

ABSTRACT
Arenaviruses are important emerging human pathogens maintained by noncytolytic persistent infection in their rodent reservoir hosts. Despite high levels of viral replication, persistently infected carrier hosts show only mildly elevated levels of type I interferon (IFN-I). Accordingly, the arenavirus nucleoprotein (NP) has been identified as a potent IFN-I antagonist capable of blocking activation of interferon regulatory factor 3 (IRF3) via the retinoic acid inducible gene (RIG)-I/mitochondrial antiviral signaling (MAVS) pathway. Another important mechanism of host innate antiviral defense is represented by virus-induced mitochondrial apoptosis via RIG-I/MAVS and IRF3. In the present study, we investigated the ability of the prototypic Old World arenavirus lymphocytic choriomeningitis virus (LCMV) to interfere with RIG-I/MAVS-dependent apoptosis. We found that LCMV does not induce apoptosis at any time during infection. While LCMV efficiently blocked induction of IFN-I via RIG-I/MAVS in response to superinfection with cytopathic RNA viruses, virus-induced mitochondrial apoptosis remained fully active in LCMV-infected cells. Notably, in LCMV-infected cells, RIG-I was dispensable for virus-induced apoptosis via MAVS. Our study reveals that LCMV infection efficiently suppresses induction of IFN-I but does not interfere with the cell's ability to undergo virus-induced mitochondrial apoptosis as a strategy of innate antiviral defense. The RIG-I independence of mitochondrial apoptosis in LCMV-infected cells provides the first evidence that arenaviruses can reshape apoptotic signaling according to their needs.
IMPORTANCE Arenaviruses are important emerging human pathogens that are maintained in their rodent hosts by persistent infection. Persistent virus is able to subvert the cellular interferon response, a powerful branch of the innate antiviral defense. Here, we investigated the ability of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) to interfere with the induction of programmed cell death, or apoptosis, in response to superinfection with cytopathic RNA viruses. Upon viral challenge, persistent LCMV efficiently blocked induction of interferons, whereas virus-induced apoptosis remained fully active in LCMV-infected cells. Our studies reveal that the persistent virus is able to reshape innate apoptotic signaling in order to prevent interferon production while maintaining programmed cell death as a strategy for innate defense. The differential effect of persistent virus on the interferon response versus its effect on apoptosis appears as a subtle strategy to guarantee sufficiently high viral loads for efficient transmission while maintaining apoptosis as a mechanism of defense.
INTRODUCTION
The arenaviruses are a large family of emerging viruses that includes several causative agents of severe viral hemorrhagic fevers with high mortality in humans (1, 2). Moreover, the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) provides a powerful experimental system for the discovery of fundamental concepts of virus-host interaction and viral immunobiology applicable to other pathogens (3, 4). Arenaviruses are enveloped negative-strand RNA viruses whose nonlytic life cycle is restricted to the cytoplasm (1). The viral genome is comprised of two RNA segments that code for two proteins each, using an ambisense coding strategy. The small (S) RNA segment encodes the envelope glycoprotein precursor (GPC) and the nucleoprotein (NP), and the L segment encodes the matrix protein (Z) and the viral polymerase (L).
In their natural reservoir hosts, arenaviruses are maintained by persistent infection via vertical transmission from infected mothers to offspring in utero (1). Infection with LCMV of most mouse strains within 24 h of birth, prior to negative selection of T cell and B cell repertoires, results in tolerance and the establishment of a largely asymptomatic carrier state (3). Despite extensive viral replication and high viral loads throughout organs and tissues (5), LCMV carrier mice show only a modest type I interferon (IFN-I) response (6), suggesting that arenaviruses evade or actively suppress, or both, innate immunity (7). Major pathogen recognition receptors (PRRs) implicated in innate detection of arenaviruses in many cell types are the cytosolic RNA helicases (RLHs) retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) (8–11), whereas Toll-like receptor 7 (TLR7) has been implicated in recognition of arenaviruses in plasmacytoid dendritic cells (12, 13). Upon activation, RIG-I and MDA5 induce downstream signaling by binding to the mitochondrial adaptor mitochondrial antiviral signaling (MAVS) protein, also known as IFN-β promoter stimulator 1 (IPS-1), CARD adaptor inducing IFN-β (Cardif), and virus-induced signaling adaptor (VISA) (14–17). Activation of MAVS leads to the assembly of a signaling complex, including tumor necrosis factor receptor-associated factors (TRAFs), classical IκB kinases IKKα/IKKβ/NEMO involved in activation of nuclear factor κB (NF-κB), and the IKK-related TANK-binding kinase (TBK1) and IKKε (14–18) that activate interferon regulatory factor 3(IRF3) and IRF7 (19). The arenavirus NP was identified as an IFN antagonist viral factor able to block the induction of IFN-I in the host cell by preventing the activation of the transcription factors IRF3 and NF-κB (20, 21). The IFN-I-counteracting activity of NP has been linked to a 3′–5′ exoribonuclease activity located within the C-terminal region of NP (22–27). Interestingly, infection with different strains of the New World arenavirus Junin (JUNV) induced IFN-I in a RIG-I-dependent manner (28), suggesting that arenavirus species differ in their ability to evade innate immunity.
Another important mechanism of innate antiviral defense is the induction of programmed cell death, or apoptosis, in response to viral infection (29). Apoptosis is a fundamental process essential for normal development and health and can be triggered via an extrinsic pathway involving death receptors of the tumor necrosis factor receptor (TNFR) family as well as an intrinsic pathway induced at the mitochondrion (30). Many DNA and RNA viruses can induce and influence apoptosis in the host cell by affecting pro- and antiapoptotic signaling pathways (31, 32). Notably, RNA and DNA viruses can trigger mitochondrial apoptosis via a RIG-I/MAVS-dependent pathway involving IRF3 (33, 34). In contrast to the IFN-I response that requires translocation of IRF3 into the nucleus, apoptotic activation of IRF3 via MAVS results in its association with the proapoptotic protein Bax in the cytosol (33). Subsequent translocation of the IRF3/Bax complex to the mitochondrion induces cytochrome c release from the inner mitochondrial membrane, which leads to oligomerization of apoptotic protease-activating factor 1 (APAF-1), followed by autocatalytic cleavage of procaspase 9 and activation of the effector caspase 3.
