

Abstract
We present an imaging and image reconstruction pipeline that captures the dynamic three-dimensional beating motion of the live embryonic zebrafish heart at subcellular resolution. Live, intact zebrafish embryos were imaged using 2-photon light sheet microscopy, which offers deep and fast imaging at 70 frames per second, and the individual optical sections were assembled into a full 4D reconstruction of the beating heart using an optimized retrospective image registration algorithm. This imaging and reconstruction platform permitted us to visualize protein expression patterns at endogenous concentrations in zebrafish gene trap lines.
OCIS codes: (170.1420) Biology, (170.3010) Image reconstruction techniques, (180.0180) Microscopy, (190.0190) Nonlinear optics
1. Introduction
The vertebrate heart is the first organ to form and function during development [1,2]. As the heart undergoes morphogenesis toward its mature 3-dimensional (3D) shape, its beating is critical for the survival of the embryo and the further development of the heart. The intrinsically dynamic and 3D nature of the embryonic heart presents a challenge to any study of its biophysics and biology, as it necessitates a means to capture fast, high 3D-resolution images as it beats. The zebrafish is an ideal model organism for studying vertebrate embryonic cardiac development owing to its optical accessibility, robust transgenic techniques, fast development, and small size - its embryonic heart begins beating as a tube about 200 µm long and 100 µm in diameter [2]. Because imaging modalities typically capture 2D images at only one instant of time, it is a challenge to capture the 3D structure of the zebrafish heart at sub-cellular resolution as it beats quasi-periodically a few beats per second and amplitude of fifty to a hundred µm [3].
The challenge of imaging a moving 3D biological structure has been addressed previously by prospective synchronization approaches, using an independent information channel (e.g. cardiac conduction or respiratory motion) to gate the 2D image acquisitions [4–7]. Alternatively, retrospective temporal registration of image stacks has been used to reconstruct the dynamic motion of cardiac tissues, taking advantage of the quasi-periodic nature of the heart motion [8]. This latter approach, relying on post-acquisition image processing, does not require the sophistication of real-time image processing and specialized hardware of the former prospective gating approach.
In continuation of our previous efforts [8–10] in capturing the 4D (3D plus time) dynamics of the developing vertebrate heart, we have combined an optimized retrospective image registration protocol with two-photon excitation selective plane illumination microscopy (2p-SPIM) [10]. SPIM, also known as light sheet microscopy, combines excitation of an optical section from the side of the sample and a camera in orthogonal direction to collect light in order to generate fast optical sectioning, while minimizing photo-damage. By using two- photon excitation in light sheet mode (Fig. 1(A)), we achieve a balance of high penetration depth and high imaging speed [10] (also see Section 5). At successive optical sections (z-planes), covering the entire depth of the organ, we acquired short movies of the beating heart, spanning several cardiac cycles. Temporal registration of the image sequences acquired from consecutive z-planes were retrospectively registered in time to produce a 4D movie of the beating heart, using a temporal registration algorithm based on maximizing the mutual information of the adjacent z-slices [8] (Fig. 1(B), 1(C)). This created an imaging and image reconstruction pipeline that can generate 4D image series of the beating heart, which we have applied to imaging zebrafish protein trap lines, FlipTraps [11], in which full-length proteins are fused to fluorescent proteins (Fig. 1(D)). The combination of FlipTraps lines that express in cardiac tissue and our imaging pipeline allowed us to capture for the first time endogenous protein expressions in a beating vertebrate heart.
Fig. 1.

Live 4D imaging of the embryonic heart with 2-photon light sheet microscopy. (A) Schematic of the orthogonal illumination and detection pathways of the imaging setup. (B) Successive movies of the beating heart were collected with the light sheet parked at different z positions. (C) Schematic overview of the retrospective synchronization algorithm using the movies collected at successive z positions. Time points t1, t2…, tM denote distinct states of contractions of the heart as captured by our imaging protocol. (D) The FlipTrap vector inserts a Citrine coding sequence (green) into the intron of genes randomly. Active expression of a trapped gene leads to splicing of the citrine exon, yielding full-length fluorescent fusion protein expressed at endogenous level in the embryo. Bottom: wide field fluorescent image of Gt(Tpm4-citrine)ct31a /+. (E) 3D-rendered view of a single time point in the 4D reconstructed image series of the beating heart (transgenic line Gt(Tpm4-citrine)ct31a /+ at 84 hpf). See Media 1 (4MB, MOV) and Media 2 (1.9MB, MOV) for the full 4D depiction of the reconstructed image series. The fluorescent fusion construct labels the cytoplasm of myocardial cells. The arrows indicate nuclei visible by negative contrast, in different cross-sections, demonstrating the quality of resolution achieved with our 2p light sheet imaging. Trabeculating myocardium (arrowhead) can be easily visualized owing to the image quality and synchronization efficiency.
