brandt CR. 2004. Virulence genes in herpes simplex virus type 1 corneal infection. Curr Eye Res 29:103–117. doi: 10.1080/02713680490504533. [DOI] [PubMed] [Google Scholar]
- 4.Brandt CR. 2005. The role of viral and host genes in corneal infection with herpes simplex virus type 1. Exp Eye Res 80:607–621. doi: 10.1016/j.exer.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Doymaz MZ, Rouse BT. 1992. Immunopathology of herpes simplex virus infections. Curr Top Microbiol Immunol 179:121–136. [DOI] [PubMed] [Google Scholar]
- 6.Kastrukoff LF, Lau AS, Puterman ML. 1986. Genetics of natural resistance to herpes simplex virus type 1 latent infection of the peripheral nervous system in mice. J Gen Virol 67(Pt 4):613–621. doi: 10.1099/0022-1317-67-4-613. [DOI] [PubMed] [Google Scholar]
- 7.Lopez C. 1975. Genetics of natural resistance to herpesvirus infections in mice. Nature 258:152–153. doi: 10.1038/258152a0. [DOI] [PubMed] [Google Scholar]
- 8.Lundberg P, Welander P, Openshaw H, Nalbandian C, Edwards C, Moldawer L, Cantin E. 2003. A locus on mouse chromosome 6 that determines resistance to herpes simplex virus also influences reactivation, while an unlinked locus augments resistance of female mice. J Virol 77:11661–11673. doi: 10.1128/JVI.77.21.11661-11673.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollara G, Katz DR, Chain BM. 2004. The host response to herpes simplex virus infection. Curr Opin Infect Dis 17:199–203. doi: 10.1097/00001432-200406000-00005. [DOI] [PubMed] [Google Scholar]
- 10.Streilein JW, Dana MR, Ksander BR. 1997. Immunity causing blindness: five different paths to herpes stromal keratitis. Immunol Today 18:443–449. doi: 10.1016/S0167-5699(97)01114-6. [DOI] [PubMed] [Google Scholar]
- 11.Stulting RD, Kindle JC, Nahmias AJ. 1985. Patterns of herpes simplex keratitis in inbred mice. Invest Ophthalmol Vis Sci 26:1360–1367. [PubMed] [Google Scholar]
- 12.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L, Leonard J, Hamilton C, Lebon P, Heron B, Vallee L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova JL. 2007. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 13.Thompson RL, Williams RW, Kotb M, Sawtell NM. 2014. A forward phenotypically driven unbiased genetic analysis of host genes that moderate herpes simplex virus virulence and stromal keratitis in mice. PLoS One 9:e92342. doi: 10.1371/journal.pone.0092342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dix RD, McKendall RR, Baringer JR. 1983. Comparative neurovirulence of herpes simplex virus type 1 strains after peripheral or intracerebral inoculation of BALB/c mice. Infect Immun 40:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wander AH, Centifanto YM, Kaufman HE. 1980. Strain specificity of clinical isolates of herpes simplex virus. Arch Ophthalmol 98:1458–1461. doi: 10.1001/archopht.1980.01020040310020. [DOI] [PubMed] [Google Scholar]
- 16.Richards JT, Kern ER, Overall JCJ, Glasgow LA. 1981. Differences in neurovirulence among isolates of herpes simplex virus types 1 and 2 in mice using four routes of infection. J Infect Dis 144:464–471. doi: 10.1093/infdis/144.5.464. [DOI] [PubMed] [Google Scholar]
- 17.Kintner RL, Allan RW, Brandt CR. 1995. Recombinants are isolated at high frequency following in vivo mixed ocular infection with two avirulent herpes simplex virus type 1 strains. Arch Virol 140:231–244. doi: 10.1007/BF01309859. [DOI] [PubMed] [Google Scholar]
- 18.Kolb AW, Adams M, Cabot EL, Craven M, Brandt CR. 2011. Multiplex sequencing of seven ocular herpes simplex virus type-1 genomes: phylogeny, sequence variability and SNP distribution. Invest Ophthalmol Vis Sci 52:9061–9073. doi: 10.1167/iovs.11-7812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szpara ML, Gatherer D, Ochoa A, Greenbaum B, Dolan A, Bowden RJ, Enquist LW, Legendre M, Davison AJ. 2014. Evolution and diversity in human herpes simplex virus genomes. J Virol 88:1209–1227. doi: 10.1128/JVI.01987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roizman B, Jacob RJ, Knipe DM, Morse LS, Ruyechan WT. 1979. On the structure, functional equivalence, and replication of the four arrangements of herpes simplex virus DNA. Cold Spring Harbor Symp Quant Biol 43(Pt 2):809–826. [DOI] [PubMed] [Google Scholar]
