

ABSTRACT
The diversity of influenza A viruses in swine (swIAVs) presents an important pandemic threat. Knowledge of the human-swine interface is particularly important for understanding how viruses with pandemic potential evolve in swine hosts. Through phylogenetic analysis of contemporary swIAVs in the United States, we demonstrate that human-to-swine transmission of pandemic H1N1 (pH1N1) viruses has occurred continuously in the years following the 2009 H1N1 pandemic and has been an important contributor to the genetic diversity of U.S. swIAVs. Although pandemic H1 and N1 segments had been largely removed from the U.S. swine population by 2013 via reassortment with other swIAVs, these antigens reemerged following multiple human-to-swine transmission events during the 2013-2014 seasonal epidemic. These findings indicate that the six internal gene segments from pH1N1 viruses are likely to be sustained long term in the U.S. swine population, with periodic reemergence of pandemic hemagglutinin (HA) and neuraminidase (NA) segments in association with seasonal pH1N1 epidemics in humans. Vaccinating U.S. swine workers may reduce infection of both humans and swine and in turn limit the role of humans as sources of influenza virus diversity in pigs.
IMPORTANCE Swine are important hosts in the evolution of influenza A viruses with pandemic potential. Here, we analyze influenza virus sequence data generated by the U.S. Department of Agriculture's national surveillance system to identify the central role of humans in the reemergence of pandemic H1N1 (pH1N1) influenza viruses in U.S. swine herds in 2014. These findings emphasize the important role of humans as continuous sources of influenza virus diversity in swine and indicate that influenza viruses with pandemic HA and NA segments are likely to continue to reemerge in U.S. swine in association with seasonal pH1N1 epidemics in humans.
INTRODUCTION
Swine are considered an important reservoir for the evolution of influenza A viruses (IAVs) with pandemic potential, owing to their high capacity to generate novel viruses via reassortment between cocirculating lineages (1). However, in recent years it has become apparent that humans frequently transmit IAVs to swine, and how important this process of “reverse zoonosis” is in the long-term evolution of IAV genetic diversity in swine (2). Of particular note is the number of times humans have transmitted pandemic H1N1 influenza A (pH1N1) viruses to swine on multiple continents since 2009 (3–8). A global estimate of 49 discrete introductions of pH1N1 viruses from humans to swine from 2009 to 2012 is likely an underestimate (9), and increased surveillance efforts in swine continue to identify additional introductions of pH1N1 viruses from humans to swine globally (10).
The genetic diversity of swine influenza A viruses (swIAVs) is monitored continuously in the United States by voluntary and anonymous submissions to the U.S. Department of Agriculture (USDA). Currently, there are at least 7 antigenically distinct hemagglutinin (HA) segment lineages circulating in U.S. swine: H3, H1-α, H1-β, H1-γ, H1-δ1, H1-δ2, and H1-pdm (the HA segment of pH1N1 viruses) (11). At least 4 distinct neuraminidase (NA) segment lineages are found in U.S. swine: N2-1998, N2-2002, N1-classical, and N1-pdm (the NA segment of pH1N1 viruses). During 2011, there was a marked drop in the proportion of USDA submissions of H1-pdm and N1-pdm. From the fourth quarter of 2009 (2009-Q4) to 2011-Q1, ∼20% (88/422) and ∼23% (89/376) of sequenced HA and NA segments were H1-pdm and N1-pdm, respectively, whereas these proportions dropped to <4% (25/749 for HA; 25/658 for NA) from 2011-Q2 to 2012-Q4 (11).
However, in 2014, there was a noticeable uptick in H1-pdm and N1-pdm submissions to the USDA. A possible explanation for the increase could involve fitness gains associated with balancing immune selection or reassortment between H1-pdm and N1-pdm and other genome segments. Alternatively, it was asked whether humans could be the source of the 2014 H1-pdm and N1-pdm segments in swine. To further understand the evolution of pH1N1 viruses in U.S. swine, and to resolve the origins of the 2014 H1-pdm and N1-pdm segments in particular, we conducted a large-scale phylogenetic analysis of sequence data from human and swine pH1N1 viruses collected from 2009 to 2014. We found that the 2014 H1-pdm and N1-pdm segments represent new introductions of pH1N1 viruses from humans, emphasizing the continued importance of human-to-swine transmission in the evolution of IAV in U.S. swine herds.
MATERIALS AND METHODS
Influenza virus sample preparation and sequencing.
Three genes (HA, NA, and M) of the swine isolates were sequenced by the contributing USDA National Animal Health Laboratory Network veterinary diagnostic laboratory and submitted to GenBank (accession numbers for HA and NA are listed in Table 1 and in Tables S2 and S3 in the supplemental material). Following the identification of HAs that were pH1N1, a subsample of the swine isolates were subjected to whole-genome sequencing. The whole-genome next-generation sequencing was performed using the Ion 316 v2 chip and Ion PGM 200 v2 sequencing kit (Life Technologies, Carlsbad, CA) as previously described (12).
TABLE 1.