Recent studies addressed the capacity of the New World arenavirus JUNV to influence apoptotic signaling in the host cell. Infection of human and primate cells with JUNV induced apoptosis in a RIG-I-dependent, IFN-I-independent manner (35). Moreover, JUNV NP was found to act as a decoy substrate for activated caspase 3 (36), providing the first evidence that arenaviruses can counteract proapoptotic signaling. In the present study, we sought to extend these studies by investigating the interplay of the prototypic Old World arenavirus LCMV with RIG-I/MAVS-dependent mitochondrial apoptosis. We found that, in contrast to JUNV, LCMV does not induce apoptosis at any time during infection. While LCMV efficiently blocked RIG-I/MAVS-dependent induction of IFN-I in response to superinfection with cytolytic RNA viruses, MAVS-dependent mitochondrial apoptosis remained fully active in LCMV-infected cells. Remarkably, virus-induced mitochondrial apoptosis became independent of RIG-I in LCMV-infected cells, suggesting that LCMV persistence reshapes the host cell program that contributes to virus-induced proapoptotic signaling.
MATERIALS AND METHODS
Cells and viruses.
Human lung adenocarcinoma epithelial cells (A549), human fibrosarcoma cells (HT-1080), mouse embryonic fibroblasts (MEF), and human hepatocarcinoma Huh7 and Huh7.5 cells (37) were maintained in Dulbecco's modified Eagle medium (DMEM) (Gibco BRL) supplemented with 10% fetal calf serum (FCS) at 37°C and 5% CO2. Primary mouse cardiac fibroblasts (MCF) isolated from day 2 mouse heart were purchased from ScienCell Research Laboratories (Carlsbad, CA) and cultured according to the manufacturer's instructions. Cells were expanded for five doubling times under the conditions recommended by the provider, and their differentiated phenotype was verified by immunofluorescence staining for fibronectin expression.
To infect cells, we used LCMV ARM53b variant clone 13, vesicular stomatitis virus (VSV) serotype Indiana, and the Cantell strain of Sendai virus (SeV), purchased from Charles River Laboratories (Wilmington, MA). For the use of VSV at the Institute of Microbiology of the Lausanne University Hospital, we obtained permission number A141238 from the Office Fédéral de la Securité Alimentaire et des Affaires Veterinaries (OSAV). The viruses were diluted in 10% FCS-DMEM and added to the cells for 1 h at 37°C. The inoculum was then removed and replaced by fresh cell medium. Detection of LCMV infection by flow cytometry was performed via intracellular staining for LCMV NP as described previously (38). Samples were analyzed with a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA) using Cell Quest software. Infectious virus titers were determined by immunofocus assay (IFA) on fresh monolayers of Vero E6 cells as described previously (39).
Antibodies and reagents.
Guinea pig serum anti-LCMV and mouse monoclonal antibody (MAb) 113 anti-LCMV NP have been previously described (40). Monoclonal antibody anti-α-tubulin was from Sigma-Aldrich (St. Louis, MO). Rabbit MAb directed against the cleaved form of poly(ADP-ribose) polymerase (clPARP), rabbit polyclonal antibody (pAb) anti-Akt, and rabbit MAb anti-phospho-Akt were from Cell Signaling Technology (Danvers, MA). Mouse MAb against RIG-I protein was purchased from Adipogen International (Liestal, Switzerland). Mouse MAb anti-MAVS and mouse MAb anti-cytochrome c were from Enzo Life Sciences (Farmingdale, NY) and Life Technologies (Carlsbad, CA), respectively. Mouse MAb 23H12 to the M protein of VSV (41) was kindly provided by Douglas S. Lyles (Wake Forest University School of Medicine, Winston-Salem, NC). Alexa Fluor 488 F(ab′)2 fragment of goat anti-mouse IgG and Alexa Fluor 594 goat anti-mouse IgG2a were purchased from Life Technologies (Carlsbad, CA). Staurosporine used as an inducer of mitochondrial apoptosis and the inhibitor of phosphatidylinositol 3-kinase (PI3K) LY294002 (25 μM) were from a kinase inhibitor library from Enzo Life Sciences (Farmingdale, NY).
Detection of caspase activity.
To monitor activation of caspases 9 and 3/7, we used Caspase-Glo 9 and 3/7 assay kits from Promega (Madison, WI). Briefly, 2 × 104 cells per well were seeded in 96-well white, clear-bottom plates on the day prior to the experiment. Cells were then infected with viruses or treated with drugs for the time points indicated on the figures. Cells were processed according to the manufacturer's protocol, and caspase activity was measured by luminescence detection in a TriStar LB 941 multimode microplate reader from Berthold Technologies (Bad Wildbad, Germany).
Microscopy.
To visualize the release of cytochrome c from mitochondria, A549 cells were seeded on glass eight-well LabTek tissue culture chamber slides (Nunc) and infected with viruses or treated with drugs for different time periods as indicated on the figures. Specimens were fixed with 2% (wt/vol) formaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature and washed with PBS. Cells were then permeabilized for 30 min at room temperature with 0.1% (wt/vol) saponin, 10% (vol/vol) goat serum, and 100 mM glycine-PBS. Primary and secondary antibodies were diluted in 0.1% (wt/vol) saponin–1% (vol/vol) goat serum in PBS and incubated overnight at 4°C and for 1 h at room temperature, respectively. Specimens were mounted using Mowiol (Sigma-Aldrich, St. Louis, MO). Image acquisition was performed with a Zeiss LSM710 Quasar confocal microscope equipped with a Plan Apochromat lens (40× or 20× objective; 1.2 numerical aperture [NA]), a 405-nm diode laser, argon lasers (458, 476, 488, and 514 nm), and a 561-nm diode-pumped solid-state (DPSS) laser. To quantify cytochrome c release, the nucleoprotein (NP) of LCMV was detected using guinea pig anti-LCMV serum using an Alexa Fluor 594-conjugated goat anti-guinea pig IgG secondary antibody. Cytochrome c was detected with mouse MAb anti-cytochrome c and an Alexa Fluor 488 F(ab′)2 fragment of goat anti-mouse IgG. Cell nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI), and specimens were examined under a fluorescence microscope equipped with a narrow band-pass filter. In three visual fields, 100 NP-positive cells (red) were counted, and cells showing release of cytochrome c (green) were scored. In the staurosporine-treated specimens, 3 × 100 cells were examined, and redistribution of cytochrome c was scored.
Western blotting.
Cells were lysed using sodium dodecyl sulfate (SDS) sample buffer, and the lysates were boiled for 5 min at 95°C before being processed by SDS-polyacrylamide gel electrophoresis (PAGE). Proteins were transferred to nitrocellulose membranes and then detected using specific primary antibodies and species-specific secondary antibodies. Protein bands were visualized using a chemiluminescence detection kit (GE Health Care, United Kingdom) and a charge-coupled-device (CCD) camera (ImageQuant LAS4000; GE Health Care) according to the manufacturer's instructions.
