

ABSTRACT
Human metapneumovirus (hMPV) is a member of the Pneumovirinae subfamily in the Paramyxoviridae family that causes respiratory tract infections in humans. Unlike members of the Paramyxovirinae subfamily, the polymerase complex of pneumoviruses requires an additional cofactor, the M2-1 protein, which functions as a transcriptional antitermination factor. The M2-1 protein was found to incorporate zinc ions, although the specific role(s) of the zinc binding activity in viral replication and pathogenesis remains unknown. In this study, we found that the third cysteine (C21) and the last histidine (H25) in the zinc binding motif (CCCH) of hMPV M2-1 were essential for zinc binding activity, whereas the first two cysteines (C7 and C15) play only minor or redundant roles in zinc binding. In addition, the zinc binding motif is essential for the oligomerization of M2-1. Subsequently, recombinant hMPVs (rhMPVs) carrying mutations in the zinc binding motif were recovered. Interestingly, rhMPV-C21S and -H25L mutants, which lacked zinc binding activity, had delayed replication in cell culture and were highly attenuated in cotton rats. In contrast, rhMPV-C7S and -C15S strains, which retained 60% of the zinc binding activity, replicated as efficiently as rhMPV in cotton rats. Importantly, rhMPVs that lacked zinc binding activity triggered high levels of neutralizing antibody and provided complete protection against challenge with rhMPV. Taken together, these results demonstrate that zinc binding activity is indispensable for viral replication and pathogenesis in vivo. These results also suggest that inhibition of zinc binding activity may serve as a novel approach to rationally attenuate hMPV and perhaps other pneumoviruses for vaccine purposes.
IMPORTANCE The pneumoviruses include many important human and animal pathogens, such as human respiratory syncytial virus (hRSV), hMPV, bovine RSV, and avian metapneumovirus (aMPV). Among these viruses, hRSV and hMPV are the leading causes of acute respiratory tract infection in infants and children. Despite major efforts, there is no antiviral or vaccine to combat these diseases. All known pneumoviruses encode a zinc binding protein, M2-1, which is a transcriptional antitermination factor. In this work, we found that the zinc binding activity of M2-1 is essential for virus replication and pathogenesis in vivo. Recombinant hMPVs that lacked zinc binding activity were not only defective in replication in the upper and lower respiratory tract but also triggered a strong protective immunity in cotton rats. Thus, inhibition of M2-1 zinc binding activity can lead to the development of novel, live attenuated vaccines, as well as antiviral drugs for pneumoviruses.
INTRODUCTION
Human metapneumovirus (hMPV), first characterized in 2001 in the Netherlands (1), belongs to the Metapneumovirus genus within the Pneumovirinae subfamily of the Paramyxoviridae family. Since its discovery, hMPV has been isolated from individuals of all ages with acute respiratory tract infection, especially from infants, children, the elderly, and immunocompromised individuals (2). hMPV infection is currently recognized as a leading cause of respiratory tract infection in the first years of life, with symptoms similar to those of human respiratory syncytial virus (hRSV) infection (3–6). Epidemiological studies suggest that 5 to 15% of all respiratory tract infections in infants and young children are caused by hMPV, a proportion second only to that of hRSV (7). Other important pneumoviruses include avian metapneumovirus (aMPV), pneumonia virus of mice (PVM), and bovine RSV, which cause respiratory tract infections in animals. Together, pneumoviruses are the major causative agents of respiratory tract infection in humans and animals. Currently, there is no vaccine for most pneumoviruses, particularly hRSV and hMPV.
The genome of hMPV is a nonsegmented negative-sense (NNS) RNA, with a size ranging from 13,280 to 13,378 nucleotides, and contains 8 genes which encode 9 proteins in the order of 3′-N-P-M-F-M2-SH-G-L-5′ (4, 6). The M2 gene of hMPV contains two overlapping open reading frames (ORFs), M2-1 and M2-2. Like all NNS RNA viruses, the genomic RNA is completely encapsidated by nucleoprotein (N), forming the N-RNA complex that serves as a template for genome replication and mRNA transcription (8). During replication, the RNA-dependent RNA polymerase (RdRp) enters at the extreme 3′ end of the genome and synthesizes full-length complementary antigenome, which in turn serves as the template for synthesis of the full-length progeny genome. During transcription, RdRp copies the genomic RNA template to synthesize a short, uncapped leader RNA and capped, methylated, and polyadenylated mRNAs that encode all the viral proteins (8). The components of RdRp of the Paramyxovirinae subfamily (family Paramyxoviridae), Rhabdoviridae, and Bornaviridae include the large (L) protein catalytic subunit and phosphoprotein (P) cofactor (8). The RdRp of the Pneumovirinae subfamily of the Paramyxoviridae family requires an additional cofactor, the M2-1 protein (9, 10), whereas the polymerase of Ebola virus within the Filoviridae family requires VP30 as an additional cofactor (11–13). Interestingly, both the M2-1 protein of pneumoviruses and VP30 protein of filoviruses are typical zinc binding proteins that are thought to play many critical regulatory roles in RNA synthesis and processing via poorly understood mechanisms (11, 13–16).
The M2-1 protein is unique to all known pneumoviruses. Our current understanding of the functions of M2-1 proteins comes predominantly from studies of the hRSV M2-1 protein. The hRSV M2-1 functions as a transcriptional elongation factor and antiterminator that enhances readthrough of intergenic junctions (14, 15, 17). Thus, M2-1 is essential for the synthesis of full-length mRNAs and polycistronic mRNAs (14, 15, 17). The hRSV M2-1 was found to be an RNA binding protein, although RNA binding specificity is controversial (18). Recent nuclear magnetic resonance (NMR) studies showed that the RSV M2-1 core domain preferentially recognizes poly(A) tails of viral mRNAs (19). Thus, M2-1 likely binds nascent mRNA transcripts, preventing premature termination through stabilization of the transcription complex and inhibition of RNA secondary structure formation (15, 19, 20). In addition, hRSV M2-1 was shown to interact with the P protein and colocalize with the N and L proteins (21, 22). Extensive deletions in M2-1, as well as single amino acid (aa) mutations in the zinc binding motif, were lethal to hRSV (9, 23, 24), suggesting that the M2-1 protein is absolutely essential for the hRSV life cycle.
The hMPV M2-1 protein is a basic protein of 187 amino acids, with a theoretical pI of 9.14 (http://web.expasy.org/compute_pi/). In contrast to hRSV, the M2-1 protein of the Metapneumovirus genus might play different roles in the virus life cycle. Unlike hRSV, hMPV M2-1 was found to be dispensable for virus viability or replication in tissue culture (25). Specifically, recombinant hMPV (rhMPV) lacking the M2-1 gene was recovered and highly attenuated in replication in vitro and in vivo but was not immunogenic and not capable of inducing protective immunity in animal models (25). This suggests that rhMPV lacking the M2-1 gene was overly attenuated in vivo such that it failed to induce a robust immune response. The M2-1 protein of aMPV, which shares 85% amino acid identity with that of hMPV M2-1, was shown to increase minigenome expression by at least 100-fold (26). Thus, unlike the hRSV M2-1 protein, it appears that hMPV M2-1 is not absolutely essential for transcriptional processivity (25, 27).
Sequence alignment found that M2-1 proteins of all known pneumoviruses possess a putative zinc binding motif (CCCH) (Fig. 1). Most recently, the crystal structures of both the hRSV and hMPV M2-1 proteins have been solved. The M2-1 protein of hRSV forms a disk-like symmetrical tetramer, which is driven by a long helix forming a four-helix bundle at its center and stabilized by contact between the zinc-binding domain and adjacent protomers (Fig. 2A) (28). Each M2-1 monomer forms three distinct regions, including the zinc binding domain, the tetramerization helix, and the core domain, which are linked by unstructured or flexible sequences (Fig. 2B). In contrast to the hRSV M2-1 structure, the hMPV M2-1 protein forms an asymmetric tetramer, in which three of the protomers exhibit a closed conformation and one forms an open conformation (Fig. 2C) (29). Each protomer is composed of an N-terminal zinc finger domain and an α-helical tetramerization motif forming a rigid unit, followed by a flexible linker and an α-helical core domain (Fig. 2D). Despite these significant discoveries, the biological roles of the zinc binding motif in the maintenance of M2-1 functions, viral replication, and pathogenesis in vivo are still poorly understood.
FIG 1.

Sequence alignment of pneumovirus M2-1 proteins. Representative members of pneumoviruses include human metapneumovirus subtype A (hMPV-A; gi 215794520), hMPV-B (gi 215794505), avian metapneumovirus subtype A (aMPV-A; gi 49823139), aMPV-B (gi 310772463), aMPV-C (gi 237847064), human respiratory virus type A (hRSV-A; gi 333959), hRSV-B (gi 2582031), bovine respiratory syncytial virus (BRSV; gi 210823), and pneumonia virus of mice (PVM; GenInfo Identifier [gi] 56900724). Fully conserved residues are highlighted by red boxes, conserved substitutions are indicated by red letters, and black letters mean no match. The dot indicates the number of amino acids. Zinc binding domain (aa 7 to 31) and oligomerization domain (aa 32 to 53) are indicated by an underline. Zinc binding motif (CCCH) is indicated by an arrow.
FIG 2.