Characteristics of 20 H1-pdm viruses from U.S. swine collected during 2014a
| GenBank accession no. | Virus name | Collection date (mo/day/yr) | Evolutionary origin |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PB2 | PB1 | PA | HA | NP | NA | MP | NS | |||
| KJ437560 | A/swine/Illinois/A01411340/2014/H1N1 | 1/−/14 | pdm | pdm | ||||||
| KJ493356 | A/swine/Illinois/A01411419/2014/H1N1 | 2/−/14 | pdm | pdm | ||||||
| KJ206094 | A/swine/Illinois/A01490609/2014 H1N1 | 1/8/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ528259 | A/swine/Illinois/A01492501/2014 H1N1 | 2/12/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ701784 | A/swine/Illinois/A01493472/2014 H1N1 | 3/26/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ701853 | A/swine/Iowa/A01410472/2014 H1N1 | 3/3/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ907733 | A/swine/Kansas/A01377299/2014 H1N1 | 4/30/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ605091 | A/swine/Kansas/A01410327/2014 H1N1 | 2/7/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ417899 | A/swine/Minnesota/A01491447/2014 H1N1 | 1/27/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ417893 | A/swine/Minnesota/A01491451/2014/H1N1 | 1/27/14 | pdm | pdm | ||||||
| KJ417884 | A/swine/Minnesota/A01491464/2014/H1N1 | 1/27/14 | pdm | pdm | ||||||
| KJ493336 | A/swine/Minnesota/A01491704/2014 H1N1 | 2/4/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ667964 | A/swine/Missouri/A01492887/2014 H1N1 | 2/10/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ206223 | A/swine/Nebraska/A01366774/2014 H1N1 | 1/17/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ417890 | A/swine/Nebraska/A01491300/2014 H1N1 | 1/27/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ417881 | A/swine/Nebraska/A01491382/2014/H1N1 | 1/27/14 | pdm | pdm | ||||||
| KJ493342 | A/swine/Nebraska/A01491508/2014/H1N1 | 1/30/14 | pdm | pdm | ||||||
| KJ588390 | A/swine/Nebraska/A01492657/2014 H1N1 | 2/27/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
| KJ809095 | A/swine/Nebraska/A01509523/2014/H1N1 | 4/24/14 | pdm | pdm | ||||||
| KJ739422 | A/swine/North Carolina/A01410573/2014 H1N1 | 3/21/14 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | pdm |
The virus name, collection date, and GenBank accession number (HA segment) are provided for the 20 HA and NA segments of pandemic origin (pdm) that were collected from swine in the United States in 2014. The evolutionary origins of HA and NA for all 20 viruses and the evolutionary origins of all eight segments for the 13 viruses that were whole-genome sequenced are provided.
Phylogenetic analysis of global pandemic H1N1 viruses in swine.
As a global context for this study, we first inferred a maximum-likelihood (ML) phylogenetic tree for 2,347 HA (H1-pdm) sequences from human and swine IAVs collected globally from 2009 to 2014. The data set included all available swine H1-pdm sequences (n = 548) and, as background, human H1-pdm sequences that were subsampled from oversampled years (e.g., 2009) (n = 1,799 total human sequences). Since relatively few human viruses from more recent years were available for 2014 from the Influenza Virus Resource at GenBank (13), 100 sequences from human viruses collected during 2014 were obtained from the GISAID Epiflu database (http://platform.gisaid.org) and used as background (see Table S1 in the supplemental material). Each segment was aligned separately using MUSCLE v3.8.1 (14). The phylogenetic relationships of these data were inferred using the ML method available in the program RAxML v7.2.6 (15), incorporating a general time-reversible (GTR) model of nucleotide substitution with a gamma-distributed (Γ) rate variation among sites. To assess the robustness of each node, a bootstrap resampling process was performed (500 replicates), again using the ML method available in RAxML v7.2.6.
Phylogenetic analysis of pandemic H1N1 viruses in swine in the United States.
To explore the evolution of pH1N1 viruses in U.S. swine in greater detail, we generated data sets of nucleotide sequences from humans and swine collected in the United States from 2009 to 2014 for three segments: H1-pdm (n = 173 swine and 760 human sequences) (see Table S2 in the supplemental material), N1-pdm (n = 178 swine and 688 human sequences) (see Table S3 in the supplemental material), and NP-pdm (n = 206 swine and 221 human sequences) (see Table S4 in the supplemental material). Phylogenetic relationships were inferred for each of the three data sets using the time-scaled Bayesian approach using the Markov chain Monte Carlo (MCMC) method available in the BEAST v1.8.0 package (16) and the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). A relaxed uncorrelated lognormal (UCLN) molecular clock was used, with a flexible Bayesian skyline plot (BSP) demographic prior (10 piecewise constant groups), and a general-time reversible (GTR) model of nucleotide substitution with gamma-distributed rate variation among sites. The MCMC chain was run separately three times for each of the data sets for at least 100 million iterations with subsampling every 10,000 iterations, using the BEAGLE library to improve computational performance (17). All parameters reached convergence, as assessed visually using Tracer v.1.6, with statistical uncertainty reflected in values of the 95% highest posterior density (HPD). At least 10% of the chain was removed as burn in, and runs for the same lineage and segment were combined using LogCombiner v1.8.0 and downsampled to generate a final posterior distribution of 1,000 trees that was used in subsequent analyses.
To more quantitatively characterize the direction and timing of the exchange of pandemic viruses between humans and swine in the United States, we adapted a phylogeographic approach, using host and temporal information as our “location states.” We assigned each virus to one of 12 categories, based on the host and the epidemic season (which was defined as September to August): (i) human 2008-2009, (ii) human 2009-2010, (iii) human 2010-2011, (iv) human 2011-2012, (v) human 2012-2013, (vi) human 2013-2014, (vii) swine 2008-2009, (viii) swine 2009-2010, (ix) swine 2010-2011, (x) swine 2011-2012, (xi) swine 2012-2013, and (xii) swine 2013-2014. Each tip on the tree was assigned to one of these categories, and transitions from one category to another (e.g., human 2008-2009 to swine 200-2009) were inferred along the internal branches representing the ancestral history of the viral population. These transitions are referred to as Markov jump counts (18) and have commonly been used in the context of phylogeography to identify discrete viral movements between spatial locations. Here, we extend this application to estimate successful introductions of virus from one host to another. For computational efficiency, the phylogeographic analysis was run using an empirical distribution of 1,000 trees, allowing the MCMC chain to be run for 25 million iterations, sampling every 1,000. A Bayesian stochastic search variable selection (BSSVS) was employed to improve statistical efficiency for all data sets containing more than four location states. Maximum clade credibility (MCC) trees were summarized using TreeAnnotator v1.8.0, and the trees were visualized in FigTree v1.4.2.
Reassortment of pandemic viruses in U.S. swine.