- 21.Mahiet C, Ergani A, Huot N, Alende N, Azough A, Salvaire F, Bensimon A, Conseiller E, Wain-Hobson S, Labetoulle M, Barradeau S. 2012. Structural variability of the herpes simplex virus 1 genome in vitro and in vivo. J Virol 86:8592–8601. doi: 10.1128/JVI.00223-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stow ND. 1982. Localization of an origin of DNA replication within the TRS/IRS repeated region of the herpes simplex virus type 1 genome. EMBO J 1:863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weller SK, Spadaro A, Schaffer JE, Murray AW, Maxam AM, Schaffer PA. 1985. Cloning, sequencing, and functional analysis of oriL, a herpes simplex virus type 1 origin of DNA synthesis. Mol Cell Biol 5:930–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacob RJ, Morse LS, Roizman B. 1979. Anatomy of herpes simplex virus DNA. XII. Accumulation of head-to-tail concatemers in nuclei of infected cells and their role in the generation of the four isomeric arrangements of viral DNA. J Virol 29:448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Severini A, Scraba DG, Tyrrell DL. 1996. Branched structures in the intracellular DNA of herpes simplex virus type 1. J Virol 70:3169–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin DW, Weber PC. 1996. The a sequence is dispensable for isomerization of the herpes simplex virus type 1 genome. J Virol 70:8801–8812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dutch RE, Bianchi V, Lehman IR. 1995. Herpes simplex virus type 1 DNA replication is specifically required for high-frequency homologous recombination between repeated sequences. J Virol 69:3084–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dutch RE, Bruckner RC, Mocarski ES, Lehman IR. 1992. Herpes simplex virus type 1 recombination: role of DNA replication and viral a sequences. J Virol 66:277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Umene K. 1991. Recombination of the internal direct repeat element DR2 responsible for the fluidity of the a sequence of herpes simplex virus type 1. J Virol 65:5410–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parris DS, Dixon RA, Schaffer PA. 1980. Physical mapping of herpes simplex virus type 1 ts mutants by marker rescue: correlation of the physical and genetic maps. Virology 100:275–287. doi: 10.1016/0042-6822(80)90519-X. [DOI] [PubMed] [Google Scholar]
- 31.Roizman B. 1979. The structure and isomerization of herpes simplex virus genomes. Cell 16:481–494. doi: 10.1016/0092-8674(79)90023-0. [DOI] [PubMed] [Google Scholar]
- 32.Umene K. 1985. Intermolecular recombination of the herpes simplex virus type 1 genome analysed using two strains differing in restriction enzyme cleavage sites. J Gen Virol 66(Pt 12):2659–2670. doi: 10.1099/0022-1317-66-12-2659. [DOI] [PubMed] [Google Scholar]
- 33.Grau DR, Visalli RJ, Brandt CR. 1989. Herpes simplex virus stromal keratitis is not titer-dependent and does not correlate with neurovirulence. Invest Ophthalmol Vis Sci 30:2474–2480. [PubMed] [Google Scholar]
- 34.Kintner RL, Brandt CR. 1994. Rapid small-scale isolation of herpes simplex virus DNA. J Virol Methods 48:189–196. doi: 10.1016/0166-0934(94)90118-X. [DOI] [PubMed] [Google Scholar]
- 35.Szpara ML, Parsons L, Enquist LW. 2010. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol 84:5303–5313. doi: 10.1128/JVI.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu K, Raghavan S, Nelesen S, Linder CR, Warnow T. 2009. Rapid and accurate large-scale coestimation of sequence alignments and phylogenetic trees. Science 324:1561–1564. doi: 10.1126/science.1171243. [DOI] [PubMed] [Google Scholar]