Real-time RT-qPCR and detection of IFN-α protein.
Total RNA was purified with an RNeasy minikit (Qiagen, Chatsworth, CA), and cDNA was synthesized using a high-capacity cDNA reverse transcription kit from Applied Biosystems (Foster City, CA). TaqMan probes specific for human IFN-β (Hs01077958_s1), mouse IFN-β (Mm00439552_s1), MDA5 (IFIH1; Hs01070332_m1), NOD2 (Hs00223394_m1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Hs99999905_m1) were obtained from Applied Biosystems. Reverse transcription-quantitative PCR (RT-qPCR) was performed using a StepOne RT-qPCR system (Applied Biosystems), and gene expression levels relative to GAPDH were determined according to the 2−ΔΔCT (where CT is threshold cycle) method (42).
For the detection of IFN-α protein, cell culture supernatants of infected cells were harvested, centrifuged for 5 min at 12,000 rpm, and kept on ice. IFN-α protein was detected using a multisubtype enzyme-linked immunosorbent assay (ELISA) kit for the quantitative determination of human IFN-α in cell culture supernatant samples (PBL Interferon Source, Piscataway, NJ) in a 96-well plate format according to the manufacturer's recommendations.
RNAi.
RNA interference (RNAi) was performed using validated small interfering RNAs (siRNAs) for RIG-I (DDX58; SI03019646), MDA5 (IFIH1; SI03648981), MAVS (VISA; SI04190368), and NOD2 (CARD15; SI00133049) and a scrambled siRNA (1027280) as a control from Qiagen (Chatsworth, CA). Briefly, 5 × 105 cells in six-well plates were first reverse transfected and then forward transfected 24 h later with 2.5 nM siRNA and Lipofectamine RNAiMAX (Invitrogen, Paisley, United Kingdom) as described previously (43). At 24 h after the second transfection, cells were replated and, 8 h later, infected. In parallel samples, the efficiency of knockdown was verified by Western blotting or RT-qPCR detection of mRNA at 72 h after the first transfection, as indicated on the figures.
Statistical analysis.
Caspase assay data were analyzed using one-way analysis of variance (ANOVA), followed by a Bonferroni posttest, as indicated in the figures and figure legends.
RESULTS
LCMV does not induce mitochondrial apoptosis.
Infection of cells with cytolytic RNA viruses such as Sendai virus (SeV) and vesicular stomatitis virus (VSV) can induce mitochondrial apoptosis via a RIG-I/MAVS-dependent pathway involving IRF3 and Bax (33, 34), and, recently, RIG-I-dependent apoptosis was observed in cells infected with the New World arenavirus JUNV (35). Here, we extended these studies investigating a possible role of this pathway in the infection with the prototypic Old World arenavirus LCMV. Human lung epithelial cells (A549) and the fibrosarcoma cell line HT-1080, originally used to elucidate the pathway (33), were infected with LCMV at a multiplicity of infection (MOI) of 3, resulting in >95% infected cells after 24 h, as assessed by flow cytometry (data not shown). Activation of caspase 9 and caspase 3/7 was monitored over time using a robust biochemical assay, as detailed in Materials and Methods. Neither caspase 9 nor caspase 3/7 underwent significant activation during up to 3 days of LCMV infection (Fig. 1A). In contrast, infection with SeV and VSV, as well as exposure to the proapoptotic drug staurosporine, induced robust caspase 9 and 3/7 activity (Fig. 1A). In line with the lack of caspase activation, we did not observe cytochrome c leaking from mitochondria in LCMV-infected cells, whereas exposure to staurosporine resulted in marked cytochrome c release (Fig. 1B and C). Next, we examined cleavage of poly(ADP-ribose) polymerase (PARP), a substrate of caspase 3 and a validated marker of apoptosis. Infection with SeV and VSV resulted in extensive PARP cleavage, as reported previously (33, 34), whereas no PARP processing was detected in LCMV-infected cells (Fig. 1D), consistent with the lack of caspase activation and absence of cytochrome c release (Fig. 1A).
FIG 1.

LCMV infection does not induce mitochondrial apoptosis. (A) No activation of caspases 9 and 3/7 in LCMV-infected cells. A549 and HT-1080 cells were infected with LCMV (MOI of 3) or left uninfected (u), and activation of caspases 9 and 3/7 was measured at the indicated time postinfection (hpi) using Caspase-Glo luminescence assays as described in Materials and Methods. In the positive controls, cells were exposed to 3 μM staurosporine (St) for 8 h or infected with SeV (80 hemagglutinin units/ml) or VSV (MOI of 3) for 16 h. Data presented are normalized to relative luminescence units (RLU) detected in uninfected cells (n = 3; means ± standard deviations). Statistical significance was assessed by one-way ANOVA as follows: *, P < 0.0001; n.s., not significant (P > 0.05). (B) Cytochrome c release in LCMV infected cells. A549 cells were infected with LCMV, left uninfected, or exposed to staurosporine as described for panel A. At 24 and 48 h postinfection cells were fixed, permeabilized, and stained for cytochrome c (green), and nuclei were visualized with DAPI (blue). Specimens were examined by confocal laser scanning microscopy. Scale bar, 20 μm. (C) Quantification of cytochrome c release in LCMV-infected cells. A549 cells were infected with LCMV for 48 and 72 h, left uninfected, or exposed to staurosporine. At the indicated time points, cells were fixed, permeabilized, and stained for LCMV NP using a guinea pig serum anti-LCMV and an Alexa Fluor 594-conjugated secondary antibody. Cytochrome c was detected as described for panel B. In LCMV-infected specimens, 100 NP-positive cells were examined, and cells showing release of cytochrome c were scored. In the staurosporine-treated specimens, cytochrome c release of 100 cells was assessed. Data represent the means of triplicates ± standard deviations. (D) PARP cleavage in LCMV-infected cells. A549 cells were infected with LCMV (MOI of 3), SeV (80 hemagglutinin units /ml), or VSV (MOI of 3) or left uninfected (U). At 48 h after LCMV infection and 16 h after VSV and SeV infections, cells were lysed, and the cleavage of PARP was assessed by Western blotting using an antibody specific for cleaved PARP (clPARP). Tubulin expression was used as a loading control.
