

Abstract
Introduction
An individual’s genetic background plays a significant role in his or her chances of developing an abdominal aortic aneurysm (AAA). This risk is likely to be due to a combination of multiple small effect genetic factors acting together, resulting in considerable difficulty in the identification of these factors.
Methods
Methods for the identification of genetic factors associated with disease are usually based on the analysis of genetic variants in case-control studies. Over the last decade, owing to advances in bioinformatics and laboratory technology, these studies have progressed from focusing on the examination of a single genetic variant in each study to the examination of many millions of variants in a single experiment. We have conducted a series of such experiments using these methods.
Results
Our original methods using candidate gene approaches led to the initial identification of a genetic variant in the interleukin-10 gene associated with AAA. However, further studies failed to confirm this association and highlighted the necessity for adequately powered studies to be conducted, as well as the need for confirmatory studies to be performed, prior to the acceptance of a variant as a risk for disease. The subsequent application of genomic techniques to our sample set, in a global collaboration, has led to the identification of three robustly verified risk loci for AAA in the LRP1, LDLR and SORT1 genes.
Conclusions
Genomic studies of AAA have led to the identification of new pathways involved in the pathogenesis of AAA. The exploration of these pathways has the potential to unlock new avenues for therapeutic intervention to prevent the development and progression of AAA.
Keywords: Aneurysm, Genomics, LRP1, LDLR, SORT1
Abdominal aortic aneurysm (AAA) is a chronic disease, predominantly affecting men over the age of 65 years. In the UK, AAAs are responsible for over 4,500 deaths per annum and over 8,500 surgical AAA repairs are carried out every year.1 The total economic cost of AAA rupture and surgery to prevent rupture is around £140 million per year.2 There has been a decline in AAA related deaths over the last decade in the UK and some other countries but this phenomenon is not global, and in some countries the prevalence of disease is increasing.3 In addition, in several countries such as the UK, community screening for AAA has been introduced and the burden of disease is likely to increase in the short to medium term.
The principal risk associated with an AAA is rupture, an event that is usually fatal. The risk of rupture is related to aneurysm size. Evidence from randomised trials has demonstrated that a strategy of surveillance for patients with AAAs under 55mm is safe and surgery is usually reserved for those patients with AAAs larger than this threshold. The majority of AAAs diagnosed are small (<55mm), particularly in community screening programmes such as the National Health Service AAA Screening Programme. However, these small AAAs usually grow over time until they reach a point where surgery is required to prevent rupture.2 This growth occurs over many years, providing a window of opportunity to intervene to prevent AAA growth but, to date, no therapeutic strategies or pharmacotherapeutics have been discovered that can retard AAA growth. Trials of beta-blockers, statins, antibiotics, anti-inflammatory agents and antihypertensives have all been performed with no benefit shown.4–8
Established risk factors for AAA are smoking, male sex and a family history of disease. Interestingly, diabetes, a risk factor for all other cardiovascular diseases, is actually protective against AAA and those patients with AAA who have diabetes exhibit reduced AAA growth rates.2 Owing to the difficulties in modifying these risk factors, there are limited opportunities to develop strategies to prevent AAA or retard AAA growth over and above routine public health measures such as smoking cessation campaigns and secondary cardiovascular risk prevention. Nevertheless, familial clustering of disease suggests a genetic contribution to the development of disease.
The discovery of the genetic traits underlying the pathogenesis of AAA may permit insights to be made into the pathways and mechanisms of disease, and as a consequence, new strategies to treat AAA can be developed. This article describes the author’s personal contribution to the progress made in the last decade in unravelling the genetic factors underlying AAA. The aim was not to perform a systematic review of the genetic aetiology of AAA as this has been addressed recently by other authors.
AAA is a genetic disease
In order to identify the genetic traits underlying a disease, it is important to determine that a disease is truly genetic in origin. Familial clustering of cases of AAA was first noted by Clifton in 1977.9 Following on from this work, multiple authors attempted to determine the mode of inheritance of AAA and identify the ‘aneurysm gene’ but no single causal genetic variant has been identified, and, furthermore, the pattern of disease inheritance does not appear to be a classical Mendelian mode of inheritance.10 What has been established is that there is a significant genetic component in the aetiology of disease. The measurement used to quantify the contribution of an individual’s genetic makeup to the chance of that individual developing a disease is ‘heritability’. This is strictly defined as the proportion of an individual’s disease phenotype that is a result of their genotype.