2. Imaging setup and procedure
Our custom-built 2p-SPIM setup, which could also be used in linear 1-photon (1p) excitation mode, has been described in detail previously [10]. Briefly, femtosecond-pulsed near-infrared laser light (Chameleon Ultra II, Coherent) was spherically focused by illumination objectives (Plan-Apochromat, 10X, 0.3 NA, water immersion, Nikon) from both sides of the specimen; the positions of the beams were scanned at 1KHz by galvanometers (H6215, Cambridge Technologies) and aligned to create a bidirectional Gaussian-beam excitation light sheet. This provided a thin excited optical section through the specimen, which was mounted in a liquid-filled sample chamber (Fig. 1(A)). The sample could alternately be excited in 1p-SPIM mode, utilizing a continuous-wave visible-wavelength laser system (Sole-6, Omicron), aligned along the same excitation light paths. The fluorescence signal from the excited optical section was captured by a high numerical aperture (NA) water-immersion detection objective (Plan-Apochromat 20X, 1.0 NA, Carl Zeiss; or Plan-Apochromat 25X, 1.1 NA, Nikon), tube lens (focal length = 164.5 mm, Carl Zeiss; or focal length = 200 mm, Mitutoyo), optional 0.63X de-magnifying image adapter, and camera (iXON 885 or Zyla, Andor). The sample position is controlled by stages (Sutter Instrument, Newport) that allow adjustments in x (optical axis of excitation light), y, z (optical axis of detection direction), and theta (rotation angle about the y axis).
Software control of imaging setup for standard tasks (e.g. view finding, z-stacks) was done through Micro-Manager [12]. Image collection for retrospective reconstruction was performed using the Solis software (Andor) with a custom routine, written within the Solis scripting feature, that collected short 70-frames-per-second movies of the beating heart at each z-plane (duration of 4 seconds; ~10 heart beats). The procedure was repeated approximately 120-140 times, collecting image sequences from the entire z-depth of the heart, with step size of 1 µm. 125 mW of laser light from each side provided the bidirectional illumination at 920 nm light. We found photobleaching of the specimen from the excitation; there was a decrease of the average signal intensity, typically by 30-40%, over the first 0.5-1 second at each z-position, after which the signal intensity remained nearly constant (Fig. 2). We used the near-constant intensity portion of each movie for subsequent image processing and reconstruction. Standard z-stack
Fig. 2.
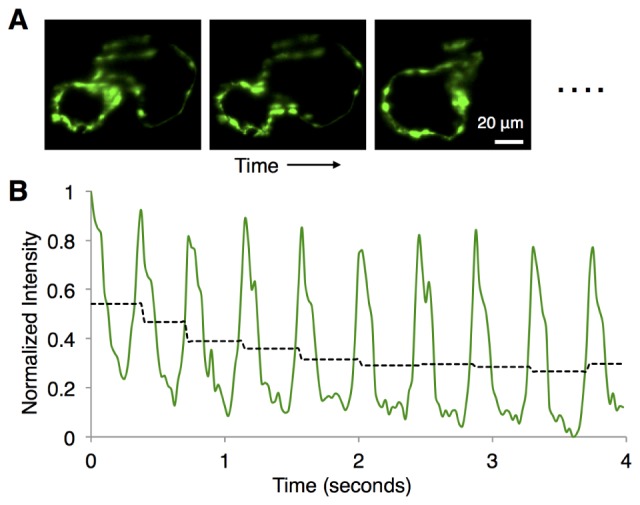
Photobleaching of the sample. For a typical movie of the beating heart at a certain z-depth (A), the normalized signal intensity integrated over the entire image is plotted as a function of time (B). The periodic time-structure of the signal intensity comes from the beating of the heart, as different parts of the heart periodically crossed the imaging focal plane. Dashed black line segments show the signal intensity averaged over the time window of one local maximum to the next, revealing a decrease of ~35% over the first ~1 second, and remaining nearly constant after that. We use the near-constant intensity portion of each movie for subsequent image processing and reconstruction.
