

Abstract
Methotrexate (MTX) has been used to treat rheumatoid arthritis at low doses and leukemia at high doses; however, this drug can produce severe side effects. Our hypothesis is that MTX side effects can be attenuated by directing the drug to the target cells (i.e., leukocytes) using cIBR peptide. To test this hypothesis, MTX was conjugated to the N-terminus of cIBR peptide to give MTX-cIBR conjugate. MTX-cIBR (5.0 mg/kg) suppressed joint arthritis in adjuvant arthritis rats and prevented periarticular inflammation and bone resorption of the limb joints. In vitro, the toxicity of MTX-cIBR peptide against Molt-3 T cells was inhibited by anti-LFA-1 antibody and cIBR peptide in a concentration-dependent manner, suggesting that the uptake of MTX-cIBR was partially mediated by LFA-1. Chemical stability studies indicated that MTX-cIBR was most stable at pH 6.0. The MTX portion of MTX-cIBR was unstable under acidic conditions whereas the cIBR portion was unstable under basic conditions. In biological media, MTX-cIBR had short half-lives in rat plasma (44 min) and homogenized rat heart tissue (38 min). This low plasma stability may contribute to the low in vivo efficacy of MTX-cIBR; therefore, there is a need to design a more stable conjugate to improve the in vivo efficacy.
Keywords: targeted delivery, ICAM-1, cIBR, MTX-cIBR, LFA-1, methotrexate, in vitro, in vivo, chemical stability, enzymatic stability, arthritis
INTRODUCTION
ICAM-1/LFA-1 interactions play an important role in the process of T-cell activation and vascular extravasation of leukocytes through vascular endothelium for recruitment to sites of infection and inflammation.1,2 Inhibition of ICAM-1/LFA-1 interactions by antibodies and peptides has been shown to suppress autoimmune diseases as well as allograft rejections.1–3 cIBR peptide (cyclo(1,12)PenPRGGSVLVTGC) derived from the domain-1 (D1) of ICAM-1 binds to the I-domain of LFA-1 and inhibits homotypic and heterotypic T-cell adhesion.1,4–9 Uptake of cIBR peptide by T cells occurs via receptor-mediated endocytosis;4 thus, it is an attractive molecule for selective delivery of cytotoxic drugs to leukocytes. The uptake of cIBR was determined using FITC conjugate with cIBR (FITC-cIBR), and FITC-cIBR was internalized by Molt-3 and HL-60 cells via receptor-mediated endocytosis.4,10,11 As a negative control, FITC-cIBR was not internalized by human umbilical vein endothelial cells (HUVEC), which do not express lymphocyte function-associated antigen-1 (LFA-1) receptors.
Methotrexate (MTX) is used to treat leukemia at high doses12,13 and autoimmune diseases such as rheumatoid arthritis at low doses.14,15 The normal cellular uptake of MTX is mediated by reduced folate carriers (RFC) and membrane folate-binding proteins (mFBP); thus, these cellular uptake processes contribute to non-selectivity for different cells, which causes side effects.16 MTX may also generate drug resistance due to (a) changes in RFC expression level and altered transport kinetics, and (b) increased dihydrofolate reductase (DHFR) expression with continued use of the drug.12,17–19 Several other reasons for MTX resistance have also been proposed in the literature.18,20 MTX has been effectively delivered to cells using its conjugates with different carrier molecules, including peptides,20 proteins,21,22 and polymers.23,24 Using target receptors other than RFC for cellular entry, the conjugate may possibly avoid cellular drug resistance. Normally, MTX is conjugated to the carrier peptides or proteins via the γ-carboxylic acid of MTX to the amino group in the carrier because the α-carboxylic acid group of MTX is necessary for binding to DHFR.4
In this work, MTX was conjugated to the cIBR peptide to produce MTX-cIBR (Figure 1A) for selective delivery of MTX to leukocytes for potential treatment of rheumatoid arthritis and leukemia. The hypothesis is that MTX-cIBR is directed toward LFA-1-expressing leukocytes over cells that do not express LFA-1, so the conjugate would have lower side effects than MTX alone. In vivo activity of the conjugate was determined in the rat adjuvant arthritic model. To study the in vitro selectivity, the activity of the conjugate to kill human Molt-3 T-cells was assessed in the presence and absence of cIBR peptide and anti-LFA-1 antibodies. Chemical stability study of MTX-cIBR was conducted under accelerated conditions at different pH values. Finally, the enzymatic stability determination was carried out in plasma and liver homogenates to determine the dosing regimen during the in vivo study and for future optimization of the physicochemical properties of the conjugate.
Figure 1.
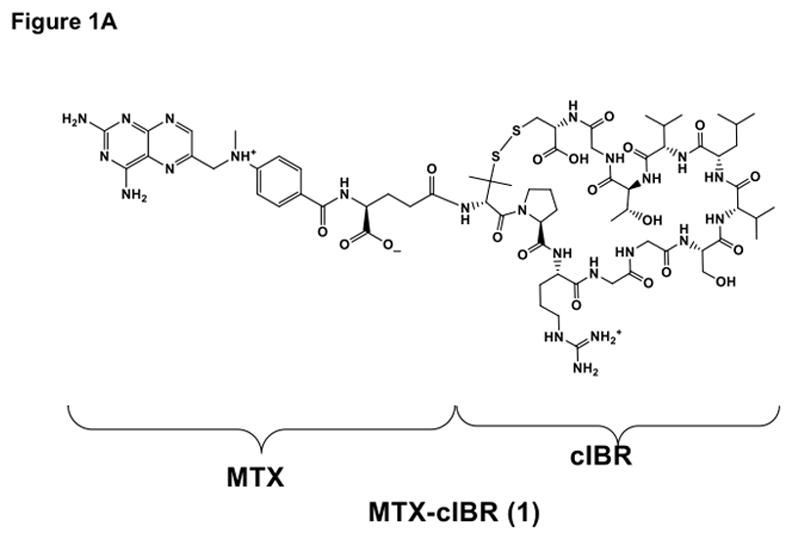
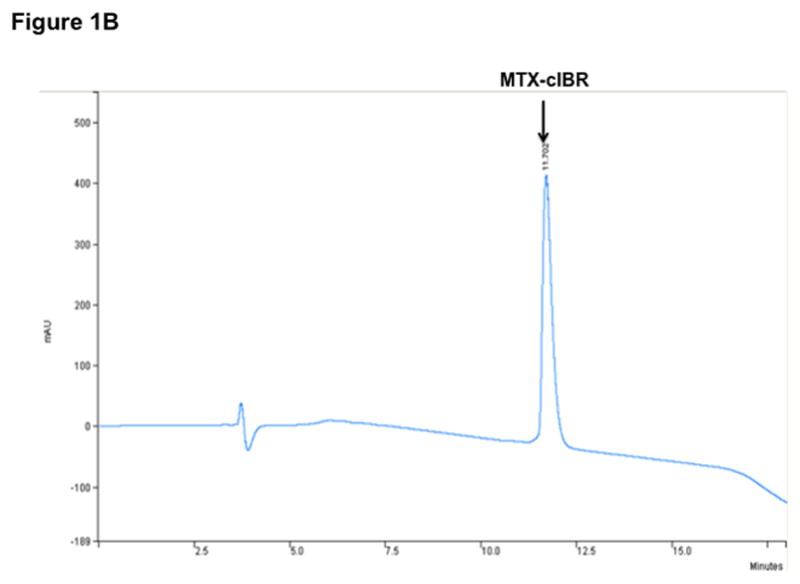
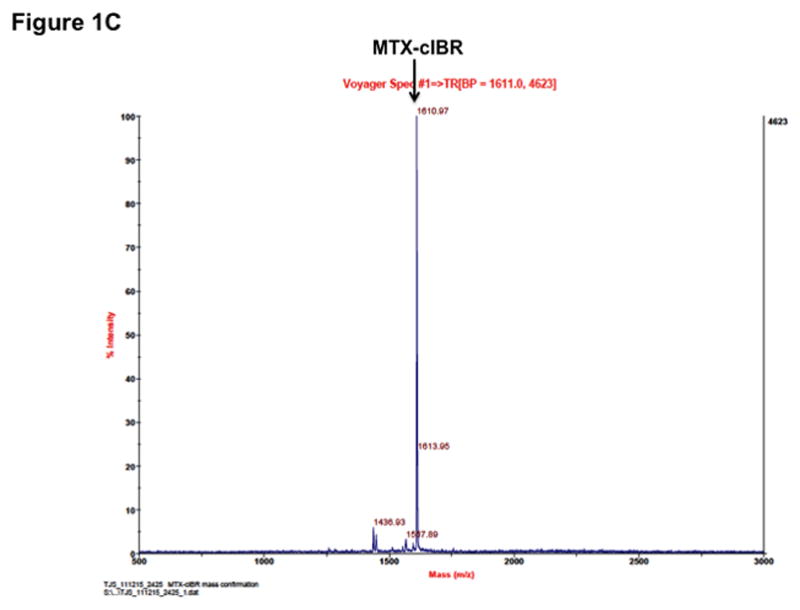
Structure and characterization of MTX-cIBR: (A) chemical structure of MTX-cIBR, (B) analytical reversed-phase HPLC chromatogram of pure MTX-cIBR, and (C) mass spectrometry spectrum of pure MTX-cIBR.
EXPERIMENTAL METHODS
Materials
All reagents for peptide synthesis and stability study were purchased from Peptides International (Louisville, KY), Advanced ChemTech (Louisville, KY), Applied Biosystems (Foster City, CA), Sigma (St. Louis, MO), and Fisher Chemical (Fair Lawn, NJ). All solvents for peptide synthesis were of analytical grade. Amber-colored 1.0 mL ampoules were from Wheaton Science Products (Millville, NJ). FITC-labeled and unlabeled monoclonal anti-human CD11a antibody (clone 38) was purchased from Ancell (Bayport, MN). MOLT-3 cells, a leukemia-derived human T-cell line, were purchased from ATCC (Rockville, MD). These cells were propagated in RPMI-1640 medium (Sigma, St. Louis, MO) containing 10% v/v fetal bovine serum and penicillin/streptomycin (100 mg/L medium) and incubated at 37 °C with 95% humidity and 5% CO2.