This can be assessed by examining the presence and absence of disease in extended families, and two early estimates found consistent estimates of 70% and 72%.11,12 A more powerful method to estimate heritability is through the analysis of disease in twins and it is remarkable that when such a study was performed approximately 20 years after the initial estimates by Powell and Greenhalgh11 and Majumder et al,12 the estimate of heritability found was 71%,13 adding further weight to the initial findings. However, all of the above studies identified evidence that AAA is not a disease due to a single genetic defect in the majority of cases. Instead, the genetic component is a result of multiple genetic variants.
Methods
Genetic association studies
There are multiple techniques used in studies of genetic association. Family-based approaches (where entire family pedigrees are assessed for the presence of a disease and this is compared with genetic variants) are powerful but difficult in the case of AAA since AAAs do not become evident until older age so collecting multigenerational pedigrees is difficult.
Analysis of sibling pairs is a strategy that avoids this problem but has no advantage in terms of statistical power over unrelated case-controlled approaches so the latter are usually preferred by investigators for logistical purposes. These case-controlled studies form the majority of studies in the field of AAA. The underlying principle for such studies is to ascertain whether each individual is a case or a control (phenotyping), to assess each individual for the presence or absence of the genetic variant of interest (genotyping) and to then statistically compare the distribution of genotypes in the cases and in the controls. Differences in the distribution of genotypes between the cases and controls can be expressed as an odds ratio (OR) and a p-value.
Candidate gene approaches to AAA
The genetic predisposition to AAA has led to multiple groups examining variants in candidate genes of interest using classical hypothesis testing approaches. In these studies, a known genetic variant in or around a gene of interest is tested for association with disease. An example of this (and the potential for misinterpretation of small scale studies) is the examination of a genetic variant in the interleukin-10 (IL10) gene. IL-10 is a potent anti-inflammatory cytokine and genetic variants in the IL10 promoter region have been shown to affect cellular IL-10 production. Owing to the heavy inflammatory infiltrate seen in the aneurysmal aortic wall, it can be hypothesised that reduced IL-10 production may predispose an individual to AAA. In 2003 we analysed one such functional variant in the IL10 promoter region and identified that this was associated with AAA with an OR of 1.8.14 However, this analysis was based on a case-controlled study with only 100 individuals in each group. When this finding was taken forward and tested, the IL10 variant for association with AAA in a larger, independent study (389 AAA cases, 404 controls), adjusted for demographic covariates, no association was found.15
This scenario of positive identification of a variant in a small pilot study followed by failure to replicate the observed association in a larger study has been a consistent theme for hypothesis driven research studies focusing on a single gene or small number of genes. Candidate gene approaches have reported on nearly 100 different genes16 without identifying any associations convincingly worthy of extended study. This is likely to be due to the fact that candidate genes for analysis are chosen on the basis of our current knowledge of the pathobiology of AAA. The pathways and genes analysed are already implicated in AAA and so new genetic associations identified in these studies do not add to our knowledge to any significant extent.
An alternative approach to selecting candidate genes for analysis in genetic studies of AAA is to select targets for analysis based on risk loci for diseases that share the similar risks as AAA. The prime candidate diseases for shared pathways with AAA are hyperlipidaemias and atherosclerosis. Both choices raise issues since the evidence for the role of lipids in aneurysmogenesis is inconsistent and diabetes, while an important risk factor for atherosclerosis, is protective against AAA. Nevertheless, there is some evidence from such studies and some interesting observations.
We have conducted two studies examining risk loci for coronary artery disease in patients with AAA. In the first, we examined a risk locus for coronary artery disease on chromosome 9 (9p21), and demonstrated an association between this risk locus and AAA.17 In the second study, because of the sexual dimorphism exhibited by AAA, we examined Y-chromosome haplogroups associated with coronary artery disease. In white European males, haplogroup I is associated with an increased risk of coronary artery disease. When we examined Y-chromosome haplogroups in men with AAA, no association was found between haplogroup I and AAA, suggesting a difference in the genetic aetiology between coronary artery disease and AAA (unpublished data, courtesy of Dr LDS Bloomer, Postdoctoral Fellow at the University of Toronto). Taken together, these two studies suggest that there is some shared genetic risk between AAA and atherosclerosis but also some distinction between the two diseases.
Genomics
The emergence of new laboratory techniques, analytical technologies and knowledge at the start of this century enabled a total shift in the study of genetic diseases. Over a short period of time, the limitations of only being able to study a single gene (genetics) were overcome and it became possible to study the entire genome (genomics) in a single experiment.
