

Abstract
Hydrogen peroxide (H2O2) is frequently used in combination with ultraviolet (UV) light to treat trace organic contaminants in advanced oxidation processes (AOPs). In small-scale applications, such as wellhead and point-of-entry water treatment systems, the need to maintain a stock solution of concentrated H2O2 increases the operational cost and complicates the operation of AOPs. To avoid the need for replenishing a stock solution of H2O2, a gas diffusion electrode was used to generate low concentrations of H2O2 directly in the water prior to its exposure to UV light. Following the AOP, the solution was passed through an anodic chamber to lower the solution pH and remove the residual H2O2. The effectiveness of the technology was evaluated using a suite of trace contaminants that spanned a range of reactivity with UV light and hydroxyl radical (HO•) in three different types of source waters (i.e., simulated groundwater, simulated surface water, and municipal wastewater effluent) as well as a sodium chloride solution. Irrespective of the source water, the system produced enough H2O2 to treat up to 120 L water d–1. The extent of transformation of trace organic contaminants was affected by the current density and the concentrations of HO• scavengers in the source water. The electrical energy per order (EEO) ranged from 1 to 3 kWh m–3, with the UV lamp accounting for most of the energy consumption. The gas diffusion electrode exhibited high efficiency for H2O2 production over extended periods and did not show a diminution in performance in any of the matrices.
Introduction
Distributed water treatment systems offer a potential means of exploiting alternative water sources, including municipal wastewater effluent, roof water, stormwater, and water from shallow aquifers.1 Unfortunately, alternative water sources often contain trace concentrations of organic contaminants (e.g., pesticides, solvents, pharmaceuticals, disinfection byproducts).2 As a result, distributed treatment is often seen as an impractical means of providing potable water. Previous attempts to develop point-of-use treatment systems capable of removing trace organic contaminants prior to nonpotable reuse have employed electrochemical processes,3,4 but these systems suffer from limitations including the production of toxic byproducts, an inability to remove recalcitrant compounds and high cost of treatment.5
Trace organic contaminants can be removed from water by exposure to hydroxyl radicals (HO•) in advanced oxidation processes (AOPs).6−9 In full-scale potable water reuse systems, trace organic contaminants are frequently removed by addition of a modest concentration of H2O2 (e.g., 3 mg/L) followed by exposure to ultraviolet (UV) light. This approach offers numerous benefits over other AOPs in terms of energy consumption, reliability, and production of toxic byproducts.10 Although UV/H2O2 is a well established technology in centralized treatment facilities, challenges associated with the transport and storage of H2O2 make it an impractical solution for distributed treatment systems.11 Electrochemical production of H2O2 from O2 is an attractive alternative means of producing H2O2 if it can be achieved without the consumption of large amounts of energy or the formation of toxic byproducts.11
Electrochemical production of low concentrations of H2O2 can be achieved by several different approaches. Systems in which oxygen is bubbled into a solution prior to reduction on an electrode surface consume a considerable amount of energy due to the low solubility of oxygen and the need to ensure that it reaches the electrode surface. Bubbling air or pure oxygen into a solution is also an impractical approach for H2O2 production in decentralized systems because it requires pumps and controllers. Furthermore, the yield of H2O2 from reduction of O2 is often quite low, which greatly increases electricity consumption.11−14 Recently, gas diffusion electrodes have been used to generate H2O2 without a need to bubble air or oxygen into a solution.11,13,14 Most of the research in this area has been focused on producing concentrated H2O2 solutions by using highly conductive solutions or organic solvents.12,13 Cathodic production of H2O2 for the removal of organics has typically been used for electro-Fenton treatment; however, differences in pH needed for the optimal kinetics of the two reactions (i.e., production of H2O2 is most efficient at basic pH values and Fenton’s reaction is more effective at acidic pH values) results in inefficient oxidation of trace organics if pH correction is not performed.11,15 To produce low concentrations of H2O2 in water immediately prior to an AOP, the cathode must be capable of producing H2O2 in a low ionic strength, poorly buffered solution at circumneutral pH values. Furthermore, increases in pH that occur in the cathode chamber due to consumption of protons must be compensated for by a subsequent treatment process if the water is to be sent into a water distribution system.
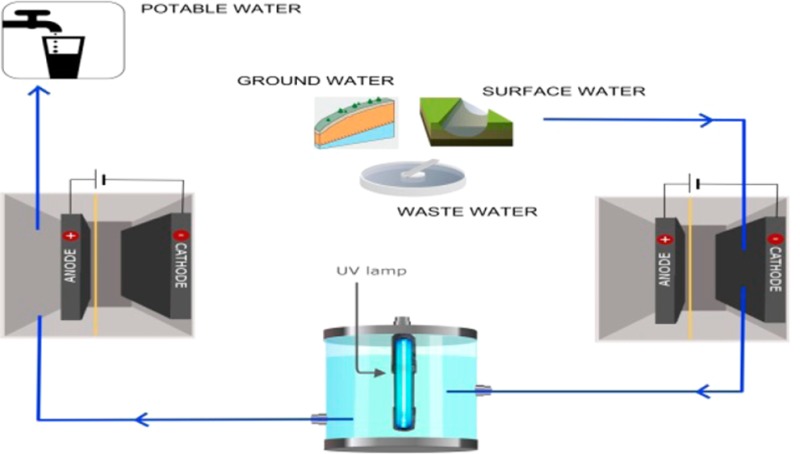
The purpose of this study was to evaluate a system that combines in situ electrochemical production of H2O2 followed by UV irradiation and anodic pH adjustment as a cost-effective means of removing trace organic contaminants from water. This new system, which also inactivates waterborne pathogens and transforms photolabile contaminants through exposure to UV light, can be controlled by varying the production of H2O2 through adjustment of the applied current. To provide insight into the performance of the system under conditions likely to be encountered in distributed water treatment systems, three representative source waters (i.e., synthetic surface water, synthetic groundwater, and municipal wastewater effluent) were tested and compared to an electrolyte solution consisting of dilute sodium chloride. The performance of the system was investigated in terms of contaminant removal and energy consumption.
Materials and Methods
Materials
All experiments were performed at room temperature (23 ± 2 °C) with chemicals of reagent grade or higher (Sigma-Aldrich, St. Louis, MO). The composition of the waters used is summarized in Table S1, Supporting Information.