Syntheses of cIBR and MTX-cIBR
The synthesis of cIBR peptide (cyclo-1,12-PenPRGGSVLVTGC) was accomplished using an Fmoc solid-phase strategy. The crude product was purified using a semi-preparative HPLC equipped with C18 reversed-phase column. The molecular weight of cIBR peptide was determined by electrospray ionization mass spectrometry (M + 1 = 1174.5 amu).
The N-terminus of cIBR was conjugated to MTX-(OtBu) via its γ-carboxylic acid group on the Glu residue to make MTX-(OtBu)-cIBR. In this case, MTX-(OtBu) was synthesized by adding a solution of L-Glu(OH)-OtBu (0.132 mmol) in 5 mL of DMF dropwise into a mixture of 4-[N-(2,4-diamino-6-pteridinylmethyl)-N-methylamino] benzoic acid hemihydrochloride dihydrate (0.132 mmol), HBTU (0.132 mmol), and DIEA (0.132 mmol) in 5 mL of DMF. The reaction mixture was stirred under nitrogen atmosphere at room temperature for 2 h. The crude product was concentrated under reduced pressure to yield MTX-(OtBu) as yellow oil. The product was further purified by semi-preparative HPLC to give 87% yield. Mass spectroscopy (FAB) analysis indicated that the product had the expected MW of 511 (M+1).
To make MTX-cIBR, a solution of cIBR peptide (0.098 mmol) was added dropwise to a solution mixture of HBTU (0.098 mmol), MTX-(OtBu) (0.098 mmol), and DIEA (0.098 mmol) in 5 mL of DMF followed by stirring under nitrogen for 3 h at room temperature. The reaction mixture was concentrated under reduced pressure to give an oily residue of MTX(OtBu)-cIBR. The resulting residue was dissolved in 3 mL of CH2Cl2 followed by addition of 3 mL of TFA. After the solution was stirred for 1 h at room temperature, the solvents were evaporated and the crude product was purified by semi-preparative HPLC using a C-18 column to give MTX-cIBR in 60–70% yield. The final products were analyzed by analytical HPLC (Figure 1B) and mass spectrometry (M + 1 = 1611, Figure 1C).
Suppression of RA in Rat Adjuvant Model by MTX-cIBR
The in vivo activity of MTX-cIBR was also determined in the rat adjuvant arthritis model via intravenous (i.v.) injections. Animals (7/group for adjuvant, 4/group for normal controls) were anesthetized with isoflurane and injected with 100 μL of Freund’s complete adjuvant (FCA, Sigma)/lipoidal amine (LA) at the base of the tail on day 0. MTX-cIBR (0.05, 0.2, 1, and 5 mg/kg) and MTX (0.05 mg/kg) were dosed i.v. once a day from day 0 to day 14. The animals were weighed on days 0, 4, 8, 9, 10, 11, 12, 13, and 14, at which times the dose volumes were adjusted accordingly. The ankle joints were measured with a caliper on day 7 prior to the swelling onset but after establishment of systemic disease. The ankles were measured again on days 8, 9, 10, 11, 12, 13, and 14. Finally, the body weights were measured on day 14. The animals were euthanized on day 14; hind paws, liver, and spleen were removed and weighed. Paws and spleen were placed in formalin and then processed for Hematoxylin and Eosin (H&E) staining for microscopy evaluation. All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC). Some of the animal and pathology studies were done through a sub-contract with Bolder BioPath Inc., Boulder, CO following the approved institutional animal protocols.
MTX-cIBR Uptake by LFA-1 Receptor
The involvement of LFA-1 in the uptake of MTX-cIBR was studied by evaluating its toxicity in Molt-3 T-cells in the presence of increasing concentrations of cIBR peptide (10, 100, 1000 μM) or an anti-LFA-1 antibody (clone 38) at 40 and 80 μL/mL. Molt-3 T-cells (2 × 104 cell/mL) were incubated in a 96-well microtiter plate in the presence of either cIBR peptide or an anti-LFA-1 antibody (clone 38) at various concentrations. As a control, some cells were left untreated. The MTX-cIBR was then added to each well to a final concentration of 1 μM. As a reference for minimal metabolic activity, 10 mM of the succinate dehydrogenase inhibitor iodoacetamide (IAA) was added to untreated wells. After 72 h of continuous exposure, the relative number of viable cells was determined using an MTT assay. After 4 h of incubation with a 5.0 mg/mL solution of MTT, the contents of each well were transferred to microcentrifuge tubes. The tubes were spun to pellet the cells, and the supernatant was carefully removed. The formazan crystals were dissolved in 200 μL of 0.04 N HCl in isopropanol; the tubes were sonicated for 5 min to completely dissolve the crystals and then re-centrifuged to pelletize the cell debris. 100 μL aliquots of the supernatant solutions were removed and transferred to a 96-well microtiter plate. Then, a UV-Vis plate reader was used to measure the optical density at 570 nm.
Binding of MTX-cIBR to LFA-1 in response to LFA-1 activation was evaluated. Cells were activated with 10% v/v phorbol 12-myristate-13-acetate (PMA)-containing medium to a final concentration of 2 μM PMA and incubated for 16 h. 200 μL aliquots of Molt-3 T-cells (1 × 106 cells/mL in 1% BSA/PBS) were added to 48-well plates and treated with either cIBR peptide or MTX-cIBR at concentrations of 1, 10, or 100 μM for 45 min at 4 °C. The cells were then washed to remove unbound cIBR or MTX-cIBR. T-cells were centrifuged for 3 min at 1800 rpm, the supernatant was decanted by flicking off excess liquid, and the cells were re-suspended in 500 μL of PBS. The cells were then re-centrifuged, supernatant was removed, and cells were re-suspended again in 150 μL of 1% BSA/PBS. Next, 50 μL of an FITC-labeled anti-CD11a antibody (clone 38, 10 μg/L) was added; the cells were incubated for 45 min at 4 °C followed by washing. After 45-min incubation with FITC-labeled antibody, cell samples were transferred to Eppendorf tubes and centrifuged at 3000 g for 3 min. The supernatant was decanted, and the pellet was washed twice with 10 mM HEPES/PBS. The cells were then fixed with ice-cold 2% w/v paraformaldehyde/PBS for 20 min. The cells were analyzed using a Becton-Dickinson FACScan flow cytometer with 3.2.1f1 software for data analysis and acquisition. Reduction in binding of the FITC-labeled antibody was calculated as a fraction of fluorescence remaining after incubation with cIBR or MTX-cIBR compared to the fluorescence of FITC-antibody binding untreated cells.
Chemical Stability Study of MTX-cIBR
The stability of MTX-cIBR was determined at 70 °C. 100 μM of MTX-cIBR was prepared in suitable buffer and 130 μL of conjugate solution was transferred to 1 mL amber, flame-sealed ampoules. Triplicate samples were removed from the oven and analyzed immediately at certain time points. A calibration curve was prepared for the conjugate, and the concentration range for the calibration curve was maintained wide enough to follow two half-lives for the degradation of the conjugate. For the calibration curve, each concentration value was determined in triplicate.
All samples were analyzed for degradation using a Hewlett Packard 1050 reversed-phase HPLC system with UV absorbance detection (220 nm). A Zorbax C18 column (2.1 × 50 mm, 5 μm particle size) was used as the stationary phase, and the column temperature was maintained constant throughout the analysis. Samples were removed from the oven at each time point and transferred to an HPLC sample vial. 10 μL of the sample was injected each time using a gradient program that started with 100% solvent A (94.9% water, 5% acetonitrile and 0.1% formic acid) and was increased to 16% solvent B (94.9% acetonitrile, 5% water and 0.1% formic acid) over 12 min followed by a ramp to increase the composition of solvent B to 100% by 14 min. The system was maintained at 100% B for 2 min followed by a gradient change to 100% A over 4 min. All the concentrations were converted to Log concentration (Log C). Mean and standard deviation were measured for each time point for each pH value. Plots of Log C with time (h) were generated in SigmaPlot (version 9.01, Systat Software, Inc., Chicago, IL) and rate constants for degradation and half-lives were calculated from the plots. A plot of kobs with different pH values was used to create a pH rate profile for the conjugate at 70 °C. Ion-product of water (Kw) was calculated at 70 °C for the pH rate profile determination.
Degradation products were identified using a Q-Tof (Waters Micromass, Milford, MA) system equipped with electrospray ionization capability. For data collection and analysis, Masslynx™ (version 4.0) software was used. For the LC analysis, a Zorbax C18 column (5 cm × 1 mm, 3.5 μm, Micro-Tech Scientific, Vista, CA) was used. Mobile phase composition was kept at 99% A (98.92% water, 1% acetonitrile, and 0.08% formic acid) for 1 min (flow rate 0.11 mL/min), reduced to 90% A over the next 1 min and to 80% A over the next 6 min (flow rate 0.11 mL/min). Solvent composition was changed to 95% B (98.94% acetonitrile, 1% water, and 0.06% formic acid) over the next 7 min to flush the column (flow rate 0.15 mL/min), and then the composition was changed back to 99% A over 2 min (flow rate 0.13 mL/min). The injection volume for each sample was kept at 5 μL.
Stability of MTX-cIBR in Rat Plasma
The stability of MTX-cIBR was evaluated in rat plasma by preparing a stock solution of 100 μM MTX-cIBR in DMSO. 10 μL of stock solution was mixed with 990 μL of rat plasma and incubated at 37 °C in a water bath (Precision reciprocal shaking bath, Model 2870, Thermo Electron Corporation, Waltham, MA). 100 μL samples were drawn after 0, 10, 20, 30, 45, 60, 90, and 120 min of incubation. Samples were immediately treated with 500 μL of chilled acetonitrile containing internal standard alprenolol (1 μg/mL). All samples were vortexed for 10 s and incubated on ice, followed by centrifugation at 14,000 rpm for 15 min (Eppendorf Refrigerated Microcentrifuge, Model 5417R). Aliquots (100 μL) of the supernatant were injected into the mass spectrometer. The standard curve was prepared by adding known concentrations of MTX-cIBR solution to rat plasma followed by immediate addition of cold acetonitrile containing internal standard. Solutions for the standard curve were treated in the same way as the samples. Each time point was measured in triplicate, and the data are represented as percent concentration remaining at each time point.