The knowledge that enabled this transition was the mapping of common variation in the human genome in the International HapMap Project. By sequencing the entire genomes of a series of individuals, common (present in over 1% of individuals) variants in the genome were catalogued. In the majority, these variants were single nucleotide polymorphisms (SNPs – pronounced ‘snips’) (Fig 1). A SNP is a genetic variant where a single nucleotide is substituted for another. The vast majority of these SNPs are non-functional (ie they have no effect on the structure or function of known gene products). However, SNPs are fairly frequent in the human genome, occurring every 100 to 300 base pairs with nearly 10 million having now been catalogued. Since SNPs are inherited, they can be used as markers of an individual’s genetic variation.
Figure 1.
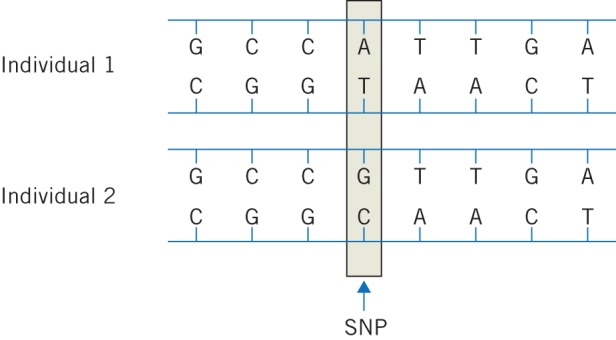
Diagrammatic representation of a single nucleotide polymorphism. The two individuals have identical deoxyribonucleic acid (DNA) sequences at this representative section of the genome with the exception of a single base change in the fourth base from the left, together with a corresponding change in the complementary strand of DNA.
The early mapping of SNPs occurred concurrently with advances in silicon chip-based laboratory genotyping assays that permitted many thousands of SNPs to be genotyped in an individual at one time, using low sample volumes and at relatively low cost. Combined with advancing computer power and storage solutions, these factors enabled the conduct of studies assessing a large proportion of common variation across the entire human genome for association with disease. This approach is termed a genome-wide association study (GWAS), and significant novel findings have been made in the understanding of the genetic aetiology of common diseases such as coronary artery disease, inflammatory bowel disease and diabetes.
Methodological aspects of genomic studies
The promise of GWAS is not without significant practical considerations to be overcome. Diseases that are chosen for GWAS are those, like AAA, where the genetic aetiology is multifactorial and where single gene defects have not usually been identified. This implies that the variants associated with disease are likely to be multiple and each will have a very modest effect on the risk of disease. Conducting a GWAS is essentially performing many hundreds of thousands of individual hypothesis tests (one for each SNP analysed) for association with the disease under study. Modern bioinformatic techniques permit the addition of computationally determined SNP genotypes to a set of SNP genotypes determined by direct laboratory testing (imputation), and modern GWASs usually analyse well in excess of 1 million SNPs and commonly up to 4 million.
Performing 1 million hypothesis tests based on a p-value threshold of 0.05 would result in 50,000 significant results to follow up, which is neither of practical use nor realistic, and many of these ‘significant’ findings will be false positives. The application of a simple Bonferroni correction for 1 million tests to a p-value threshold of 0.05 results in only p-values of <5 × 10-8 being considered statistically significant and it is this threshold that is usually considered to indicate ‘genome-wide significance’. In order to achieve such levels of significance given the low effect sizes of the causal variants for multifactorial genetic diseases, large sample sizes are required and case-controlled studies for GWAS usually analyse many thousands of individuals in both the case and control groups.
The necessity to analyse large samples in GWASs results in two main practical issues. The first is funding such studies. While genotyping costs per individual are relatively cheap (around £100 or less for 500,000 SNPs), the large sample size (thousands) necessary results in high overall costs. These costs can be limited through the use of publically available control sample data from previous studies such as the Wellcome Trust Case Control Consortium and the adoption of multistage experiments where an initial screen of a subset of individuals with whole-genome genotyping is followed up by taking limited numbers of positive findings through into a second stage replication experiment in which limited genotyping is performed.
The second main practical issue is cohort assembly. In order to obtain sample sizes of many thousands of cases and controls, it is usually necessary to work in collaborative groups, often over a wide geographical area, and the logistics of sample delivery to the genotyping centre and research agreements are not inconsiderable.