To identify patterns of reassortment between pH1N1 and other viruses circulating in U.S. swine, 470 whole-genome swIAV sequences from the United States from 2009 to 2014 downloaded from the Influenza Virus Resource at GenBank (13) were used in the analysis (viruses collected by surveillance targeting U.S. agricultural fairs were removed from the analysis due to the high number of identical sequences). Trees were inferred for each segment, and for the H1, H3, N1, and N2 subtypes separately, using the ML methods described above (15). For the PB2, PB1, PA, NP, MP, and NS internal gene segments, viruses were categorized as triple reassortant (trig) or pandemic (pdm). The HA segments were categorized as H1-γ, H1-δ1, H1-δ2, H1-pdm, or H3. The NA segments were categorized as N2-1998, N2-2002, N1-classical, or N1-pdm. Of the 470 viruses, 26 had eight pH1N1 segments and were not reassortants, and they were removed from subsequent analyses of reassortment.
Human influenza virus surveillance.
Influenza virus surveillance reports in the United States are prepared by the Centers for Disease Control and Prevention (CDC) and are publically available at FluView (http://www.cdc.gov/flu/weekly). Virological surveillance reports from 2009 to 2014 were downloaded from FluView (accessed 17 November 2014), and the proportion of total influenza virus specimens that were positive for pandemic influenza virus H1 was calculated for each week from the beginning of the pandemic in the United States (week 35 of 2009) to the most recent report (week 39 of 2014).
Nucleotide sequence accession numbers.
The sequences described were submitted to GenBank under the accession numbers given in Table 1.
RESULTS
Evolution of H1-pdm in swine globally from 2009 to 2014.
A phylogenetic tree was inferred using ML methods for the 2,347 H1-pdm segments collected in humans and swine globally (see Fig. S1 in the supplemental material). The tree is consistent with global phylogenies inferred previously with fewer swIAV data (9). Human-to-swine transmission of pandemic H1N1 influenza viruses occurred an estimated 133 times on a global scale from 2009 to 2014. This estimate is based on a definition of a transmission event as a monophyletic cluster of swine viruses with bootstrap values of ≥70 and singleton viruses, using methods similar to those used previously to estimate the number of introductions of human pH1N1 viruses into swine (9). This estimate is conservative, and numerous additional putative human-to-swine transmission events were apparent on the tree that did not qualify by our strict definition. A lack of spatial structuring of pH1N1 viruses in humans is evidence of extensive global migration of pH1N1 viruses via human movement. In contrast, the 133 defined clusters of pH1N1 viruses in swine remain strongly spatially structured by country, indicating that, to date, pH1N1 viruses have not disseminated via swine across national boundaries. Rather, separate human introductions into swine were observed on six continents in countries including Argentina, Australia, Brazil, Canada, Cameroon, China, Colombia, Costa Rica, Cuba, England, Finland, France, Germany, Hungary, Japan, Mexico, Nigeria, Russia, Spain, Sri Lanka, South Korea, Thailand, the United States, and Vietnam. The fact that pH1N1 viruses found in U.S. swine were not related to pH1N1 viruses found in swine in other countries, including Canada, allowed us to restrict subsequent analyses to U.S. data only.
Evolution of the H1-pdm and N1-pdm segments in U.S. swine from 2009 to 2014.
For a more refined understanding of pH1N1 evolution in U.S. swine herds, MCC trees were inferred for the H1-pdm and N1-pdm segments of viruses collected from swine and humans in the United States (the HA tree is shown in Fig. 1, the HA tree with tip labels is available in Fig. S2 in the supplemental material, and the NA tree with tip labels in Fig. S3 in the supplemental material; accession numbers are listed in Tables S2 and S3 in the supplemental material). The human pH1N1 viruses are strongly structured temporally (ladderlike), in the HA and NA phylogenies. Human viruses from the 2009 pandemic reside in the lower left portion of the phylogeny, and human viruses from the 2013-2014 epidemic are positioned at the upper right (Fig. 1, orange box), with viruses from intervening years positioned in the middle of the tree. This structured pattern of human viruses greatly facilitates inference of the timing of each human-to-swine transmission event, even in the case of long branch lengths between human and swine viruses. For example, all 2014 swIAVs on the H1-pdm tree (n = 20) (Fig. 1, purple, and Table 1) are positioned within the clade of human viruses collected during the 2013-2014 U.S. epidemic and are most likely to represent IAV transmission events from humans to swine that occurred during the 2013-2014 epidemic (Fig. 1; see Fig. S2 in the supplemental material). The number of times that the virus was transmitted from humans to U.S. swine during the 2013-2014 epidemic is estimated to be ∼10 to 12, with the lower end of the range based on clades supported by high bootstrap values and singletons on the ML tree (see Fig. S1 in the supplemental material) and the higher end of the range based on Markov jumps on the MCC tree (Fig. 1; see Fig. S2 in the supplemental material). Of the 20 HA-pdm swIAV sequences available for 2014, whole-genome sequences were also available for 13. All 13 wholly sequenced viruses had 8 pH1N1 segments, with no evidence of reassortment with other swIAVs (Table 1). This pattern is consistent with the recent introduction of these 2014 H1-pdm segments from humans, as the full pH1N1 genome has not been conserved long term in U.S. swine owing to frequent reassortment (see below).
FIG 1.

Phylogenetic relationships between pandemic H1 segments. The time-scaled Bayesian MCC tree was inferred for the HA (H1) sequences of 933 viruses collected from swine and humans in the United States from 2009 to 2014, including 173 viruses from swine and 760 viruses from humans. The branches are color coded by host and time (U.S. influenza epidemic) of collection (September to August). sw09-10, swine viruses collected during the 2009-2010 epidemic; sw10-11, swine viruses collected during the 2010-2011 epidemic; sw11-12, swine viruses collected during the 2011-2012 epidemic; sw12-13, swine viruses collected during the 2012-2013 epidemic; sw13-14, swine viruses collected during the 2013-2014 epidemic. The clade of human viruses from the 2013-2014 epidemic is highlighted in the box shaded orange. Posterior probabilities of >0.90 are provided for key nodes. An identical phylogeny with tip strain name labels is provided in Fig. S2 in the supplemental material.