Stability of MTX-cIBR in Homogenized Rat Heart
Tissue stability of MTX-cIBR was evaluated by incubating the conjugate with homogenized rat heart. 6.28 mL of Dulbecco’s PBS (1X) solution without Ca2+ and Mg2+ (Mediatech, Inc. Cellgro®, Manassas, VA) was added to 3.18 g of rat heart tissue in a test tube. The tissues in different tubes were homogenized using a sonifier (Branson Sonifier, Branson Ultrasonic Corp., Danbury, CT) for 3–5 min. From the stock solution of MTX-cIBR used in the plasma stability studies, 10 μL of the compound solution was mixed with 990 μL of tissue homogenate and incubated at 37 °C in a water bath. At different time points (0, 10, 20, 30, 45, 60, 90, 120 min) after incubation, triplicate samples were drawn and treated in the same way as described above for plasma stability studies.
LC-MS Analysis of MTX-cIBR Degradation in Biological Matrices
An Integrated Cohesive Technologies LX-2 series liquid chromatography system (comprised of pump, autosampler, valve interface module) coupled with a 4000 Q Trap triple quadrupole mass spectrometer (Applied Biosystems MDS-SCIEX) was used to quantify the compound extracted from the biological matrices. The mass spectrometer was equipped with a Turbo ion-spray ionization source, and was operated in the positive mode. Samples were detected by multiple-reaction monitoring (MRM) scan mode. For chromatographic separation, an Agilent Technologies Eclipse XDB-C18 column (4.6 × 15 mm, 3.5 μm particle diameter) was used with solvent A (10 mM ammonium formate buffer at pH 3.5) and solvent B (acetonitrile with 0.1% formic acid) with a flow rate of 1.0 mL/min. A combination of linear and step gradients was used for the liquid chromatography separation with the following solvent composition changes: (a) 90% A to 10% A over 17 sec, (b) 10% A maintained for 2 min, (c) changed to 90% A over 50 s, and (d) maintained at 90% A for 17 s.
RESULTS
In Vivo Efficacy of MTX-cIBR in Arthritis Animals
The in vivo efficacies of MTX-cIBR at different doses (0.05, 0.2, 1.0, and 5.0 mg/kg) were determined using the rat adjuvant arthritis model generated via intravenous (i.v.) injections compared to vehicle-treated arthritis rats, 0.05 mg/kg MTX-treated arthritis rats, and normal control rats. The efficacy of MTX-cIBR in arthritis animals was monitored over 14 days by comparing the observed parameters such as ankle diameter, paw weight, inflammation, and bone resorption (Table 1). The paw weight of the arthritis animals injected with vehicle was approximately double that of the normal animals at day 14. There was a significant reduction (73% reduction) in the paw weight in rats treated with 5 mg/kg of MTX-cIBR compared to vehicle-treated animals (0% reduction as control). Only a small reduction in paw weight (7–11%) was observed on the animals treated with 0.5–1.0 mg/kg of MTX-cIBR (Table 1). Injection of 5.0 mg/kg MTX-cIBR significantly suppressed the increase in ankle diameter over 14 days compared to animals treated with vehicle (Figure 2, Table 1). For lower doses (0.05, 0.2, and 1.0 mg/kg), MTX-cIBR-treated animals had a significant reduction (17–23%) in ankle diameter compared to that of vehicle-treated animals, especially on days 13 and 14 (Figure 2, Table 1). Animals treated with 5.0 mg/kg of MTX-cIBR had a 56% reduction in joint inflammation and a 71% reduction in bone resorption scores compared to vehicle-treated animals (Table 1); at lower doses of MTX-cIBR, the differences in inflammation and bone resorption were not observed (Table 1).
Table 1.
The effect of MTX-cIBR on suppressing arthritis in the limbs of rats using different parameters at Day 14.
| Treatment | Paw Weight ± SE (g) | AUC Paw Ankle Diameter ± SE | Inflammation Score ± SE | Bone Resorption Score ± SE |
|---|---|---|---|---|
| Normal + Vehicle | 1.7761 ± 0.0262 (100%) * | 1.927 ± 0.005 (100%) * | 0.00 ± 0.0 (100%) * | 0.0 ± 0.0 (100%) * |
| Arthritis + Vehicle | 3.5825 ± 0.0866 (0%) | 2.420 ± 0.028 (0%) | 5.0 ± 0.0 (0%) | 3.9 ± 0.4 (0%) |
| Arthritis + 0.05 mg/kg MTX-cIBR | 3.3780 ± 0.1104 (11%) | 2.305 ± 0.036 (23%) ** | 4.6 ± 0.2 (9%) ** | 4.0 ± 0.4 (−1%) |
| Arthritis + 0.2 mg/kg MTX-cIBR | 3.4321 ± 0.1212 (8%) | 2.336 ± 0.035 (17%) | 4.7 ± 0.2 (6%) | 4.5 ± 0.2 (−15%) |
| Arthritis + 1 mg/kg MTX-cIBR | 3.4582 ± 0.0850 (7%) | 2.309 ± 0.030 (22%)** | 4.9 ± 0.1 (1%) | 3.6 ± 0.4 (8%) |
| Arthritis + 5 mg/kg MTX-cIBR | 2.2680± 0.2227 (73%) * | 2.050 ± 0.049 (75%) ** | 2.2 ± 0.5 (56%) * | 1.1 ± 0.5 (71%) * |
| Arthritis + 0.05 mg/kg MTX | 3.5790 ± 0.0877 (0%) | 2.388 ± 0.028 (6%) | 4.9 ± 0.1 (3%) | 3.1 ± 0.4 (22%) |
p-value < 0.01 when compared to Arthritis + Vehicle.
p-value < 0.05 when compared to Arthritis + Vehicle.
Figure 2.

The efficacy of MTX-cIBR at different concentrations (0.05–5.0 mg/kg) in suppressing the increase in ankle diameters of arthritis rats compared to arthritis rats treated with vehicle, 0.05 mg/kg MTX, and normal rats. The results were from measuring two paws per rat with n = 7 for arthritis rats and n = 4 for normal rats. * = p-value < 0.05 when compared to vehicle-treated arthritis rats.
The effect of MTX-cIBR was also evaluated by ankle histology compared to controls (Figure 3). The ankle histology of a normal rat showed normal synovium (S) and periarticular tissue, articular cartilage (blue arrows), epiphyseal bone (E), and distal tibia growth plate (black arrow) (Figure 3A). At a magnification of 100x, the distal tibia has a normal epiphyseal bone (E) and growth plate (black arrow) (Figure 3B). The ankle of a vehicle-treated rat had severe synovitis (S) and periarticular inflammation due to edema and neutrophil infiltration (Figures 3C and 3D). The black arrow indicates the presence of bone resorption across the physis with fragments of bone (green arrow) undergoing resorption in the medullary cavity (Figure 3D). Minor bone resorption was present in epiphyseal bone (E) while the articular cartilage was unaffected (blue arrow). The growth plate (Figure 3D) had bone loss associated with pyogranulomatous marrow inflammation and numerous osteoclasts. Fragmented bone trabeculae (green arrow) had undergone osteoclastic destruction with only minor changes in eiphyseal (E) bone.
Figure 3.
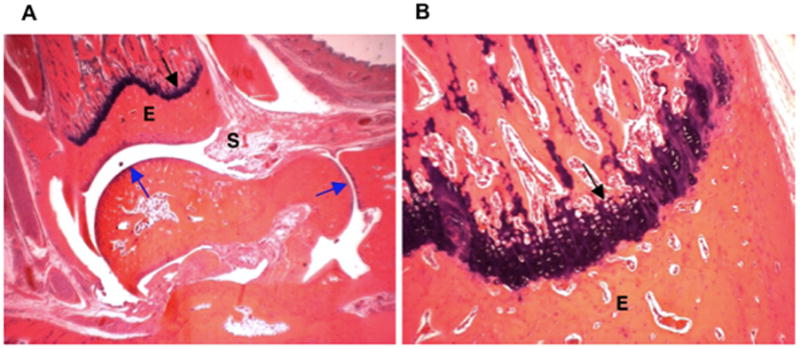
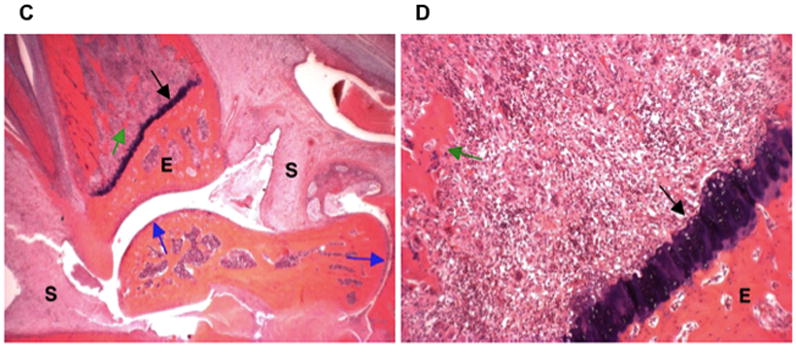
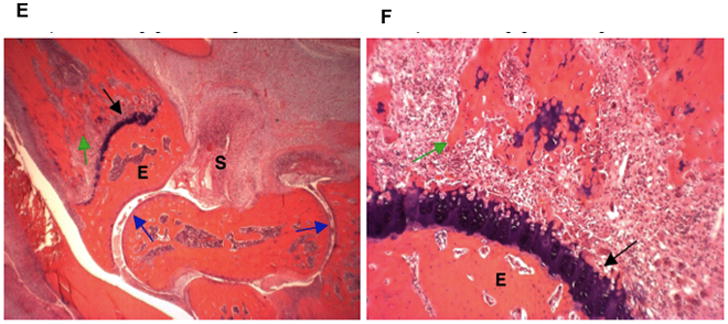
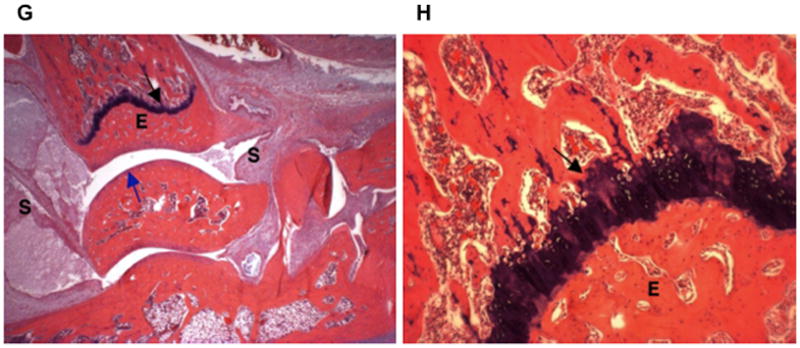
Histology photomicrographs of the left ankle joints at low (16x) and high (100x) magnifications. The photomicrographs of a representative ankle joint of a normal rat at (A) 16x and (B) 100x; a diseased rat treated with vehicle at (C) 16x and (D) 100x; a disease rat treated with 0.05 mg/kg MTX at (E) 16x and (F) 100x; a diseased rat treated with 5 mg/kg MTX-cIBR at (G) 16x and (H) 100x. Synovium (S); articular cartilage (blue arrows); fragment of bone (green arrow); epiphyseal bone (E); and distal tibial growth plate (black arrow).