AAA GWASs
In Leicester, as a result of collecting our bank of deoxyribonucleic acid (DNA) samples that were used for our candidate gene studies, it became clear that we had developed a resource that could potentially be used for a GWAS of AAA. So as to obtain funding for such a study, it was necessary to form a collaborative group and demonstrate an overall sample resource of adequate size to enable a GWAS. This early group became known as the Aneurysm Consortium and following on from the initial GWASs funded by the Wellcome Trust, we were able to bid successfully for funding to conduct a GWAS of AAA in the second round of GWAS funding from the Wellcome Trust. Members and additional samples were added to the consortium through word of mouth and through publicising the project.18
In 2009 the first phase of our GWAS was initiated: the genotyping of 535,296 SNPs in 1,921 individuals with AAA. This was performed using SNP genotyping arrays covering between 80% and 90% of known genes. The data from these individuals were subjected to an extensive quality control process that removed data of 55 individuals owing to inadequate genotyping, sample duplication or failed quality checks such as sex mismatches, resulting in data being available from 1,866 individuals for analysis.
These data were compared with genotype data from 5,435 unscreened controls provided by the Wellcome Trust Case Control Consortium. The results of this primary analysis are shown in Figure 2. This analysis failed to demonstrate any genome-wide significant associations with AAA (those with p-values <5 × 10-8). However, there were nine regions where a SNP (or a group of SNPs) demonstrated a degree of association with AAA at a level that could be considered above the general background ‘noise’ at a p-value threshold of <1 × 10-5.
Figure 2.
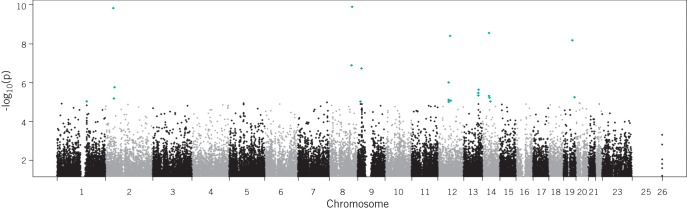
A Manhattan plot displaying the results of a genome-wide association study of abdominal aortic aneurysm (AAA). Each point represents a single nucleotide polymorphism (SNP) tested for association with AAA in a case-controlled study of 1,866 individuals with AAA and 5,435 healthy controls. The horizontal axis represents the position of the SNP on the chromosome indicated on the axis label. The vertical axis is the negative logarithmic value of the p-value for the association test. Higher values indicate a stronger degree of association in terms of p-value. Points in red are those SNPs with p-values of <1 × 10-5.
In order to confirm or refute the associations seen in our primary analysis, the 9 putative associations with AAA were taken, and the lead SNP was tested at each locus for association with AAA in a case-controlled study of 2,871 AAA cases and 32,687 controls from a variety of sources. Adjusting the results of this experiment using a Bonferroni correction for the nine hypothesis tests performed, we found that one SNP was associated significantly with AAA in this ‘replication’ study. The significant SNP was located on chromosome 12, in the gene encoding low density lipoprotein receptor related protein 1 (LRP1) (Fig 3).
Figure 3.

A ‘zoomed in’ Manhattan plot based on the data shown in Figure 2. The region shown is that region of chromosome 12 containing the LRP1 gene. The other genes in the region are shown below the horizontal axis and the regional recombination rate is shown on the right vertical axis. Below the main plot is the regional linkage disequilibrium (LD) plot. LD is a measure of the likelihood that SNPs are inherited together in blocks and this likelihood (LD) between two SNPs is displayed as the intensity of colour in the plot at the diagonal intersection of lines drawn down from pairs of SNPs.
In a further confirmatory follow-up study, the SNP identified in LRP1 was tested for association with AAA in an additional set of samples from 1,491 patients with AAA and 11,060 controls, and confirmed the association in this additional group of samples. When analysed together, data from the primary study, the replication study and the follow-up study (6,228 cases and 49,182 controls) demonstrated that LRP1 was associated with AAA at a genome-wide significant p-value of 4.5 × 10-10, with an OR of 1.15 (95% confidence interval: 1.10–1.21) per risk allele present, a finding reported nearly three years after starting work on the study.19 As part of this piece of work, data were also obtained from other GWASs relating to other cardiovascular diseases and found that the association in LRP1 was specific to AAA, with no association seen in studies of hyperlipidaemia, diabetes, hypertension or coronary artery disease.