Some RNA viruses are able to activate phosphatidylinositol 3-kinase (PI3K) early in infection, resulting in a delay of mitochondrial apoptosis via phosphorylation and activation of the antiapoptotic kinase Akt/protein kinase B (PKB) (44). To address this possibility, we examined the activation of PI3K/Akt during the course of LCMV infection. Briefly, A549 cells were infected with LCMV, and Akt phosphorylation was detected with phospho-Akt-specific antibodies at different time points postinfection. During infection with LCMV, we observed enhanced phosphorylation of Akt in a biphasic manner, with an early activation at around 4 h postinfection, followed by a later and more sustained activation at 24 h (Fig. 2A). Inhibition of PI3K by the inhibitor LY294002 abolished virus-induced phosphorylation of Akt (Fig. 2A). To address a possible role of early virus-induced PI3K/Akt activation in preventing apoptosis, cells were infected with LCMV or SeV in the presence of LY294002, and activation of caspase 3/7 was monitored over time. The presence of the PI3K inhibitor did not accelerate or enhance the induction of caspase 3/7 after challenge with LCMV (Fig. 2B), but it did so after challenge with SeV (Fig. 2C), as reported earlier (44), suggesting that activation of PI3K is not responsible for the absence of apoptosis during LCMV infection.
FIG 2.
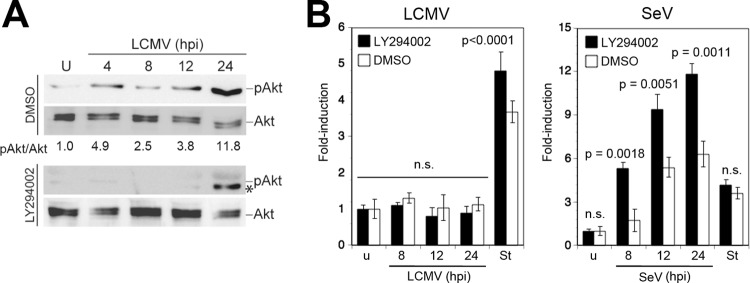
LCMV activates the PI3K/Akt pathway. (A) LCMV infection induces Akt phosphorylation. A549 cells were infected with LCMV (MOI of 3) or left uninfected (U) in the presence of the PI3K inhibitor LY294002 or vehicle alone (dimethyl sulfoxide [DMSO]). Cells were lysed at the indicated time points, and phosphorylation of Akt (pAkt) was assessed by Western blotting using a phosphospecific antibody. Expression of Akt was detected with a phosphorylation-insensitive antibody. Signals were analyzed by densitometry, and the ratio of phospho-Akt/Akt calculated with uninfected cells was defined as 1. At 24 h postinfection an additional band (*), distinct from phosphorylated Akt (pAkt), was detected in the presence of LY294002. (B) Inhibition of PI3K activation does not induce apoptosis in LCMV-infected cells. A549 cells were infected with LCMV (MOI of 3), SeV (80 hemagglutinin units/ml), or left uninfected (u) in the presence or absence of LY294002 as described for panel A. Activation of caspase 3/7 was assessed at the indicated time postinfection as described in the legend of Fig. 1A. Staurosporine (St) treatment (3 μM for 8 h) was used as a positive control. Data presented are normalized to values of uninfected (u) cells (n = 3; means ± standard deviations). Statistical analysis for SeV-infected specimens was performed by one-way ANOVA comparing results for specimens treated with LY294002 with those for specimens treated with vehicle (DMSO). For LCMV, we compared results for LCMV-infected cells with those for staurosporine-treated cells. P values are indicated. n.s., not significant (P > 0.05).
LCMV blocks RIG-I/MAVS-dependent virus-induced IFN-I production but not apoptosis.
Next, we compared the effect of LCMV on the RIG-I/MAVS-dependent signaling pathways involved in the induction of IFN-I (14–17) and virus-activated mitochondrial apoptosis in response to challenge with cytolytic RNA viruses (33, 34). First, we confirmed the role of RIG-I in both pathways in our system. To this end, we depleted RIG-I from A549 cells by RNA interference (RNAi). Cells treated with an siRNA to RIG-I showed >95% reduction in RIG-I protein after 72 h compared to the level of the control, as assessed by densitometric analysis of the Western blot (Fig. 3A). Consistent with earlier reports, depletion of RIG-I prevented the induction of IFN-β mRNA (14, 33) (Fig. 3B) and markedly reduced virus-induced mitochondrial apoptosis in response to infection with SeV or VSV (33) (Fig. 3C).
FIG 3.
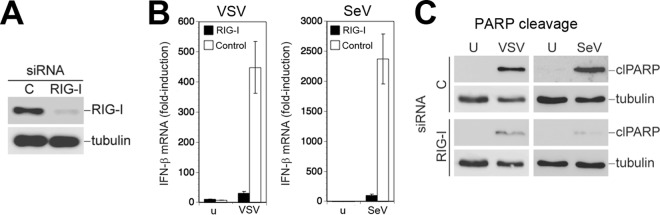
Virus-induced IFN-I response and mitochondrial apoptosis are RIG-I dependent. (A) Depletion of RIG-I by RNAi. A549 cells were first reverse transfected with siRNA for RIG-I or control siRNA (C), followed by forward transfection 24 h later. Efficiency of depletion was assessed 72 h after the first transfection by Western blotting using specific antibody against RIG-I. (B) Depletion of RIG-I prevents induction of IFN-I by VSV and SeV. Cells depleted of RIG-I, as described for panel A, were infected with VSV (MOI of 3) and SeV (80 hemagglutinin units/ml) for 16 h or left uninfected (u). Cells were lysed, total RNA was extracted, and cDNA was synthesized by reverse transcription. IFN-β mRNA was detected by RT-qPCR using specific TaqMan probes for IFN-β and GAPDH. Gene expression levels relative to GAPDH were determined according to the 2−ΔΔCT method. Data represent fold induction relative to levels in uninfected cells (n = 3; means ± standard deviations). (C) Induction of apoptosis by SeV and VSV is dependent on RIG-I. Cells depleted for RIG-I, as described for panel A, were infected with VSV (MOI of 3) or SeV (80 hemagglutinin units/ml). Cleavage of PARP was assessed by Western blotting using a specific antibody. Tubulin expression was used as a loading control.
We next examined whether LCMV interfered with these two pathways. For this, A549 cells were infected with LCMV for 48 h, and the extent of infection was verified by detection of NP in flow cytometry, revealing >95% infection (Fig. 4A). LCMV- and mock-infected A549 cells were then superinfected with SeV and VSV, and the induction of IFN-β mRNA was detected by RT-qPCR. In line with previous reports (20), LCMV infection drastically reduced the induction of IFN-β mRNA upon SeV and VSV superinfection (Fig. 4B). When cell culture supernatants were probed for IFN-α protein by ELISA, significantly reduced levels were found in LCMV-infected cells upon challenge with VSV or SeV (Fig. 4C). In contrast to the marked suppression of the IFN-I response, LCMV infection could not prevent activation of caspases 9 and 3/7 (Fig. 4D). Consistent with the high levels of caspase activation, we detected extensive release of cytochrome c (Fig. 4E and F) and PARP cleavage (Fig. 4G) in LCMV-infected cells challenged with VSV. Superinfection with SeV gave similar results (data not shown). In sum, the data indicated that LCMV differentially affected the RIG-I/MAVS-induced type I IFN response and mitochondrial apoptosis.