The ankle joints of animals treated with MTX (0.05 mg/kg) had severe synovitis (S) and periarticular inflammation due to edema and neutrophil infiltration (Figures 3E and 3F). Moderate bone resorption was present across the physis (black arrow), with fragments of bone (green arrow) undergoing resorption in the medullary cavity (Figure 3F). Minor bone resorption also occurred in epiphyseal bone (E) with unaffected articular cartilage (blue arrows). Moderate bone loss was found associated with pyogranulomatous marrow inflammation and numerous osteoclasts. Some fragmented bone trabeculae (green arrow) were undergoing osteoclastic destruction, but maintained connectivity (Figure 3F).
The ankle of a rat treated with 5 mg/kg of MTX-cIBR had moderate synovitis (S) and periarticular inflammation due to edema and neutrophil infiltration (Figure 3G). No bone resorption was observed at the physis (black arrow), and trabeculae filled the medullary cavity. No bone resorption was present in epiphyseal bone (E), and the articular cartilage (blue arrow) was unaffected. At high magnification (100x, Figure 3H), the growth plate showed normal bone and growth plate cartilage.
Effects of MTX-cIBR on Body Weight and Spleen and Liver Inflammation
The effects of MTX-cIBR on the animal body, spleen, and liver weights were measured. As expected, the normal animals gained weight throughout the study. The arthritis animals treated with 5.0 mg/kg MTX-cIBR gained weight on days 0–8 and their bodyweights remained constant after day 8 (Figure 4). In contrast, animals treated with vehicle, MTX (0.05 mg/kg), and low doses of MTX-cIBR (0.05–1.0 mg/kg) lost weight after day 8.
Figure 4.

The effect of increasing doses of MTX-cIBR (0.05–5.0 mg/kg) on the increase in animal bodyweight compared to controls (i.e., vehicle and 0.05 mg/kg MTX). The animals treated with 5.0 mg/kg MTX-cIBR maintained their bodyweight significantly compared to vehicle-treated animals. n = 7 for arthritis rats and n = 4 for normal rats. * = p-value < 0.05 when compared to vehicle-treated arthritis rats.
MTX-cIBR had an effect on the spleens of treated rats. The spleen of a normal rat had large areas of lymphoid white pulp (W) and surrounding red pulp (R) with reticuloendothelial cells; small fuci of extramedullary hematopoiesis (EMH) were not visible at this magnification (Figure 5A). On the other hand, the spleen of an arthritis rat treated with vehicle had a large area of pyogranulomatous inflammation (black outline), decreased lymphoid white pulp (W), and increased EMH as red pulp (R) (Figure 5B). Treatment of the arthritis rat with 0.05 mg/kg MTX (Figure 5C) and 0.05 mg/kg of MTX-cIBR (data not shown) indicated that the spleen had multi-focal small areas of pyogranulomatous inflammation (black tracing) and mildly decreased white pulp (W) due to lymphoid atrophy with moderate EMH in the red pulp (R). It was interesting to find that the spleens of rats treated with of MTX-cIBR at a dose of 5 mg/kg (Figure 5D) as well as 1.0 and 0.2 mg/kg (data not shown) had no areas of pyogranulamatous inflammation. The spleens of rats treated with 5 mg/kg had normal white pulp (W) and red pulp (R). The 0.2 and 1 mg/kg dose-treated rats had spleens with mildly decreased white pulp (W) due to lymphoid atrophy and moderate extramedullary hematopoiesis in the red pulp (R) (data not shown).
Figure 5.
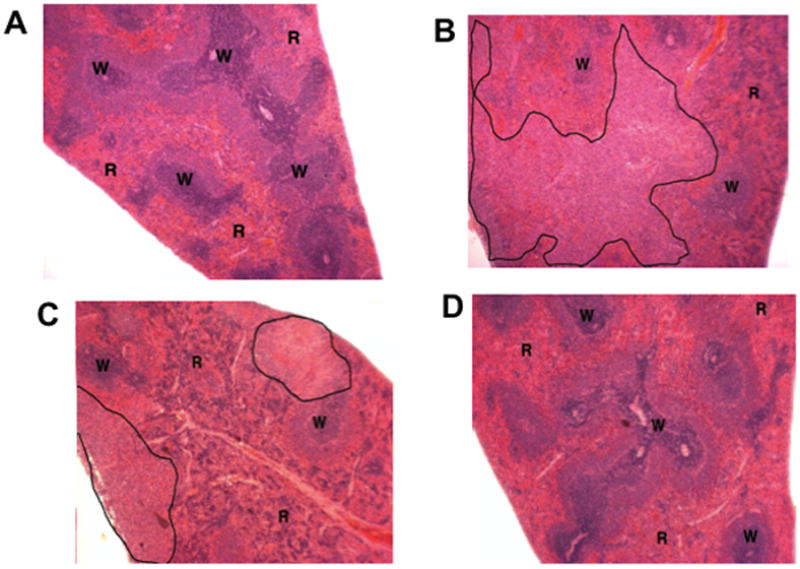
Photomicrograph of spleen dissection from (A) normal rat treated with vehicle, (B) arthritis rat treated with vehicle, (C) arthritis rat treated with 0.05 mg/kg of MTX, and (D) arthritis rat treated with MTX-cIBR (5 mg/kg). W = lymphoid white pulp. R = red pulp with reticuloendothelial cells.
The differences in spleen histopathology from normal, untreated arthritis, and drug-treated arthritis rats were quantified using the following scoring scale: 0 = normal, 1 = minimal, 2 = mild, 3 = moderate, 4 = marked, 5 = severe (Figure 6). The inflammation was significantly decreased in a dose-dependent manner, and no inflammation was found in the spleens of rats treated with 1 and 5 mg/kg MTX-cIBR. Remarkably, the spleen of rats treated with 0.05 mg/kg of MTX-cIBR had less inflammation than those treated with 0.05 mg/kg MTX (Figure 6). The lymphoid atrophy decreased as the dose of MTX-cIBR increased, with the lowest lymphoid atrophy in animals treated with 5.0 mg/kg of MTX-cIBR. Only the spleens from animals treated with 5.0 mg/kg of MTX-cIBR had significantly lower EMH compared to vehicle-treated animals, and there was no observable EMH difference in animals treated with 0.05 to 2.0 mg/kg MTX-cIBR and those treated with vehicle. There were increases in the spleen and liver weights in arthritic rats compared to normal rats (Table 2). Treatment of arthritic rats with 5.0 mg/kg MTX-cIBR suppressed the increases in spleen and liver weights by 78%. Lowering the dose of MTX-cIBR to 1.0 mg/kg lowered its ability to suppress the increase in spleen and liver weights by 49% and 30%, respectively.
Figure 6.
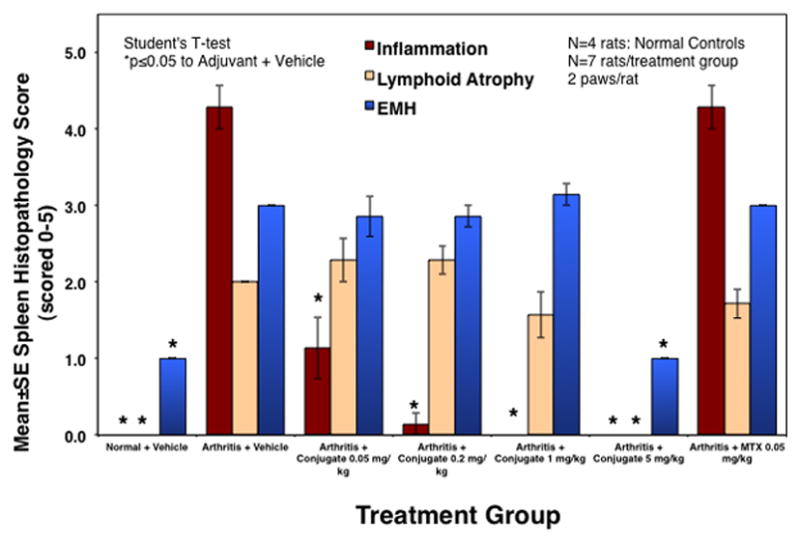
The scores of inflammation, lymphoid atrophy, and extramedullary hemotopoiesis (EMH) of normal and arthritis rat treated with vehicle, MTX (0.05 mg/kg), and MTX-cIBR (0.05. 0.2, 1, and 5 mg/kg). * indicates the statistical significance with p value ≤ 0.05.
Table 2.
The effect of MTX-cIBR on spleen and liver weights at Day 14.
| Treatment | Rat Spleen Weight ± SE (g) | Rat Liver Weight ± SE (g) |
|---|---|---|
| Normal + Vehicle | 0.2139 ± 0.0060 (100%) * | 4.3689 ± 0.1475 (100%) * |
| Arthritis + Vehicle | 0.5823 ± 0.0175 (0%) | 6.6191 ± 0.0858 (0%) |
| Arthritis + 0.05 mg/kg MTX-cIBR | 0.4524 ± 0.0139 (35%) * | 5.8817 ± 0.0690 (33%) * |
| Arthritis + 0.2 mg/kg MTX-cIBR | 0.4822 ± 0.0248 (27%) | 6.1970 ± 0.1721 (19%) ** |
| Arthritis + 1 mg/kg MTX-cIBR | 0.4027 ± 0.0202 (49%) * | 5.9485 ± 0.1947 (30%) * |
| Arthritis + 5 mg/kg MTX-cIBR | 0.2953 ± 0.017 (78%) * | 4.8620 ± 0.2053 (78%) * |
| Arthritis + 0.05 mg/kg MTX | 0.500 ± 0.000 (11%) | 6.1289 ± 0.1451 (22%) ** |
p-value < 0.01 when compared to Arthritis + Vehicle.
p-value < 0.05 when compared to Arthritis + Vehicle.