Structural comparison of hMPV and RSV M2-1 protein. (A) hRSV M2-1 tetramer. Structure was generated using PDB identifier (ID) 4C3D. (B) hRSV M2-1 monomer. Oligomerization domain is highlighted in red. (C) hMPV M2-1 tetramer structure was generated using PDB ID 4CS7. (D) hMPV M2-1 monomer. The predicted oligomerization domain is highlighted in red.
In this study, we found that the last two amino acid residues (C21 and H25) in the zinc binding motif in hMPV M2-1 are essential for zinc binding activity and that the amino acids in the zinc binding motif are essential for oligomerization of the M2-1 protein. Unlike RSV, rhMPVs lacking zinc binding activity were successfully recovered from infectious cDNA clones. Interestingly, rhMPVs lacking zinc binding activity were attenuated not only in an in vitro cell culture system but also in viral replication in the upper and lower respiratory tract of cotton rats. Importantly, unlike rhMPV lacking the entire M2-1 gene, cotton rats immunized with rhMPVs lacking zinc binding activity triggered a high level of neutralizing antibody and were completely protected from challenge with wild-type (WT) rhMPV.
MATERIALS AND METHODS
Ethics statement.
The animal study was conducted in strict accordance with USDA regulations and the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council (30) and was approved by The Ohio State University Institutional Animal Care and Use Committee (IACUC; animal protocol no. 2009A0221). The animal care facilities at The Ohio State University are AAALAC accredited. Every effort was made to minimize potential distress, pain, or discomfort to the animals throughout all experiments.
Cell lines.
LLC-MK2 (ATCC CCL-7) cells were maintained in Opti-MEM medium (Life Technologies, Bethesda, MD) supplemented with 2% fetal bovine serum (FBS). Vero E6 cells (ATCC CRL-1586) and BHK-SR19-T7 cells (kindly provided by Apath, LLC, Brooklyn, NY) were grown in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% FBS. The medium for the BHK-SR19-T7 cells was supplemented with 10 μg/ml puromycin (Life Technologies) during every other passage to select for T7 polymerase-expressing cells.
Plasmids and site-directed mutagenesis.
Plasmids encoding the hMPV minigenome, the full-length genomic cDNA of hMPV strain NL/1/00, and support plasmids expressing the hMPV N protein (pCITE-N), P protein (pCITE-P), L protein (pCITE-L), and M2-1 protein (pCITE-M2-1) were kindly provided by Ron A. M. Fouchier at the Department of Virology, Erasmus Medical Center, Rotterdam, the Netherlands. The F cleavage site in the genome of hMPV NL/1/00 was modified to a trypsin-independent F cleavage site, as described previously (31, 32). The M2-1 mutants of hMPV were generated by site-directed mutagenesis using the QuikChange methodology (Stratagene, La Jolla, CA). All constructs were sequenced at The Ohio State University Plant Microbe Genetics Facility to confirm the presence of the introduced mutations.
Expression and purification of recombinant hMPV M2-1 protein from Escherichia coli.
The hMPV M2-1 gene was PCR amplified from a cDNA clone of strain NL/1/00 and was inserted into the Escherichia coli expression system pGEX-4T-1 vector at BamHI and NotI sites. To facilitate the protein purification, a glutathione S-transferase (GST) fusion protein was fused to the N terminus of the M2-1 gene. The resulting plasmids were transformed into E. coli Rosetta(DE3) and grown at 37°C until the absorbance at 600 nm reached 0.6 to 0.8. Then, the cells were chilled on ice for 10 min and protein expression was induced by the addition of 20 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) and 75 μM ZnSO4. The cells were grown for an additional 20 h at 25°C and harvested by centrifugation at 5,000 × g for 10 min. Bacterial pellets were resuspended in lysis buffer (40 mM Tris-HCl [pH 7.4], 1.0 M NaCl, 0.5 mM dithiothreitol [DTT], and 20 μM ZnSO4) supplemented with 1 mg/ml of lysozyme and protease inhibitor cocktail (Roche, Mannheim, Germany). After 30 min of incubation on ice, the cells were lysed using sonication, followed by centrifugation at 15,000 × g at 4°C for 40 min. The supernatant was collected and loaded into a column containing 5 ml of glutathione HiCap matrix (Qiagen). The G resins were washed with 150 ml of resuspension buffer, followed by being washed with 100 ml of the cleavage buffer (50 mM Tris-HCl [pH 7.6], 2.5 mM CaCl2, 150 mM NaCl, and 0.2 mM DTT). To isolate GST-free M2-1, 20 ml of the cleavage buffer containing 2 units/ml of thrombin (Sigma, St. Louis, MO) was loaded into the column and incubated at room temperature for 8 h. Cleavage was seized by adding 4-amidinophenylmethanesulfonyl fluoride hydrochloride (APMSF) (Sigma). Similarly, the GST fusion hMPV M2-1 mutants C7S, C15S, C21S, and H25L were generated. The purified hMPV M2-1 proteins were dialyzed against phosphate-buffered saline (PBS) buffer containing 300 mM NaCl and 10 μM ZnSO4. Protein concentration was determined by the Bradford assay (Sigma).
Circular dichroism (CD) spectroscopy.
Recombinant hMPV M2-1 protein was dissolved in buffer (40 mM Tris-HCl [pH 7.4], 10 μM ZnCl2) and dialyzed against this same buffer at 4°C overnight. A total of 2 μM M2-1 protein was incubated with increasing concentrations of EDTA and analyzed by an AVIV far-UV spectropolarimeter at 20°C using a quartz microcell of 0.1-cm path length with a wavelength range of 190 to 260 nm at a scanning speed of 50 nm per minute. Ten individual readings were recorded and averaged. After subtraction of the baseline, the spectra were mathematically transformed into molar ellipticity.
Colorimetric determination of the zinc content.
Purified hMPV M2-1 was dialyzed against 50 mM PBS (pH 7.0) containing 0.3 M NaCl at 4°C overnight. M2-1 proteins at concentrations of 2 μM, 3 μM, and 4 μM in 1 ml of solution containing 100 μM 4-(2-pyridylazo)resorcinol (PAR) were incubated at 25°C for 20 min, and the absorbance at 500 nm was monitored for 5 min. Upon addition of 100 μM p-chloromercuriphenylsulfonic acid (PMPS), the release of strongly bound Zn2+ was monitored. The amount of Zn2+ bound to hMPV M2-1 was quantified by comparing the sample readings to a standard curve generated using 2 μM, 3 μM, and 4 μM ZnSO4 (Sigma). Buffer containing 50 mM PBS (pH 7.0), 0.3 M NaCl, and 100 μM PAR was used as the blank control. The zinc ion content of GST–M2-1 and mutants were measured at 2 μM and compared with rM2-1 at the same zinc concentration.
Glutaraldehyde cross-linking.
A total of 1.2 μg of rM2-1, GST–M2-1, or mutant proteins was diluted in 20 μl of reaction buffer (50 mM PBS [pH 7.0], 300 mM NaCl) and incubated with increasing amounts of glutaraldehyde from 0 to 2% at 25°C for 30 s. The reaction was stopped by the addition of Tris-HCl (pH 7.4) at a final concentration of 50 mM. Subsequently, the cross-linked products were analyzed by 12% SDS-PAGE, and the proteins were visualized by Coomassie blue staining.
Recovery of rhMPVs from the full-length cDNA clones.
rhMPVs were rescued using a reverse genetics system as described previously (27, 31). Briefly, BHK-SR19-T7 cells (kindly provided by Apath LLC), which stably express T7 RNA polymerase, were transfected with 5.0 μg of plasmid phMPV carrying the full-length hMPV genome, 2.0 μg of pCITE-N, 2.0 μg of pCITE-P, 1.0 μg of pCITE-L, and 1.0 μg of pCITE-M2-1 using Lipofectamine 2000 (Life Technologies). At day 6 posttransfection, the cells were harvested using scrapers and were cocultured with LLC-MK2 cells at 50 to 60% confluence. When extensive cytopathic effects (CPE) were observed, the cells were subjected to three freeze-thaw cycles, followed by centrifugation at 3,000 × g for 10 min. The supernatant was subsequently used to infect new LLC-MK2 cells. The successful recovery of the rhMPVs was confirmed by immunostaining, agarose overlay plaque assay, and reverse transcription (RT)-PCR.
Immunostaining plaque assay.
Vero E6 cells were seeded in 24-well plates and infected with serial dilutions of rhMPV. At day 6 postinfection, the supernatant was removed and cells were fixed in a prechilled acetone-methanol solution at room temperature for 15 min. Cells were permeabilized in PBS containing 0.4% Triton X-100 at room temperature for 10 min and blocked at 37°C for 1 h using 1% bovine serum albumin (BSA) in PBS. The cells were then labeled with an anti-hMPV N protein primary monoclonal antibody (Millipore, Billerica, MA) at a dilution of 1:1,000, followed by incubation with horseradish peroxidase (HRP)-labeled rabbit anti-mouse secondary antibody (Thermo Scientific, Waltham, MA) at a dilution of 1:5,000. After incubation with 3-amino-9-ethylcarbazole (AEC) chromogen substrate (Sigma, St. Louis, MO), positive cells were visualized under a microscope. The viral titer was calculated as the number of PFU per milliliter.
Viral replication kinetics in LLC-MK2 cells.