Electrochemical Cell and UV Reactor
Experiments were carried out in a two-chambered parallel plate electrochemical cell consisting of two square Perspex frames (internal dimensions: 8 × 8 × 1.9 cm3) separated by a cation exchange membrane (Ultrex CMI-7000, Membranes International Inc., Ringwood, NJ). The frames were bolted together between two square Perspex side plates, creating anode and cathode compartments that each had effective volumes of 122 mL (Figure S1, Supporting Information). A solid plate was used for the anode frame, while the cathode chamber was bolted with a hollow side plate allowing for one side of the gas diffusion cathode to be exposed to air. A Ti mesh electrode coated with an Ir mixed-metal oxide was used as the anode (dimensions: 7.8 × 7.8 cm; 1 mm thickness; specific surface area 1.0 m2 m–2, Magneto Special Anodes, Netherlands). The anode and cathode had projected electrode surface areas of 64 cm2. The UV reactor consisted of a 1 L brown glass bottle (Veffective = 925 mL) containing a low-pressure UV lamp (arc length = 16.5 cm, optical path length = 4.3 cm) used typically for swimming pool disinfection (G23 Odyssea Pool Lamp, 9W, Odyssea Aquarium Appliance Co., Ltd., Guangdong, China; Figure S1, Supporting Information).
Gas Diffusion Cathode Fabrication
The gas diffusion cathode was created by modifying carbon fiber paper (AvCarb P75T, 10 × 10 cm2, Fuel Cell Store, College Station, TX) with a conductive, hydrophobic support layer and a carbon catalyst.14 The air-facing side of the cathode was prepared by coating a mixture of 60 wt % PTFE and 30 wt % graphite powder (200 mesh, Alfa Aesar, Ward Hill, MA) onto one side of the carbon base layer. The cathode was then air-dried at room temperature, followed by sintering at 350 °C for 40 min. The liquid-facing side was prepared by applying a mixture of 3 mL of propanol with 150 mg of carbon black (Cabot Black Pearls 2000, Cabot, Boston, MA) and 50 mg of PTFE onto the other side of the carbon base layer. The cathode was again air-dried at room temperature, followed by sintering at 350 °C for 40 min.
Experimental Approach
Electrolysis experiments were performed at fixed currents controlled by a multichannel potentiostat (Gamry Instruments Inc., Warminster, PA). Water entered the cathode compartment operating in a flow-through mode with hydraulic residence times (HRT) ranging from 1.5 to 5.0 min (120–35 L d–1). Cathode effluent was supplied to the UV reactor and then passed through the anode of the electrochemical cell. The applied charge density (ρq, C L–1) was expressed as a product of the current density (I, A m–2), electrode surface area (A, m2), and the hydraulic residence time (t, s) normalized by the half chamber reactor volume (V, L):
| 1 |
Source waters were amended with a mixture of ten test compounds each at a concentration of 10 μg L–1. For each experiment, samples were collected prior to the electrochemical cell, after passing through the cathode chamber, after the UV reactor, and after passing through the anode. At least 3.5 L of the test solution was passed through the system prior to collection of a sample. Samples were analyzed for H2O2, trace organic compounds, and pH. 1.9 mL subsamples to be analyzed for trace organic compounds were mixed with 0.1 mL of methanol to quench radical reactions that could occur prior to analysis. H2O2 and pH were measured within 5 min, whereas trace contaminants were stored for a maximum of 8 h. Experiments quantifying H2O2 production in the varying waters were performed with applied cathodic current densities from 0 to 30 A m–2 under varying flow regimes (35–120 L d–1). To assess the long-term cathode performance, 6000 L of 5 mM Na2SO4 in tap water (alkalinity = 0.34 mM, [Ca2+] = 0.2 mM) was run through the cell continuously at an applied current density of 15 A m–2 at a fixed flow rate of 120 L d–1. Samples were collected daily and analyzed for H2O2. The effects of dissolved oxygen concentration on H2O2 production were evaluated by sparging source water with N2 to remove O2. The rate of production of H2O2 remained unchanged under N2-sparged conditions.
Analytical Methods
H2O2 and free chlorine were measured with a Shimadzu UV-2600 spectrophotometer with the titanium(IV) sulfate method at 405 nm and the N,N-diethyl-p-phenylenediamine (DPD) method at 515 nm, respectively.16,17 Determination of free chlorine was performed in the absence of H2O2 to eliminate the positive interference of H2O2 with DPD.18 The UV absorbance of the four source waters was measured with a Shimadzu UV-2600 spectrophotometer. Dissolved organic carbon (DOC) and dissolved inorganic carbon (DIC) were measured using a Shimadzu TOC-V analyzer. NO3–, Cl–, and SO42– were analyzed using a Dionex DX-120 ion chromatograph with an AS19G column. K+, Na+, Ca2+, and Mg2+ were analyzed using a Dionex ICS-2000 ion chromatograph with a CS12A column. Fluence rate values were determined by chemical actinometry using 10 μM atrazine as an actinometer (ε254 = 3860 M–1 cm–1, ϕ254= 0.046 mol Ei–1, buffered at pH = 8 using a borate buffer; details of the calculation in the Supporting Information).19,20 Conductivity was measured with an Ultrameter II 4P (Myron L Company, Carlsbad, CA). Test compounds were quantified in multiple reaction monitoring (MRM) mode with an Agilent 1200 series HPLC system coupled to a 6460 triple quadrupole tandem mass spectrometer (HPLC-MS/MS), as described previously.21 Analytical details and compound specific parameters are provided in the Supporting Information text and Tables S2 and S3, respectively.