3. FlipTrap lines
One of the major challenges in understanding morphogenetic events during embryonic development is the identification of key genetic players and their roles in the 3D context of the developing tissue. Most studies of the genetic basis of morphogenesis utilize in situ hybridization to define the regional distribution of mRNA expression for candidate genes; the location of the encoded proteins are defined through the use of antibody straining, both of which require that the specimen be chemically fixed. To study protein localization in living specimens, over-expression approaches are typically used, through injecting a synthetic RNA that fuses the coding sequence of the protein(s) under study with the sequence of a fluorescent protein such as GFP. To permit proteins to be studied at their normal expression levels and patterns, various gene-trapping approaches have been developed; in these, the sequence encoding a fluorescent protein is randomly inserted into the genome, in some cases creating fusion proteins that provide information on both spatiotemporal expression of the gene [13–15]. Our lab has pioneered the use of “FlipTraps” [11], a gene trap that integrates an internal exon for the fluorescent protein Citrine, which in some cases generates full-length fluorescent fusion proteins (Fig. 1(D)). Coupled with 2p-SPIM, usage of the FlipTrap lines that express Citrine in the heart allows us for the first time to visualize protein expression at endogenous levels in a beating heart.
Adult fish and embryos were raised and maintained as described in [16] with recommendations in the Guide for the Care and Use of Laboratory Animals at the California Institute of Technology. The protocol was approved by Institutional Animal Care and Use Committee (IACUC) (Permit Number: 1227, Caltech). Wild-type embryos were obtained from AB strains. Here we employ two FlipTrap lines, Gt(tpm4-citrine)ct31a and Gt(ctnna-citrine)ct3a, that were obtained from a previously described screen [11]. Transgenic line Tg(gata5:dsRed) was obtained from Zebrafish International Resource Center (ZIRC, Eugene, OR), and line Tg(kdrl:eGFP) was provided by the Stainier lab. Embryos obtained from crossing appropriate adult lines were raised in Egg Water (60 µg/ml of stock salts in distilled water) at 28.5°C with addition of 0.003% (w/v) 1-phenyl-2-thiourea (PTU) at around 18 hpf to reduce pigmentation [16].
4. Sample preparation
The samples were immobilized in 0.85mm-inner-diameter glass capillaries (Wiretrol I, Drummond) with 1% UltraPure Low Melting Point Agarose (Catalog No. 16520-050, Invitrogen) solution prepared in 30% Danieau (17.4mM NaCl, 210µM KCl, 120µM MgSO4•7H2O, 180µM Ca(NO3)2, 0.1mM HEPES buffer in water, pH 7.6) with 0.003% PTU and 0.01% (100mg/L) Tricaine. The embryos were transferred to the agarose solution, and gently swirled to bring them into direct contact with the agarose, and then pulled into the capillary with a stainless steel plunger (VWR). Following solidification of agarose at room temperature (1-2 minutes), the capillary was transferred to the sample chamber filled with 30% Danieau solution and 0.01% Tricaine, at 24.0°C. After being mounted in the imaging chamber, the embryo embedded in agarose, was extruded out of the capillary and allowed to equilibrate for 15 minutes before imaging. The extrusion allows full optical access for imaging without lensing effects/reflections from light passing through the glass capillary.
5. Imaging setup characterization
We estimated the resolution performance of our imaging setup in both 1p- and 2p-SPIM modes, by imaging 170-nm fluorescent beads (Invitrogen) embedded in 1.5% agarose (Fig. 3(A)). The light sheet thickness was adjusted for the 1p and 2p laser light to achieve the same axial resolution of ~1.5 µm full width half maximum (FWHM). Lateral resolution was found to be ~0.5 µm FWHM for both imaging modes, as expected from the image pixel size of 260 nm and Nyquist’s criterion (which states that the effective resolution is at best equal to 2 times the pixel size). Note that to make full use of the detection NA of ~1, the pixel size would have to be smaller than ~140 µm. For imaging the cardiac samples, to enhance signal to noise ratio (SNR) and reduce image size to maximize imaging speed, we used pixel size of 0.520 or 0.635 µm, which then should yield lateral resolution of ~1 to 1.3 µm. Summary of imaging parameters of all presented data is provided in Table 1 (in Appendix).
Fig. 3.