Confluent LLC-MK2 cells in 35-mm dishes were infected with wild-type (WT) rhMPV or mutant rhMPV at a multiplicity of infection (MOI) of 0.01. After 1 h of adsorption, the inoculum was removed and the cells were washed three times with PBS. Fresh DMEM (supplemented with 2% FBS) was added, and the infected cells were incubated at 37°C. At different time points postinfection, the supernatant and cells were harvested by three freeze-thaw cycles, followed by centrifugation at 1,500 × g at room temperature for 15 min. The virus titer was determined by an immunostaining assay in Vero E6 cells.
Replication and pathogenesis of rhMPV in cotton rats.
Twenty five 4-week-old female specific-pathogen-free (SPF) cotton rats (Harlan Laboratories, Indianapolis, IN) were randomly divided into five groups (5 cotton rats per group). Prior to virus inoculation, the cotton rats were anesthetized with isoflurane. The cotton rats in group 1 were inoculated with 1.0 × 106 PFU of WT rhMPV and served as positive controls. The cotton rats in groups 2 to 5 were inoculated with 1.0 × 106 PFU of four rhMPV mutants (rhMPV-C7S, -C15S, -C21S, and -H25L strains). The cotton rats in group 6 were mock infected with 0.1 ml of Opti-MEM medium and served as uninfected controls. Each cotton rat was inoculated intranasally with a volume of 100 μl. After inoculation, the animals were evaluated on a daily basis for mortality and the presence of any respiratory symptoms. At day 4 postinfection, the cotton rats were sacrificed, and lungs and nasal turbinates were collected for both virus isolation and histological analysis.
Immunogenicity of rhMPVs in cotton rats.
Twenty five cotton rats (Harlan Laboratories, Indianapolis, IN) were randomly divided into five groups (5 cotton rats per group). The cotton rats in group 1 were mock infected with Opti-MEM medium and used as an infected control, and those in groups 2 to 4 were intranasally inoculated with 1.0 × 106 PFU of the WT rhMPV, rhMPV-C21S, or rhMPV-H25L strain in 0.1 ml Opti-MEM medium. The cotton rats in group 5 were inoculated with DMEM and served as the unimmunized challenged control. After immunization, the cotton rats were evaluated daily for mortality and the presence of any symptoms of hMPV infection. Blood samples were collected from each rat weekly by facial vein retro-orbital bleeding, and serum was isolated for neutralizing antibody detection. At week 4 postimmunization, the cotton rats in groups 1 to 4 were challenged intranasally with wild-type rhMPV at a dose of 1.0 × 106 PFU per cotton rat. After challenge, the animals were evaluated twice every day for mortality and the presence of any symptoms of hMPV infection. At day 4 postchallenge, all cotton rats from each group were euthanized by CO2 asphyxiation. The lungs and nasal turbinates from each cotton rat were collected for virus isolation and histological evaluation. The immunogenicity of rhMPV mutants was evaluated using the following methods: (i) humoral immunity was determined by analysis of serum neutralization of virus using an endpoint dilution plaque reduction assay; (ii) viral titers in the nasal turbinates and lungs were determined by an immunostaining plaque assay; and (iii) pulmonary histopathology and viral antigen distribution were determined using the procedure described below. The protection was evaluated with respect to viral replication, antigen distribution, and pulmonary histopathology.
Pulmonary histology.
After sacrifice, the right lung of each animal was removed, inflated, and fixed with 4% neutral buffered formaldehyde. Fixed tissues were embedded in paraffin and sectioned at 5 μm. Slides were then stained with hematoxylin-eosin (H&E) for the examination of histological changes by light microscopy. The pulmonary histopathological changes were reviewed by 2 or 3 independent pathologists. Histopathological changes were scored to include the extent of inflammation (focal or diffuse), the pattern of inflammation (peribronchiolar, perivascular, interstitial, alveolar), and the nature of the cells making up the infiltrate (neutrophils, eosinophils, lymphocytes, macrophages).
Immunohistochemical (IHC) staining.
The right lung of each animal was fixed in 10% neutral buffered formaldehyde and embedded in paraffin. Five-micrometer sections were cut and placed onto positively charged slides. After deparaffinization, sections were incubated with target retrieval solution (Dako, Carpinteria, CA) for antigen retrieval. After antibody block, a primary mouse anti-hMPV monoclonal antibody (Virostat, Portland, ME) was added for 30 min at room temperature, followed by incubation with a biotinylated horse anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA). Slides were further incubated with ABC Elite complex to probe biotin (Vector Laboratories) and then developed using a 3,3′-diaminobenzidine (DAB) chromogen kit (Dako) and hematoxylin as a counterstain. Lung sections from hMPV-infected and uninfected samples were used as positive and negative controls, respectively.
Determination of hMPV neutralizing antibody.
hMPV-specific neutralizing antibody titers were determined using a plaque reduction neutralization assay. Briefly, cotton rat sera were collected by retro-orbital bleeding weekly until challenge. The serum samples were heat inactivated at 56°C for 30 min. Twofold dilutions of the serum samples were mixed with an equal volume of DMEM containing approximately 100 PFU/well hMPV NL/1/00 in a 96-well plate, and the plate was incubated at room temperature for 1 h with constant rotation. The mixtures were then transferred to confluent Vero E6 cells in a 96-well plate in triplicate. After 1 h of incubation at 37°C, the virus-serum mixtures were removed and the cells were overlaid with 0.75% methylcellulose in DMEM and incubated for another 4 days before virus plaque titration. The plaques were counted, and 50% plaque reduction titers were calculated as the hMPV-specific neutralizing antibody titers.
Determination of viral titer in lung and nasal turbinate.
The nasal turbinate and the left lung from each cotton rat were removed, weighed, and homogenized in 1 ml of PBS solution using a Precellys 24 tissue homogenizer (Bertin, MD) by following the manufacturer's recommendations. The presence of infectious virus was determined by an immunostaining plaque assay in Vero cells, as described above.
Statistical analysis.
Quantitative analysis was performed by either densitometric scanning of autoradiographs or by using a phosphorimager (Typhoon; GE Healthcare, Piscataway, NJ) and ImageQuant TL software (GE Healthcare, Piscataway, NJ). Statistical analysis was performed by one-way multiple comparisons using SPSS (version 8.0) statistical analysis software (SPSS Inc., Chicago, IL). A P value of <0.05 was considered statistically significant.
RESULTS
Identification of amino acid residues in hMPV M2-1 protein essential for zinc binding activity.
To begin to explore the biological roles of the M2-1 protein in the hMPV life cycle, we first attempted to characterize its zinc binding activity. To do this, we developed a time course colorimetric assay that can directly and quantitatively measure zinc binding. Briefly, various amounts of highly purified M2-1 protein were incubated with 100 μM 4-(2-pyridylazo)resorcinol (PAR), the metallochromic indicator, and the release of Zn2+ bound by M2-1 protein was monitored upon addition of 100 μM PMPS, which is a thiol oxidizer for the sulfhydryl group (11, 33, 34). Subsequently, the amounts of zinc ion release were determined using standard curves generated by ZnSO4. At the concentration of 2, 3, and 4 μM Zn2+, the absorbance at 500 nm was 0.14, 0.20, and 0.27, respectively (Fig. 3A). At a concentration of 2, 3, and 4 μM hMPV M2-1, the absorbance at 500 nm was 0.14, 0.20, and 0.26 upon addition of PMPS (Fig. 3B). Therefore, the molar ratio between hMPV M2-1 and zinc ion release was 1:1; namely, one M2-1 molecule binds to one zinc ion.
FIG 3.

Identification of amino acids in the M2-1 protein essential for zinc binding activity. (A) Standard curve of zinc ion release. Standard curve generated using 2 μM, 3 μM, and 4 μM ZnSO4. (B) Measurement of zinc binding of the hMPV M2-1 protein by a colorimetric assay. The amount of Zn2+ bound to the hMPV M2-1 protein was quantified by comparing the sample readings to a standard curve. (C) Expression and purification of recombinant hMPV M2-1 protein. The GST-tagged hMPV M2-1 protein was expressed in E. coli Rosetta(DE3) and purified using a column containing glutathione HiCap matrix. Where indicated, GST was cleaved from M2-1 by thrombin. Proteins were analyzed by SDS-PAGE. (D) The effect of mutations to the zinc binding motif of the M2-1 protein on zinc binding activity. Zinc binding activity was measured by a colorimetric assay. Percent of wild-type M2-1 binding for each M2-1 mutant was shown. Data are the averages from three independent experiments ± standard deviations.
Sequence alignment found that M2-1 proteins of all known pneumoviruses possess a putative zinc binding motif (CCCH) (Fig. 1). Next, we determine whether the zinc binding motif is, indeed, essential for zinc binding activity. To do this, the cysteine residues at positions 7, 15, and 21 were changed to serine individually, and the histidine at position 25 was changed to leucine. To enhance the solubility of mutant M2-1 proteins, a GST tag was fused to the N terminus of M2-1. All mutant M2-1 proteins were expressed and purified (Fig. 3C). As shown in Fig. 3D, GST-fused rM2-1 (GST–rM2-1) was found to bind the same amount of zinc as rM2-1, demonstrating that the GST tag did not affect the zinc binding activity of the M2-1 protein. Subsequently, the impact of the M2-1 mutations on zinc binding activity was determined. As shown in Fig. 3D, the C7S and C15S mutants impaired zinc binding activity by 58% and 62%, respectively. Notably, the C21S and H25L mutants almost completely abolished zinc binding ability. These results demonstrated that C21 and H25 in the zinc binding motif are essential for zinc binding activity, whereas C7 and C15 play minor or redundant roles in zinc binding.