Electrical Power Calculations
The gas diffusion electrode was polarized cathodically against a Ag/AgCl reference electrode (+0.197 V vs SHE; BASi, USA). The full cell potential between the working (i.e., cathode) and counter (i.e., anode) electrodes was measured in a two-electrode setup. The total system power (Ptotal, W) is a combination of the UV lamp power (Plamp, W) and the electrochemical cell power, which can be expressed as a product of the current density (I, A m–2), cell potential (Vcell), and the electrode surface area (A, m2):
| 2 |
Results
Hydrogen Peroxide Production as a Function of Current in Varying Source Waters
Hydrogen peroxide concentrations (Figure 1A) and production rates (Figure S2, Supporting Information) were determined for an array of charge densities that were achieved from combinations of current densities (0–30 A m–2) and cathode chamber retention times (1.5–5 min). A linear relationship between H2O2 production and applied charge density was observed at charge densities greater than 50 C L–1, independent of how the charge was obtained (i.e., high current density and short retention times or low current density and long retention times):
| 3 |
where ρq is the specific charge density applied in C L–1 and [H2O2] is the hydrogen peroxide concentration in mM. For all of the waters tested, the Coulombic efficiency of O2 reduction to H2O2 averaged 88.8 ± 1.8% at charge densities greater than 50 C L–1 (Figure 1B). At lower charge densities, lower Coulombic efficiencies were observed (note the deviation from the linear fit in Figure 1A at charge densities below 50 C L–1). The observed increased Coulombic efficiencies at higher charge densities agreed with previously published data for electrochemical synthesis of H2O2 using a gas diffusion electrode composed of a fluorocarbon binder and activated carbon catalyst fed with conductive, alkaline solutions.11,12,22
Figure 1.

Production of hydrogen peroxide (A) and Coulombic efficiency (B) as a function of applied charge density (WWTP: wastewater treatment plant).
H2O2 production was independent of the type of source water used despite the substantial variability in the composition of the matrices (Table 1). This suggests that H2O2 production was not affected by pH, the presence of natural organic matter (NOM), dissolved ions, or conductivity over the range of applied charge densities studied. Although H2O2 production was not influenced by influent water quality, the cell potential, and therefore energy consumption, was affected by the conductivity of the source waters. Higher ionic strength waters exhibited lower ohmic resistances and therefore operated at lower cell potentials, thus decreasing their energy consumption. For a given applied charge, the power required decreased with increased conductivity (synthetic surface water > synthetic groundwater > wastewater effluent ≅ electrolyte) (Figure S3, Supporting Information). Even for low conductivity surface water, however, the energy consumption for hydrogen peroxide production at a flow rate of 120 L d–1 was still relatively low (0.018–0.31 kWh m–3 for 5 < I < 30 A m–2), indicating that in situ H2O2 production required much less energy than operation of the UV lamp (1.8 kWh m–3; calculations provided in the Supporting Information).
Table 1. Composition and Properties of the Tested Waters.
| property |
scavenging compound |
|||||||
|---|---|---|---|---|---|---|---|---|
| water matrix | UV absorbance254 nm (cm–1) |
conductivity (μS cm–1) |
pHinitial | TIC (mequiv L–1) |
[DOC] (mgC L–1) |
H2O2 (mM)b |
HCO3– (mM) |
CO32– (mM) |
| electrolyte | 0 | 1515 | 5.36 | 0 | 0 | 0–0.54 | 0 | 0 |
| synthetic groundwater | 0.0027 | 440.6 | 8.69 | 3.9 | 0.1 | 0–0.54 | 3.79 | 0.09 |
| synthetic surface water | 0.131 | 360.4 | 8.55 | 2.4 | 1.6 | 0–0.54 | 2.35 | 0.04 |
| wastewater effluent | 0.137 | 2040 | 8.17 | 5.0 | 4.9 | 0–0.54 | 4.90 | 0.03 |
| rate constant | ||||||||
| kHO•, cont (M–1 s–1) | 9.8 × 103 47 a | 2.7 × 107 48 | 8.5 × 106 48 | 3.9 × 108 48 | ||||
kHO•,NOM rate constant is given in L mgC–1s–1.
[H2O2] is variable and dependent on the applied current density.
The H2O2 production rate increased linearly with applied current density, with a maximum of between 14.4 and 14.8 mg H2O2 L–1 min–1 at 30 A m–2 for all of the waters tested (Figure S2, Supporting Information). In full-scale AOP systems (e.g., the Orange County Water District’s Groundwater Replenishment System), 3 mg H2O2 L–1 (0.09 mM) is typically applied.23 This concentration can be obtained with the gas diffusion electrode at an applied current density of only 4.14 A m–2 at a hydraulic residence time of 1.5 min. Under these conditions, this benchtop system could process approximately 120 L of water per day while consuming approximately 1.7 Wh to produce H2O2.
Trace Organic Contaminant Removal by Electro-Generated H2O2 and UV Irradiation
The removal of trace organic contaminants involved direct photolysis, reactions with HO• produced by photolysis of H2O2 in the UV chamber, and direct oxidation of contaminants on the anode. At the fluence employed in the UV chamber (F0 ∼ 3000 mJ cm–2), direct photolysis only removed those compounds that exhibited high quantum yields and strong light absorbance at 254 nm (Figure S4, Supporting Information). Among the compounds tested, carbamazepine exhibited the lowest tendency for direct transformation by UV light (<30% removal for all matrices), while more photoreactive compounds, such as sulfamethoxazole, propranolol, and atrazine, displayed higher removals (55–99%). Unlike the variability of the compounds with respect to direct photolysis, the suite of trace organics all reacted with HO• at near diffusion controlled rates (109–1010 M–1 s–1; Table S3, Supporting Information). As a result of its low reactivity with UV light, carbamazepine removal in the presence of H2O2 and UV light provided useful information on the transformation of organic contaminants by HO•.
The extent of removal of carbamazepine varied among the different matrices (Figure 2). The presence of HO• scavengers explained much of the variability. In the absence of current (and therefore H2O2), the variability of carbamazepine transformation was predominately influenced by the screening of light, as accounted for by the water factor:20,24
| 4 |
where z is the mixed water body depth (m) and α is the attenuation coefficient of the water body. The water factor, which was primarily influenced by the amount of NOM, followed the trend: electrolyte (0.998) > groundwater (0.985) > synthetic surface water (0.598) > wastewater effluent (0.508). Transformation of carbamazepine solely in the presence of UV light varied from 22 ± 5% in the electrolyte solution to 15 ± 4% in the wastewater effluent. The observed removal of carbamazepine in the surface water and wastewater effluent, however, was greater than suggested from the water factor. This may be explained by the generation of HO• and 3DOM* produced from NOM sensitization by UV light, which can be significant at UV fluences employed in AOPs.25,26
Figure 2.