Characterization of imaging performance of 1p-SPIM and 2p-SPIM. (A) Resolution estimates from imaging 170nm-diameter fluorescent beads embedded in agarose. Images show the maximum intensity projection (MIP) along the xy and xz planes, for the same bead imaged by the two modalities. Plot shows the signal line profiles along the axial z-axis (solid line) and lateral x-axis (dashed line) over the bead images, showing FWHM axial resolution of ~1.5 µm and lateral resolution of ~0.5 µm. (B, C) Comparison of penetration depth of 1p-SPIM and 2p-SPIM in imaging the heart of a 60-hpf transgenic zebrafish, Tg(kdrl:GFP) s843, where the endocardium cell layer is labeled with GFP. The heart beating was stopped with a high concentration of anesthetic, allowing conventional z-stack imaging to capture the 3D structure. (B) MIPs of the 2p-SPIM data set along the three principal Cartesian planes are shown to aid the visualization of the anatomy of the heart. (C) Orthogonal views of the 3D data sets, showing single image slices at particular planes (designated by dashed green lines in the central image panels), to demonstrate the penetration depth of 1p-SPIM and 2p-SPIM. 3D images were taken with either uni-directional (light sheet coming from side 1 or side 2) or bidirectional (light sheets coming from both sides simultaneously) excitation. To facilitate fair comparison between the two imaging modalities, best effort was made to show the same planes within the heart for each data set (with the constraint that the live sample did shift slightly between data sets), with linear display contrast minimum and maximum set equal to the minimum and maximum pixel value, respectively, in each of the presented image slice. Overall, we see that 1p-SPIM and 2p-SPIM performs equally well in resolving the endocardium wall closer to the outer surface of the embryo, but 2p-SPIM performs better at capturing cardiac structures deeper inside the animal, as highlighted by structures marked with green arrowheads.
Table 1. Imaging parameters for all the presented data.
| Figure 1, Fig. 2, Fig. 4, Figure 5, Media 1 (4MB, MOV) -7 (6.5MB, MOV) | Figure 3(A) | Figure 3(B), 3(C) | |
|---|---|---|---|
| Embryonic stage (hpf) | 84 | n/a | 60 |
| Camera used | iXon 885 | Zyla | Zyla |
| Use of 0.63X adapter | Yes | No | No |
| Binning | 1 | 1 | 2 |
| Detection objective | 20X (Zeiss) | 25X (Nikon) | 25X (Nikon) |
| Imaged volume (xyz) (voxels), typical | 400 x 500 x150 | 100 x 100 x 100 | 400 x 500 x 170 |
| Lateral (x,y) pixel size (μm) | 0.635 | 0.26 | 0.52 |
| Axial section (z-step) size (μm) | 1 | 0.26 | 1 |
| Excitation wavelength (nm) | 920 | 920 for 2p 488 for 1p | 920 for 2p 488 for 1p |
| Unidirectional laser power (mW) | 125 for 2p | 40 for 2p 0.5 for 1p | 125 for 2p 2 for 1p |
| Continuous frame rate (frames per second) | 70 | n/a | n/a |
| Exposure time (ms) | 12.5 | 40 | 40 |
| Excitation rejection filter | SP 750 | SP 750 | SP 750 |
| Detection band pass filter | BP 525/50 for GFP-labeled samples; BP 535/50 for Citrine-labeled samples. | BP 525/50 | BP 525/50 |
| Number of frames imaged (for 4D reconstruction) | 22500 | n/a | n/a |
With 1p-SPIM and 2p-SPIM modes performing similarly with the ideal beads sample, we compared how they performed in imaging the live zebrafish heart sample. For the comparison to be independent from possible confounding issues related to the reconstruction algorithm, we stopped the heart beating by using a Tricaine concentration of 0.18% in the imaging chamber (while still keeping the embryo alive), to capture 3D structure via a conventional z-stack. Figure 3(B) and 3(C) show the comparison of 1p-SPIM and 2p-SPIM in imaging the non-beating heart. We see that 1p- and 2p-SPIM perform equally well in capturing the endocardium wall structures near the outer surface of the embryo, while 2p-SPIM does better at resolving the endocardium deeper into the heart. The higher penetration depth of 2p-SPIM comes at a cost: fluorescence signal rate for 2p excitation is typically lower than for 1p excitation; thus to maintain similar SNR, 2p-SPIM cannot achieve imaging speeds that could be possible with the 1p counterpart. Note that 1p-SPIM has been used to image the dynamic structure of zebrafish hearts [17]; in our study we chose to use 2p-SPIM for its higher penetration depth, and achieved imaging speed of 70 fps, which has been shown to be sufficiently fast for capturing the beating of the zebrafish heart [8].
We have used Gaussian-beam light sheets for our imaging because of its simplicity; further improvement to the sectioning capability of the 2p excitation could be achieved by utilizing Bessel-beam light sheets [18–20]. Since the side lobes of Bessel beams do not contribute to useful signal generation in 2p excitation, more average laser power would be required for using Bessel- versus Gaussian-beam 2p-SPIM – care then must be exercised to ensure that the extra laser power does not inflict unacceptable photodamage to the live samples under study.