Treatment with EDTA alters the secondary structure of hMPV M2-1.
Having demonstrated that the M2-1 protein coordinates zinc ions, we next determined whether the secondary structure of M2-1 is altered by incubating with various concentrations of EDTA, which can chelate metal ions, including zinc. As shown in Fig. 4A, α-helical signal was detected with two negative maximums at 208 nm and 222 nm using a circular dichroism (CD) spectroscopy analysis. The secondary structure of hMPV M2-1 at pH 7.4 and 20°C was dominated by the ordered α-helices (Fig. 4A). The CD spectroscopy showed that the α-helical content gradually decreased when the concentration of EDTA increased from 0 to 20 μM (Fig. 4B). At the maximum metal chelation concentration (20 μM EDTA), the CD spectra were completely altered and did not exhibit a classical α-helix curve (Fig. 4B). These observations indicate that metal coordination, most likely zinc coordination, is necessary for the proper folding of hMPV M2-1.
FIG 4.

EDTA chelation alters the secondary structure of the hMPV M2-1 protein. (A) Circular dichroism (CD) spectroscopic analysis of the M2-1 protein. A 2 μM M2-1 protein was analyzed by a UV spectropolarimeter at a wavelength range of 190 to 260 nm at a scanning speed of 50 nm per min. Data are averages from 10 individual readings. (B) Alteration of the M2-1 structure by EDTA. A 2 μM M2-1 protein was incubated with increasing concentrations of EDTA, and CD spectra were recorded.
Mutations in the zinc binding motif impair oligomerization of the M2-1 protein.
The effect of mutations in the zinc binding motif on oligomerization of M2-1 was determined by a chemical cross-linking assay. Briefly, 1.0 μg of rM2-1 protein (without any tag) was cross-linked by an increasing concentration (from 0 to 0.2%) of glutaraldehyde, and the resulting products were resolved by SDS-PAGE. As shown in Fig. 5A, protein bands with masses of approximately 40, 60, and 80 kDa were observed when the concentration of glutaraldehyde was gradually increased. Since the native hMPV M2-1 is a 21.3-kDa protein, these bands correlate with the predicted sizes of M2-1 dimers, trimers, and tetramers, respectively (Fig. 5A). No protein bands larger than tetramers were observed even when the concentration of glutaraldehyde was increased above 0.2% with a longer time for cross-linking (data not shown). This is consistent with recent structural studies, which showed that the M2-1 protein is a tetramer (29). To test whether the GST tag affects the formation of tetramers, GST–M2-1 was subjected to a cross-linking assay. The predicted product of the GST–M2-1 tetramer (196 kDa) was clearly detectable by SDS-PAGE, although the predicted dimers and trimers were difficult to visualize (Fig. 5B). This result indicates that the GST tag did not affect oligomerization of M2-1. Subsequently, we examined the effects of mutations in the zinc binding motif on oligomerization of M2-1. Interestingly, the predicted M2-1 tetramer (196-kDa protein) was not observed for C7S, C15S, C21S, and H25L (Fig. 5C). Thus, this result suggests that mutations in the zinc binding motif impair the oligomerization of the hMPV M2-1 protein.
FIG 5.
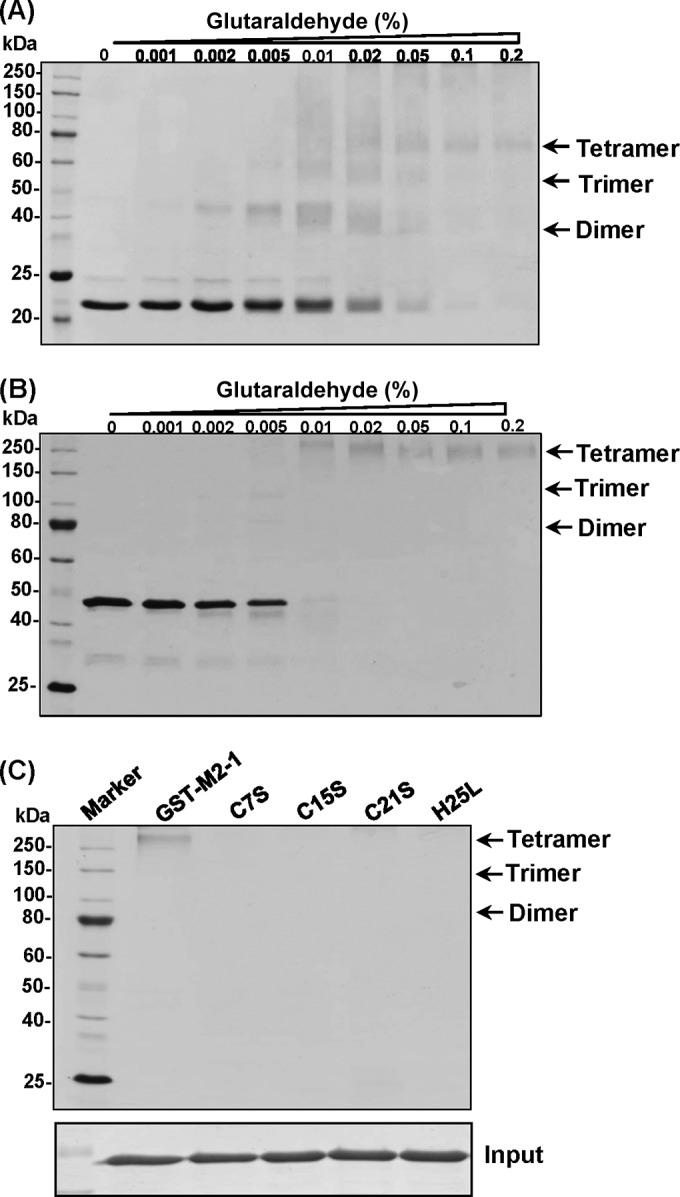
Mutations in the zinc binding motif impair the oligomerization of the M2-1 protein. (A) Glutaraldehyde cross-linking of rM2-1. A total of 1.2 μg of rM2-1 protein was incubated with increasing amounts of glutaraldehyde from 0 to 2% at 25°C for 30 s. The reaction was stopped by adding Tris-HCl (pH 7.4) at a final concentration of 50 mM. The cross-linked products were analyzed by a 12% SDS-PAGE gel, followed by Coomassie blue staining. The predicted dimer, trimer, and tetramer were indicated. (B) Glutaraldehyde cross-linking of GST–M2-1. (C) Glutaraldehyde cross-linking of GST–M2-1 mutants. A total of 1.2 μg of each GST–M2-1 mutant was incubated with 2% glutaraldehyde, and the products were analyzed by SDS-PAGE.
Recovery of recombinant hMPV (rhMPV) carrying mutations in the zinc binding motif of the M2-1 protein.
To determine the role of zinc binding activity in viral replication and pathogenesis, we mutated the zinc binding motif of M2-1 in an infectious cDNA clone of hMPV lineage A strain NL/1/00. All four recombinant viruses (rhMPV-C7S, -C15S, -C21S, and -H25L mutants) were successfully recovered. The viruses were passed three times in LLC-MK2 cells and stained by a monoclonal antibody against N protein. As shown in Fig. 6A, all rhMPV mutants were positive for viral N protein expression in the immunostaining assay at day 3 postinfection. However, the immunospots formed by these rhMPV mutants in LLC-MK2 cells were smaller than those formed by WT rhMPV, especially those of the C21S and H25L mutants. The ability of these recombinant viruses to form plaques in LLC-MK2 cells was determined by an agarose overlay plaque assay (Fig. 6B). After 7 days of incubation, the plaque sizes for rhMPV-C7S, C15S, C21S, and H25L were 1.21 ± 0.13, 1.03 ± 0.20, 0.84 ± 0.26, and 0.68 ± 0.11 mm in diameter, respectively, which were all significantly smaller than that of WT rhMPV (1.59 ± 0.31 mm) (P < 0.05). This suggests that rhMPVs carrying mutations in the zinc binding motif had defects in cell-cell spread and/or viral replication in cell culture.
FIG 6.

Recovery of recombinant hMPVs carrying mutations in the zinc binding motif. (A) Immunostaining spots formed by recombinant hMPVs. LLC-MK2 cells were infected with recombinant hMPV mutants and incubated at 37°C for 4 days. The cells were stained with an anti-hMPV N protein monoclonal antibody. (B) Plaque morphology of recombinant hMPVs. An agarose overlay plaque assay was performed in monolayer LLC-MK2 cells. Viral plaques were developed at day 6 postinfection. The plaque pictures were taken from different dilutions from each hMPV mutant. Data are the averages from 20 plaques ± standard deviations.
Subsequently, all hMPV mutants were plaque purified. The M2-1 gene of each recombinant virus was amplified by RT-PCR and sequenced, and the presence of the desired mutation was confirmed. Finally, the entire genome of each hMPV mutant was amplified and sequenced. The results showed that no additional mutations were found in the genome except for the introduced mutation in the M2-1 gene. These hMPV mutants were passed 10 times in LLC-MK2 cells, and sequencing found that all of the mutants retained the desired mutation. This suggests that these zinc binding-defective hMPV mutants are genetically stable in cell culture.
Recombinant rhMPV lacking zinc binding activity exhibited delayed replication but grew to a titer comparable to that of rhMPV.