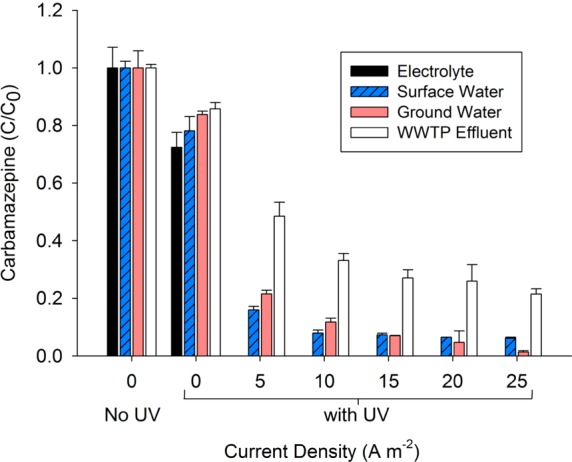
Removal of carbamazepine as a function of current density for the four types of source waters (WWTP: wastewater treatment plant).
The rate of transformation of trace contaminants increased with current density due to additional HO• production that occurred at higher H2O2 concentrations (Tables S4–S7, Supporting Information). Complete carbamazepine transformation was observed after the UV treatment chamber at an applied current density of 5 A m–2 for the electrolyte. For the three representative source waters, carbamazepine transformation increased to 98.7 ± 0.6% for groundwater, 93.3 ± 1.0% for surface water, and 78.5 ± 1.9% for wastewater effluent as the current increased to 25 A m–2. Organic compounds that have lower reaction rate constants with HO• and are not susceptible to direct photolysis will require higher current densities to achieve a similar level of treatment.
The fraction of HO• that reacted with the contaminants can be estimated by considering the concentrations and rate constants for reactions of different solutes with HO•:
| 5 |
where kOH•,S and kHO•,cont are the second order reaction rate constants of scavengers and contaminants with HO•, respectively, and [S] is the concentration of the scavenger (e.g., HCO3–, CO32–, NOM, H2O2; Tables 1 and S3, Supporting Information).
Effect of H2O2 on Treatment Efficiency
At increasing H2O2 concentrations, a trade-off exists between additional transformation of trace organics by HO• produced from H2O2 photolysis and greater radical scavenging and light screening by H2O2.27 Therefore, despite linear increases in H2O2 production with current density, there is a diminishing benefit to the treatment. As H2O2 increased from 0.09 mM (4.14 A m–2) to 0.54 mM (25 A m–2), the fraction of HO• reacting with contaminants decreased by 20%, 21%, and 10% for the surface water, groundwater, and wastewater effluent, respectively. At 0.54 mM H2O2 (25 A m–2), there was a 4.0%, 4.7%, and 3.8% reduction in direct photolysis rates of contaminants from additional light screening by H2O2 for the surface water, groundwater, and wastewater effluent, respectively (details of HO• branching ratio and direct photolysis calculations are included in the Supporting Information).
Effect of pH on Treatment Efficiency
At pH 8, approximately 6.7%, 6.5%, and 2.9% of HO• reacted with the organic contaminants in the UV reactor at an initial H2O2 concentration of 3 mg L–1 (0.09 mM) for the surface water, groundwater, and wastewater effluent, respectively. At pH 10, the fraction of HO• reacting with trace organic contaminants decreased to 0.9%, 0.6%, and 0.4% for the three source waters, respectively. The significant decrease in HO• reacting with the trace organic contaminants was due to scavenging by carbonate at the higher pH values. The product of this reaction, •CO3–, can play a significant role in the transformation of certain organic compounds (e.g., propranolol and sulfamethoxazole; Table S3, Supporting Information).28 As a result of differences in alkalinity of the different source waters, the importance of carbonate scavenging and •CO3– reactions depends on the source water composition and the applied current density (i.e., higher applied currents result in greater pH increases in the cathode chamber). Although nitrite is an effective scavenger of HO• (kHO•,NO2– = 6 × 109 M–1 s–1), less than 0.3% of the generated HO• would be scavenged by nitrite at concentrations typically found in nitrified wastewater effluent (i.e., ∼0.1 mg L–1). The formation of halogen radicals from reactions between HO• and halide ions (chloride and bromide) should only be significant at low pH values and therefore will have a negligible impact on contaminant transformation at the circumneutral and basic pH values observed in this system.29
Anodic pH Adjustment
The generation of H2O2 by the cathode consumed protons and increased the solution pH (reaction 6). In the anode, oxidation reactions produced protons and lowered the solution pH (reaction 7):
| 6 |
| 7 |
| 8 |
| 9 |
For the production of 3 mg L–1 (0.09 mM) of H2O2 at a current density of 4.14 A m–2, approximately 0.20 mequiv L–1 of protons should have been consumed or produced at the cathode and anode, respectively. To maintain electroneutrality, a net migration of protons occurred from the anode chamber to the cathode chamber via the cation exchange membrane. In addition to protons, cations that were present at higher concentrations (e.g., Na+, Ca2+, Mg2+) also carried ionic charge through the membrane satisfying electroneutrality in the cathode chamber while creating a proton deficit in the cathode chamber.30
As a result of differences in buffering among matrices, the solution pH should have increased more in the cathode chamber for waters with low alkalinity. The pH in the cathode chamber increased for each of the waters as the current increased from 0 to 25 A m–2 (Figure 3). The magnitude of pH increase was most pronounced for the electrolyte and surface water (alkalinity = 0 and 2.45 mM, respectively) with post-cathode pH values ranging from 10 to 10.5, while the pH never exceeded 9.9 and 9.2 in the groundwater (alkalinity = 3.89 mM) and the municipal wastewater effluent (alkalinity = 4.97 mM), respectively. The pH increases following the cathode resulted in supersaturation with respect to calcite (CaCO3(s)) in the groundwater (log SI > 1.31), surface water (log SI > 1.58), and wastewater effluent (log SI > 1.09) beginning at a current density of 5 A m–2. This reaction could result in scaling on the cathode or the ion exchange membrane that might eventually affect system performance. Loss of CaCO3(s) from the system could also result in an overall decrease in pH as water passed through the treatment system. For example, if the surface water solution reached equilibrium at the current density needed to produce 3 mg L–1 (0.09 mM) H2O2, 0.74 mmol (74 mg) of calcite would precipitate for each liter of water treated and the pH would have dropped from 9.82 to 7.23. On the basis of the observed pH values, it is evident that equilibrium was not achieved. However, additional research is needed to assess the importance of calcite precipitation to scaling and pH control.
Figure 3.

pH change of the source waters prior to entering the electrochemical cell, after passing through the cathode chamber, after the UV reactor, and after the anode as a function of current density (WWTP: wastewater treatment plant).