LFA-1-Mediated Uptake of MTX-cIBR
The LFA-mediated uptake of MTX-cIBR in Molt-3 T cells was evaluated by inhibiting the toxicity of 1 μM MTX-cIBR in the presence of different concentrations of anti-LFA-1 mAb and cIBR peptide (Table 3). The viability of T cells was increased upon increasing concentrations of anti-LFA-1 mAb and cIBR peptide, indicating that the MTX-cIBR uptake was partially mediated by LFA-1. Conversely, MTX-cIBR and cIBR peptide were used to inhibit anti-CD11a antibody binding (clone 38, Accurate) in both PMA-activated Molt-3 T-cells at 4 °C. MTX-cIBR and cIBR peptide inhibited binding of anti-LFA-1 mAb (clone 38) in PMA-activated cells in a concentration-dependent manner (data not shown).
Table 3.
Protective Effect of cIBR Peptide or CD11a mAb against MTX-cIBR Activity (1 μM).
| cIBR peptide [μM] | % Viable cells | CD11a antibody dose | % Viable cells |
|---|---|---|---|
| 10 | 58 ± 3 | 40 μL/mL | 35 ± 7 |
| 100 | 81 ± 7 | 80 μL/mL | 97 ± 10 |
| 1000 | 94 ± 5 |
Chemical Stability of MTX-cIBR
Degradation of MTX-cIBR was monitored at pH 1, 4, 5, 6, 7, 8, 10, and 12, and the degradation products at acidic and basic pH values were identified. Upon incubation at 70 °C, the samples were analyzed up to two half-lives, and the degradation profile for MTX-cIBR appeared to follow pseudo-first-order kinetics at all pH values. The Log C of MTX-cIBR was plotted against time for all pH values, and the half-lives of the conjugate are shown in Table 4. MTX-cIBR has the longest t1/2 at pH 6.0 (770.00 min) and the second longest at pH 7.0 (577.50 min).
Table 4.
The observed pseudo-first-order rate constants and half-lives for MTX-cIBR at different pH conditions at 70 °C.
| pH | Buffer type | kobs(h−1) | t1/2(h) |
|---|---|---|---|
| 1.0 | HCl | 5.25 × 10−1 | 1.32 |
| 4.0 | Acetate | 3.2 × 10−3 | 216.56 |
| 5.0 | Acetate | 1.3 × 10−3 | 533.08 |
| 6.0 | Phosphate | 9 × 10−4 | 770.00 |
| 7.0 | Phosphate | 1.2 × 10−3 | 577.50 |
| 8.0 | Phosphate | 3.1 × 10−3 | 223.55 |
| 10.0 | Carbonate | 2.3 × 10−2 | 30.13 |
| 12.0 | NaOH | 7.4 × 10−2 | 9.29 |
Ionic strengths of all buffer solutions were adjusted to 0.15 M with NaCl. For pH 1.0, a solution of 0.1 N HCl was used; 0.01 N NaOH was used for pH 12. For all other pH values, buffer concentration was kept at 50 mM.
The overall stability profile of MTX-cIBR was also expressed in the pH-rate profile (Figure 7), which could be used to develop a suitable condition for the formulation of this conjugate. The highest stability was shown at pH 6.0, and two inflection points were observed at pH 3.9 and 8.9, which were due to the protonation-deprotonation of the carboxylic acid and amino groups, respectively. This pH-rate profile was constructed with the following assumptions: (a) the ionization of the two carboxylic groups occurs at the same pH, (b) the tertiary amine in the MTX can be neutral or protonated, and (c) the two amino groups on the pteridine ring are considered to be highly unreactive and their contributions negligible. It has been reported previously that the amines in heterocyclic ring systems of methotrexate, purines, and pyrimidines are highly unreactive.20 Thus, MTX-cIBR was treated as a molecule with three ionizable groups (a carboxylic acid, a tertiary amine, and a guanidium group). Because the guanidium group has a pKa of 12.5 and the maximum pH value studied in this experiment was 12, the system was considered to have three species with two different equilibrium constants for the dissociation, as shown in the following equation with Ka1 and Ka2 as the equilibrium constants, respectively:
Figure 7.
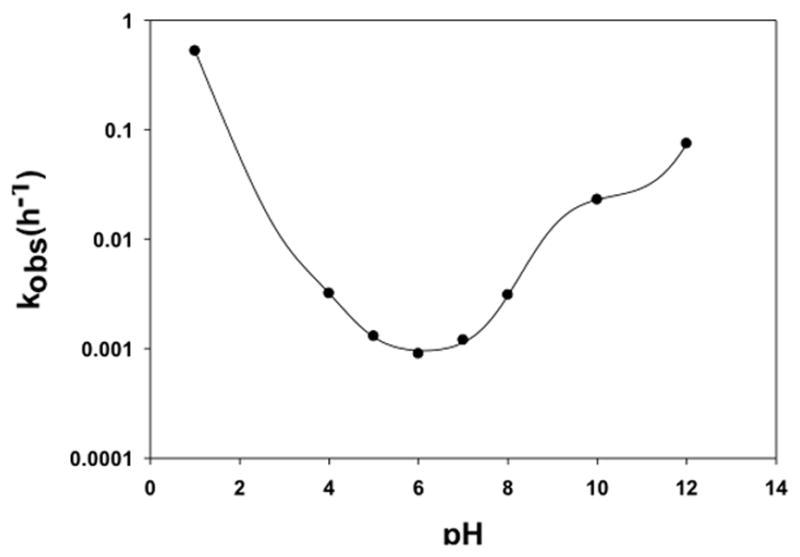
pH-rate profile for MTX-cIBR at 70 °C with buffer concentrations and ionic strength as described in Table 4. Stability of the conjugate was followed at pH values 1, 4, 5, 6, 7, 8, 10, and 12. Maximum stability of the conjugate was observed at pH 6.
The overall degradation kinetics for the conjugate is described by the following equation:
where
The observed rate constant shown below was obtained by substituting the values for the fraction of the individual species.
Because the study was conducted at 70 °C, the value of the ion product of water (pKw) was calculated using the empirical relationship below.
From the model, the values for the constants were:
where kH is the rate constant for acid-catalyzed degradation of H3A2+, and k′OH and k″OH are the rate constants for the base-catalyzed degradation of H2A+ and HA, respectively, as shown above. kO and k′O are the rate constants for water-catalyzed degradation of the H3A2+ and H2A+ species, respectively.
Degradation of MTX-cIBR under Acidic Conditions
MTX-cIBR degraded rapidly at pH 1.0 with a half-life of 1.32 h (Table 4). However, the stability of the compound increased dramatically at pH 4.0 (t1/2 = 216.56 h). The degradation products of the conjugate at acidic pH were analyzed by LC-MS. The parent MTX-cIBR (1, Figure 8) has two positive charges (m/z = 805.9), which are on the side chain of the arginine residue and the tertiary amine of MTX. Because the degradation products at pH 1.0 and 4.0 were similar, only the products at pH 1.0 were identified. There were two major degradation products of the conjugate with double charges (2, m/z = 718.8; 3, m/z = 711.8) as determined using the isotopic distribution profiles. Compound 2 is the cleavage product of the C–N bond between the 6-methylpteridine and phenyl rings (pathway a, Figure 8), and the bond cleavage may be assisted by a positively charged tertiary amine as a leaving group (Figure 8). Compound 2 degraded further to compound 3 by releasing the methyl group from the phenyl amine moiety (pathway b, Figure 8). The HPLC profile indicated that the abundance of compound 3 gradually decreased with the increase in pH value, suggesting that the reaction was pH-dependent. To confirm the identity of compounds 2 and 3, we showed that they both produced compound 4 with m/z = 1303.6 (z = +1) when they were subjected to fragmentation. This suggested that the degradation was, in fact, in the drug portion of the conjugate, as the cIBR peptide was intact even in this fragmentation product (pathway c, Figure 8).
Figure 8.
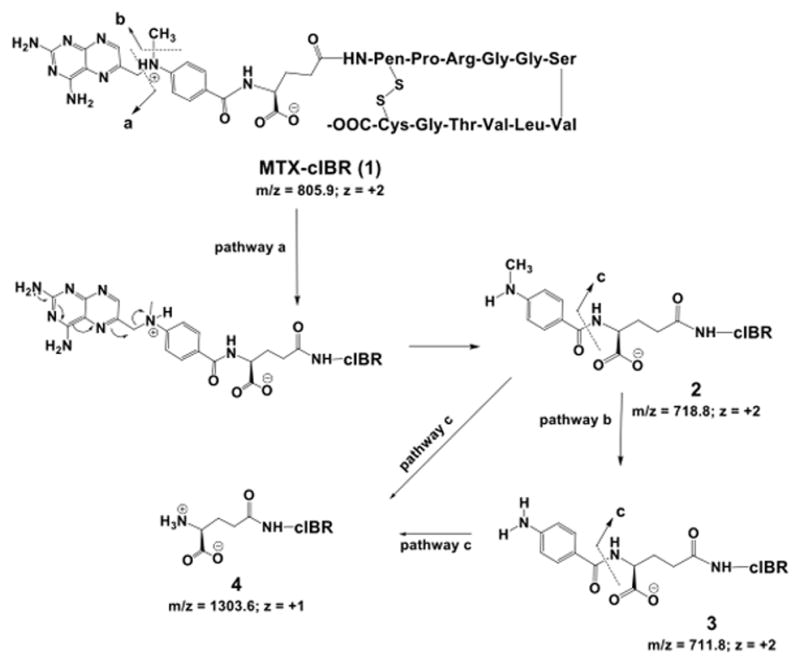
Identification of the major degradation products of MTX-cIBR at pH 1.0. Pathway a shows the degradation of the MTX portion of the conjugate by clipping the C-N bond between the pteridine ring and the p-amino-benzoyl ring to give compound 2. Pathway b represents demethylation reaction of compound 2 to give degradation product 3. Pathway c shows cleavage between the p-amino-benzoyl group and the Glu residue of the MTX fragment of the conjugate (compound 4).