The replication kinetics of recombinant hMPV carrying mutations in the zinc binding motif were determined in LLC-MK2 cells (Fig. 7). Briefly, LLC-MK2 cells were infected with each recombinant virus at an MOI of 0.01. At the indicated time points, the amount of virus in the supernatant was determined by immunostaining. WT rhMPV reached a peak titer of 106.6 PFU/ml at day 7 postinoculation. Recombinant rhMPV-C7S and -C15S mutants reached a similar titer at day 8 postinoculation. Recombinant rhMPV-C21S and -H25L mutants showed delayed replication kinetics compared to rhMPV and reached a peak titer of 106.4 and 106.1 PFU/ml at day 9 postinoculation, respectively. Recombinant rhMPV-C21S and -H25L mutants also had significant delayed cytopathic effects (CPEs), genomic RNA synthesis, and mRNA synthesis in LLC-MK2 cells (data not shown). Therefore, although rhMPV-C21S and -H25L mutants exhibited a significant delay in viral replication, they grew to a titer comparable to that of WT rhMPV.
FIG 7.

Multistep growth curve of recombinant hMPVs carrying mutations in the zinc binding site. Vero E6 cells in 35-mm dishes were infected with each recombinant hMPV at an MOI of 0.01. After adsorption for 1 h, the inoculums were removed and the infected cells were washed 3 times with Opti-MEM. Then, fresh Opti-MEM containing 2% FBS was added and cells were incubated at 37°C for various time periods. Aliquots of the cell culture fluid were removed at the indicated intervals. Viral titer was determined by an immunostaining assay in Vero-E6 cells.
Recombinant rhMPV viruses lacking zinc binding activity were highly attenuated in cotton rats.
The replication and pathogenesis of rhMPV carrying mutations in the zinc binding motif were determined in cotton rats, the best small-animal model for hMPV (35, 36). Briefly, 4-week-old SPF cotton rats were inoculated intranasally with 1.0 × 106 PFU of WT rhMPV or rhMPV mutants. At day 4 postinoculation, cotton rats were terminated and viral replication in nasal turbinate and lungs and pulmonary histology were evaluated (Table 1). As expected, rhMPV replicated efficiently in the nasal turbinates and lungs of all five cotton rats, with average viral titers of 105.68 and 104.25 PFU/g tissue, respectively. Recombinant rhMPV-C7S and -C15S mutants, which retained 60% of zinc binding activity, replicated as efficiently as rhMPV in cotton rats, producing similar titers in both the nasal turbinates and the lungs of all five cotton rats (P > 0.05). Recombinant rhMPV-C21S and -H25L mutants, which abolished zinc binding activity, were highly attenuated in the replication in cotton rats. No infectious virus was detected in either nasal turbinate or lungs in rhMPV-C21S mutant-infected cotton rats. Only one out of five cotton rats had a low level of infectious virus (2.3 log PFU/ml) in nasal turbinate, and no infectious virus was found in lungs. Pulmonary histology showed that rhMPV caused moderate histological lesions, including interstitial pneumonia, peribronchial lymphoplasmacytic infiltrates, mononuclear cell infiltrate, and edematous thickening of the bronchial submucosa (Fig. 8). Recombinant rhMPV-C7S and -C15S mutants (data not shown for the C15S mutant) caused histologic lesions similar to those of rhMPV. In contrast, rhMPV-C21S and -H25L mutants (data not shown for the H25L mutant) caused no or only mild pulmonary histological changes (Fig. 8). Immunohistobiochemistry analysis found that the lungs of cotton rats infected by rhMPV deposited a large amount of viral antigen in epithelial cells. The viral antigen expression of rhMPV-C7S and -C15S mutants in lung epithelial cells was similar to that of WT rhMPV (Fig. 9). In contrast, no viral antigen was found in the lungs of cotton rats infected by rhMPV-C21S and -H25L mutants (Fig. 9). Taken together, these results demonstrated that rhMPV-C7S and -C15S mutants replicated efficiently in cotton rats and caused similar histologic lesion as rhMPV, whereas rhMPV-C21S and -H25L mutants were highly attenuated in replication in both the upper and lower respiratory tracts of cotton rats.
TABLE 1.
Replication of rhMPV carrying mutations in the zinc binding motif in cotton ratse
| Mutanta | No. of cotton rats per group | Viral replicationb in: |
Lung histologyc | Lung IHCd | |||
|---|---|---|---|---|---|---|---|
| Nasal turbinate |
Lung |
||||||
| % of infected animals | Mean titer log (PFU/g) | % of infected animals | Mean titer log (PFU/g) | ||||
| rhMPV | 5 | 100 | 5.68 ± 0.36 A | 100 | 4.25 ± 0.43 A | 1.8 A | 3.0 A |
| rhMPV-C7S | 5 | 100 | 5.31 ± 0.60 A | 100 | 4.14 ± 0.34 A | 1.5 A | 2.5 A |
| rhMPV-C15S | 5 | 100 | 5.56 ± 0.39 A | 100 | 4.08 ± 0.47 A | 1.6 A | 2.5 A |
| rhMPV-C21S | 5 | 0 | ND | 0 | ND | 0.5 B | 0 B |
| rhMPV-H25L | 5 | 20 | 2.3 | 0 | ND | 0.8 B | 0.5 B |
Cotton rats were inoculated intranasally with DMEM or 2 × 105 PFU wild-type rhMPV or rhMPV mutants. At day 4 postimmunization, animals were euthanized for pathology study.
For rhMPV-H25L, 1 out of 5 cotton rats had detectable virus with a titer of 2.3 log PFU/g. ND indicates that infectious virus was not detectable.
The severity of lung histology was scored for each lung tissues. Average score for each group is shown. 0, no change; 1, mild change; 2, moderate change; and 3, severe change.
The amount of hMPV antigen expression in lung was scored. Average score for each group is shown. 0, no antigen; 1, small amount; 2, moderate amount; and 3, large amount.
Values within a column followed by different uppercase letters (A and B) are significantly different (P < 0.05).
FIG 8.

Lung histological changes in cotton rats infected by rhMPVs. The right lung from each cotton rat was preserved in 4% (vol/vol) phosphate-buffered paraformaldehyde. Fixed tissues were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin-eosin (HE) for the examination of histological changes by light microscopy. Lungs from rhMPV and rhMPV-C7S had moderate to severe lymphocyte infiltrates, whereas lungs from rhMPV-C21S had no to mild lymphocyte infiltrates.
FIG 9.

Immunohistochemical (IHC) staining of lungs from cotton rats infected by rhMPVs. Lung tissues were fixed in 4% (vol/vol) phosphate-buffered paraformaldehyde. Deparaffinized sections were stained with monoclonal antibody against hMPV matrix protein (Virostat, Portland, ME) to determine the distribution of viral antigen.
Recombinant rhMPVs lacking zinc binding activity were highly immunogenic and protected cotton rats from hMPV infection.
The immunogenicity of the two attenuated mutants (rhMPV-C21S and -H25L mutants) was determined by vaccination of cotton rats, followed by challenge with rhMPV. After vaccination, serum antibody was determined weekly by a plaque reduction neutralization assay. WT rhMPV infection and reinfection were used as a control. An ideal live attenuated vaccine candidate should generate immunogenicity similar to or higher than that of WT hMPV. As shown in Fig. 10, rhMPV-C21S and -H25L mutants triggered high levels of neutralizing antibody that were comparable to those generated after WT rhMPV immunization (P > 0.05). Antibody was detectable at week 1 postimmunization, and the levels gradually increased during weeks 2 to 4. However, there was no significant difference in antibody titer during a 4-week time period (P > 0.05). No hMPV-specific antibody was detected in the unvaccinated control. At week 4 postimmunization, all vaccinated cotton rats were challenged with WT rhMPV, and cotton rats were terminated at day 4 postinfection. No infectious viruses were detectable in either nasal turbinates or lungs from the animals vaccinated with rhMPV-C21S and -H25L mutants followed by challenging with rhMPV (Table 2). Pulmonary histology showed that the unvaccinated challenged control had moderate pathological changes characterized by interstitial pneumonia, mononuclear cell infiltrate, and edematous thickening of the bronchial submucosa. In contrast, no or only mild histological changes were found in the lungs of cotton rats vaccinated with rhMPV-C21S and rhMPV-H25L mutants (Table 2). No enhanced lung damage was found for the mutants. IHC showed that large numbers of viral antigens were found at the luminal surface of the bronchial epithelial cells in lung tissues from unvaccinated challenged controls (Fig. 11). In contrast, no antigens were found on the luminal surface of bronchial epithelial cells in rhMPV-C21S and rhMPV-H25L mutant groups (Fig. 11). Instead, antigen was found within bronchial tissue, which may be related to viral clearance. Collectively, these results demonstrate that rhMPVs lacking zinc binding activity are not only sufficiently attenuated but also capable of triggering high levels of antibody and providing complete protection against hMPV infection.
FIG 10.

Recombinant hMPVs triggered a high level of neutralizing antibody titer in cotton rats. Cotton rats were immunized with each recombinant hMPV intranasally at a dose of 1.0 × 106 PFU per rat. Blood samples were collected from each rat weekly by retro-orbital bleeding. The hMPV neutralizing antibody was determined using a plaque reduction neutralization assay as described in Materials and Methods. Data are the average titers from four cotton rats ± standard deviations.
TABLE 2.