To readjust the solution pH, water leaving the UV reactor was passed through the anode chamber. If no mineral precipitation occurred in the cathode and UV chambers, the final pH should have been equal to the influent pH. A slight decrease in pH was observed in all solutions to which current was applied, with greater pH decreases at higher current densities occurring in the least buffered of the three environmental matrices (i.e., surface water). Under the conditions that would likely be used for treatment (i.e., 5–10 A m–2 and short hydraulic residence times), the final pH was approximately equal to the initial pH.
Anodic Quenching of Residual Hydrogen Peroxide
Due to the relatively low molar absorptivity of H2O2 at 254 nm (ε254 = 18.6 M–1 cm–1) and the limited residence times in the UV reactor (τ = 660 s), much of the H2O2 passed through the UV chamber without undergoing photolysis. For solutions with relatively low light screening (i.e., electrolyte, synthetic groundwater, and synthetic surface water), between 40% and 50% of the H2O2 was photolyzed at current densities ranging from 5 to 25 A m–2 (Figure 4 and Figure S5, Supporting Information). As expected, less H2O2 photolysis occurred in municipal wastewater effluent due to light screening.
Figure 4.
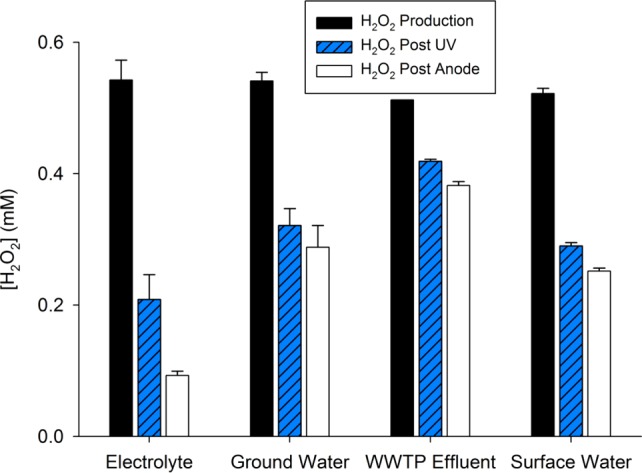
Production of H2O2 in the cathode, residual H2O2 after the UV cell, and residual H2O2 after the anode for the four types of source waters at applied current density of 25 A m–2. See Figure S5, Supporting Information, for data on the production and removal of H2O2 over the full range of current densities (5–25 A m–2) (WWTP: wastewater treatment plant).
In practice, many centralized treatment plants employ reducing agents (e.g., bisulfite), chlorine, or activated carbon to remove residual H2O2 before distribution.31 The use of activated carbon or the addition of chemicals, however, may be impractical in a distributed treatment system. Partial removal of H2O2 occurred when the solution passed through the anode, especially in the electrolyte solution (Figure 4 and Figure S5, Supporting Information). Anodic removal of H2O2 increased with increasing current density (Figure S5, Supporting Information). In the NaCl electrolyte solution, up to 0.12 mM H2O2 was removed at 25 A m–2. For the three source waters, however, the anode only removed about 25% of the amount removed in the electrolyte.
Removal of H2O2 in the anode was attributable to a combination of direct anodic oxidation and reactions with oxidants produced on the anode surface. For example, oxidation of chloride can result in the production of hypochlorous acid (HOCl; pKa = 7.6) (Reactions 8 and 9). Hypochlorite reacts rapidly with hydrogen peroxide under alkaline conditions with the bimolecular rate constant increasing from 196 to 7.5 × 103 M–1 s–1 from pH 6 to 9 (see the Supporting Information for calculation of the pH-dependent bimolecular rate constant):32−34
| 10 |
Although Ti-IrO2 electrodes have a high electrocatalytic activity with respect to chlorine evolution, only modest concentrations of chlorine were produced in control experiments at varying chloride concentrations due to the short hydraulic residence times and relatively low current densities applied35−37 (Table 2). To separate the effects of reactive halogen species from direct electrode oxidation on the removal of H2O2, experiments were repeated using an inert electrolyte (i.e., Na2SO4) (Figure S6, Supporting Information). H2O2 removal was independent of applied current density with 37 ± 2 μM H2O2 removed from 5 to 25 A m–2; a concentration equivalent to the observed H2O2 removal in the anode for the three source waters in Figure S5, Supporting Information. Given the low chloride concentrations (<1 mM) of the simulated surface water and groundwater, it is not surprising that OCl– production was low and only a small quantity of H2O2 was removed in the anode.
Table 2. Chloride-Chlorine Electrochemical Oxidationa.
| current
density (A m–2) |
|||||
|---|---|---|---|---|---|
| [Cl–] (mM) | 2.5 | 5 | 10 | 15 | 25 |
| 0.5 | 0 | 0 | 0 | 0 | 0 |
| 5 | 0 | 0 | 0.79 | 17 | 34 |
| 10 | 0 | 0 | 3.9 | 25 | 41 |
| 15 | 0 | 0 | 7.9 | 32 | 61 |
Free chlorine production (as μM Cl[I]) in the anode chamber as a function of applied current density and chloride concentration. Experiments were performed with a stainless steel cathode to prevent H2O2 formation, which interferes with the chlorine measurement.
Despite the electrolyte and municipal wastewater effluent having roughly the same concentration of chloride, H2O2 removal was significantly higher in the electrolyte control than in the wastewater effluent, suggesting that reactive halogen species did not play an important role in H2O2 removal in the wastewater effluent matrix. NOM is an effective sink of HOCl/OCl–; however, under the experimental conditions used in this study, the half-life of HOCl/OCl– with respect to its reaction with H2O2 was much shorter than that predicted for NOM (i.e., 1.39 and 49.3 s, respectively, as described in the Supporting Information).38 As a result, NOM is only a minor sink for HOCl/OCl– in the presence of H2O2. This was consistent with observations from experiments in which H2O2 removal decreased by less than 40% (58 μM) when NOM was added to a solution containing a fixed concentration of chloride at a current density of 25 A m–2, suggesting that NOM is only partially responsible for the difference in H2O2 removal between the two solutions (Figure S6, Supporting Information).