Degradation of MTX-cIBR at Basic Conditions
Under basic conditions, as expected, the conjugate had the lowest stability at pH 12 (t1/2 = 9.29 h, Table 4). The degradation characteristics at pH 10 were similar to those at pH 12; therefore, only the degradation products at pH 12 were identified. The parent MTX-cIBR showed a single peak before incubation at pH 12; upon incubation at pH 12, four peaks with the same molecular weight (i.e., 805.9, z = +2) appeared in the extracted ion chromatograms. These four products could be attributed to derivatives of compound 7 with different stereoisomers on the Ser and Thr residues of the peptide portion of the conjugate (Figure 9). The stereoisomers were generated by a racemization reaction, which occurred at the Ser6 or Thr10 residues. The Ser and Thr residues may undergo dehydration and rehydration reactions to racemize the α-carbon and give a mixture of L-/D-Ser or L-/D-Thr. This hypothesis was supported by the observation of four different dehydrated peaks with m/z 797.4 (z = +2) in the chromatograms, which corresponded to compounds 5 and 6 and their derivatives (Figure 9). The dehydrated products 5 and 6 could only be generated via β-elimination of the hydroxyl group in Ser6 and Thr10.
Figure 9.
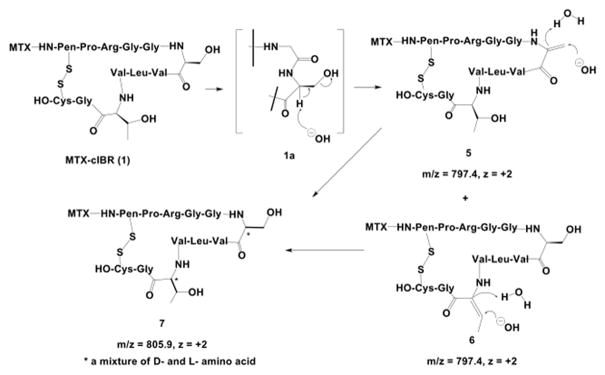
Identification of the major degradation products for MTX-cIBR at pH 12. The conjugate can undergo dehydration and racemization at the Ser6 and Thr10 residues. Compounds 5 and 6 are representative of the dehydration reaction of the Thr10 and Ser6 residues. Subsequent attack by hydroxyl ions on the alkene groups produces Ser6 and Thr10 with mixed chirality (L- and D-amino acid) in compound 7.
Another family of products was derived from the degradation of the disulfide bond of the cyclic peptide under basic conditions (Figure 10). The attack of the disulfide bond by a hydroxide ion led to the opening of the disulfide bond to form a sulfenic acid and a thiol group. One of two possible products is compound 8 (m/z = 814.9, z = +2). In this case, the sulfenic acid group is at the Pen1 residue of compound 8; however, the other possible product is the sulfenic acid group at Cys12. The thiolate group in compound 8 could react further with another molecule at the disulfide bond to give a dimer intermediate 9. The intramolecular reaction between the thiolate and sulfenic acid groups generated a cyclic dimer 10 with m/z 805.9 and quadruple charges (Figure 10).
Figure 10.
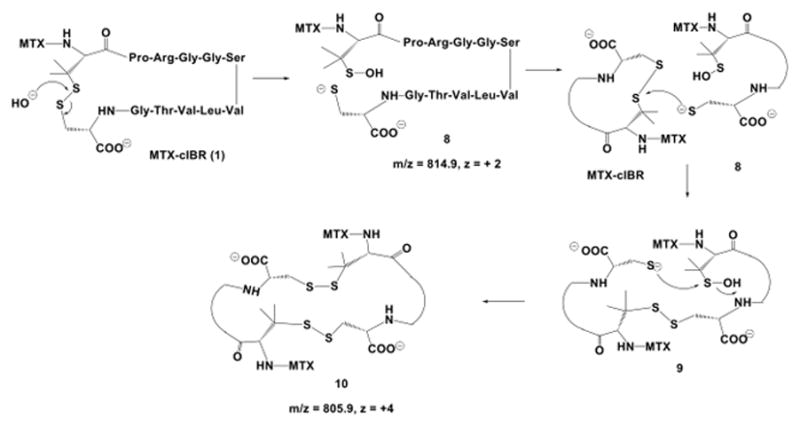
The disulfide bond degradation in MTX-cIBR at pH 12 produced thiolate-sulfenic acid 8 and cyclic dimer 10. The disulfide bond opening was via direct attack of hydroxyl anion on the sulfur of the disulfide bond. Thiolate 8 was reacted with another molecule of the conjugate to produce a linear dimer 9; intramolecular reaction of thiolate anion with the sulfur atom of sulfenic acid generated a cyclic dimer 10.
In Vitro Stability of MTX-cIBR in Plasma and Tissue Homogenate
The stability of MTX-cIBR was evaluated in rat plasma and homogenized rat heart tissue to determine its biological disposition (Figure 11). MTX-cIBR has half-lives of 43.8 and 38.1 min in rat plasma and homogenized rat heart, respectively.
Figure 11.
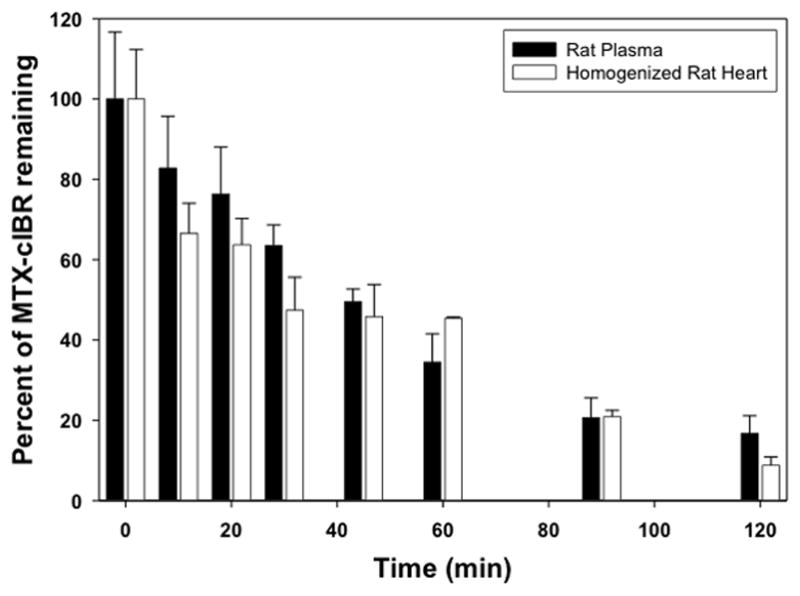
Determination of the stability of the MTX-cIBR in isolated rat plasma and homogenized rat heart tissue. Stability of the conjugate was determined over a period of 2 half-lives. Pseudo-first-order plots were generated from the degradation profiles.
DISCUSSION
MTX is widely used to treat the chronic inflammation of RA and Crohn’s disease; however, patients treated with MTX can suffer from severe side effects. To improve the efficacy profile of MTX, it has been conjugated to different carriers to increase its cellular uptake with or without targeting a specific type of cell.17,25 Because autoimmune diseases are characterized by ‘self-reactive’ T-cells, it would be ideal to target T cells that are responsible for mediating the inflammatory response. For this purpose, an integrin called LFA-1 receptor is an ideal and selective receptor for delivering MTX to activated T cells because LFA-1 is expressed exclusively on leukocytes. Selective delivery of MTX to activated inflammatory leukocytes could provide a significant reduction in the side effects of MTX treatment. As a proof-of-concept, MTX-cIBR was developed and evaluated in in vitro and in vivo assays to demonstrate that cIBR peptide could improve delivery of MTX to leukocytes.
As previously shown using FITC-cIBR, the uptake mechanism of cIBR peptide in Molt-3 T cells and HL-60 cells is via receptor-mediated endocytosis through receptor clustering.4,10,11 However, it is more difficult to evaluate the mechanism of endocytosis of MTX-cIBR compared to that of FITC-cIBR using fluorescence microscopy; this is due to the low fluorescence emission of MTX. Thus, several alternative methods were carried out to indirectly evaluate receptor-mediated endocytosis of MTX-cIBR. First, binding of MTX-cIBR to LFA-1 receptors on T cells was inhibited by anti-LFA-1 mAb, suggesting that cIBR fragment on the conjugate binds to LFA-1 receptors. MTX-cIBR was more effective in inhibiting anti-LFA-1 mAb in PMA-activated T cells than in resting T cells, suggesting that MTX-cIBR preferably binds to the activated form of LFA-1 receptors. It could also suggest that binding of MTX-cIBR to LFA-1 induces the internalization of LFA-1 receptors on the activated T cells. Second, the toxicity of the MTX-cIBR in Molt-3 T cells was also inhibited by cIBR peptide or an anti-LFA-1 monoclonal antibody in a concentration-dependent manner (Table 3). This result indicated that MTX-cIBR entered the cells partially using LFA-1 receptors to elicit MTX toxicity. It should be noted that MTX-cIBR could enter the cells via RFC and mFBP receptors upon recognition of the MTX fragment. The mechanism of entry of MTX-cIBR via LFA-1 vs. (RFC + mFBP) has not been fully elucidated at this time. Thirdly, although the cytotoxicity of MTX-cIBR (IC50 = 2.75 μM) was 45-fold lower than MTX (IC50 = 0.061 μM) against LFA-1-expressing Molt-3 T cells, MTX-IBR was not toxic up to 10 μM against non-LFA-1-expressing KB epithelial cells.26 In contrast, MTX has a similar range of toxicity (IC50 = 0.027 μM) in KB epithelial cells and in Molt-3 T cells (IC50 = 0.061 μM). These results suggest that the LFA-1-mediated endocytosis differentiates the selectivity of MTX-cIBR over MTX in T cells and KB epithelial cells. The lower cell toxicity of MTX-cIBR could be due to the structural interference of cIBR fragment on the binding of MTX fragment to DHFR. This steric interference reduced the DHFR inhibitory activity of MTX-cIBR (Km = 29.8±2.1 × 10−9 M) by 15-fold compared to MTX (Km = 1.8±0.7 × 10−9 M).26 Taken together, the results suggest that the uptake of MTX-cIBR is via receptor-mediated endocytosis and that DHFR inhibitory activity may not necessarily be due to the release of MTX from MTX-cIBR.