Immunogenicity of rhMPV lacking zinc binding activity in cotton rats
| Mutanta | No. of cotton rats per group | Nasal turbinate |
Lung |
Lung histologyc | ||
|---|---|---|---|---|---|---|
| % of infected animals | Mean titer log (PFU/g) | % of infected animals | Mean titer log (PFU/g) | |||
| DMEM | 5 | 100 | 4.82 ± 0.19 | 100 | 4.34 ± 0.20 | 2.0 A |
| rhMPV | 5 | 0 | NDb | 0 | ND | 0.8 B |
| rhMPV-C21S | 5 | 0 | ND | 0 | ND | 0.6 B |
| rhMPV-H25L | 5 | 0 | ND | 0 | ND | 0.5 B |
Animals were immunized intranasally with DMEM or 2 × 105 PFU wild-type rhMPV or rhMPV mutants. At day 28 postimmunization, animals were challenged with 106 PFU wild-type rhMPV.
ND indicates infectious virus was not detectable.
The severity of lung histology was scored for each lung tissues. Average score for each group is shown. 0, no change; 1, mild change; 2, moderate change; and 3, severe change. Values within a column followed by different uppercase letters (A and B) are significantly different (P < 0.05).
FIG 11.

Immunohistochemical (IHC) staining of lungs from cotton rats vaccinated by rhMPV mutants followed by rhMPV challenge. Lung tissues were fixed in 4% (vol/vol) phosphate-buffered paraformaldehyde. Deparaffinized sections were stained with monoclonal antibody against the hMPV matrix protein (Virostat, Portland, ME) to determine the distribution of viral antigen. Arrows indicate antigen-positive cells.
DISCUSSION
In this study, we characterized the roles of the hMPV M2-1 protein in viral replication in vitro and in vivo. Using highly purified M2-1 protein expressed from E. coli, we showed that M2-1 is a zinc binding protein that coordinates zinc ion at a molecular ratio of 1:1. Subsequent mutagenesis showed that C21 and H25 in the zinc binding motif are essential for zinc binding activity, whereas C7 and C15 play minor or redundant roles in zinc binding. Recombinant rhMPVs lacking zinc binding activity not only were highly attenuated in replication in cell culture and in cotton rats but also elicited high levels of neutralizing antibody and provided complete protection against rhMPV. In contrast, recombinant rhMPVs retaining approximately 60% of zinc binding activity replicated as efficiently as rhMPV in vitro and in vivo. Collectively, these data indicate that the zinc binding activity of M2-1 is indispensable for viral replication and pathogenesis in vivo.
Metal ions were shown to be integrated in several gene regulatory proteins as early as the 1970s (37–39). Among them, zinc is an important structural component of proteins involved in nucleic acid binding and gene regulation. Typical zinc binding motifs, including CCHH, CCHC, and CCCH, are often involved in transcriptional and translational processes (40, 41). Many viruses encode their own zinc binding proteins that regulate viral replication and/or pathogenesis. The NS5A protein of hepatitis C virus contains a CCCC zinc binding motif, and zinc coordination is required for its role in hepatitis C replication (42). All retroviral nucleocapsid proteins (with the exception of spumaretrovirus) contain one or two copies of a zinc finger motif, which is essential for viral replication (43, 44). This is based on the evidence that mutations in the zinc-coordinating residues led to the loss of viral infectivity and a significant reduction in genomic RNA packaging (44). The V proteins of many paramyxoviruses (such as simian virus 5, Sendai, mumps, measles, and Newcastle disease viruses) have been shown to bind zinc ion (45, 46). Sendai virus defective in the zinc binding activity of the V protein exhibited a WT level of viral protein synthesis and growth patterns in vitro. However, these Sendai virus mutants were strongly attenuated in mice, exhibiting earlier viral clearance from the mouse lung and less virulence to mice (46, 47). Therefore, the zinc binding capacity of the Sendai V protein is essential for viral replication in vivo but not in vitro. Interestingly, deletion of the VP30 gene from the Ebola virus genome resulted in a replication-deficient Ebola virus (EbolaΔVP30) which can replicate efficiently only if the VP30 protein is provided in trans (48). Replication-deficient EbolaΔVP30 does not cause disease or death in STAT-1-knockout mice. In addition, mice and guinea pigs vaccinated with EbolaΔVP30 virus are protected against lethal challenge with Ebola virus infection (49). In this case, VP30 is essential for Ebola virus replication both in vitro and in vivo.
It is well established that M2-1 of hRSV is a transcriptional elongation factor and antiterminator. In the RSV lacZ minigenome assay, no lacZ reporter gene expression was detected when M2-1 was omitted from the polymerase complex (50). Similarly, without M2-1, a very low level of reporter gene expression (2 to 5%) was detected in the minigenome assay of PVM, another pneumovirus (50). Consistent with the processivity function of M2-1, deletion of the M2-1 gene from the RSV genome is lethal to the virus (24). Subsequently, the cysteine residues at positions 7, 15, 21, and 96 in the M2-1 protein of the hRSV A2 strain were each individually replaced by a glycine residue. It was found that mutations in the first three cysteine residues, which are located in the zinc binding motif, were also lethal to virus (24). This suggests that the zinc binding motif in the hRSV M2-1 protein is absolutely essential for virus viability. In contrast to hRSV, the M2-1 protein of hMPV is dispensable for minigenome replication and virus viability (25). Deletion of hMPV M2-1 from the genome yielded a viable virus (rΔM2-1) that replicated less efficiently than parent hMPV in cell culture and was completely defective in replication in the lungs of cotton rats (25). Therefore, the M2-1 protein of hMPV was dispensable for efficient replication in vitro but was essential for virus replication in vivo. In this study, we found that zinc binding-deficient rhMPVs were highly attenuated in replication in in vitro cell culture and in cotton rats (in vivo) and did not cause lung histologic lesions. In contrast, rhMPVs that retained 60% of zinc binding activity were capable of replicating as efficiently as wild-type hMPV in vitro and in vivo and causing significant lung damage. Thus, the zinc binding activity of hMPV M2-1 is dispensable for viral replication in vitro but essential for viral replication and pathogenesis in vivo. In addition, our results showed that all four mutations in zinc binding motif equally affected oligomerization of M2-1 protein but exhibited the distinct ability in viral replication in vivo, suggesting that the oligomerization of M2-1 is not essential for its in vivo replication. Perhaps 60% of zinc binding activity for C7S and C15S is not sufficient for oligomerization but is sufficient for efficient viral replication in vivo.
The individual cysteine and histidine residues within the zinc finger motif contribute differently to zinc coordination. In the case of the Ebola VP30 zinc binding motif (CCCH), mutations to the first cysteine and the last histidine did not significantly impair zinc binding ability (11). However, an additional mutation of the third cysteine to serine reduced the zinc binding ability of VP30 by 80%. When all four zinc-coordinating residues were mutated, the zinc binding ability of the VP30 protein was abolished (11). Prior to our study, it was not known whether this zinc binding motif is, indeed, required for zinc coordination. We found that the third cysteine and the histidine in the zinc binding motif (CCCH) of hMPV M2-1 are crucial for zinc coordination, while the first two cysteine residues each play a minor or redundant role in zinc binding. Interestingly, all four M2-1 mutants affected the formation of M2-1 tetramers, suggesting that zinc binding residues are essential for oligomerization of M2-1. The recently solved structure of hMPV M2-1 showed that the oligomerization domain maps to amino acids 32 to 58, and this region folds as an α-helix, which is exposed on the protein surface (Fig. 2D) (29). However, the CCCH zinc binding motif is physically separated from the oligomerization domain (Fig. 2D). It is likely that mutations in the zinc binding motif affect local structure, which in turn alters the global structure of the M2-1 protein. In fact, differential scanning fluorimetry (DSF) experiment showed that GST–M2-1 had a melting point (Tm) of 43.6°C, and C7S and C15S mutants decreased the Tm by 1°C, whereas C21S and H25L mutants had dramatic alteration of Tm, decreasing by 3°C and 5°C, respectively. These results suggest that mutations in the zinc binding motif decrease the thermostability of M2-1. In the crystal structure of hMPV M2-1, the cysteine and histidine residues in the zinc binding motif form an extensive hydrogen bond network with side chain-side chain, main chain-side chain, and main chain-main chain interactions (Table 3).
TABLE 3.
Interaction of the zinc binding motif with other amino acids within the M2-1 monomer
| Amino acida | Hydrogen bond (bond length inÅ) |
Ionic interaction | ||
|---|---|---|---|---|
| Main chain-main chain | Main chain-side chain | Side chain-side chain | ||
| C7 | A5 (3.34) | Y9 (3.46) | C15 (3.80) | |
| Y9 (3.23) | E10 (3.56) | C21 (3.90) | ||
| E10 (2.99) | F23 (3.79) | C25 (3.63) | ||
| V11 (2.99) | N24 (3.58) | |||
| N24 (2.87) | ||||
| C15 | E10 (3.07) | C7 (3.80) | ||
| R17 (3.80) | C21 (3.86) | |||
| C25 (3.47) | ||||
| C21 | F23 (3.63) | C7 (3.90) | ||
| C15 (3.86) | ||||
| C25 (3.59) | ||||
| H25 | Y27 (3.43) | E10 (3.80) | C7 (3.63) | E10 (3.9) |
| F23 (3.10) | C15 (3.47) | |||
| C21 (3.59) | ||||
Data were collected using the hMPV M2-1 structure (PDB ID 4CS7). The bond length is indicated in parentheses in each column. C15 is also involved in the interprotein interaction between chains in the tetramer. There are four chains (A, B, C, and E) in the tetramer. C15 (chain E) interacts with S75 (chain A), and C15 (chain B) interacts with T76 (chain E).