- 38.Martin D, Rybicki E. 2000. RDP: detection of recombination amongst aligned sequences. Bioinformatics 16:562–563. doi: 10.1093/bioinformatics/16.6.562. [DOI] [PubMed] [Google Scholar]
- 39.Martin DP, Posada D, Crandall KA, Williamson C. 2005. A modified Bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res Hum Retroviruses 21:98–102. doi: 10.1089/aid.2005.21.98. [DOI] [PubMed] [Google Scholar]
- 40.Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 41.Wand MP, Jones MC. 1995. Kernel smoothing, 1st ed Springer Science and Business Media BV, New York, NY. [Google Scholar]
- 42.Sheather SJ, Jones MC. 1991. A reliable data-based bandwidth selection method for kernel density-estimation. J R Stat Soc Series B Stat Methodol 53:683–690. [Google Scholar]
- 43.Elemento O, Slonim N, Tavazoie S. 2007. A universal framework for regulatory element discovery across all genomes and data types. Mol Cell 28:337–350. doi: 10.1016/j.molcel.2007.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao XD, Elias P. 2001. Recombination during early herpes simplex virus type 1 infection is mediated by cellular proteins. J Biol Chem 276:2905–2913. doi: 10.1074/jbc.M005627200. [DOI] [PubMed] [Google Scholar]
- 45.Abbotts J, Nishiyama Y, Yoshida S, Loeb LA. 1987. On the fidelity of DNA replication: herpes DNA polymerase and its associated exonuclease. Nucleic Acids Res 15:1185–1198. doi: 10.1093/nar/15.3.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qin L, Evans DH. 2014. Genome scale patterns of recombination between coinfecting vaccinia viruses. J Virol 88:5277–5286. doi: 10.1128/JVI.00022-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duffy KE, Quail MR, Nguyen TT, Wittrock RJ, Bartus JO, Halsey WM, Leary JJ, Bacon TH, Sarisky RT. 2002. Assessing the contribution of the herpes simplex virus DNA polymerase to spontaneous mutations. BMC Infect Dis 2:7. doi: 10.1186/1471-2334-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kolb AW, Cécile A, Brandt CR. 2013. Using HSV-1 genome phylogenetics to track past human migrations. PLoS One 8:e76267. doi: 10.1371/journal.pone.0076267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahar JE, Bok K, Green KY, Kirkwood CD. 2013. The importance of intergenic recombination in norovirus GII.3 evolution. J Virol 87:3687–3698. doi: 10.1128/JVI.03056-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bull RA, Tanaka MM, White PA. 2007. Norovirus recombination. J Gen Virol 88:3347–3359. doi: 10.1099/vir.0.83321-0. [DOI] [PubMed] [Google Scholar]
- 51.Muggeridge MI, Fraser NW. 1986. Chromosomal organization of the herpes simplex virus genome during acute infection of the mouse central nervous system. J Virol 59:764–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marsolier-Kergoat MC, Yeramian E. 2009. GC content and recombination: reassessing the causal effects for the Saccharomyces cerevisiae genome. Genetics 183:31–38. doi: 10.1534/genetics.109.105049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fullerton SM, Bernardo Carvalho A, Clark AG. 2001. Local rates of recombination are positively correlated with GC content in the human genome. Mol Biol Evol 18:1139–1142. doi: 10.1093/oxfordjournals.molbev.a003886. [DOI] [PubMed] [Google Scholar]
- 54.Tortereau F, Servin B, Frantz L, Megens HJ, Milan D, Rohrer G, Wiedmann R, Beever J, Archibald AL, Schook LB, Groenen MA. 2012. A high density recombination map of the pig reveals a correlation between sex-specific recombination and GC content. BMC Genomics 13:586. doi: 10.1186/1471-2164-13-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Runckel C, Westesson O, Andino R, DeRisi JL. 2013. Identification and manipulation of the molecular determinants influencing poliovirus recombination. PLoS Pathog 9:e1003164. doi: 10.1371/journal.ppat.1003164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.