One of the common observations in GWASs is that despite the strong combined evidence of genetic association as a result of performing multiple confirmatory studies, the effect size (OR) of variants identified is often small, as seen here. It is therefore always necessary to explore the biological plausibility of GWAS findings, either experimentally or from the literature. In our work, we not only demonstrated the genetic association of LRP1 with AAA but also that the SNP associated with AAA has an effect of LRP1 expression, probably through altered regulation of LRP1 transcription.19
In addition, further evidence was available in the literature. Serendipitously, data from murine models published prior to our GWAS had demonstrated that if LRP1 is selectively knocked out in vascular smooth muscle cells, the mice spontaneously develop aortic aneurysms,20 a finding recently confirmed and shown to be acting via increases in HtrA1,21 a serine protease not implicated previously in AAA. Furthermore, additional evidence published since our GWAS has demonstrated that human AAA tissue samples show low LRP1 expression.22 This is consistent with these animal studies and supports the hypothesis that reduction in LRP1 activity contributes to the development of AAA.
One of the advantages of performing a GWAS is that the whole-genome genotyping data contain valuable information on the entire genome. Additional value can be obtained from such datasets through the use of alternative analysis strategies and contribution to methodologically similar studies. In Figure 2 it can be seen that there are many regions of the genome that demonstrate association with AAA at less stringent p-value thresholds than the one we used to take candidate regions forward in our first GWAS. It is impractical to follow up all of these regions in further studies.
One method to select likely regions for further study is to not simply look at the degree of association of a single SNP in a region as a threshold for further study but to examine blocks of related SNPs for association with AAA. In an analysis performed by Bradley et al using a variation of this technique, we have identified an association of low density lipoprotein receptor (LDLR) with AAA at a genome-wide significant level.23 We have also used our GWAS dataset to provide in silico genotyping results to other investigators and we have contributed to the identification of a genetic variant in the sortilin gene (SORT1) associated with AAA.24
Discussion
Our genomic studies of AAA have led to the identification of three novel, robustly identified genetic risk loci for AAA. The genes at these loci (LRP1, LDLR and SORT1) represent new candidate pathways for exploring the pathogenesis of AAA.
LRP1 is a particularly plausible candidate for contributing to the development of AAA and it would appear that loss of LRP1 function is the biological effect contributing to aneurysmogenesis, a hypothesis supported by data from human and murine studies.20–22 LRP1 is a large transmembrane receptor that binds and facilitates the endocytosis and degradation of many biological molecules implicated in extracellular matrix degradation such as matrix metalloproteinase 9 and thrombospondin 2 (Fig 4). LRP1 plays a role in the migration and proliferation of vascular smooth muscle cells, the loss of which is a hallmark of AAA.25 Loss of LRP1 may lead to reduced tissue clearance of proteolytic enzymes and an inability to institute vascular smooth muscle cell mediated reparative processes. However, both of these hypotheses remain to be explored. The observation that LRP1 is not related to other cardiovascular diseases and may be an AAA specific disease locus is of particular interest.
Figure 4.

Diagrammatic representation of low density lipoprotein receptor related protein 1 (LRP1) indicating known ligands and their binding sites (where known)
LDLR and SORT1 both play roles in lipid metabolism. The genetic variants identified to be associated with AAA at both loci are also associated with lipid traits and cardiovascular disease.26–28 Nevertheless, analysis of our data, adjusted for lipid levels, demonstrates that these loci are associated independently with AAA. While associations between lipid levels and the risk of AAA is uncertain,29 our findings provide evidence to support further clinical studies of these relationships.
Joint consideration of the three disease loci we have identified (LRP1, LDLR and SORT1) leads to some interesting observations regarding the interactions between these pathways. LRP1 and LDLR are both cell surface low density lipoprotein (LDL)-cholesterol receptors, which, taken together, suggests that aberrations in cellular LDL-cholesterol clearance/utilisation may play a role in the development of AAA. Our genomic data also suggest some intriguing pathways for exploration outside of the traditional functions of the gene products implicated in our GWAS. LRP1 and SORT1 interact with each other in the pathway responsible for the trafficking of the GLUT4 glucose transporter to the cell surface in response to insulin stimulation.30 Given the negative association between diabetes and AAA/AAA growth, it is tempting to hypothesise that this pathway may be involved in AAA development and progression.
Conclusions
Overall, genomic studies of AAA have resulted in the generation of new hypotheses for future research. It is likely that further mining of the available genomic data on AAA through meta-analysis will yield additional risk loci. The challenge for aneurysm biologists in using these data is to move forwards from the identification of genomic risk loci into mechanistic laboratory and clinical studies, thereby ensuring that the high cost of these studies results in true advances in the understanding of disease and the development of new therapeutic strategies for AAA.