6. Image reconstruction algorithm
For 4D volume reconstruction we used the method detailed in [8,21,22]. The 2p-SPIM imaging pipeline, described above, yielded the raw data consisting of a set of movies, each acquired at a different z-depth in the sample and of duration ~10 cardiac cycles. Briefly, the reconstruction method comprises the following steps. The user selects one of the movies (usually the one at mid-depth) and manually cuts out a time-subset, spanning 1 to 2 cardiac cycles, which serves as the reference movie. Next, the algorithm synchronizes this reference movie to the raw movie at the adjacent z-depth (the test movie); specifically, it determines a warping function (cardiac phase function) that pairs each frame in the reference movie with a similar frame in the test movie under the constraint that movie frames cannot be brought out of order. This warping function is computed by minimizing a cost function that has two terms: a similarity term that favors solutions in which the frames of the warped test movie match the frames of the reference movie; and a regularizing term that penalizes candidate warping functions that would excessively shrink or dilate the time-axis in the warped test movie. The optimal warping function is obtained by solving a minimization problem by use of a dynamic time warping algorithm [22]. Once the optimal warping function is determined, the test movie is interpolated (possibly at time-points in between frames if the local cardiac phase in the reference advances faster than in the test, or leaving out frames if it advances slower). This interpolated test movie then becomes the new reference movie for synchronizing the neighbor at the following z-depth. By proceeding recursively, the entire stack of movies is synchronized. The accuracy of this method is limited by the frame rate (higher frame rates lead to better registration accuracy), any measurement noise, and the recursive nature of the procedure (as discussed in [23] and [24]). Interpolation or mis-registration artifacts are most visible during the fastest phases of the cardiac cycle, and can be partially avoided by acquiring long movies (here ~10 cardiac cycles long) to offer multiple cycles as candidates for matching.
Using a mixed Matlab-C implementation within Matlab 2012b (The Mathworks, Natick, MA) we produced 4D data sets that we rendered with Imaris 7.7.2 (Bitplane AG, Zurich). Synchronization of our data set where each movie had ~200 frames of size 300 x 400 pixels and ~120 z-slices, required approximately 10 hours on a 2.3GHz Intel(R) Xeon(R) processor with 64GB RAM.
7. Results
Our imaging and image reconstruction pipeline not only enables observations of the anatomy but also of the molecular markers with the endogenous protein expression (Fig. 1(E)). The high axial resolution of the images obtained with 2p-SPIM allows us to resolve cellular structures in the beating 84 hpf heart in the FlipTrap line Gt(tpm4-citrine)ct31a/ + in 3D (Fig. 1(E), 4(A), Fig. 5(A) and Media 1 (4MB, MOV) and Media 2 (1.9MB, MOV) ). As seen in Fig. 1(E), cytoplasmic expression of the Citrine fusion protein distinctly highlights the nuclei as darkened regions. The cytoplasmic labeling allows high-resolution visualization of the convoluted spongiform tissue, called trabeculae, in the inner surface of the ventricular myocardium. Iso-surface rendering of the image data further highlights the 3D extent of these structures (Fig. 4(F), Fig. 5(B), Media 3 (2.7MB, MOV) ). Recent studies using 3D time-lapse imaging of beating zebrafish hearts have investigated the initiation of cardiac trabeculation from 60 to 78 hpf, with the novel finding of myocardial protrusions that invade luminally into the trabecular layer to form new cell-cell contacts [25]. Our results demonstrate that 4D imaging of the developing heart can be performed into later stages of trabeculation, as heart structure becomes thicker and more complex, and thus would enable investigation into the further development of the trabeculae.
Fig. 4.

Results of 4D capture of the beating heart. (A-D) Fluorescent images of the heart in different transgenic lines at representative time points during its beating cycle. Images are from a single optical section in (A) Gt(Tpm4-citrine)ct31a /+ and (C) Tg(kdrl:GFP)s843. (B) 3D-rendered images of Tg(kdrl:GFP) s843 heart, at 84 hours post-fertilization (hpf), from two viewing angles demonstrating the resolution achieved with 2p-SPIM. (D) 3D rendered image of an Ct(ctnna-citrine)ct3a/+ embryonic heart at 84 hpf permits visualization of cellular boundaries at two time points in the heart beating cycle. The high image quality allows segmentation of cells through the entire cardiac cycle. (E) Endocardial cells in Tg(kdrl:GFP) s843, when tracked over time (right), return to their initial position (due to periodicity of the heart motion), verifying the accuracy of the reconstruction algorithm. (F) Iso-surface rendering of Gt(Tpm4-citrine)ct31a /+ at 84 hpf and digitally cropped showing the heart tubular topology, shown in 2 different time points during the beating. The surface morphology of the inside of the myocardium is clearly visualized, revealing the trabeculating ventricle. Movie versions of the data sets shown in (A-F) are found in Media 1 (4MB, MOV) , Media 2 (1.9MB, MOV) , Media 3 (2.7MB, MOV) , Media 4 (720.3KB, MOV) , Media 5 (2.1MB, MOV) , Media 6 (2.4MB, MOV) , and Media 7 (6.5MB, MOV) .