FIG 4.

LCMV blocks virus-induced IFN-β production but not apoptosis. (A) A549 cells were infected with LCMV (MOI of 0.1), and NP-positive cells were detected at 48 h postinfection by flow cytometry. Solid line, MAb 113 to LCMV NP combined with anti-mouse IgG-phycoerythrin; dashed line, secondary antibody only. One representative example of three independent experiments is shown. (B) LCMV infection prevents VSV- and SeV-induced IFN-I response. A549 cells were infected with LCMV at an MOI of 0.1 or mock infected (u) for 48 h and then superinfected with VSV (MOI of 3) or SeV (80 hemagglutinin units/ml). At 16 h postinfection, levels of IFN-β mRNA were determined by RT-qPCR as described in the legend of Fig. 3B. Data represent fold induction above values for uninfected (u) cells (n = 3; means ± standard deviations). (C) Detection of IFN-α protein. Supernatants from the experiment described in panel B were analyzed in a multisubtype IFN-α ELISA. Induction of IFN-α protein was calculated over the level in mock-treated uninfected (u) cells (n = 3; means ± standard deviations). (D) LCMV infection does not prevent virus-induced caspase activation. A549 cells were infected with LCMV (MOI of 0.1) or mock infected for 48 h, followed by superinfection with VSV (MOI of 3) (C) or SeV (80 hemagglutinin units/ml) (D) as described for panel B. At 16 h postinfection, activity of caspases 9 and 3/7 was measured as described in the legend of Fig. 1A. Data presented are normalized to levels of uninfected (u) cells (n = 3; means ± standard deviations). For statistical analysis, LCMV-infected and mock-infected specimens were compared at each time point by one-way ANOVA. P values are indicated. n.s., not significant (P > 0.05). (E) LCMV infection does not prevent VSV-induced cytochrome c release. A549 cells were infected with LCMV or mock infected, followed by superinfection with VSV. At the indicated time points, cells were stained for cytochrome c (green) with a specific antibody, and nuclei were visualized with DAPI (blue). (F) Cytochrome c release in LCMV-infected and staurosporine-treated cells was quantified as described in the legend of Fig. 1C. (G) LCMV does not prevent VSV-induced PARP cleavage. Cells were LCMV infected or mock infected and superinfected with VSV for 16 h. PARP cleavage was assessed by Western blotting. Tubulin was used as a loading control. The increased PARP cleavage in LCMV-infected versus mock-infected cells was consistently observed.
A hallmark of arenaviruses is their ability to establish persistent infection in cells in vitro and in vivo (1). To address the effect of persistent LCMV infection on the host cell's ability to undergo virus-induced apoptosis, we established LCMV persistently infected (LCMV-pi) A549 and HT-1080 cells, as well as LCMV-pi mouse embryonic fibroblasts (MEF). In LCMV-pi cultures >95% of cells expressed LCMV NP, as detected by flow cytometry (data not shown). LCMV-pi cells were challenged with VSV and SeV, and virus-induced apoptosis was assessed by detecting the activation of caspases 9 and 3/7. As shown in Fig. 5, superinfection with both VSV and SeV resulted in moderately enhanced activation of both caspase 9 and caspase 3/7 in all three LCMV-pi cell types compared to levels in mock-infected controls.
FIG 5.
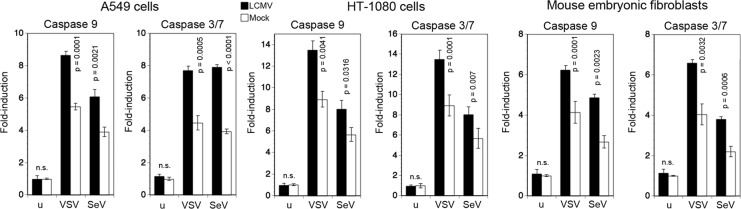
LCMV-pi cells remain sensitive to virus-induced mitochondrial apoptosis. A549 cells, HT-1080 cells, and MEF were either persistently infected with LCMV or mock infected. Cells were then superinfected with SeV (80 hemagglutinin units/ml) and VSV (MOI of 3) for 16 and 20 h. Activation of caspases 9 and 3/7 was measured as described in the legend of Fig. 1A. Data presented are normalized to levels of mock-treated, uninfected (u) cells (n = 3; means ± standard deviations). Statistical analysis was performed by one-way ANOVA, comparing results for LCMV-infected and noninfected specimens at each time point. P values are indicated. n.s., not significant (P > 0.05).
So far, our studies on the effects of LCMV on virus-induced apoptosis had been performed in cell lines (A549 and HT-1080) or MEF. In a next step we sought to validate our findings using a suitable primary murine cell type. For our studies, we chose primary mouse cardiac fibroblasts (MCF) isolated from postnatal day 2 mouse heart. We found this cell type to maintain a stable phenotype in vitro for up to five population doublings and to be highly susceptible to LCMV infection. When MCF were infected with LCMV at an MOI of 0.1, >98% of MCF were NP positive after 48 h (Fig. 6A). Infection with LCMV at a high MOI (MOI of 3) did not induce activation of caspase 9 or caspase 3/7 over time (Fig. 6B), and no signs of cytopathic effect (CPE) were observed. In contrast, infection of MCF with VSV or treatment with staurosporine caused induction of caspases 9 and 3/7 (Fig. 6B). Similar to results with the cell lines studied before, LCMV infection in MCF did not result in cytochrome release (Fig. 6C) or PARP cleavage (Fig. 6D). As observed in our cell lines, infection with LCMV prevented induction of IFN-I in MCF upon superinfection with VSV (Fig. 6E), whereas virus-induced apoptosis remained active, as evidenced by induction of caspases (Fig. 6F), cytochrome c release (Fig. 6G), and PARP cleavage (Fig. 6H). Together, the data provided the first evidence that acute and persistent LCMV infection efficiently blocks virus-induced IFN-I expression but retains the host cell's ability to undergo efficient mitochondrial apoptosis in response to superinfection with lytic RNA viruses.
FIG 6.