An important application of this study is to develop rhMPVs that lack zinc binding activity as potential live vaccine candidates for hMPV. It is well known that inactivated vaccines are not suitable for hRSV, hMPV, and parainfluenza virus type 3 (PIV3), all of which cause acute respiratory tract infection in the same populations (infants, children, and the elderly), since an inactivated vaccine causes enhanced lung damage upon reinfection by the same virus (51–53). Therefore, live attenuated vaccines are the most promising vaccine candidates for these viruses. However, it has been a challenge to identify a vaccine candidate that has an optimal balance between attenuation and immunogenicity. For decades, approaches to generate attenuated vaccines by employing chemical mutagens or deleting nonessential genes led to recombinant viruses that were genetically unstable, attenuated too much or too little, exhibited impaired virus titer, and/or lacked sufficient immunogenicity (54, 55). From these perspectives, zinc binding-deficient rhMPVs are ideal live vaccine candidates. These two rhMPV mutants grow to high titers in cell culture, making it economically feasible for vaccine production. No revertants or additional mutations were detected after 10 passages of these mutants in cell culture, suggesting that they are genetically stable. Importantly, these two rhMPV mutants were sufficiently attenuated yet retained high immunogenicity in cotton rats. Previously, it was shown that rhMPV lacking the entire M2-1 gene (rΔM2-1) was overly attenuated so that it failed to trigger either an hMPV-specific antibody response or protective immunity in animals (25). In contrast to rΔM2-1, rhMPVs lacking zinc binding activity were completely defective in replication in the upper and lower respiratory tracts of cotton rats but were capable of inducing high levels of neutralizing antibody and providing complete protection against the challenge of rhMPV. Previously, we showed that rhMPV replicated to a peak titer in cotton rats at day 4 postinoculation (56). At this time point, no infectious virus was detected in the nasal turbinate and lungs of cotton rats inoculated with rhMPV-C21S and -H25L mutants. However, these tissues were positive for hMPV RNA (data not shown), as determined by real-time RT-PCR, which is not surprising, because real-time RT-PCR was much more sensitive than plaque assay. Similar results were observed for other hMPV mutants, such as receptor binding-defective rhMPVs and methyltransferase-defective rhMPVs, which were defective in receptor binding activity in fusion (F) protein and mRNA cap methyltransferase activity in the large (L) polymerase protein, respectively (31, 35). Another possibility is that infectious hMPV has been cleared from nasal turbinate and lungs for these mutants at day 4 postinfection. Perhaps this low level of replication of hMPV mutants in vivo was sufficient to trigger a high level of neutralizing antibody and protective immunity. Thus, the inhibition of zinc binding activity carries great potential as a novel approach to rationally attenuate hMPV for vaccine purposes. The phenotypes (growth kinetics, attenuation, genetic stability, and immunogenicity) of zinc binding-defective rhMPVs in vitro and in vivo were similar to those that have been observed for receptor binding-defective rhMPV and methyltransferase-defective rhMPVs (35, 56). Future experiments are required to determine whether cellular immunity is involved in immune protection and to test the safety and immunogenicity of these promising vaccine candidates in nonhuman primates and humans.
In conclusion, this work highlights a major gap in our understanding of the biological roles of the zinc binding activity of the M2-1 protein in the replication and pathogenesis of hMPV in vivo. Our findings also suggest that inhibition of M2-1 zinc binding activity can potentially lead to the development of novel live attenuated vaccines, as well as antiviral drugs, for pneumoviruses.
ACKNOWLEDGMENTS
This study was supported by grants from the NIH (R01AI090060) to J.L.
We thank Ron A. M. Fouchier for the infectious cDNA clone of hMPV. We thank Steven Krakowka for his help on histology. We are grateful to Ronald Iorio and members of the J. Li laboratory for critical readings of the manuscript.
REFERENCES
- 1.van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, Osterhaus AD. 2001. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med 7:719–724. doi: 10.1038/89098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Principi N, Bosis S, Esposito S. 2006. Human metapneumovirus in paediatric patients. Clin Microbiol Infect 12:301–308. doi: 10.1111/j.1469-0691.2005.01325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams JV, Harris PA, Tollefson SJ, Halburnt-Rush LL, Pingsterhaus JM, Edwards KM, Wright PF, Crowe JE Jr. 2004. Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N Engl J Med 350:443–450. doi: 10.1056/NEJMoa025472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van den Hoogen BG, Bestebroer TM, Osterhaus AD, Fouchier RA. 2002. Analysis of the genomic sequence of a human metapneumovirus. Virology 295:119–132. doi: 10.1006/viro.2001.1355. [DOI] [PubMed] [Google Scholar]
- 5.Falsey AR, Erdman D, Anderson LJ, Walsh EE. 2003. Human metapneumovirus infections in young and elderly adults. J Infect Dis 187:785–790. doi: 10.1086/367901. [DOI] [PubMed] [Google Scholar]
- 6.Schildgen V, van den Hoogen B, Fouchier R, Tripp RA, Alvarez R, Manoha C, Williams J, Schildgen O. 2011. Human metapneumovirus: lessons learned over the first decade. Clin Microbiol Rev 24:734–754. doi: 10.1128/CMR.00015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feuillet F, Lina B, Rosa-Calatrava M, Boivin G. 2012. Ten years of human metapneumovirus research. J Clin Virol 53:97–105. doi: 10.1016/j.jcv.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Whelan SP, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immunol 283:61–119. [DOI] [PubMed] [Google Scholar]
- 9.Collins PL, Camargo E, Hill MG. 1999. Support plasmids and support proteins required for recovery of recombinant respiratory syncytial virus. Virology 259:251–255. doi: 10.1006/viro.1999.9762. [DOI] [PubMed] [Google Scholar]
- 10.Collins PL, Hill MG, Cristina J, Grosfeld H. 1996. Transcription elongation factor of respiratory syncytial virus, a nonsegmented negative-strand RNA virus. Proc Natl Acad Sci U S A 93:81–85. doi: 10.1073/pnas.93.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Modrof J, Becker S, Muhlberger E. 2003. Ebola virus transcription activator VP30 is a zinc-binding protein. J Virol 77:3334–3338. doi: 10.1128/JVI.77.5.3334-3338.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biedenkopf N, Hartlieb B, Hoenen T, Becker S. 2013. Phosphorylation of Ebola virus VP30 influences the composition of the viral nucleocapsid complex impact on viral transcription and replication. J Biol Chem 288:11165–11174. doi: 10.1074/jbc.M113.461285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez MJ, Biedenkopf N, Volchkova V, Hartlieb B, Alazard-Dany N, Reynard O, Becker S, Volchkov V. 2008. Role of Ebola virus VP30 in transcription reinitiation. J Virol 82:12569–12573. doi: 10.1128/JVI.01395-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy RW, Wertz GW. 2000. The Cys(3)-His(1) motif of the respiratory syncytial virus M2-1 protein is essential for protein function. J Virol 74:5880–5885. doi: 10.1128/JVI.74.13.5880-5885.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fearns R, Collins PL. 1999. Role of the M2-1 transcription antitermination protein of respiratory syncytial virus in sequential transcription. J Virol 73:5852–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esperante SA, Noval MG, Altieri TA, de Oliveira GA, Silva JL, de Prat-Gay G. 2013. Fine modulation of the respiratory syncytial virus M2-1 protein quaternary structure by reversible zinc removal from its Cys(3)-His(1) motif. Biochemistry 52:6779–6789. doi: 10.1021/bi401029q. [DOI] [PubMed] [Google Scholar]
- 17.Hardy RW, Wertz GW. 1998. The product of the respiratory syncytial virus M2 gene ORF1 enhances readthrough of intergenic junctions during viral transcription. J Virol 72:520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuesta I, Geng X, Asenjo A, Villanueva N. 2000. Structural phosphoprotein M2-1 of the human respiratory syncytial virus is an RNA binding protein. J Virol 74:9858–9867. doi: 10.1128/JVI.74.21.9858-9867.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blondot ML, Dubosclard V, Fix J, Lassoued S, Aumont-Nicaise M, Bontems F, Eleouet JF, Sizun C. 2012. Structure and functional analysis of the RNA- and viral phosphoprotein-binding domain of respiratory syncytial virus M2-1 protein. PLoS Pathog 8:e1002734. doi: 10.1371/journal.ppat.1002734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cartee TL, Wertz GW. 2001. Respiratory syncytial virus M2-1 protein requires phosphorylation for efficient function and binds viral RNA during infection. J Virol 75:12188–12197. doi: 10.1128/JVI.75.24.12188-12197.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia J, Garcia-Barreno B, Vivo A, Melero JA. 1993. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 195:243–247. doi: 10.1006/viro.1993.1366. [DOI] [PubMed] [Google Scholar]