The apparent discrepancy between municipal wastewater effluent and the sodium chloride solution may be partially attributable to differences in pH values in the anode chamber.39 At higher pH values, like those found in the municipal wastewater effluent after anodic treatment, there is a larger driving force for oxygen evolution compared to Cl2 production:
| 11 |
| 12 |
Experiments conducted in the anode chamber at different pH values confirmed that chlorine production increased substantially as pH dropped from 9 to 7 (Figure S7, Supporting Information). Due to the presence of bicarbonate in the municipal wastewater effluent, the anode pH was considerably higher (i.e., ∼8) during treatment than in the unbuffered NaCl electrolyte, where pH decreased to approximately 6 during anodic treatment. As a result, considerably more HOCl/OCl– was produced in the anode chamber when the electrolyte was treated.
Total hydrogen peroxide removal in the system (i.e., photolysis and anodic loss) was 48 ± 5% for the simulated groundwater, 49 ± 3% for the simulated surface water, and 25 ± 3% for the wastewater effluent for the array of current densities tested. At the current density required to produce 3 mg L–1 (0.09 mM) H2O2, water leaving the treatment system effluent contained 1.5–2.3 mg L–1 H2O2. Although H2O2 does not pose a health risk at these concentrations, its presence in potable water may be undesirable. In a point-of-use water treatment system, it might be possible to remove the excess H2O2 by passing it through activated carbon or a high surface area catalyst consisting of metal oxide40 or silver.41 Alternatively, the efficiency of chloride oxidation in the anode chamber might be improved through the use of three-dimensional or porous electrodes that reduce mass transfer limitations or through the use of more catalytic anode materials.5
Anodic Transformation of Trace Organic Contaminants
Despite only accounting for a small fraction of the total removal observed in the treatment system, the anode did transform some of the compounds. Experiments conducted in the absence of UV exposure with solutions amended with 10 μg L–1 of trace organic contaminants indicated that direct anodic oxidation resulted in the removal of up to 20% of certain trace organics (e.g., propranolol) at a current density of 25 A m–2 (Figure S8, Supporting Information). Oxidation of organic compounds on the anode could be increased through the use of inactive anodes (e.g., boron-doped diamond, doped-SnO2, PbO2).5,37 These electrodes, however, have higher capital and operating costs than Ti-IrO2 electrodes.
Although carbamazepine is relatively unreactive with hypochlorite, other compounds (e.g., propranolol and sulfamethoxazole) react with HOCl (Table S3, Supporting Information). However, the presence of H2O2 reduced the importance of reactions between trace organics and chlorine species generated at the anode because chlorine preferentially reacts with H2O2. As a result, nearly all of the observed loss of the test compounds in the anode chamber was due to direct oxidation on the anode surface. This observation was consistent with experiments comparing anodic removal of trace organics in the presence and absence of H2O2 (Figure S9, Supporting Information).
Long-term Cathode Performance
The performance of gas diffusion electrodes can decrease over time due to clogging of the pores by precipitates, fouling with NOM as well as charge transfer resistance attributable to the loss of conductive graphite paste.42 In a long-term trial, cathode performance (i.e., H2O2 production at a fixed current density) decreased by less than 2% after 6000 L of tap water amended with 5 mM Na2SO4 was passed through the system at an applied current density of 15 A m–2 (Figure S10, Supporting Information). Calcium carbonate scaling due to the elevated pH and migration of calcium ions in the tap water into the cathode chamber was observed on the interior of the cathode. Nonetheless, H2O2 production was unaffected during this 50 day test. Additional experiments are needed to assess the importance of scaling and the efficacy of simple descaling approaches (i.e., polarization reversal) over longer time periods and more realistic operating conditions.
System Energy Consumption
The treatment system used electricity to produce the oxidant (i.e., H2O2) and to convert it into HO• (i.e., the UV lamp). Electrical energy per order (EEO) is a useful figure of merit for comparing the efficiency and cost of the treatment system with other AOPs. EEO is the electrical energy (in kWh) required to reduce a contaminant concentration by 1 order of magnitude in 1 m3 of water:43−45
| 13 |
where P (kW) is the electrical power for the electrochemical cell and UV lamp, Q (m3 h–1) is the system flow rate, and C0 and C (M) are the initial and final contaminant concentrations. Without the production of H2O2, EEO values ranged from 16.8 ± 0.3 to 28.1 ± 0.2 kWh m–3 order –1, with larger amounts of energy needed to transform contaminants in waters that contained high concentrations of HO• scavengers and chromophores (i.e., municipal wastewater effluent and synthetic groundwater) (Figure 5). A substantial decrease in EEO occurred when current was applied to the electrochemical cell (i.e., from 0 to 5 A m–2). As current density increased from 5 to 25 A m–2, the EEO decreased by less than 11%. These data indicate that the UV–H2O2 AOP system is much more efficient than the use of UV alone and that there is a marginal benefit associated with the production of higher H2O2 concentrations in the cathode because the reactions become less efficient at higher concentrations of H2O2. Only 8–14% of the energy demand was attributable to the electrochemical production of H2O2, with the majority of the energy required for the operation of the low-pressure UV lamp. At a current density of 25 A m–2, an energy requirement of 1.08 ± 0.1 to 2.84 ± 0.1 kWh m–3 order–1 was observed, which was similar to results from previous studies of the transformation of trace organic compounds by UV/H2O2 in different source waters.10
Figure 5.
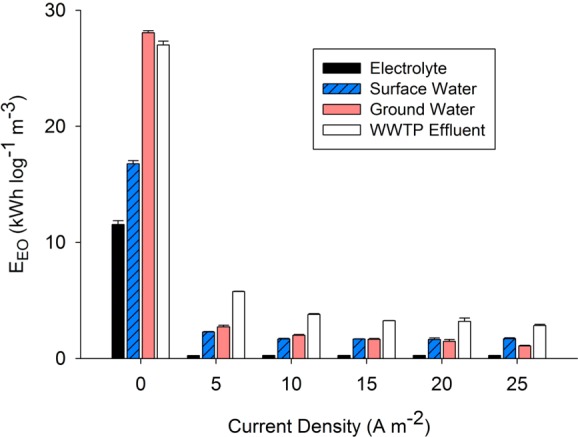
Electrical energy per order (EEO) for the removal of carbamazepine as a function of current density (WWTP: wastewater treatment plant).