Fig. 5.
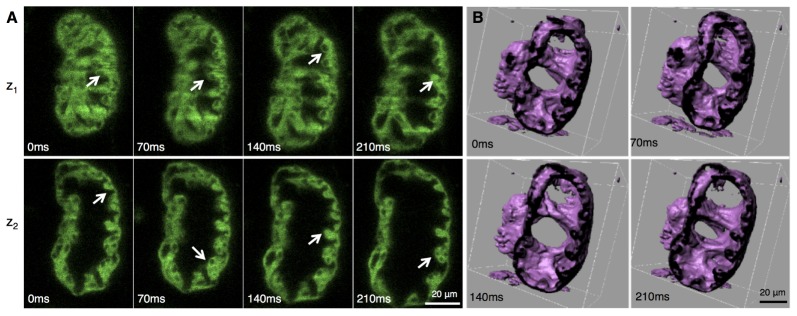
(A) Trabeculating myocardium during cardiac beating cycle. Optical sections of the reconstructed beating heart of the transgenic line Gt(Tpm4-citrine) Gt31a /+, at 84 hpf, shown at 4 distinct time points during the beating cycle, and at 2 different z-sections. The fluorescent fusion construct labels the cytoplasm of myocardial cells. The arrows indicate the spongiform structure (trabeculae) formed by the inner lining of the ventricular myocardium. The nuclei are visible by negative contrast, in different cross-sections. (B) Iso-surface rendering of the same reconstructed data set, shown at 4 distinct phase points and digitally cropped showing that the heart remains topologically equivalent to the cylindroid it started as. Not only can trabeculating ventricle be easily seen in the isosurface view but also the surface morphology of the inside of the myocardium is clearly visualized.
The myocardium generates the contraction of the heartbeat; the endocardium lining the inner surface of the heart conveys this motion to the blood, thereby pumping blood for circulation throughout the body. Applying our imaging and reconstruction pipeline to a transgenic zebrafish line that fluorescently labels the endothelial cells with eGFP, Tg(kdrl:eGFP)s843, allowed us to visualize the motion of the endocardium of the beating heart (Fig. 4(B), 4(C) and Media 4 (720.3KB, MOV) ). We characterized the endocardium motion in 3D by tracking the trajectories of individual endocardial cells during the beating cycle, the trajectories are closed paths with each cell returning to its initial positions due to the periodic nature of the heart beat (Fig. 4(E) and Media 5 (2.1MB, MOV) ).
Beyond the visualization of the myocardial and endocardial tissue layers of the beating heart, the ability to see single cardiac cells undergoing contraction in full 3D provides a needed linkage between the changes of individual cell shapes and the overall motion of the in vivo. Progress in this direction in the context of the beating murine heart is providing new windows into cardiac anatomy and physiology [26]. We report here the imaging of the cellular anatomy of myocardial cells as they undergo contractions using the transgenic line Gt(ctnna-citrine)ct3a/ct3a, which labels the endogenous expression of alpha-catenin and highlights the boundaries of all cells in the developing heart (Fig. 4(D) and Media 6 (2.4MB, MOV) and Media 7 (6.5MB, MOV) ). Our imaging of endogenous proteins expressed at their normal level expression compliments recent imaging efforts [17,25,27] where the fluorescence signal came from increasing the protein levels through the use of non-native over-expression transgenic systems. Even though our labeling is of lower intensity, the reconstructed image quality is high enough for us to carry out cell boundary segmentation of representative myocardial cells in the ventricle, over the entire beating cycle (Fig. 4(D), Media 7 (6.5MB, MOV) ). Further studies into the nature of contractions in the 3D context of developing embryos will facilitate better understanding of forces and motions involved in cardiac function, thereby opening avenues for studying diseases and possible therapeutic interventions.