- 22.Tran TL, Castagne N, Dubosclard V, Noinville S, Koch E, Moudjou M, Henry C, Bernard J, Yeo RP, Eleouet JF. 2009. The respiratory syncytial virus M2-1 protein forms tetramers and interacts with RNA and P in a competitive manner. J Virol 83:6363–6374. doi: 10.1128/JVI.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins PL, Hill MG, Camargo E, Grosfeld H, Chanock RM, Murphy BR. 1995. Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc Natl Acad Sci U S A 92:11563–11567. doi: 10.1073/pnas.92.25.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang RS, Nguyen N, Cheng X, Jin H. 2001. Requirement of cysteines and length of the human respiratory syncytial virus M2-1 protein for protein function and virus viability. J Virol 75:11328–11335. doi: 10.1128/JVI.75.23.11328-11335.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buchholz UJ, Biacchesi S, Pham QN, Tran KC, Yang L, Luongo CL, Skiadopoulos MH, Murphy BR, Collins PL. 2005. Deletion of M2 gene open reading frames 1 and 2 of human metapneumovirus: effects on RNA synthesis, attenuation, and immunogenicity. J Virol 79:6588–6597. doi: 10.1128/JVI.79.11.6588-6597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naylor CJ, Brown PA, Edworthy N, Ling R, Jones RC, Savage CE, Easton AJ. 2004. Development of a reverse-genetics system for Avian pneumovirus demonstrates that the small hydrophobic (SH) and attachment (G) genes are not essential for virus viability. J Gen Virol 85:3219–3227. doi: 10.1099/vir.0.80229-0. [DOI] [PubMed] [Google Scholar]
- 27.Herfst S, de Graaf M, Schickli JH, Tang RS, Kaur J, Yang CF, Spaete RR, Haller AA, van den Hoogen BG, Osterhaus AD, Fouchier RA. 2004. Recovery of human metapneumovirus genetic lineages A and B from cloned cDNA. J Virol 78:8264–8270. doi: 10.1128/JVI.78.15.8264-8270.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanner SJ, Ariza A, Richard CA, Kyle HF, Dods RL, Blondot ML, Wu W, Trincao J, Trinh CH, Hiscox JA, Carroll MW, Silman NJ, Eleouet JF, Edwards TA, Barr JN. 2014. Crystal structure of the essential transcription antiterminator M2-1 protein of human respiratory syncytial virus and implications of its phosphorylation. Proc Natl Acad Sci U S A 111:1580–1585. doi: 10.1073/pnas.1317262111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leyrat C, Renner M, Harlos K, Huiskonen JT, Grimes JM. 2014. Drastic changes in conformational dynamics of the antiterminator M2-1 regulate transcription efficiency in Pneumovirinae. eLife 3:e02674. doi: 10.7554/eLife.02674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 31.Zhang Y, Wei Y, Zhang X, Cai H, Niewiesk S, Li J. 2014. Rational design of human metapneumovirus live attenuated vaccine candidates by inhibiting viral mRNA cap methyltransferase. J Virol 88:11411–11429. doi: 10.1128/JVI.00876-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Wei Y, Li J. 2012. Development and optimization of a direct plaque assay for human and avian metapneumoviruses. J Virol Methods 185:61–68. doi: 10.1016/j.jviromet.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunt JB, Neece SH, Ginsburg A. 1985. The use of 4-(2-pyridylazo)resorcinol in studies of zinc release from Escherichia coli aspartate transcarbamoylase. Anal Biochem 146:150–157. doi: 10.1016/0003-2697(85)90409-9. [DOI] [PubMed] [Google Scholar]
- 34.Pfister T, Jones KW, Wimmer E. 2000. A cysteine-rich motif in poliovirus protein 2C(ATPase) is involved in RNA replication and binds zinc in vitro. J Virol 74:334–343. doi: 10.1128/JVI.74.1.334-343.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei Y, Zhang Y, Cai H, Mirza AM, Iorio RM, Peeples ME, Niewiesk S, Li J. 2014. Roles of the putative integrin-binding motif of the human metapneumovirus fusion (F) protein in cell-cell fusion, viral infectivity, and pathogenesis. J Virol 88:4338–4352. doi: 10.1128/JVI.03491-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams JV, Tollefson SJ, Johnson JE, Crowe JE Jr. 2005. The cotton rat (Sigmodon hispidus) is a permissive small animal model of human metapneumovirus infection, pathogenesis, and protective immunity. J Virol 79:10944–10951. doi: 10.1128/JVI.79.17.10944-10951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lohmar PH, Toft DO. 1975. Inhibition of the binding of progesterone receptor to nuclei: effects of o-phenanthroline and rifamycin AF/013. Biochem Biophys Res Commun 67:8–15. doi: 10.1016/0006-291X(75)90275-2. [DOI] [PubMed] [Google Scholar]
- 38.Shyamala G. 1975. Is the estrogen receptor of mammary glands a metallo-protein? Biochem Biophys Res Commun 64:408–415. doi: 10.1016/0006-291X(75)90268-5. [DOI] [PubMed] [Google Scholar]
- 39.Hanas JS, Hazuda DJ, Bogenhagen DF, Wu FY, Wu CW. 1983. Xenopus transcription factor A requires zinc for binding to the 5 S RNA gene. J Biol Chem 258:14120–14125. [PubMed] [Google Scholar]
- 40.Berg JM. 1990. Zinc fingers and other metal-binding domains. Elements for interactions between macromolecules. J Biol Chem 265:6513–6516. [PubMed] [Google Scholar]
- 41.Kelly SM, Pabit SA, Kitchen CM, Guo P, Marfatia KA, Murphy TJ, Corbett AH, Berland KM. 2007. Recognition of polyadenosine RNA by zinc finger proteins. Proc Natl Acad Sci U S A 104:12306–12311. doi: 10.1073/pnas.0701244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM. 2004. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J Biol Chem 279:48576–48587. doi: 10.1074/jbc.M407787200. [DOI] [PubMed] [Google Scholar]
- 43.South TL, Blake PR, Sowder RC, Arthur LO, Henderson LE, Summers MF. 1990. The nucleocapsid protein isolated from HIV-1 particles binds zinc and forms retroviral-type zinc fingers. Biochemistry 29:7786–7789. doi: 10.1021/bi00486a002. [DOI] [PubMed] [Google Scholar]
- 44.Gorelick RJ, Nigida SM Jr, Bess JW Jr, Arthur LO, Henderson LE, Rein A. 1990. Noninfectious human immunodeficiency virus type 1 mutants deficient in genomic RNA. J Virol 64:3207–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paterson RG, Leser GP, Shaughnessy MA, Lamb RA. 1995. The paramyxovirus SV5 V-protein binds to atoms of zinc and is a structural component of virions. Virology 208:121–131. doi: 10.1006/viro.1995.1135. [DOI] [PubMed] [Google Scholar]
- 46.Huang C, Kiyotani K, Fujii Y, Fukuhara N, Kato A, Nagai Y, Yoshida T, Sakaguchi T. 2000. Involvement of the zinc-binding capacity of Sendai virus V protein in viral pathogenesis. J Virol 74:7834–7841. doi: 10.1128/JVI.74.17.7834-7841.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kato A, Kiyotani K, Sakai Y, Yoshida T, Shioda T, Nagai Y. 1997. Importance of the cysteine-rich carboxyl-terminal half of V protein for Sendai virus pathogenesis. J Virol 71:7266–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halfmann P, Kim JH, Ebihara H, Noda T, Neumann G, Feldmann H, Kawaoka Y. 2008. Generation of biologically contained Ebola viruses. Proc Natl Acad Sci U S A 105:1129–1133. doi: 10.1073/pnas.0708057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Halfmann P, Ebihara H, Marzi A, Hatta Y, Watanabe S, Suresh M, Neumann G, Feldmann H, Kawaoka Y. 2009. Replication-deficient ebolavirus as a vaccine candidate. J Virol 83:3810–3815. doi: 10.1128/JVI.00074-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou H, Cheng X, Jin H. 2003. Identification of amino acids that are critical to the processivity function of respiratory syncytial virus M2-1 protein. J Virol 77:5046–5053. doi: 10.1128/JVI.77.9.5046-5053.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim HW, Canchola JG, Brandt CD, Pyles G, Chanock RM, Jensen K, Parrott RH. 1969. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol 89:422–424. [DOI] [PubMed] [Google Scholar]
- 52.Yim KC, Cragin RP, Boukhvalova MS, Blanco JCG, Hamlin ME, Boivin G, Porter DD, Prince GA. 2007. Human metapneumovirus: enhanced pulmonary disease in cotton rats immunized with formalin-inactivated virus vaccine and challenged. Vaccine 25:5034–5040. doi: 10.1016/j.vaccine.2007.04.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ottolini MG, Porter DD, Hemming VG, Prince GA. 2000. Enhanced pulmonary pathology in cotton rats upon challenge after immunization with inactivated parainfluenza virus 3 vaccines. Viral Immunol 13:231–236. doi: 10.1089/vim.2000.13.231. [DOI] [PubMed] [Google Scholar]
- 54.Collins PL, Murphy BR. 2002. Respiratory syncytial virus: reverse genetics and vaccine strategies. Virology 296:204–211. doi: 10.1006/viro.2002.1437. [DOI] [PubMed] [Google Scholar]
- 55.Karron RA, Buchholz UJ, Collins PL. 2013. Live-attenuated respiratory syncytial virus vaccines. Curr Top Microbiol 372:259–284. doi: 10.1007/978-3-642-38919-1_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Niewiesk S, Li J. 2014. Small animal models for human metapneumovirus: cotton rat is more permissive than hamster and mouse. Pathogens 3:633–655. doi: 10.3390/pathogens3030633. [DOI] [PMC free article] [PubMed] [Google Scholar]