The high Coulombic efficiency and low current densities result in a low-cost means of H2O2 production even for treatment of water with low conductivity. For comparison, electrochemically produced H2O2 costs between 0.1 and 0.3 $ kg–1, while H2O2 produced by the anthraquinone process typically costs between 1 and 2 $ kg–1.46 When considering the lack of a need to transport, store, and handle H2O2 as well as modest capital and operational costs, the modular AOP treatment system can be a competitive technology for point-of-use treatment at a household and community level or even for wellhead treatment of trace organic contaminants present in potable water sources. As H2O2 production was directly proportional to current density, the treatment system could be scaled up by using faster flow rates accompanied by higher applied current densities or by increasing surface area of the cathode. To obtain the equivalent contaminant removal after scale-up, optimization of the UV reactor geometry would be required. Additional research is needed to assess long-term system performance under realistic operating conditions.
Acknowledgments
This study was supported by the U.S. National Institute for Environmental Health Sciences (NIEHS) Superfund Research Program (Grant P42 ES004705) and the Superfund Research Center at University of California, Berkeley. T.H. was also supported by a postdoctoral fellowship from the Research Foundation Flanders (FWO Vlaanderen). The authors thank Dr. Oskar Modin for his guidance with the fabrication of the gas diffusion electrode and Dr. Jelena Radjenovic for her insightful comments.
Supporting Information Available
Details of the calculations for fluence rate, photolysis rates, H2O2 half-life, oxidant branching ratios, EEO, and methods for test compound analysis in addition to supporting tables, schematics, and figures referenced in this work. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.5b01254.
Author Contributions
# J.M.B. and T.H. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Larsen A. T.; Udert K. M.; Lienert J.. Source separation and decentralization for wastewater management; IWA Publishing: London, 2013; p 491. [Google Scholar]
- Benotti M. J.; Trenholm R. A.; Vanderford B. J.; Holady J. C.; Stanford B. D.; Snyder S. A. Pharmaceuticals and endocrine disrupting compounds in US drinking water. Environ. Sci. Technol. 2009, 433597–603. [DOI] [PubMed] [Google Scholar]
- Butkovskyi A.; Jeremiasse A. W.; Leal L. H.; van der Zande T.; Rijnaarts H.; Zeeman G. Electrochemical conversion of micropollutants in gray water. Environ. Sci. Technol. 2014, 4831893–1901. [DOI] [PubMed] [Google Scholar]
- Hernandez-Leal L.; Temmink H.; Zeeman G.; Buisman C. J. N. Removal of micropollutants from aerobically treated grey water via ozone and activated carbon. Water Res. 2011, 4592887–2896. [DOI] [PubMed] [Google Scholar]
- Chaplin B. P. Critical review of electrochemical advanced oxidation processes for water treatment applications. Environ. Sci.: Processes Impacts 2014, 1661182–1203. [DOI] [PubMed] [Google Scholar]
- Shu Z. Q.; Bolton J. R.; Belosevic M.; El Din M. G. Photodegradation of emerging micropollutants using the medium-pressure UV/H2O2 advanced oxidation process. Water Res. 2013, 4782881–2889. [DOI] [PubMed] [Google Scholar]
- Rosario-Ortiz F. L.; Wert E. C.; Snyder S. A. Evaluation of UV/H2O2 treatment for the oxidation of pharmaceuticals in wastewater. Water Res. 2010, 4451440–1448. [DOI] [PubMed] [Google Scholar]
- Pereira V. J.; Weinberg H. S.; Linden K. G.; Singer P. C. UV degradation kinetics and modeling of pharmaceutical compounds in laboratory grade and surface water via direct and indirect photolysis at 254 nm. Environ. Sci. Technol. 2007, 4151682–1688. [DOI] [PubMed] [Google Scholar]
- Pereira V. J.; Linden K. G.; Weinberg H. S. Evaluation of UV irradiation for photolytic and oxidative degradation of pharmaceutical compounds in water. Water Res. 2007, 41194413–4423. [DOI] [PubMed] [Google Scholar]
- Katsoyiannis I. A.; Canonica S.; von Gunten U. Efficiency and energy requirements for the transformation of organic micropollutants by ozone, O-3/H2O2 and UV/H2O2. Water Res. 2011, 45133811–3822. [DOI] [PubMed] [Google Scholar]
- Campos-Martin J. M.; Blanco-Brieva G.; Fierro J. L. G. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem., Int. Ed. 2006, 45426962–6984. [DOI] [PubMed] [Google Scholar]
- Yan H.; Fangong K.; Shoujuan W.; Guihua Y. Novel gas diffusion electrode system for effective production of hydrogen peroxide. Appl. Mech. Mater. 2014, 496–500, 159–162. [Google Scholar]
- Rozendal R. A.; Leone E.; Keller J.; Rabaey K. Efficient hydrogen peroxide generation from organic matter in a bioelectrochemical system. Electrochem. Commun. 2009, 1191752–1755. [Google Scholar]
- Modin O.; Fukushi K. Production of high concentrations of H2O2 in a bioelectrochemical reactor fed with real municipal wastewater. Environ. Technol. 2013, 34192737–2742. [DOI] [PubMed] [Google Scholar]
- Brillas E.; Sires I.; Oturan M. A. Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem. Rev. 2009, 109126570–6631. [DOI] [PubMed] [Google Scholar]
- Eisenberg G. M. Colorimetric determination of hydrogen peroxide. Ind. Eng. Chem. 1943, 155327–328. [Google Scholar]
- American Public Health Association; Eaton A. D.; American Water Works Association; Water Environment Federation. Standard methods for the examination of water and wastewater; APHA-AWWA-WEF: Washington, D.C., 2005. [Google Scholar]
- Dotson A. D.; Keen V. S.; Metz D.; Linden K. G. UV/H2O2 treatment of drinking water increases post-chlorination DBP formation. Water Res. 2010, 44123703–3713. [DOI] [PubMed] [Google Scholar]
- Canonica S.; Meunier L.; Von Gunten U. Phototransformation of selected pharmaceuticals during UV treatment of drinking water. Water Res. 2008, 421–2121–128. [DOI] [PubMed] [Google Scholar]