The evolutionary dynamics of the H1-pdm and N1-pdm segments prior to 2014 are more complex, representing a combination of human-to-swine transmission events that occurred during the initial waves of the 2009 H1N1 pandemic but did not sustain onward transmission in swine, as well as a number of human-to-swine transmission events from the same period that continued to be transmitted in swine for at least several years. As an example of a pH1N1 virus that was not transmitted onward in pigs following its introduction from humans during the 2009 fall pandemic wave, the singleton virus A/swine/Minnesota/A01076188/2010/H1N1 (collected 24 January 2010) is closely related to the human virus, A/California/VRDL101/2009/H1N1 (collected 5 November 2009), with posterior support of 90% on the H1-pdm tree (see Fig. S2, purple arrow, in the supplemental material). In contrast, swIAVs in this lower section of the tree that were collected in later years (e.g., A/swine/Illinois/A01395201/2013/H1N1, collected 18 November 2013) represent human-to-swine transmission events that also occurred during the major U.S. fall/winter pandemic wave of 2009 and 2010 but continued to be successfully transmitted onward in swine (see Fig. S2, orange arrow, in the supplemental material). Onward transmission in swine is evidenced by highly supported monophyletic clades containing only swine viruses (100% posterior support in the case of A/swine/Illinois/A01395201/2013/H1N1) and long branch lengths. Onward virus transmission in swine is distinguishable from multiple independent introductions of similar human viruses, which would be represented on the tree by clades containing a mixture of human and swine viruses.
Markov jump counts representing host switches on the tree were used to summarize the proportion of all swIAVs that were recently introduced from humans, distinguishable from viruses that represent onward transmission in pigs across seasons. Annotating each virus by host (swine or human), as well as by season (2009-2010, 2010-2011, 2011-2012, 2012-2013, and 2013-2014 seasonal epidemics, defined as September to August), provides additional information about the likely timing of each human-to-swine transmission event, which may have occurred many years prior to the sampling of the virus in pigs. The patterns of inferred Markov jump counts were similar for the H1-pdm and N1-pdm segments, with higher estimates of human-to-swine transmission than onward swine-to-swine transmission for both of the segments. Markov jump counts for the representative N1-pdm are visualized in Fig. 2a, and the H1-pdm heat map is provided in Fig. S4 in the supplemental material. Based on these counts, it is estimated that almost 100 human-to-swine transmission events occurred in the United States from 2009 to 2014 (see Tables S5 and S6 in the supplemental material).
FIG 2.

Heat map of virus transmission between humans and swine. Markov jump counts measure the number of inferred location state transitions, modeled by a continuous-time Markov chain process, that occur along the branches of the phylogeny. The intensity of the color (red, high; white, low) reflects the number of Markov jump counts between one temporally defined host population (y axis) and another (x axis), which have been abbreviated (e.g., hu08-09, viruses collected in humans during the 2008-2009 epidemic; sw08-09, swine viruses collected during the 2008-2009 epidemic). The results are presented separately for the NA (a) and NP (b) segments. For clarity, each heat map has been divided into four quadrants that represent linkages between human viruses from a seasonal epidemic and human viruses from a different (usually preceding) epidemic (i.e., human-to-human transmission) (I), linkages between swine viruses from a seasonal epidemic and human viruses from a different (usually preceding) epidemic (i.e., human-to-swine transmission) (II), linkages between human viruses and swine viruses (i.e., swine-to-human transmission) (III), and linkages between swine viruses and swine viruses from a different (usually preceding) epidemic (i.e., swine-to-swine transmission) (IV).
Each heat map is divided into quadrants that represent four routes of transmission. Quadrant I of the N1-pdm (Fig. 2a) and H1-pdm (see Fig. S4 in the supplemental material) heat maps represents human-to-human transmission, which follows a diagonal pattern, as expected, with each epidemic's viral population reseeded primarily from the preceding year's epidemic in humans. Although only U.S. data were used in the analysis, these data should be considered to represent a global gene pool for human viruses, with viruses in the United States reseeded by global diversity in each epidemic (19). Quadrant II represents virus transmission from humans to swine. Quadrant II also has a diagonal pattern that reflects how most human and swine viruses associated with a human-to-swine transmission event are collected during the same season, which again is expected. Due to the somewhat arbitrary definition of influenza season in the Northern Hemisphere (September to August), some swine viruses were related to human viruses from what was defined as the previous season. For example, a large number of swIAVs from the 2010-2011 season were related to human viruses from the 2009-2010 season. As expected, very little swine-to-human transmission was observed in these data (Fig. 2, quadrant III). Surprisingly, however, relatively little onward swine-to-swine transmission of the N1-pdm or H1-pdm segment was observed, particularly after 2011 (Fig. 2a, quadrant IV; see Fig. S4 in the supplemental material).
Evolution of NP-pdm segments in U.S. swine from 2009 to 2014.
The pandemic internal genes were found to be evolving differently from the H1-pdm and N1-pdm segments, as reflected by the much higher rates of onward swine-to-swine transmission evident on the representative NP heat map (Fig. 2b; see Fig. S5 and Table S7 in the supplemental material). It is difficult to draw direct comparisons between the internal gene segments and H1-pdm and N1-pdm, because not all viruses are whole-genome sequenced by USDA surveillance. Therefore, the lower absolute number of human-to-swine transmission events inferred on the NP phylogeny (Fig. 2b, quadrant II) compared to the N1-pdm (Fig. 2a) and H1-pdm (see Fig. S4 in the supplemental material) phylogenies, respectively, more likely reflects the lower number of available NP sequences from swine (see Tables S2 to S4 in the supplemental material). However, a more meaningful comparison is the number of onward transmissions in swine (quadrant IV) compared to human-to-swine transmissions (quadrant II). This proportion is much higher for the NP-pdm segment than for H1-pdm and N1-pdm, reflecting the higher onward swine-to-swine transmission of NP-pdm in swine via reassortment with endemic swine viruses (described below).