Acknowledgements
MJB is funded by the Circulation Foundation and holds a Higher Education Funding Council for England clinical senior lecturer fellowship. Parts of the work reported in this article were funded by the Dunhill Medical Trust, the Garfield Weston Trust for Research into Heart Surgery, the National Health and Medical Research Council (Australia) and the EU’s Seventh Framework Programme Fighting Aneurysmal Disease project grant agreement. The Aneurysm Consortium GWAS makes use of data generated by the Wellcome Trust Case Control Consortium. A full list of the investigators who contributed to the generation of the data is available from http://www.wtccc.org.uk/. Further funding for the GWAS was provided by the Wellcome Trust under awards 076113 and 085475.
Much of the work reported in this article is based on worldwide collaboration between multiple investigators. A list of individuals who contributed to the generation of some of the data presented above is given in Appendix 1 (available online only).
Appendix 1.
List of individuals who contributed to the generation of some of the data presented in this Hunterian lecture
| DT Bradley; Centre for Public Health, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, UK |
| AE Hughes; Centre for Public Health, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, UK |
| SA Badger; Centre for Public Health, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, UK |
| GT Jones; Department of Surgical Sciences, University of Otago, Dunedin, New Zealand |
| SC Harrison; Centre for Cardiovascular Genetics, Institute of Cardiovascular Science, University College London, UK |
| BJ Wright; Department of Cardiovascular Sciences, University of Leicester, UK, and Leicester Cardiovascular Biomedical Research Unit, Glenfield Hospital, Leicester, UK |
| S Bumpstead; Genetics of Complex Traits in Humans Group, Wellcome Trust Sanger Institute, Cambridge, UK |
| AF Baas; Research Section, Department of Medical Genetics, University Medical Centre Utrecht, Netherlands |
| S Gretarsdottir; deCODE Genetics, Reykjavik, Iceland, and Faculty of Medicine, University of Iceland, Reykjavik, Iceland |
| K Burnand; Academic Department of Surgery, King’s College London, UK, British Heart Foundation Centre of Research Excellence, London, UK, and Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London, UK |
| AH Child; Department of Cardiac and Vascular Sciences, St George’s, University of London, UK |
| RE Clough; Academic Department of Surgery, King’s College London, UK, British Heart Foundation Centre of Research Excellence, London, UK, and Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London, UK |
| G Cockerill; Department of Vascular Surgery, St George’s, University of London, UK |
| H Hafez; Department of Vascular Surgery, St Richard’s Hospital, Chichester, UK |
| DJA Scott; Division of Cardiovascular and Diabetes Research, Leeds Institute of Genetics, Health and Therapeutics, University of Leeds, UK |
| RAS Ariëns; Division of Cardiovascular and Diabetes Research, Leeds Institute of Genetics, Health and Therapeutics, University of Leeds, UK |
| A Johnson; Division of Cardiovascular and Diabetes Research, Leeds Institute of Genetics, Health and Therapeutics, University of Leeds, UK |
| S Sohrabi; Division of Cardiovascular and Diabetes Research, Leeds Institute of Genetics, Health and Therapeutics, University of Leeds, UK |
| A Smith; Academic Department of Surgery, King’s College London, UK, British Heart Foundation Centre of Research Excellence, London, UK, and Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London, UK |
| MM Thompson; St George’s Healthcare Vascular Institute, St George’s Hospital, London, UK |
| FM van Bockxmeer; School of Surgery, University of Western Australia, Crawley, Australia |
| M Waltham; Academic Department of Surgery, King’s College London, UK, British Heart Foundation Centre of Research Excellence, London, UK, and Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London, UK |
| SE Matthiasson; Laekning Medical Clinics, Reykjavik, Iceland |
| G Thorleifsson; deCODE Genetics, Reykjavik, Iceland, and Faculty of Medicine, University of Iceland, Reykjavik, Iceland |
| U Thorsteinsdottir; deCODE Genetics, Reykjavik, Iceland, and Faculty of Medicine, University of Iceland, Reykjavik, Iceland |