LCMV infection in primary MCF blocks virus-induced IFN-I production but not apoptosis. (A) Primary MCF were infected with LCMV at an MOI of 0.1. At the indicated time points, cells were fixed, and LCMV NP was detected by immunofluorescence. One hundred cells were counted, and NP-positive cells were scored (n = 3; means ± standard deviations). (B) MCF were infected with LCMV as described for panel A, and activation of caspases 9 and 3/7 was monitored over time as described in the legend of Fig. 1A. VSV (MOI of 3; 16 h of infection) and staurosporine (St) (3 μM; 8 h) were used as positive controls. (n = 3; means ± standard deviations). Data were analyzed by one-way ANOVA comparing values for each specimen with those of the uninfected cells. *, P < 0.0001; n.s., not significant (P > 0.05). (C) MCF infected with LCMV for the indicated time points were examined for cytochrome c release as described in the legend of Fig. 1C (n = 3; means ± standard deviations). (D) Detection of cleaved PARP (clPARP) in MCF infected with LCMV (MOI of 0.1) for 48 and 72 h or with VSV (MOI of 3) for 16 h or left uninfected (u). (E) LCMV prevents induction of IFN-β mRNA in MCF. MCF were infected with LCMV (MOI of 0.1) or mock infected for 48 h, followed by challenge with VSV (MOI of 3; 16 h of infection). IFN-β mRNA was detected by RT-qPCR as described in the legend of Fig. 3B (n = 3; means ± standard deviations). (F to H) LCMV infection in MCF does not prevent apoptosis upon challenge with VSV. MCF infected with LCMV as described for panel E were superinfected with VSV (MOI of 3). At the indicated time points, activation of caspases 9 and 3/7 (F) and cytochrome c release (G) were detected as described in the legend of Fig. 1A and C, respectively (n = 3; means ± standard deviations). Cleavage of PARP (H) was assessed after 20 h by Western blotting as described in the legend of Fig. 1D. Statistical analysis in panel F was performed by one-way ANOVA comparing values for LCMV- and mock-infected cells for each time point. P values are indicated. n.s., not significant (P > 0.05).
Virus-induced mitochondrial apoptosis in LCMV-infected cells is independent of RIG-I, MDA5, and NOD2 but requires signaling via MAVS.
Our data revealed that LCMV efficiently blocked RIG-I-mediated induction of IFN-I without perturbing mitochondrial apoptosis triggered by superinfection with a different RNA virus. Since induction of mitochondrial apoptosis by SeV and VSV critically depends on RIG-I and MAVS (33, 34), we examined the role of these signaling molecules in the observed mitochondrial apoptosis in LCMV-pi cells. For this purpose, we used RNAi to deplete RIG-I and MAVS from LCMV-pi and mock-infected A549 cells. Treatment of cells with specific siRNAs to RIG-I and MAVS, but not control siRNAs, resulted in a reduction of protein expression of >95%, as assessed by Western blotting (Fig. 7A and B). As expected, mock-infected cells depleted of RIG-I and MAVS were unable to upregulate IFN-β mRNA after challenge with VSV (Fig. 7C and D). LCMV blocked VSV-mediated induction of IFN-β mRNA to a similar extent as knockdown of RIG-I and MAVS (Fig. 7C and D). Remarkably, LCMV-pi cells depleted of RIG-I were still able to undergo efficient apoptosis in response to VSV challenge, as assessed by PARP cleavage (Fig. 7E). In contrast, depletion of MAVS abolished VSV-induced apoptosis in both LCMV-pi and mock-infected cells (Fig. 7F), indicating that apoptosis in the presence of LCMV still requires signaling through MAVS.
FIG 7.

Virus-induced mitochondrial apoptosis in LCMV-pi cells is RIG-I independent but requires MAVS. (A and B) LCMV-pi A549 cells were depleted of RIG-I and MAVS using specific siRNAs, and efficiency of knockdown was verified by Western blotting using tubulin as a loading control. (C and D) Persistent LCMV prevents induction of IFN-β via RIG-I and MAVS. LCMV-pi and mock-infected control cells depleted of RIG-I or MAVS were superinfected with VSV (MOI of 3) for 16 h. The induction of IFN-β mRNA was assessed by RT-qPCR as described in the legend of Fig. 3B. Data represent fold induction above the level seen in uninfected (u) and control siRNA (C)-treated cells (n = 3; means ± standard deviations). (E and F) Virus-induced apoptosis in LCMV-pi cells is independent of RIG-I but requires MAVS. Cells were depleted of RIG-I and MAVS and then superinfected with VSV as described for panels C and D. PARP cleavage was detected in cell lysates by Western blotting as described in the legend of Fig. 1D, using tubulin as a loading control.
The RIG-I-independent virus-induced mitochondrial apoptosis in the presence of LCMV suggested detection of the challenge virus by another cytosolic PRR linked to MAVS. To assess a possible role of MDA5 in this context, we used RNAi to deplete LCMV-pi and mock-infected A549 cells of MDA5 (Fig. 8A). Silencing of MDA5 had no effect on VSV-induced apoptosis in either LCMV-pi or mock-infected cells (Fig. 8B), making a role of MDA5 in the process rather unlikely. Recent studies implicated nucleotide-binding oligomerization domain 2 (NOD2) in detection of viral RNA derived from the paramyxovirus respiratory syncytial virus (RSV) (45). Upon recognition of virus-derived single-stranded RNA (ssRNA), NOD2 associates with MAVS, resulting in activation of IRF3. To address a possible function of NOD2 in virus-induced mitochondrial apoptosis in LCMV-pi cells, NOD2 was depleted by RNAi (Fig. 8C), which was followed by challenge with VSV. Despite marked depletion of NOD2 mRNA, virus-induced mitochondrial apoptosis remained intact in LCMV-pi cells (Fig. 8D), arguing against a major role of NOD2 as a PRR. Together, our data indicate that virus-induced mitochondrial apoptosis in LCMV-infected cells is independent of RIG-I, MDA5, and NOD2 but requires signaling via MAVS, suggesting that persistent virus alters the molecular mechanism of virus-induced apoptosis.
FIG 8.