Reassortment between pH1N1 viruses and other cocirculating swIAVs.
The relatively low levels of onward transmission of H1-pdm and N1-pdm, paired with higher levels for onward transmission of pandemic internal genes in swine, indicate that the H1-pdm and N1-pdm segments are frequently being replaced by reassortment with other HA and NA antigens circulating in U.S. swine (e.g., H3, H1-γ, H1-δ1, N1-classical, and N2-2002) (Fig. 3; see Table S8 in the supplemental material). USDA conducts swIAV surveillance through a voluntary and passive syndromic system that is not necessarily an unbiased representation of swIAV diversity in the United States at the population level, making it difficult to estimate the prevalence of segments with different evolutionary origins. Still, among 444 whole-genome sequences from U.S. swIAVs collected from 2009 to 2014, no viruses were identified with a H1-pdm segment and at least one nonpandemic segment, and only one virus was identified that contained an N1-pdm segment and another segment of nonpandemic origin (see Table S8 in the supplemental material). This pattern suggests that the H1-pdm and N1-pdm segments were rarely retained following reassortment events between pH1N1 and other swine viruses. In contrast, the pH1N1 internal gene segments are frequently retained following reassortment events, as reflected by the large number and diversity of reassortant viruses containing pH1N1 internal gene segments, particularly PA-pdm, NP-pdm, and MP-pdm (Fig. 3; see Table S8 in the supplemental material). There is evidence that viruses with all eight genome segments of pH1N1 origin may be transmitted transiently in swine following human-to-swine transmission (e.g., A/sw/Minnesota/165A/2009/H1N1 and A/sw/Minnesota/074A/2009/H1N1, indicated by a black arrow in Fig. S5 in the supplemental material). However, over time, the eight-segment pH1N1 genotype is rapidly broken up by reassortment with other swIAVs, in almost all cases losing the H1-pdm and N1-pdm segments. Although global reassortment patterns were not formally analyzed in our U.S.-focused study, higher onward transmission of pH1N1 internal gene segments was also observed in other countries, including China, relative to H1-pdm and N1-pdm (representative PB2 phylogeny is shown in Fig. S6 in the supplemental material).
FIG 3.

Numbers of viruses with each of the eight pandemic segments. Of 444 viruses with whole-genome sequences (not including fair pigs or viruses with all eight segments of pandemic origin), the numbers that contain PB2, PB1, PA, HA, NP, NA, MP, and NS segments of pandemic origin are shown. The frequency of each segment indicates the extent of reassortment and onward maintenance of the pH1N1 internal genes in U.S. swine, with the loss of the HA and NA genes outside wholly pandemic viruses.
Human-to-swine transmission is associated with pH1N1 activity in humans.
The frequency of human-to-swine transmission was found to be temporally associated with pH1N1 activity in humans in the United States, as measured by the proportion of respiratory samples tested by the U.S. CDC that were pH1N1 positive from 2009 to 2014 (Fig. 4). Major peaks in pH1N1 activity in humans in the United States occurred in late 2009 and again during the winter of 2013-2014, which corresponds to seasons with high numbers of human-to-swine transmission events (Fig. 4). Deviations from the apparent association, such as the high number of human-to-swine transmission events, inferred during 2010 and 2011, when pH1N1 activity in humans in the United States was low, are explained by deeper examination of the data. A heat map of the H1-pdm segment with greater temporal resolution (see Fig. S4 in the supplemental material) indicates that many of the swIAVs collected in 2010 and 2011 are likely to have been transmitted from humans during the 2009-2010 season (see Table S5 in the supplemental material), with a year-long lag in detection owing to gaps in surveillance in swine that are apparent in the phylogeny (see Fig. S2 in the supplemental material). As a case in point, a clustered pair of viruses, A/swine/Iowa/A01049077/2010/H1N1 and A/swine/Iowa/A01049195/2010/H1N1, were collected during the 2010-2011 season (collected 17 November 2010 and 1 December 2010, respectively) but are actually likely to have been transmitted from humans to swine during the fall of 2009 based on their genetic relationship with viruses sampled from humans during 2009 (e.g., A/Texas/45034157/2009/H1N1, indicated with a black arrow in Fig. S2 in the supplemental material).
FIG 4.
