

Abstract
Lymphangiomas are rare benign neoplasms derived from portions of lymph sacs. They most often occur in children in the cervical region and 90% have manifested by the end of the second year of life. An isolated mediastinal lymphangioma in an adult is an uncommon disease entity. We report the case of a mediastinal lymphangioma in a 29-year-old man presenting as a suprasternal lump, which was resected through a partial sternotomy/hemiclamshell thoracotomy.
Keywords: Lymphangioma, Mediastinum, Adult, Case report
Lymphangiomas are uncommon benign tumours that present primarily in children in the cervical region. We present a rare case of an adult patient with a mediastinal lymphangioma, followed by a concise reflection on the physiopathology and occurrence of lymphangiomas in adults as well as current knowledge about treatment options.
Case History
A 29-year-old man presented with a soft lump just cranially of the sternum, which he had noticed two months earlier. He experienced difficulty in swallowing with gradually progressive retrosternal discomfort. On physical examination, there was a jugular tumour, protruding upwards from behind the sternum. This was very soft on palpation and not moving on swallowing.
On ultrasonography, the mass was seen from the thyroid level extending caudally and retrosternally, and consisting of fluid pockets of varying size. Thoracic computed tomography (CT) showed a mass next to and beneath the thyroid gland extending into the anterior mediastinum as well as into the paratracheal and left paravertebral regions, encasing the aortic arch and branching vessels and the left brachiocephalic vein. No cysts or calcifications were seen (Fig 1).
Figure 1.
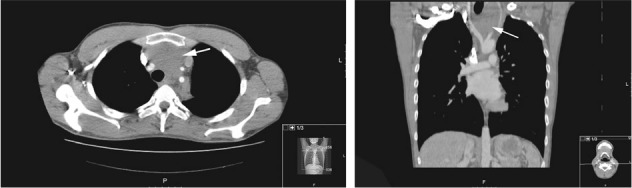
Axial and coronal computed tomography with arrow indicating a large mediastinal mass encasing branching vessels of the aorta
Cytological examination of fluid aspirate from multiple ultrasonography guided punctures was inconclusive, showing erythrocytes and some pigmented macrophages. Incisional surgical biopsy through a jugular incision showed connective tissue with cavities lined with endothelium and focal lymphocytic clusters without Hassall’s bodies (Fig 2). Immunohistochemistry showed positive CD31 and CD34 staining, indicating endothelial origin of tissue. These pathological findings suggested the diagnosis of lymphangioma.
Figure 2.
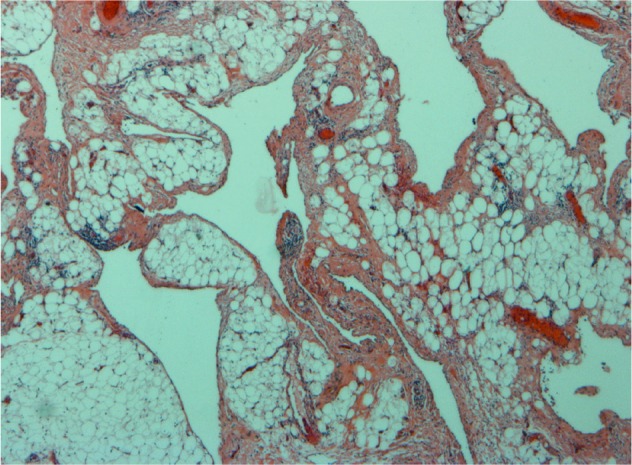
Histology of incisional biopsy showing cavities lined with flattened endothelium and vascular structures
The patient underwent a partial sternotomy/hemiclamshell thoracotomy, and the tumour was dissected sharply of the aortic arch, left brachiocephalic vein, superior vena cava, right subclavian artery, left carotid and subclavian artery. A macroscopically radical resection of the tumour was reached. The left laryngeal nerve could not be identified during surgery because of encasement of the tumour. Pathological examination confirmed the diagnosis of mediastinal lymphangioma.