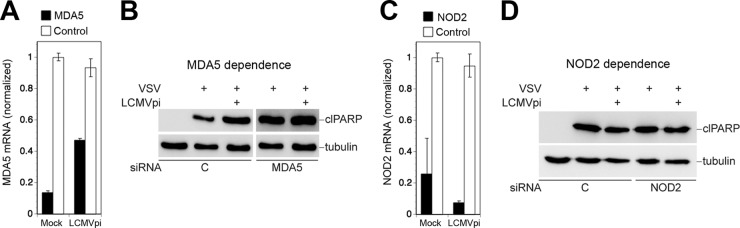
Virus-induced mitochondrial apoptosis in LCMV-pi cells is independent of MDA5 and NOD2. LCMV-pi A549 cells were depleted of MDA5 (A) and NOD2 (C) by transfection of specific siRNAs and control siRNAs. Efficiency of knockdown was verified by RT-qPCR. LCMV-pi and mock-infected control cells depleted of MDA5 (B) and NOD2 (D) were superinfected with VSV (MOI of 3). After 16 h, cells were lysed, and PARP cleavage was detected by Western blotting using tubulin as a loading control.
DISCUSSION
Many viruses are capable of delaying or preventing apoptosis to maintain a favorable cellular environment for productive infection (31, 32, 46). This is of particular importance in persistent viral infections, where efficient virus replication and gene expression must occur without causing overt cytopathic effects. With a life cycle confined to the cytoplasm, arenaviruses are unable to integrate into the genome of the host cell and to adopt a latent state (1). As a consequence, arenavirus persistence is maintained by productive infection, characterized by high levels of viral replication and gene expression (5, 47). Recent studies revealed that infection with the New World arenavirus JUNV induces apoptosis in an IFN-I-independent manner via RIG-I (35) and that JUNV NP is able to block apoptosis by acting as a decoy substrate for the effector caspase 3 (36). In mice persistently infected with LCMV, apoptotic cells are scarce (3), despite high viral load in many organs and tissues, suggesting that the virus can evade or actively suppress proapoptotic signaling. First, we infected different human and murine cells with LCMV and found no evidence for the induction of mitochondrial apoptosis, as assessed by activation of caspases 9 and 3/7, cytochrome c release, and PARP cleavage. Similar to other RNA viruses, including SeV (44, 48), influenza virus (49), and JUNV (50), LCMV was found to activate the anti-apoptotic PI3K/Akt pathway early in infection. However, inhibition of PI3K did not accelerate apoptosis in LCMV-infected cells, arguing against a crucial antiapoptotic function of the virus-induced activation of PI3K/Akt.
Consistent with previous studies, we found that LCMV efficiently suppressed induction of IFN-I in response to superinfection by SeV and VSV (20). However, challenge of LCMV-infected cells with SeV and VSV resulted in efficient induction of mitochondrial apoptosis. In LCMV-pi cells, virus-induced apoptosis seemed to occur even more efficiently than in uninfected cells, suggesting that the prolonged presence of LCMV somehow sensitized proapoptotic pathways. Findings obtained in human cell lines (A549 and HT-1080) and MEF were confirmed in primary mouse cells, excluding cell type-specific artifacts. Our data provided the first evidence that LCMV differentially affects the virus-induced RIG-I/MAVS-mediated IFN-I response, on the one hand, and mitochondrial apoptosis, on the other hand (Fig. 9). This appears distinct from the effects of JUNV and other RNA viruses, like VSV and SeV, that induce both IFN-I and apoptosis via RIG-I/MAVS (28, 35).
FIG 9.
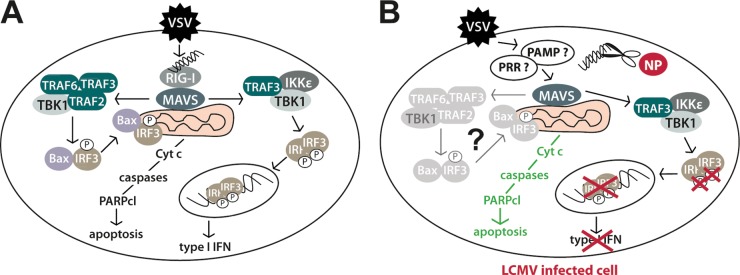
Model of the differential subversion of type I IFN and mitochondrial apoptotic pathway by LCMV. (A) In uninfected cells, a challenging RNA virus (VSV) induces IFN-I and mitochondrial apoptosis via RIG-I/MAVS involving TRAF2, -3, and -6, TBK1, Bax, and IRF3 (33). (B) In LCMV-infected cells, the IFN-I response, but not mitochondrial apoptosis, is blocked in response to superinfection. LCMV NP's 3′–5′ exoribonuclease activity may remove free viral RNA (scissors), preventing IRF3 phosphorylation, nuclear translocation, and, hence, IFN-I production. The ability of LCMV-infected cells to undergo rapid mitochondrial apoptosis via MAVS suggests that in the presence of LCMV, an unknown PRR (?) recognizes a yet undefined virus-derived pathogen-associated molecular pattern (PAMP) (?). Activation of MAVS may then result in mitochondrial apoptosis via TRAF2, -3, and -6, TBK1, Bax, and IRF3. Cyt c, cytochrome c.
The ability of LCMV to block RIG-I/MAVS-mediated induction of IFN-I but not apoptosis seemed contradictory to recent studies by Chattopadhyay and colleagues that revealed a lower activation threshold of IRF3 for the IFN-I response than for apoptosis (51). To address this apparent contradiction, we evaluated the role of RIG-I and MAVS in virus-induced apoptosis in LCMV-pi cells. Silencing of RIG-I and MAVS by RNAi revealed that RIG-I was dispensable for induction of apoptosis in LCMV-pi cells upon challenge with RNA viruses, whereas MAVS was still required. This finding suggested that in LCMV-pi cells, incoming challenge virus may be sensed via a distinct PRR (Fig. 9). Evaluation of MDA5 and NOD2 excluded these candidates, and the nature of the MAVS-linked PRR involved in virus-induced apoptosis in LCMV-pi cells remains unknown. In sum, our study reveals a differential effect of LCMV on the virus-induced IFN response versus apoptosis. In the context of the coevolution of the virus with its natural reservoir species, this appears as a subtle strategy to guarantee sufficiently high viral loads for efficient transmission within the host population while maintaining a basic mechanism of antiviral defense against other pathogens.
ACKNOWLEDGMENTS
We thank Laurent Roux and Dominique Garcin (both, University of Geneva) and Margot Thome Miazza and Pascal Schneider (both, University of Lausanne) for valuable reagents.
This research was supported by Swiss National Science Foundation grant number 310030_149746 (S.K.), funds from the University of Lausanne (S.K.), and NIH grant RO1 AI077719 (JC.D.L.T. and LM.-S.). LM.-S was further supported by the NIH grants R03AI099681-01A1.