8. Summary
In our work, we optimized image collection and processing to permit the normal levels of protein expression provided by FlipTraps to be studied. Such studies of both the 4D structure of the developing heart and gene expression patterns at endogenous proteins concentration are of critical importance in the pursuit of understanding of heart development. This molecular and structural imaging with sub-cellular resolution provides a natural system for exploring how the genetic program shapes the development of the heart. This provides the necessary groundwork to begin deciphering the interplay between the dynamic changes in cellular shapes, genetic programs, and cellular physical properties in the development of the vertebrate heart.
Acknowledgments
This work was funded by Rosen Center for Bioengineering, Caltech, Center of Excellence in Genomic Science (Caltech), NIH/NHGRI Grant # P50HR004071 and FaceBase, NIH Grant # U01DE020063. We thank Leigh Ann Fletcher, Colleen Paquette and Andrey Andreev for fish care.
9. Appendix
References and links
- 1.Hove J. R., Köster R. W., Forouhar A. S., Acevedo-Bolton G., Fraser S. E., Gharib M., “Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis,” Nature 421(6919), 172–177 (2003). 10.1038/nature01282 [DOI] [PubMed] [Google Scholar]
- 2.Stainier D. Y., “Zebrafish genetics and vertebrate heart formation,” Nat. Rev. Genet. 2(1), 39–48 (2001). 10.1038/35047564 [DOI] [PubMed] [Google Scholar]
- 3.Forouhar A. S., Liebling M., Hickerson A., Nasiraei-Moghaddam A., Tsai H. J., Hove J. R., Fraser S. E., Dickinson M. E., Gharib M., “The embryonic vertebrate heart tube is a dynamic suction pump,” Science 312(5774), 751–753 (2006). 10.1126/science.1123775 [DOI] [PubMed] [Google Scholar]
- 4.Markl M., Chan F. P., Alley M. T., Wedding K. L., Draney M. T., Elkins C. J., Parker D. W., Wicker R., Taylor C. A., Herfkens R. J., Pelc N. J., “Time-resolved three-dimensional phase-contrast MRI,” J. Magn. Reson. Imaging 17(4), 499–506 (2003). 10.1002/jmri.10272 [DOI] [PubMed] [Google Scholar]
- 5.Jerecic R., Bock M., Nielles-Vallespin S., Wacker C., Bauer W., Schad L. R., “ECG-gated 23Na-MRI of the human heart using a 3D-radial projection technique with ultra-short echo times,” MAGMA 16(6), 297–302 (2004). 10.1007/s10334-004-0038-8 [DOI] [PubMed] [Google Scholar]
- 6.Taylor J. M., Saunter C. D., Love G. D., Girkin J. M., Henderson D. J., Chaudhry B., “Real-time optical gating for three-dimensional beating heart imaging,” J. Biomed. Opt. 16(11), 116021 (2011). 10.1117/1.3652892 [DOI] [PubMed] [Google Scholar]
- 7.Taylor J. M., “Optically gated beating-heart imaging,” Front Physiol 5, 481 (2014). 10.3389/fphys.2014.00481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liebling M., Forouhar A. S., Gharib M., Fraser S. E., Dickinson M. E., “Four-dimensional cardiac imaging in living embryos via postacquisition synchronization of nongated slice sequences,” J. Biomed. Opt. 10(5), 054001 (2005). 10.1117/1.2061567 [DOI] [PubMed] [Google Scholar]
- 9.Ohn J., Tsai H. J., Liebling M., “Joint dynamic imaging of morphogenesis and function in the developing heart,” Organogenesis 5(4), 248–255 (2009). 10.4161/org.5.4.10568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Truong T. V., Supatto W., Koos D. S., Choi J. M., Fraser S. E., “Deep and fast live imaging with two-photon scanned light-sheet microscopy,” Nat. Methods 8(9), 757–760 (2011). 10.1038/nmeth.1652 [DOI] [PubMed] [Google Scholar]
- 11.Trinh A., Hochgreb T., Graham M., Wu D., Ruf-Zamojski F., Jayasena C. S., Saxena A., Hawk R., Gonzalez-Serricchio A., Dixson A., Chow E., Gonzales C., Leung H. Y., Solomon I., Bronner-Fraser M., Megason S. G., Fraser S. E., “A versatile gene trap to visualize and interrogate the function of the vertebrate proteome,” Genes Dev. 25(21), 2306–2320 (2011). 10.1101/gad.174037.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edelstein A. D., Tsuchida M. A., Amodaj N., Pinkard H., Vale R. D., Stuurman N., “Advanced methods of microscope control using μManager software,” J. Biol. Methods 1(2), 10 (2014). 10.14440/jbm.2014.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skarnes W. C., von Melchner H., Wurst W., Hicks G., Nord A. S., Cox T., Young S. G., Ruiz P., Soriano P., Tessier-Lavigne M., Conklin B. R., Stanford W. L., Rossant J., International Gene Trap Consortium , “A public gene trap resource for mouse functional genomics,” Nat. Genet. 36(6), 543–544 (2004). 10.1038/ng0604-543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanford W. L., Cohn J. B., Cordes S. P., “Gene-trap mutagenesis: past, present and beyond,” Nat. Rev. Genet. 2(10), 756–768 (2001). 10.1038/35093548 [DOI] [PubMed] [Google Scholar]