The in vivo efficacy of MTX-cIBR was evaluated in the rat adjuvant arthritis model; it should be noted that cIBR peptide has a similar amino acid sequence in human, mouse, or rat ICAM-1. The efficacy was determined by evaluating the joint inflammation and the inhibition of periarticular inflammation and bone resorption of developing adjuvant arthritis. The secondary endpoints of this conjugate efficacy include the inhibition of splenomegaly, hepatomegaly, and body weight change of adjuvant disease. Treatment of arthritic rats with 5.0 mg/kg of MTX-cIBR was effective in suppressing the progress of arthritis with an increase in animal body weight over time. The animals had moderate synovium and no bone resorption at the physis. In contrast, the adjuvant rats treated with vehicle had severe synovitis and periarticular inflammation due to edema and neutrophil infiltration. In addition, these animals had bone resorption across the physis with the presence of bone fragments. Due to the toxicity of MTX, the activity of MTX-cIBR could not be compared to that of MTX at a high dose (i.e., 5.0 mg/kg). Although animals treated daily with 0.2 mg/kg had excellent suppression of RA, the animals did not show an increase in body weight, possibly due to the toxicity of MTX at a high dose.
Unfortunately, MTX-cIBR was not as effective at a 1-mg/kg dose. One possible explanation is that the conjugate is not very stable in the systemic circulation. Clearly, this molecule was rapidly eliminated in the blood upon i.v. injections in the animal model of RA as indicated by low plasma stability (t1/2 = 43.8 min). This may explain the need to deliver a higher dose (5 mg/kg) of the conjugate before observing significant efficacy compared to vehicle. However, the low dose (0.2–1.0 mg/kg) significantly suppressed the spleen inflammation (Figure 6). Even at a 0.05-mg/kg dose, MTX-cIBR suppressed spleen inflammation better than 0.05 mg/kg of MTX alone (Figure 6, Table 2). The rapid peptide metabolism of MTX-cIBR could be attributed to the presence of the Arg residue that makes the peptide prone to trypsin digestion. The disulfide bond in the cyclic peptide could be opened by reaction with glutathione in plasma and tissue. In addition, the MTX portion of the molecule could be metabolized to form 2,4-diamino-7-hydroxy-pteridine.27 Although the disulfide bond is unstable, several peptide drugs such oxytocin28 and integrilin29 are currently being used for inducing labor and for treating myocardial infarction, respectively. These cyclic peptides have been successfully developed with sufficient in vivo stability to produce in vivo biological activity. In the case of MTX-cIBR, the rationales to select cIBR for conjugation were (a) its mechanism of uptake of cIBR has been well studied,4,10,11 (b) the N-terminal of cIBR is available for conjugation to MTX, and (c) cIBR peptide has also been shown to suppress RA in the collagen-induced arthritis (CIA) mouse model.30
Conjugation of MTX to cIBR maintains the binding properties of cIBR peptide to LFA-1 because MTX-cIBR can inhibit binding of anti-LFA-1 mAb to LFA-1 on Molt-3 T-cells. These results suggest that conjugation of MTX to cIBR does not alter the binding characteristics of the cIBR-peptide fragment to LFA-1. MTX-cIBR preferably binds to the activated state of LFA-1 on activated Molt-3 T cells. LFA-1 recognition of the cIBR-peptide fragment within the MTX-conjugate may be due to the preservation of the peptide conformation after conjugation. The NMR structure of the conjugate showed that the β-turn conformation of the important PRG sequence in the cIBR peptide of MTX-cIBR was not altered by conjugation to MTX (data not shown). This PRG motif in cIBR peptide is essential for binding LFA-1 and inhibiting T-cell adhesion.6 However, the NMR structure showed that the turn around the Val-Thr-Gly sequence on the MTX-cIBR was altered upon conjugation by moving it to the opposite side compared to the cIBR peptide (data not shown). Because the conjugate demonstrated binding characteristics to LFA-1 similar to those of cIBR, this suggests that the conformational difference in the Val-Thr-Gly sequence may not influence the binding properties of MTX-cIBR.
It is also important to determine the chemical stability of the conjugate for future development of its formulation. Furthermore, degradation mechanisms of the conjugate can be utilized to design a more stable molecule. Interestingly, the MTX portion of the conjugate is more prone to degradation in acidic conditions than in basic conditions. There are two degradation pathways of the MTX segment of the conjugate. The first is the release of the pteridine ring from the conjugate (pathway a, Figure 8) to give compound 2. This release is due to the cleavage of the C-N bond between the pteridine ring and the p-amino-benzoyl ring. This reaction is assisted by the presence of the protonated tertiary amine of the p-amino-benzoyl group, which acts as a good leaving group. The second major degradation product was produced by demethylation of the phenylamine moiety (pathway b, Figure 8) of compound 2 to give compound 3.
The major degradation sites for the peptide portion of the conjugate are at the Ser6 and Thr10 residues and at the disulfide bond. The Ser6 and Thr10 residues degrade via β-elimination under both acidic and basic conditions. Under basic conditions, the dehydration reaction is also observed as the major degradation pathway of the peptide segment. After incubation in basic conditions, four peaks with the same molecular weight as the parent conjugate were observed. These compounds are due to racemization at the α-carbon of Ser6 and Thr10 to give four possible conjugates with (1) L-Ser6 and L-Thr10 (the parent compound), (2) L-Ser6 and D-Thr10, (3) D-Ser and L-Thr10, and (4) D-Ser6 and D-Thr10. Racemization of the Ser residue in peptides has been observed previously in RS-26306 decapeptide and the LHRH analog histrelin.31,32 The mechanism of racemization is supported by the presence of four different dehydration products (e.g., compounds 5 and 6) of the conjugate. It was not possible to identify each of these peaks individually due to the lack of separation and their co-elution with the dimer products. Our attempts to improve the separation of these molecules were unsuccessful. In addition, the fragmentation of the dehydrated species failed to yield additional information because the 6-methylpteridine-2,4-diamine ring carried most of the ion current that led to the suppression of the signal from the peptide fragments.
Finally, the disulfide bond can be cleaved directly by hydroxide ion to produce thiolate-sulfenic acid 8; this type of reaction has been observed previously in cyclic peptides.33,34 The thiolate anion can attack a disulfide bond in another molecule of conjugate to produce a linear dimer 9. The intramolecular reaction of the thiolate ion on the sulfur of sulfenic acid generates the cyclic dimer 10 and releases hydroxyl anion.
The fact that MTX-cIBR is most stable at pH 6.0 is advantageous for formulation development of the conjugate as it is close to physiological pH (pH 7.4). Peptides and proteins with higher stability in highly acidic or alkaline pH often present a challenge in formulation development. Indications that MTX-cIBR is unstable in the presence of biological matrices combined with the identification of the chemical degradation mechanisms will help to design a more stable conjugate. Identification of the in vivo degradation products as well as the pharmacokinetic profile will be evaluated in the future.
One way to improve the in vivo efficacy of MTX-cIBR is by stabilizing the cIBR peptide via elimination of the disulfide bond. In this case, the peptide can be cyclized via N-to-C-terminus peptide bond. To achieve this goal, an initial effort has been made to design a cyclic hexapeptide (CH7, cyclo(1,6)PRGGSV); this peptide has activity comparable to that of cIBR in inhibiting ICAM-1/LFA-1-mediated heterotypic T-cell adhesion upon binding to LFA-1.6 Because CH7 does not have a free amino group for conjugation to MTX, CHK7 peptide (cyclo(1,6)PRGGKV) peptide was synthesized by replacing Ser-5 with Lys-5 to provide the side chain amino group. Unfortunately, CHK7 has lower cell adhesion inhibitory activity than the parent CH7, indicating lower LFA-1 binding affinity. Therefore, further Lys mutation studies are being carried out to find a good position for the lysine residue in CHK7 that does not lower binding affinity of CHK7 to LFA-1. Once a CHK7 derivative with similar LFA-1 binding affinity with cIBR peptide is found, MTX can be conjugated to CHK7 derivative to make MTX-CHK7 for in vitro and in vivo evaluations. Another way to improve the efficacy of the MTX-peptide conjugate is by increasing the separation between MTX and the peptide. Molecular modeling studies indicated that cIBR peptide could interfere with MTX binding to DHFR; this is one possible reason that MTX-cIBR has lower DHFR inhibitory activity than MTX. In this case, different lengths of spacer or linker between the peptide and MTX can be introduced to reduce peptide interference during binding of MTX fragment to DHFR. Alternatively, the MTX can be conjugated to the peptide via a cleavable linker that is sensitive to enzymatic cleavage (i.e., esterase) or pH change in lysosomes to release MTX in the intracellular space. Finally, a full comparison of dose-dependent efficacy and toxicity of MTX-cIBR, MTX, and cIBR can be carried out to determine the potential advantages of the conjugate.
In conclusion, cIBR has a potential to deliver MTX to leukocytes for suppressing rheumatoid arthritis in the animal model. The cIBR peptide improves the delivery of MTX to LFA-1-expressing leukocytes, and the in vitro results suggest that the conjugate binds to LFA-1 on T cells. However, the contributions of RFC and mFBP to the uptake of MTX-cIBR compared to those of LFA-1 should be evaluated in the future. The conjugate also had short half-lives in plasma and tissue homogenates, which could explain why the conjugate was not effective in vivo at low doses. Therefore, there is an effort our laboratory to design a conjugate that is more stable than MTX-cIBR in biological systems. Finally, the mechanism of action of MTX-cIBR in suppressing inflammatory T cells (i.e., Th1 and Th17) and possibly enhancing suppressor/regulatory T cells (i.e., Th2 and Treg) will be investigated.
Acknowledgments
This study was supported by grants from National Institutes of Health (R01-AI-063002 and R43-AI-052556). We are grateful for the help of Nancy Harmony in proofreading the manuscript.