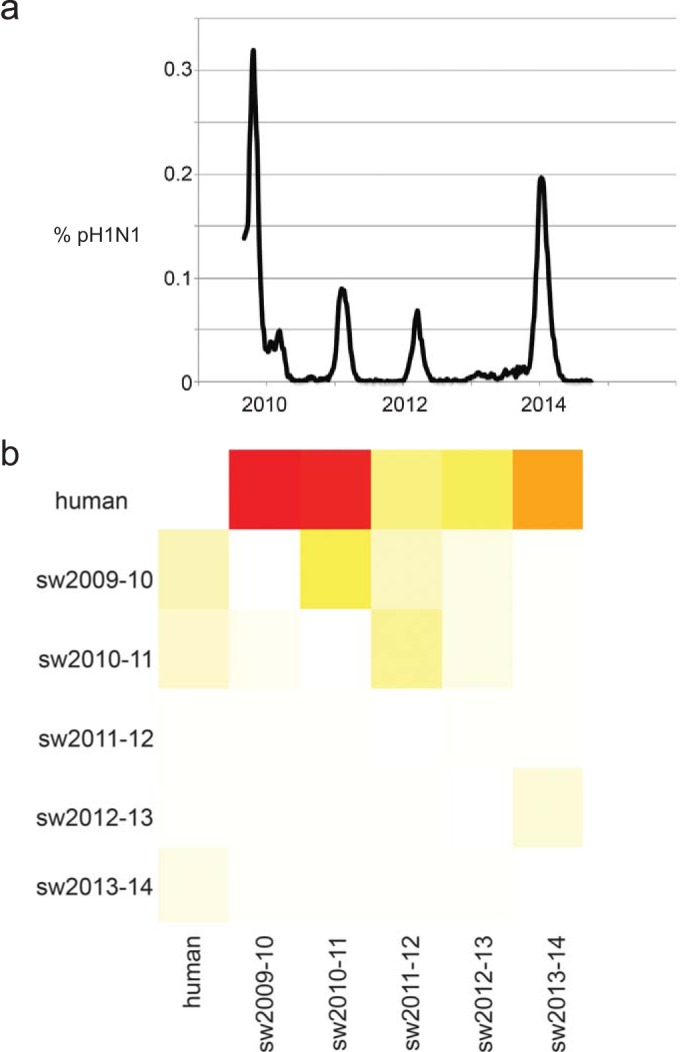
Association between human-to-swine virus transmission and pH1N1 activity in humans. (a) Proportion of total influenza virus specimens that were positive for pH1N1 virus calculated for each week from the beginning of the pandemic in the United States (week 35 of 2009) to the most recent report (week 39 of 2014), as reported by CDC FluView (http://www.cdc.gov/flu/weekly). (b) Heat map of the number of Markov jump counts between one temporally defined host population (y axis) and another (x axis). The labeling of the heat map is similar to that in Fig. 2 except that, for simplicity, all human viruses from 2009 to 2014 were consolidated into a single category, “human.”
DISCUSSION
The dissemination of pH1N1 viruses from humans to swine has been a global phenomenon and arguably represents the largest-scale reverse zoonosis of pathogens from humans to animals documented to date. Since 2009, pH1N1 viruses and pH1N1 reassortants (i.e., viruses with at least one segment of pH1N1 origin) have been identified in swine populations in the Americas (United States [20], Canada [21], Mexico [22], Argentina [6], Brazil [23], and Colombia [24]), Europe (United Kingdom [25], Germany [26], Norway [8], and Italy [27]), Asia (China [3], South Korea [28], Sri Lanka [10], Thailand [29], and Vietnam [7]), Africa (4), and Australia (5). We previously estimated that the pH1N1 virus was introduced at least 49 times from humans to swine on a global scale (9). The earlier global estimate was based on limited availability of swIAV sequence data that were restricted to the period 2009 to 2012 and that underrepresented many regions. The methods used were conservative and based on high bootstrap values, and the resulting estimate was admittedly a lower bound. Using similarly conservative methods but including a newer data set that contained recently obtained sequences from 2013 and 2014, our analysis of the H1-pdm phylogeny, as expected, identified a larger number of virus introductions. In fact, we estimated that 38 human-to-swine transmissions occurred from 2009 to 2014 in the United States alone. Our Bayesian analysis using Markov jumps suggests that this number could be considerably higher. Critically, our investigation demonstrates that human-to-swine transmission of pH1N1 has continued to occur in the United States during recent seasonal epidemics of pH1N1 and is likely to continue to recur as long as pH1N1 circulates as a seasonal influenza virus in humans.
The large-scale introduction of pH1N1 virus from humans to swine has had a major impact on the diversity of IAVs in swine globally. The pH1N1 viruses have reassorted extensively with other cocirculating swIAVs, generating a diversity of new reassortant viruses with novel combinations of genome segments (3, 20, 26). In the United States, one of the new genotypes that resulted from reassortment with pH1N1 viruses is H3N2v, with a high profile in 2011 and 2012, which was a triple-reassortant H3N2 virus that acquired the MP-pdm segment. This particular H3N2v genotype is notable because it has been associated with >330 human infections since 2011, mostly in children attending agricultural fairs (30). Although the largest number of cases occurred during the summer of 2012, with fewer cases observed during 2013 and 2014, the equivalent H3N2 virus continues to circulate in U.S. swine populations, and the risk of another major outbreak in the future remains.
The high onward transmission of pandemic internal genes in U.S. swine populations is notable, given the previous decades of strong dominance of the triple-reassortant internal-gene “TRIG” constellation since their emergence in the mid-1990s. The high level of detection of the MP-pdm segment, which is originally of Eurasian avian-like virus origin and highly genetically divergent from the TRIG MP, is particularly notable in the United States, both for the expansion of genetic diversity and for the evolution of the H3N2v variants isolated in humans that contain this segment (31). The population dynamics of swIAVs remain poorly understood, including why certain segments and segment combinations are transmitted more efficiently than others in pigs and how this relates to viral fitness and severity of clinical illness. Particularly perplexing is why the H1-pdm and N1-pdm segments do not appear to sustain transmission in swine. The fact that eight-segment pH1N1 viruses do not sustain transmission in U.S. swine herds raises important questions about where and how a virus with unclear fitness in pigs in field settings could have caused a pandemic in humans. Whole-genome sequencing and population-based surveillance with minimized bias are central to understanding viral fitness at a genomic level, along with experimental studies of transmission and pathology.