- 15.Lisette A., Maddison L., Wenbiao C., “Conditional gene-trap mutagenesis in Zebrafish methods,” Mol. Biol. 1101, 393–411 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westerfield M., “The zebrafish book,” University of Oregon Press, Eugene, OR: (1994) [Google Scholar]
- 17.Mickoleit M., Schmid B., Weber M., Fahrbach F. O., Hombach S., Reischauer S., Huisken J., “High-resolution reconstruction of the beating zebrafish heart,” Nat. Methods 11(9), 919–922 (2014). 10.1038/nmeth.3037 [DOI] [PubMed] [Google Scholar]
- 18.Planchon T. A., Gao L., Milkie D. E., Davidson M. W., Galbraith J. A., Galbraith C. G., Betzig E., “Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination,” Nat. Methods 8(5), 417–423 (2011). 10.1038/nmeth.1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olarte O. E., Licea-Rodriguez J., Palero J. A., Gualda E. J., Artigas D., Mayer J., Swoger J., Sharpe J., Rocha-Mendoza I., Rangel-Rojo R., Loza-Alvarez P., “Image formation by linear and nonlinear digital scanned light-sheet fluorescence microscopy with Gaussian and Bessel beam profiles,” Biomed. Opt. Express 3(7), 1492–1505 (2012). 10.1364/BOE.3.001492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fahrbach F. O., Gurchenkov V., Alessandri K., Nassoy P., Rohrbach A., “Light-sheet microscopy in thick media using scanned Bessel beams and two-photon fluorescence excitation,” Opt. Express 21(11), 13824–13839 (2013). 10.1364/OE.21.013824 [DOI] [PubMed] [Google Scholar]
- 21.Liebling M., Forouhar A. S., Wolleschensky R., Zimmermann B., Ankerhold R., Fraser S. E., Gharib M., Dickinson M. E., “Rapid three-dimensional imaging and analysis of the beating embryonic heart reveals functional changes during development,” Dev. Dyn. 235(11), 2940–2948 (2006). 10.1002/dvdy.20926 [DOI] [PubMed] [Google Scholar]
- 22.Liebling M., Vermot J., Forouhar A. S., Gharib M., Dickinson M. E., Fraser S. E., “Nonuniform temporal alignment of slice sequences for four-dimensional imaging of cyclically deforming embryonic structures,” Proceedings of the 3rd IEEE International Symposium on Biomedical Imaging: Macro to Nano (ISBI'06), Arlington, VA, USA, April 6–9, 1156–1159 (2006) 10.1109/ISBI.2006.1625128 [DOI] [Google Scholar]
- 23.Bhat S., Larina I. V., Larin K. V., Dickinson M. E., Liebling M., “4D reconstruction of the beating embryonic heart from two orthogonal sets of parallel optical coherence tomography slice-sequences,” IEEE Trans. Med. Imaging 32(3), 578–588 (2013). 10.1109/TMI.2012.2231692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larina I. V., Larin K. V., Dickinson M. E., Liebling M., “Sequential turning acquisition and reconstruction (STAR) method for four-dimensional imaging of cyclically moving structures,” Biomed. Opt. Express 3(3), 650–660 (2012). 10.1364/BOE.3.000650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Staudt D. W., Liu J., Thorn K. S., Stuurman N., Liebling M., Stainier D. Y. R., “High-resolution imaging of cardiomyocyte behavior reveals two distinct steps in ventricular trabeculation,” Development 141(3), 585–593 (2014). 10.1242/dev.098632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aguirre A. D., Vinegoni C., Sebas M., Weissleder R., “Intravital imaging of cardiac function at the single-cell level,” Proc. Natl. Acad. Sci. U.S.A. 111(31), 11257–11262 (2014). 10.1073/pnas.1401316111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahou P., Vermot J., Beaurepaire E., Supatto W., “Multicolor two-photon light-sheet microscopy,” Nat. Methods 11(6), 600–601 (2014). 10.1038/nmeth.2963 [DOI] [PubMed] [Google Scholar]