Abbreviations
- ICAM-1
intercellular adhesion molecule-1
- LFA-1
lymphocyte function-associated antigen-1
- FITC
fluorescein isothiocyanate
- HUVEC
human umbilical vein endothelial cells
References
- 1.Anderson ME, Siahaan TJ. Targeting ICAM-1/LFA-1 interaction for controlling autoimmune diseases: Designing peptide and small molecule inhibitors. Peptides. 2003;24(3):487–501. doi: 10.1016/s0196-9781(03)00083-4. [DOI] [PubMed] [Google Scholar]
- 2.Yusuf-Makagiansar H, Anderson ME, Yakovleva TV, Murray JS, Siahaan TJ. Inhibition of LFA-1/ICAM-1 and VLA-4/VCAM-1 as a therapeutic approach to inflammation and autoimmune diseases. Med Res Rev. 2002;22(2):146–167. doi: 10.1002/med.10001. [DOI] [PubMed] [Google Scholar]
- 3.Majumdar S, Siahaan TJ. Peptide-mediated targeted drug delivery. Med Res Rev. 2010 doi: 10.1002/med.20225. [DOI] [PubMed] [Google Scholar]
- 4.Anderson ME, Siahaan TJ. Mechanism of binding and internalization of ICAM-1-derived cyclic peptides by LFA-1 on the surface of T cells: A potential method for targeted drug delivery. Pharm Res. 2003;20(10):1523–1532. doi: 10.1023/a:1026188212126. [DOI] [PubMed] [Google Scholar]
- 5.Anderson ME, Tejo BA, Yakovleva T, Siahaan TJ. Characterization of binding properties of ICAM-1 peptides to LFA-1: Inhibitors of T-cell adhesion. Chem Biol Drug Des. 2006;68(1):20–28. doi: 10.1111/j.1747-0285.2006.00407.x. [DOI] [PubMed] [Google Scholar]
- 6.Anderson ME, Yakovleva T, Hu Y, Siahaan TJ. Inhibition of ICAM-1/LFA-1-mediated heterotypic T-cell adhesion to epithelial cells: Design of ICAM-1 cyclic peptides. Bioorg Med Chem Lett. 2004;14(6):1399–1402. doi: 10.1016/j.bmcl.2003.09.100. [DOI] [PubMed] [Google Scholar]
- 7.Barnard AL, Igakura T, Tanaka Y, Taylor GP, Bangham CR. Engagement of specific T-cell surface molecules regulates cytoskeletal polarization in HTLV-1-infected lymphocytes. Blood. 2005;106(3):988–995. doi: 10.1182/blood-2004-07-2850. [DOI] [PubMed] [Google Scholar]
- 8.Zimmerman T, Oyarzabal J, Sebastian ES, Majumdar S, Tejo BA, Siahaan TJ, Blanco FJ. ICAM-1 peptide inhibitors of T-cell adhesion bind to the allosteric site of LFA-1. An NMR characterization. Chem Biol Drug Des. 2007;70(4):347–353. doi: 10.1111/j.1747-0285.2007.00566.x. [DOI] [PubMed] [Google Scholar]
- 9.Iskandarsyah, Tejo BA, Tambunan US, Verkhivker G, Siahaan TJ. Structural modifications of ICAM-1 cyclic peptides to improve the activity to inhibit heterotypic adhesion of T cells. Chem Biol Drug Des. 2008;72(1):27–33. doi: 10.1111/j.1747-0285.2008.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majumdar S, Kobayashi N, Krise JP, Siahaan TJ. Mechanism of internalization of an ICAM-1-derived peptide by human leukemic cell line HL-60: Influence of physicochemical properties on targeted drug delivery. Mol Pharm. 2007;4(5):749–758. doi: 10.1021/mp0700458. [DOI] [PubMed] [Google Scholar]
- 11.Majumdar S, Tejo BA, Badawi AH, Moore D, Krise JP, Siahaan TJ. Effect of modification of the physicochemical properties of ICAM-1-derived peptides on internalization and intracellular distribution in the human leukemic cell line HL-60. Mol Pharm. 2009;6(2):396–406. doi: 10.1021/mp800120t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorlick R, Goker E, Trippett T, Waltham M, Banerjee D, Bertino JR. Intrinsic and acquired resistance to methotrexate in acute leukemia. New Engl J Med. 1996;335(14):1041–1048. doi: 10.1056/NEJM199610033351408. [DOI] [PubMed] [Google Scholar]
- 13.Pui CH. Childhood leukemias. New Engl J Med. 1995;332(24):1618–1630. doi: 10.1056/NEJM199506153322407. [DOI] [PubMed] [Google Scholar]
- 14.Andersson SE, Johansson LH, Lexmuller K, Ekstrom GM. Anti-arthritic effect of methotrexate: is it really mediated by adenosine? Eur J Pharm Sci. 2000;9(4):333–343. doi: 10.1016/s0928-0987(99)00073-1. [DOI] [PubMed] [Google Scholar]
- 15.Williams HJ, Willkens RF, Samuelson CO, Jr, Alarcon GS, Guttadauria M, Yarboro C, Polisson RP, Weiner SR, Luggen ME, Billingsley LM, Dahl SL, Egger MJ, Reading JC, Ward JR. Comparison of low-dose oral pulse methotrexate and placebo in the treatment of rheumatoid arthritis. A controlled clinical trial. Arthritis Rheum. 1985;28(7):721–730. doi: 10.1002/art.1780280702. [DOI] [PubMed] [Google Scholar]
- 16.Westerhof GR, Rijnboutt S, Schornagel JH, Pinedo HM, Peters GJ, Jansen G. Functional activity of the reduced folate carrier in KB, MA104, and IGROV-I cells expressing folate-binding protein. Cancer Res. 1995;55(17):3795–3802. [PubMed] [Google Scholar]
- 17.Ryser HJ, Shen WC. Conjugation of methotrexate to poly(L-lysine) increases drug transport and overcomes drug resistance in cultured cells. P Natl Acad Sci USA. 1978;75(8):3867–3870. doi: 10.1073/pnas.75.8.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Heijden JW, Dijkmans BA, Scheper RJ, Jansen G. Drug Insight: resistance to methotrexate and other disease-modifying antirheumatic drugs--from bench to bedside. Nature clinical practice. 2007;3(1):26–34. doi: 10.1038/ncprheum0380. [DOI] [PubMed] [Google Scholar]
- 19.Zeng H, Chen Z-S, Belinsky MG, Rea PA, Kruh GD. Transport of Methotrexate (MTX) and Folates by Multidrug Resistance Protein (MRP) 3 and MRP1: Effect of Polyglutamylation on MTX Transport. Cancer Res. 2001;61(19):7225–7232. [PubMed] [Google Scholar]
- 20.Kuefner U, Lohrmann U, Montejano Y, Vitols KS, Huennekens FM. Chemotherapeutic potential of methotrexate peptides. Adv Enz Regulation. 1988;27:3–13. doi: 10.1016/0065-2571(88)90005-2. [DOI] [PubMed] [Google Scholar]
- 21.Wolff D, Frei E, Hofmeister N, Steiner B, Kleine HD, Junghanss C, Sievert K, Terpe H, Schrenk HH, Freund M, Hartung G. Methotrexate-albumin and aminopterin-albumin effectively prevent experimental acute graft-versus-host disease. Transplantation. 2006;82(4):527–533. doi: 10.1097/01.tp.0000229451.40286.64. [DOI] [PubMed] [Google Scholar]
- 22.Fiehn C, Muller-Ladner U, Gay S, Krienke S, Freudenberg-Konrad S, Funk J, Ho AD, Sinn H, Wunder A. Albumin-coupled methotrexate (MTX-HSA) is a new anti-arthritic drug which acts synergistically to MTX. Rheumatology. 2004;43(9):1097–1105. doi: 10.1093/rheumatology/keh254. [DOI] [PubMed] [Google Scholar]
- 23.N’Da DD, Neuse E, Nell M, Van Rensburg CEJ. Carrier-bound Methotrexate. III Antiproliferative Activity of Macromolecular MTX Conjugates Against the Human HeLa and Colo Carcinoma Cell Lines South African. J Chem. 2006;59:33–42. [Google Scholar]
- 24.Gurdag S, Khandare J, Stapels S, Matherly LH, Kannan RM. Activity of dendrimer-methotrexate conjugates on methotrexate-sensitive and -resistant cell lines. Bioconjug Chem. 2006;17(2):275–283. doi: 10.1021/bc0501855. [DOI] [PubMed] [Google Scholar]
- 25.Shen WC, Ryser HJ, LaManna L. Disulfide spacer between methotrexate and poly(D-lysine). A probe for exploring the reductive process in endocytosis. J Biol Chem. 1985;260(20):10905–10908. [PubMed] [Google Scholar]
- 26.Siahaan TJ, Yusuf-Makagiansar H, Anderson M, Xu RC. Leukocyte Internalized Peptide-Drug Conjugates. 2004/0037775 US Patent Application. 2004 Feb 26;
- 27.Dhondt JL, Hayte JM, Millot F, Klein R, Blais JC, Pfleiderer W. 2,4-diamino-7- hydroxy-pteridines, a new class of catabolites of methotrexate. Eur J Biochem. 1991;200(1):237–244. doi: 10.1111/j.1432-1033.1991.tb21072.x. [DOI] [PubMed] [Google Scholar]
- 28.Viero C, Shibuya I, Kitamura N, Verkhratsky A, Fujihara H, Katoh A, Ueta Y, Zingg HH, Chvatal A, Sykova E, Dayanithi G. REVIEW: Oxytocin: Crossing the bridge between basic science and pharmacotherapy. CNS Neurosci Ther. 2010;16(5):e138–156. doi: 10.1111/j.1755-5949.2010.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shah I, Khan SO, Malhotra S, Fischell T. Eptifibatide: The evidence for its role in the management of acute coronary syndromes. Core Evid. 2010;4:49–65. doi: 10.2147/ce.s6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benedict S, Siahaan TJ, Chan MA, Tibbetts SA. Peptide compositions which induce immune tolerance and methods of use. 006075004A US Patent. 2000
- 31.Avis KE, Lieberman HA, Lachman L, editors. Pharmaceutical Dosage Forms: Parenteral Medications. 2. New York: M. Dekker; 1992. p. 295. [Google Scholar]
- 32.Oyler AR, Naldi RE, Lloyd JR, Graden DA, Shaw CJ, Cotter ML. Characterization of the solution degradation products of histrelin, a gonadotropin releasing hormone (LH/RH) agonist. J Pharm Sci. 1991;80(3):271–275. doi: 10.1002/jps.2600800316. [DOI] [PubMed] [Google Scholar]
- 33.Bogdanowich-Knipp SJ, Chakrabarti S, Williams TD, Dillman RK, Siahaan TJ. Solution stability of linear vs. cyclic RGD peptides. J Pept Res. 1999;53(5):530–541. doi: 10.1034/j.1399-3011.1999.00052.x. [DOI] [PubMed] [Google Scholar]
- 34.He HT, Gursoy RN, Kupczyk-Subotkowska L, Tian J, Williams T, Siahaan TJ. Synthesis and chemical stability of a disulfide bond in a model cyclic pentapeptide: cyclo(1,4)-Cys-Gly-Phe-Cys-Gly-OH. J Pharm Sci. 2006;95(10):2222–2234. doi: 10.1002/jps.20701. [DOI] [PubMed] [Google Scholar]