The CDC recommends that all healthy people over the age of 6 months receive the seasonal influenza vaccine. However, despite the clear relationship between human and swine IAVs and the dramatic role that human seasonal IAV has played in driving the genetic diversity of IAV in swine (32), swine workers and veterinarians are not considered to be a priority for human seasonal or pandemic vaccines (33). The swine industry recommends vaccination of swine workers, as well as sick leave policies for workers with influenza-like illness (34), but there are no public reports available to indicate compliance with these recommendations. Since human pH1N1 virus in its entirety is virulent in pigs (35, 36) and would more likely be captured by the USDA syndromic surveillance than an avirulent or weakly pathogenic virus, such as a human-adapted seasonal H3N2 or pre-2009 H1N1 virus, the pH1N1 virus human-to-swine transmission episodes we describe here provide an optimal example to reinforce the human influenza vaccine recommendations within the pork industry. Although gaps in sequence data from the U.S. swine population are apparent, surveillance has been greatly intensified since 2009, allowing these important observations in IAV evolution and population dynamics. The results reported here and elsewhere (11) underscore the value of the national surveillance system and the need to sustain these efforts beyond the initial 2009 H1N1 pandemic response.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the Multinational Influenza Seasonal Mortality Study (MISMS), an ongoing international collaborative effort to understand influenza epidemiology and evolution, led by the Fogarty International Center, National Institutes of Health, with funding from the Office of Global Affairs at the Department of Health and Human Services (M.I.N.). Funding was provided by USDA-ARS (J.S. and A.L.V.) and by USDA-APHIS (A.J.-M. and M.L.K.).
Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00459-15.
REFERENCES
- 1.Scholtissek C. 1990. Pigs as the “mixing vessel” for the creation of new pandemic influenza A viruses. Med Princ Pract 2:65–71. [Google Scholar]
- 2.Nelson MI, Wentworth DE, Culhane MR, Vincent AL, Viboud C, LaPointe MP, Lin X, Holmes EC, Detmer SE. 2014. Introductions and evolution of human-origin seasonal influenza A viruses in multinational swine populations. J Virol 88:10110–10119. doi: 10.1128/JVI.01080-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vijaykrishna D, Poon LLM, Zhu HC, Ma SK, Li OTW, Cheung CL, Smith GJD, Peiris JSM, Guan Y. 2010. Reassortment of pandemic H1N1/2009 influenza A virus in swine. Science 328:1529. doi: 10.1126/science.1189132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Njabo KY, Fuller TL, Chasar A, Pollinger JP, Cattoli G, Terregino C, Monne I, Reynes J-M, Njouom R, Smith TB. 2012. Pandemic A/H1N1/2009 influenza virus in swine, Cameroon, 2010. Vet Microbiol 156:189–192. doi: 10.1016/j.vetmic.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holyoake PK, Kirkland PD, Davis RJ, Arzey KE, Watson J, Lunt R, Wang J, Wong F, Moloney BJ, Dunn SE. 2011. The first identified case of pandemic H1N1 influenza in pigs in Australia. Aust Vet J 89:427–431. doi: 10.1111/j.1751-0813.2011.00844.x. [DOI] [PubMed] [Google Scholar]
- 6.Pereda A, Cappuccio J, Quiroga M, Baumeister E, Insarralde L, Ibar M, Sanguinetti R, Cannilla ML, Franzese D, Escobar Cabrera OE, Craig MI, Rimondi A, Machuca M, Debenedetti RT, Zenobi C, Barral L, Balzano R, Capalbo S, Risso A, Perfumo CJ. 2010. Pandemic (H1N1) 2009 outbreak on pig farm, Argentina. Emerg Infect Dis 16:304–307. doi: 10.3201/eid1602.091230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trevennec K, Leger L, Lyazrhi F, Baudon E, Cheung CY, Roger F, Peiris M, Garcia J-M. 2012. Transmission of pandemic influenza H1N1 (2009) in Vietnamese swine in 2009–2010. Influenza Other Respir Viruses 6:348–357. doi: 10.1111/j.1750-2659.2011.00324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofshagen M, Gjerset B, Er C, Tarpai A, Brun E, Dannevig B, Bruheim T, Fostad IG, Iversen B, Hungnes O, Lium B. 2009. Pandemic influenza A(H1N1)v: human to pig transmission in Norway? Euro Surveill 14(45):pii=19406. http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19406. [DOI] [PubMed] [Google Scholar]
- 9.Nelson MI, Gramer MR, Vincent AL, Holmes EC. 2012. Global transmission of influenza viruses from humans to swine. J Gen Virol 93:2195–2203. doi: 10.1099/vir.0.044974-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perera HKK, Wickramasinghe G, Cheung CL, Nishiura H, Smith DK, Poon LLM, Perera AKC, Ma SK, Sunil-Chandra NP, Guan Y, Peiris JSM. 2013. Swine influenza in Sri Lanka. Emerg Infect Dis 19:481–484. doi: 10.3201/eid1903.120945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson TK, Nelson MI, Kitikoon P, Swenson SL, Korslund JA, Vincent AL. 2013. Population dynamics of cocirculating swine influenza A viruses in the United States from 2009 to 2012. Influenza Other Respir Viruses 7(Suppl 4):S42–S51. doi: 10.1111/irv.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowman AS, Sreevatsan S, Killian ML, Page SL, Nelson SW, Nolting JM, Cardona C, Slemons RD. 2012. Molecular evidence for interspecies transmission of H3N2pM/H3N2v influenza A viruses at an Ohio agricultural fair, July 2012. Emerg Microbes Infect 1:e33. doi: 10.1038/emi.2012.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. 2008. The influenza virus resource at the National Center for Biotechnology Information. J Virol 82:596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 16.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suchard MA, Rambaut A. 2009. Many-core algorithms for statistical phylogenetics. Bioinformatics 25:1370–1376. doi: 10.1093/bioinformatics/btp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minin VN, Suchard MA. 2008. Counting labeled transitions in continuous-time Markov models of evolution. J Math Biol 56:391–412. doi: 10.1007/s00285-007-0120-8. [DOI] [PubMed] [Google Scholar]