| JD Blankensteijn; Department of Vascular Surgery, VU University Medical Centre, Amsterdam, Netherlands |
| JAW Teijink; Department of Vascular Surgery, Catharina Hospital, Eindhoven, Netherlands |
| C Wijmenga; Department of Genetics, University Medical Centre Groningen, Netherlands |
| J de Graaf; Department of Internal Medicine, Radboud University Medical Centre, Nijmegen, Netherlands |
| LA Kiemeney; Department for Health Evidence and Department of Urology, Radboud University Medical Centre, Nijmegen, Netherlands |
| JB Wild; Department of Cardiovascular Sciences, University of Leicester, UK, and Leicester Cardiovascular Biomedical Research Unit, Glenfield Hospital, Leicester, UK |
| S Edkins; Genetics of Complex Traits in Humans Group, Wellcome Trust Sanger Institute, Cambridge, UK |
| R Gwilliam; Genetics of Complex Traits in Humans Group, Wellcome Trust Sanger Institute, Cambridge, UK |
| SE Hunt; Genetics of Complex Traits in Humans Group, Wellcome Trust Sanger Institute, Cambridge, UK |
| S Potter; Genetics of Complex Traits in Humans Group, Wellcome Trust Sanger Institute, Cambridge, UK |
| JS Lindholt; Department of Vascular Surgery, Viborg Hospital, Denmark |
| J Golledge; Queensland Research Centre for Peripheral Vascular Disease, James Cook University, Townsville, Australia |
| PE Norman; School of Surgery, University of Western Australia, Crawley, Australia |
| A van Rij; Department of Surgical Sciences, University of Otago, Dunedin, New Zealand |
| JT Powell; Department of Surgery and Cancer, Imperial College London, UK |
| P Eriksson; Atherosclerosis Research Unit, Centre for Molecular Medicine, Karolinska Institutet, Stockholm, Sweden |
| K Stefansson; deCODE Genetics, Reykjavik, Iceland, and Faculty of Medicine, University of Iceland, Reykjavik, Iceland |
| JR Thompson; Department of Health Sciences, University of Leicester, UK |
| SE Humphries; Centre for Cardiovascular Genetics, Institute of Cardiovascular Science, University College London, UK |
| RD Sayers; Department of Cardiovascular Sciences, University of Leicester, UK, and Leicester Cardiovascular Biomedical Research Unit, Glenfield Hospital, Leicester, UK |
| P Deloukas; Genetics of Complex Traits in Humans Group, Wellcome Trust Sanger Institute, Cambridge, UK |
| NJ Samani; Department of Cardiovascular Sciences, University of Leicester, UK, and Leicester Cardiovascular Biomedical Research Unit, Glenfield Hospital, Leicester, UK |
References
- 1.Stather PW, Sidloff DA, Rhema IA et al. A review of current reporting of abdominal aortic aneurysm mortality and prevalence in the literature. Eur J Vasc Endovasc Surg 2014; 47: 240–242. [DOI] [PubMed] [Google Scholar]
- 2.Thompson SG, Brown LC, Sweeting MJ et al. Systematic review and meta-analysis of the growth and rupture rates of small abdominal aortic aneurysms: implications for surveillance intervals and their cost-effectiveness. Health Technol Assess 2013; 17: 1–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sidloff D, Stather P, Dattani N et al. Aneurysm global epidemiology study: public health measures can further reduce abdominal aortic aneurysm mortality. Circulation 2014; 129: 747–753. [DOI] [PubMed] [Google Scholar]
- 4.Karlsson L, Gnarpe J, Bergqvist D et al. The effect of azithromycin and Chlamydophilia pneumonia infection on expansion of small abdominal aortic aneurysms – a prospective randomized double-blind trial. J Vasc Surg 2009; 50: 23–29. [DOI] [PubMed] [Google Scholar]
- 5.Propanolol Aneurysm Trial Investigators. Propranolol for small abdominal aortic aneurysms: results of a randomized trial. J Vasc Surg 2002; 35: 72–79. [DOI] [PubMed] [Google Scholar]
- 6.Sweeting MJ, Thompson SG, Brown LC et al. Use of angiotensin converting enzyme inhibitors is associated with increased growth rate of abdominal aortic aneurysms. J Vasc Surg 2010; 52: 1–4. [DOI] [PubMed] [Google Scholar]
- 7.Twine CP, Williams IM. Systematic review and meta-analysis of the effects of statin therapy on abdominal aortic aneurysms. Br J Surg 2011; 98: 346–353. [DOI] [PubMed] [Google Scholar]
- 8.CRD007 for the Treatment of Abdominal Aorta Aneurysm (The AORTA Trial). ClinicalTrials.gov. http://clinicaltrials.gov/show/NCT01354184 (cited May 2014).