- Bolton J. R.; Linden K. G. Standardization of methods for fluence (UV dose) determination in bench-scale UV experiments. J. Environ. Eng.-ASCE 2003, 1293209–215. [Google Scholar]
- Jasper J. T.; Jones Z. L.; Sharp J. O.; Sedlak D. L. Biotransformation of trace organic contaminants in open-water unit process treatment wetlands. Environ. Sci. Technol. 2014, 4895136–5144. [DOI] [PubMed] [Google Scholar]
- Foller P. C.; Bombard R. T. Processes for the production of mixtures of caustic soda and hydrogen-peroxide via the reduction of oxygen. J. Appl. Electrochem. 1995, 257613–627. [Google Scholar]
- DDB Engineering, Inc. Groundwater Replenishment System 2013 Annual Report; DDB Engineering, Inc.: Irvine, CA, 2013. [Google Scholar]
- Schwarzenbach R. P.; Gschwend P. M.; Imboden D. M.. Environmental Organic Chemistry; John Wiley & Sons: New York, 2002. [Google Scholar]
- Dong M. M.; Rosario-Ortiz F. L. Photochemical formation of hydroxyl radical from effluent organic matter. Environ. Sci. Technol. 2012, 4673788–3794. [DOI] [PubMed] [Google Scholar]
- Lester Y.; Sharpless C. M.; Mamane H.; Linden K. G. Production of photo-oxidants by dissolved organic matter during UV water treatment. Environ. Sci. Technol. 2013, 472011726–11733. [DOI] [PubMed] [Google Scholar]
- Sharpless C. M.; Linden K. G. Experimental and model comparisons of low- and medium-pressure Hg lamps for the direct and H2O2 assisted UV photodegradation of N-nitrosodimethylamine in simulated drinking water. Environ. Sci. Technol. 2003, 3791933–1940. [DOI] [PubMed] [Google Scholar]
- Jasper J. T.; Sedlak D. L. Phototransformation of wastewater-derived trace organic contaminants in open-water unit process treatment wetlands. Environ. Sci. Technol. 2013, 471910781–10790. [DOI] [PubMed] [Google Scholar]
- Grebel J. E.; Pignatello J. J.; Mitch W. A. Effect of halide ions and carbonates on organic contaminant degradation by hydroxyl radical-based advanced oxidation processes in saline waters. Environ. Sci. Technol. 2010, 44176822–6828. [DOI] [PubMed] [Google Scholar]
- Rozendal R. A.; Hamelers H. V. M; Buisman C. J. N. Effects of membrane cation transport on pH and microbial fuel cell performance. Environ. Sci. Technol. 2006, 40175206–5211. [DOI] [PubMed] [Google Scholar]
- Watts M. J.; Hofmann R.; Rosenfeldt E. J. Low-pressure UV/Cl2 for advanced oxidation of taste and odor. J. - Am. Water Works Assoc. 2012, 104147–48. [Google Scholar]
- Held A. M.; Halko D. J.; Hurst J. K. Mechanisms of chlorine oxidation of hydrogen peroxide. J. Am. Chem. Soc. 1978, 100185732–5740. [Google Scholar]
- Connick R. E. The interaction of hydrogen peroxide and hypochlorous acid in acidic solution containing chloride ion. J. Am. Chem. Soc. 1947, 6961509–1514. [Google Scholar]
- VonGunten U.; Oliveras Y. Kinetics of the reaction between hydrogen peroxide and hypobromous acid: Implication on water treatment and natural systems. Water Res. 1997, 314900–906. [Google Scholar]
- Jeong J.; Kim C.; Yoon J. The effect of electrode material on the generation of oxidants and microbial inactivation in the electrochemical disinfection processes. Water Res. 2009, 434895–901. [DOI] [PubMed] [Google Scholar]
- Bagastyo A. Y.; Radjenovic J.; Mu Y.; Rozendal R. A.; Batstone D. J.; Rabaey K. Electrochemical oxidation of reverse osmosis concentrate on mixed metal oxide (MMO) titanium coated electrodes. Water Res. 2011, 45164951–4959. [DOI] [PubMed] [Google Scholar]
- Szpyrkowicz L.; Kaul S. N.; Neti R. N.; Satyanarayan S. Influence of anode material on electrochemical oxidation for the treatment of tannery wastewater. Water Res. 2005, 3981601–1613. [DOI] [PubMed] [Google Scholar]
- Zhai H.; Zhang X.; Zhu X.; Liu J.; Ji M. Formation of brominated disinfection byproducts during chloramination of drinking water: New polar species and overall kinetics. Environ. Sci. Technol. 2014, 4852579–2588. [DOI] [PubMed] [Google Scholar]
- Consonni V.; Trasatti S.; Pollak F.; Ogrady W. E. Mechanism of chlorine evolution on oxide anodes - Study of pH effects. J. Electroanal. Chem. 1987, 2281–2393–406. [Google Scholar]
- Salem I. A.; El-Maazawi M.; Zaki A. B. Kinetics and mechanisms of decomposition reaction of hydrogen peroxide in presence of metal complexes. Int. J. Chem. Kinet. 2000, 3211643–666. [Google Scholar]
- Anipsitakis G. P.; Dionysiou D. D. Radical generation by the interaction of transition metals with common oxidants. Environ. Sci. Technol. 2004, 38133705–3712. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Pant D.; Logan B. E. Long-term performance of activated carbon air cathodes with different diffusion layer porosities in microbial fuel cells. Biosens. Bioelectron. 2011, 30149–55. [DOI] [PubMed] [Google Scholar]
- Bolton J. R.; Bircher K. G.; Tumas W.; Tolman C. A. Figures-of-merit for the technical development and application of advanced oxidation technologies for both electric- and solar-driven systems - (IUPAC Technical Report). Pure Appl. Chem. 2001, 734627–637. [Google Scholar]
- Bolton J. R.; Stefan M. I. Fundamental photochemical approach to the concepts of fluence (UV dose) and electrical energy efficiency in photochemical degradation reactions. Res. Chem. Intermed. 2002, 287–9857–870. [Google Scholar]
- Cater S. R.; Stefan M. I.; Bolton J. R.; Safarzadeh-Amiri A. UV/H2O2 treatment of methyl tert-butyl ether in contaminated waters. Environ. Sci. Technol. 2000, 344659–662. [Google Scholar]
- Jones C. W.Applications of hydrogen peroxide and derivates; Royal Society of Chemistry: Cambridge, U.K., 1999. [Google Scholar]
- Appiani E.; Page S. E.; McNeill K. On the use of hydroxyl radical kinetics to assess the number-average molecular weight of dissolved organic matter. Environ. Sci. Technol. 2014, 482011794–11802. [DOI] [PubMed] [Google Scholar]
- Buxton G. V.; Greenstock C. L.; Helman W. P.; Ross A. B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms, and hydroxyl radicals (·OH/·O–) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 172513–886. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.