- 19.Nelson MI, Simonsen L, Viboud C, Miller MA, Holmes EC. 2007. Phylogenetic analysis reveals the global migration of seasonal influenza A viruses. PLoS Pathog 3:1220–1228. doi: 10.1371/journal.ppat.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ducatez MF, Hause B, Stigger-Rosser E, Darnell D, Corzo C, Juleen K, Simonson R, Brockwell-Staats C, Rubrum A, Wang D, Webb A, Crumpton J-C, Lowe J, Gramer M, Webby RJ. 2011. Multiple reassortment between pandemic (H1N1) 2009 and endemic influenza viruses in pigs, United States. Emerg Infect Dis 17:1624–1629. doi: 10.3201/eid1709.110338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasma T, Joseph T. 2010. Pandemic (H1N1) 2009 infection in swine herds, Manitoba, Canada. Emerg Infect Dis 16:706–708. doi: 10.3201/eid1604.091636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Escalera-Zamudio M, Cobián-Güemes G, de los Dolores Soto-del Río M, Isa P, Sánchez-Betancourt I, Parissi-Crivelli A, Martínez-Cázares MT, Romero P, Velázquez-Salinas L, Huerta-Lozano B, Nelson M, Montero H, Vinuesa P, López S, Arias CF. 2012. Characterization of an influenza A virus in Mexican swine that is related to the A/H1N1/2009 pandemic clade. Virology 433:176–182. doi: 10.1016/j.virol.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 23.Schaefer R, Zanella JRC, Brentano L, Vincent AL, Ritterbusch GA, Silveira S, Caron LNM. 2011. Isolation and characterization of a pandemic H1N1 influenza virus in pigs in Brazil. Pesqui Vet Bras 31:761–767. doi: 10.1590/S0100-736X2011000900007. [DOI] [Google Scholar]
- 24.Ramirez-Nieto GC, Rojas CAD, Alfonso VJ, Correa JJ, Galvis JD. 2012. First isolation and identification of H1N1 swine influenza viruses in Colombian pig farms. Health 4:983–990. doi: 10.4236/health.2012.430150. [DOI] [Google Scholar]
- 25.Howard W, Essen S. 2011. Reassortant pandemic (H1N1) 2009 virus in pigs, United Kingdom. Emerg Infect Dis 17:1049–1052. doi: 10.3201/eid/1706.101886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Starick E, Lange E, Fereidouni S, Bunzenthal C, Höveler R, Kuczka A, grosse Beilage E, Hamann H-P, Klingelhöfer I, Steinhauer D, Vahlenkamp T, Beer M, Harder T. 2011. Reassorted pandemic (H1N1) 2009 influenza A virus discovered from pigs in Germany. J Gen Virol 92:1184–1188. doi: 10.1099/vir.0.028662-0. [DOI] [PubMed] [Google Scholar]
- 27.Guercio A, Purpari G, Conaldi P, Pagano V. 2012. Pandemic influenza A/H1N1 virus in a swine farm house in Sicily, Italy. J Environ Biol 33:155–157. [PubMed] [Google Scholar]
- 28.Han J, Park SJ, Kim HK, Rho S, Nguyen Van G, Song D, Kang BK, Moon HJ, Yeom MJ, Park BK. 2012. Identification of reassortant pandemic H1N1 influenza virus in Korean pigs samples for virus isolation. J Microbiol Biotechnol 22:699–707. doi: 10.4014/jmb.1106.05062. [DOI] [PubMed] [Google Scholar]
- 29.Sreta D, Tantawet S, Na Ayudhya SN, Thontiravong A, Wongphatcharachai M, Lapkuntod J, Bunpapong N, Tuanudom R, Suradhat S, Vimolket L, Poovorawan Y, Thanawongnuwech R, Amonsin A, Kitikoon P. 2010. Pandemic (H1N1) 2009 virus on commercial swine farm, Thailand. Emerg Infect Dis 16:1587–1590. doi: 10.3201/eid1610.100665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Epperson S, Jhung M, Richards S, Quinlisk P, Ball L, Moll M, Boulton R, Haddy L, Biggerstaff M, Brammer L, Trock S, Burns E, Gomez T, Wong KK, Katz J, Lindstrom S, Klimov A, Bresee JS, Jernigan DB, Cox N, Finelli L. 2013. Human infections with influenza A(H3N2) variant virus in the United States, 2011-2012. Clin Infect Dis 57:S4–S11. doi: 10.1093/cid/cit272. [DOI] [PubMed] [Google Scholar]
- 31.Nelson MI, Vincent AL, Kitikoon P, Holmes EC, Gramer MR. 2012. Evolution of novel reassortant A/H3N2 influenza viruses in North American swine and humans, 2009–2011. J Virol 86:8872–8878. doi: 10.1128/JVI.00259-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson MI, Vincent AL. 2015. Reverse zoonosis of influenza to swine: new perspectives on the human-animal interface. Trends Microbiol 23:142–153. doi: 10.1016/j.tim.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.CDC. 2014. Vaccination: who should do it, who should not and who should take precautions. http://www.cdc.gov/flu/protect/whoshouldvax.htm Accessed 15 January 2015.
- 34.National Pork Board. 2015. Pork checkoff recommends producers and workers get flu vaccination. http://www.pork.org/pork-checkoff-recommends-producers-workers-get-flu-vaccination-2/ Accessed 15 January 2015.
- 35.Weingartl HM, Berhane Y, Hisanaga T, Neufeld J, Kehler H, Emburry-Hyatt C, Hooper-McGreevy K, Kasloff S, Dalman B, Bystrom J, Alexandersen S, Li Y, Pasick J. 2010. Genetic and pathobiologic characterization of pandemic H1N1 2009 influenza viruses from a naturally infected swine herd. J Virol 84:2245–2256. doi: 10.1128/JVI.02118-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brookes SM, Núñez A, Choudhury B, Matrosovich M, Essen SC, Clifford D, Slomka MJ, Kuntz-Simon G, Garcon F, Nash B, Hanna A, Heegaard PMH, Quéguiner S, Chiapponi C, Bublot M, Garcia JM, Gardner R, Foni E, Loeffen W, Larsen L, Van Reeth K, Banks J, Irvine RM, Brown IH. 2010. Replication, pathogenesis and transmission of pandemic (H1N1) 2009 virus in non-immune pigs. PLoS One 5:e9068. doi: 10.1371/journal.pone.0009068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.