- 9.Clifton MA. Familial abdominal aortic aneurysms. Br J Surg 1977; 64: 765–766. [DOI] [PubMed] [Google Scholar]
- 10.Kuivaniemi H, Shibamura H, Arthur C et al. Familial abdominal aortic aneurysms: collection of 233 multiplex families. J Vasc Surg 2003; 37: 340–345. [DOI] [PubMed] [Google Scholar]
- 11.Powell JT, Greenhalgh RM. Multifactorial inheritance of abdominal aortic aneurysm. Eur J Vasc Surg 1987; 1: 29–31. [DOI] [PubMed] [Google Scholar]
- 12.Majumder PP, Jean PL St, Ferrell RE et al. On the inheritance of abdominal aortic aneurysm. Am J Hum Genet 1991; 48: 164–170. [PMC free article] [PubMed] [Google Scholar]
- 13.Wahlgren CM, Larsson E, Magnusson PK et al. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J Vasc Surg 2010; 51: 3–7. [DOI] [PubMed] [Google Scholar]
- 14.Bown MJ, Burton PR, Horsburgh T et al. The role of cytokine gene polymorphisms in the pathogenesis of abdominal aortic aneurysms: a case-control study. J Vasc Surg 2003; 37: 999–1,005. [DOI] [PubMed] [Google Scholar]
- 15.Bown MJ, Lloyd GM, Sandford RM et al. The interleukin-10–1082 ‘A’ allele and abdominal aortic aneurysms. J Vasc Surg 2007; 46: 687–693. [DOI] [PubMed] [Google Scholar]
- 16.Hinterseher I, Tromp G, Kuivaniemi H. Genes and abdominal aortic aneurysm. Ann Vasc Surg 2011; 25: 388–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bown MJ, Braund PS, Thompson J et al. Association between the coronary artery disease risk locus on chromosome 9p21.3 and abdominal aortic aneurysm. Circ Cardiovasc Genet 2008; 1: 39–42. [DOI] [PubMed] [Google Scholar]
- 18.Aneurysm Consortium. Genome wide association studies: identifying the genes that determine the risk of abdominal aortic aneurysm. Eur J Vasc Endovasc Surg 2008; 36: 395–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bown MJ, Jones GT, Harrison SC et al. Abdominal aortic aneurysm is associated with a variant in low-density lipoprotein receptor-related protein 1. Am J Hum Genet 2011; 89: 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boucher P, Gotthardt M, Li WP et al. LRP: role in vascular wall integrity and protection from atherosclerosis. Science 2003; 300: 329–332. [DOI] [PubMed] [Google Scholar]
- 21.Muratoglu SC, Belgrave S, Hampton B et al. LRP1 protects the vasculature by regulating levels of connective tissue growth factor and HtrA1. Arterioscler Thromb Vasc Biol 2013; 33: 2,137–2,146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan CY, Chan YC, Cheuk BL, Cheng SW. A pilot study on low-density lipoprotein receptor-related protein-1 in Chinese patients with abdominal aortic aneurysm. Eur J Vasc Endovasc Surg 2013; 46: 549–556. [DOI] [PubMed] [Google Scholar]
- 23.Bradley DT, Hughes AE, Badger SA et al. A variant in LDLR is associated with abdominal aortic aneurysm. Circ Cardiovasc Genet 2013; 6: 498–504. [DOI] [PubMed] [Google Scholar]
- 24.Jones GT, Bown MJ, Gretarsdottir S et al. A sequence variant associated with sortilin-1 (SORT1) on 1p13.3 is independently associated with abdominal aortic aneurysm. Hum Mol Genet 2013; 22: 2,941–2,947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wild JB, Stather PW, Sylvius N et al. Low density lipoprotein receptor related protein 1 and abdominal aortic aneurysms. Eur J Vasc Endovasc Surg 2012; 44: 127–132. [DOI] [PubMed] [Google Scholar]
- 26.Schunkert H, König IR, Kathiresan S et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 2011; 43: 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teslovich TM, Musunuru K, Smith AV et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010; 466: 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willer CJ, Sanna S, Jackson AU et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 2008; 40: 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golledge J, van Bockxmeer F, Jamrozik K et al. Association between serum lipoproteins and abdominal aortic aneurysm. Am J Cardiol 2010; 105: 1,480–1,484. [DOI] [PubMed] [Google Scholar]
- 30.Bogan JS. Regulation of glucose transporter translocation in health and diabetes. Annu Rev Biochem 2012; 81: 507–532. [DOI] [PubMed] [Google Scholar]