In the postoperative course, laryngoscopy demonstrated paralysis of the left vocal cord, indicating left recurrent laryngeal nerve damage. There were no clinical or radiological signs of recurrence at the four-year follow-up appointment.
Discussion
Lymphangiomas (also known as cavernous lymphangiomas, cystic hygromas, lymphatic cysts, cystic lymphangiomas, multiloculated hygromas and chylus cysts) most probably represent embryological remnants of lymphatic tissues that either arose from portions of lymph sacs that were sequestered during development or failed to connect to efferent channels.1 Morphologically, they are composed of thin-walled, cystically dilated vascular channels, lined by endothelial cells. Damage from tissue handling during surgery or processing can result in haemorrhage that can make the diagnosis of lymphangioma challenging.
Owing to its derivation from a primitive lymphatic sac, lymphangioma is more common in childhood and typically located cervically. About 90% of lymphangiomas present in the first two years of life.1 The majority appear in the neck (75%) and axillary regions (20%).
There is an increased recognition of lymphangiomas presenting later in life. Acquired or secondary lymphangiomas can develop at sites of chronic lymphatic obstruction, for example after radiotherapy, chronic infection or surgery.2 Still, fewer than 200 cases of lymphangiomas in adults have been reported in the English literature in the 20th century.3 Extracervical locations have been described, including mesenteric, retroperitoneal, orbital, pancreatic and mediastinal sites.3
Thoracic lymphangiomas may remain asymptomatic for many years because of their slow growth and only become apparent when a patient develops problems related to compression of vital structures. Alternatively, they are discovered as incidental findings on radiography. If symptomatic, patients present most commonly with a lump. Suggestive for the diagnosis is detection of a cystic lesion on a chest x-ray or CT. Even if initially asymptomatic, the majority of patients eventually need treatment owing to (progression of) symptoms.
Standard treatment has been surgical excision. Overall recurrence after resection is reported to be between 10% and 50%.4 In a series of 12 cases of mediastinal lymphangiomas, Park et al reported recurrence in 4 out of 5 cases in which initial excision was not complete and no recurrence after radical resection.5 Additionally, Stromberg et al reported curation after partial excision to be only 12%.6
Various alternative treatments for lymphangioma have been attempted including chemotherapy, radiotherapy, sclerosing agents and laser therapy. The efficacy of these treatments is highly variable in incidental reports.
Conclusions
Mediastinal lymphangioma is a rare condition, especially in adults, and extracervical location is even more rare. Only limited data regarding choice and timing of treatment are available. Cure seems most likely after radical resection and early stage surgical intervention therefore seems to be warranted.
Acknowledgement
The authors would like to thank Guy Brutel de la Riviere for his contribution to the histological details in this report.
References
- 1.Hilliard RI, McKendry JB, Phillips MJ. Congenital abnormalities of the lymphatic system: a new clinical classification. Pediatrics 1990; 86: 988–994. [PubMed] [Google Scholar]
- 2.Faul JL, Berry GJ, Colby TV et al Thoracic lymphangiomas, lymphangiectasis, lymphangiomatosis, and lymphatic dysplasia syndrome. Am J Respir Crit Care Med 2000; 161: 1,037–1,046. [DOI] [PubMed] [Google Scholar]
- 3.Kransdorf MJ. Benign soft-tissue tumors in a large referral population. Am J Roentgenol 1995; 164: 395–402. [DOI] [PubMed] [Google Scholar]
- 4.Adams MT, Saltzman B, Perkins JA. Head and neck lymphatic malformation treatment: a systematic review. Otolaryngol Head Neck Surg 2012; 147: 627–639. [DOI] [PubMed] [Google Scholar]
- 5.Park JG, Aubry MC, Godfrey JA, Midthun DE. Mediastinal lymphangioma: Mayo Clinic experience of 25 cases. Mayo Clin Proc 2006; 81: 1,197–1,203. [DOI] [PubMed] [Google Scholar]
- 6.Stromberg BV, Weeks PM, Wray RC. Treatment of cystic hygroma. South Med J 1976; 69: 1,333–1,335. [DOI] [PubMed] [Google Scholar]