REFERENCES
- 1.Buchmeier MJ, de la Torre JC, Peters CJ. 2007. Arenaviridae: the viruses and their replication, p 1791–1828. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Geisbert TW, Jahrling PB. 2004. Exotic emerging viral diseases: progress and challenges. Nat Med 10:S110–S121. doi: 10.1038/nm1142. [DOI] [PubMed] [Google Scholar]
- 3.Oldstone MB. 2002. Biology and pathogenesis of lymphocytic choriomeningitis virus infection. Curr Top Microbiol Immunol 263:83–118. [DOI] [PubMed] [Google Scholar]
- 4.Zinkernagel RM. 2002. Lymphocytic choriomeningitis virus and immunology. Curr Top Microbiol Immunol 263:1–5. [DOI] [PubMed] [Google Scholar]
- 5.Fazakerley JK, Southern P, Bloom F, Buchmeier MJ. 1991. High resolution in situ hybridization to determine the cellular distribution of lymphocytic choriomeningitis virus RNA in the tissues of persistently infected mice: relevance to arenavirus disease and mechanisms of viral persistence. J Gen Virol 72:1611–1625. doi: 10.1099/0022-1317-72-7-1611. [DOI] [PubMed] [Google Scholar]
- 6.Saron MF, Riviere Y, Hovanessian AG, Guillon JC. 1982. Chronic production of interferon in carrier mice congenitally infected with lymphocytic choriomeningitis virus. Virology 117:253–256. doi: 10.1016/0042-6822(82)90524-4. [DOI] [PubMed] [Google Scholar]
- 7.Borrow P, Martinez-Sobrido L, de la Torre JC. 2010. Inhibition of the type I interferon antiviral response during arenavirus infection. Viruses 2:2443–2480. doi: 10.3390/v2112443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou S, Cerny AM, Zacharia A, Fitzgerald KA, Kurt-Jones EA, Finberg RW. 2010. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J Virol 84:9452–9462. doi: 10.1128/JVI.00155-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habjan M, Andersson I, Klingstrom J, Schumann M, Martin A, Zimmermann P, Wagner V, Pichlmair A, Schneider U, Muhlberger E, Mirazimi A, Weber F. 2008. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS One 3:e2032. doi: 10.1371/journal.pone.0002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marq JB, Kolakofsky D, Garcin D. 2010. Unpaired 5′ ppp-nucleotides, as found in arenavirus double-stranded RNA panhandles, are not recognized by RIG-I. J Biol Chem 285:18208–18216. doi: 10.1074/jbc.M109.089425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marq JB, Hausmann S, Veillard N, Kolakofsky D, Garcin D. 2011. Short double-stranded RNAs with an overhanging 5′ ppp-nucleotide, as found in arenavirus genomes, act as RIG-I decoys. J Biol Chem 286:6108–6116. doi: 10.1074/jbc.M110.186262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walsh KB, Teijaro JR, Zuniga EI, Welch MJ, Fremgen DM, Blackburn SD, von Tiehl KF, Wherry EJ, Flavell RA, Oldstone MB. 2012. Toll-like receptor 7 is required for effective adaptive immune responses that prevent persistent virus infection. Cell Host Microbe 11:643–653. doi: 10.1016/j.chom.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macal M, Lewis GM, Kunz S, Flavell R, Harker JA, Zuniga EI. 2012. Plasmacytoid dendritic cells are productively infected and activated through TLR-7 early after arenavirus infection. Cell Host Microbe 11:617–630. doi: 10.1016/j.chom.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 15.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 17.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. 2005. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell 19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 18.Michallet MC, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M, Curran J, Poeck H, Bscheider M, Hartmann G, Konig M, Kalinke U, Pasparakis M, Tschopp J. 2008. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity 28:651–661. doi: 10.1016/j.immuni.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 19.Hiscott J. 2007. Convergence of the NF-kappaB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev 18:483–490. doi: 10.1016/j.cytogfr.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Sobrido L, Zuniga EI, Rosario D, Garcia-Sastre A, de la Torre JC. 2006. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 80:9192–9199. doi: 10.1128/JVI.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodrigo WW, Ortiz-Riano E, Pythoud C, Kunz S, de la Torre JC, Martinez-Sobrido L. 2012. Arenavirus nucleoproteins prevent activation of nuclear factor kappa B. J Virol 86:8185–8197. doi: 10.1128/JVI.07240-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Sobrido L, Emonet S, Giannakas P, Cubitt B, Garcia-Sastre A, de la Torre JC. 2009. Identification of amino acid residues critical for the anti-interferon activity of the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 83:11330–11340. doi: 10.1128/JVI.00763-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hastie KM, Bale S, Kimberlin CR, Saphire EO. 2012. Hiding the evidence: two strategies for innate immune evasion by hemorrhagic fever viruses. Curr Opin Virol 2:151–156. doi: 10.1016/j.coviro.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hastie KM, Kimberlin CR, Zandonatti MA, MacRae IJ, Saphire EO. 2011. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc Natl Acad Sci U S A 108:2396–2401. doi: 10.1073/pnas.1016404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi X, Lan S, Wang W, Schelde LM, Dong H, Wallat GD, Ly H, Liang Y, Dong C. 2010. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 468:779–783. doi: 10.1038/nature09605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang X, Huang Q, Wang W, Dong H, Ly H, Liang Y, Dong C. 2013. Structures of arenaviral nucleoproteins with triphosphate dsRNA reveal a unique mechanism of immune suppression. J Biol Chem 288:16949–16959. doi: 10.1074/jbc.M112.420521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carnec X, Baize S, Reynard S, Diancourt L, Caro V, Tordo N, Bouloy M. 2011. Lassa virus nucleoprotein mutants generated by reverse genetics induce a robust type I interferon response in human dendritic cells and macrophages. J Virol 85:12093–12097. doi: 10.1128/JVI.00429-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang C, Kolokoltsova OA, Yun NE, Seregin AV, Poussard AL, Walker AG, Brasier AR, Zhao Y, Tian B, de la Torre JC, Paessler S. 2012. Junin virus infection activates the type I interferon pathway in a RIG-I-dependent manner. PLoS Negl Trop Dis 6:e1659. doi: 10.1371/journal.pntd.0001659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barber GN. 2001. Host defense, viruses and apoptosis. Cell Death Differ 8:113–126. doi: 10.1038/sj.cdd.4400823. [DOI] [PubMed] [Google Scholar]
- 30.Boatright KM, Salvesen GS. 2003. Mechanisms of caspase activation. Curr Opin Cell Biol 15:725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Herold S, Ludwig S, Pleschka S, Wolff T. 2012. Apoptosis signaling in influenza virus propagation, innate host defense, and lung injury. J Leukoc Biol 92:75–82. doi: 10.1189/jlb.1011530. [DOI] [PubMed] [Google Scholar]
- 32.Best SM. 2008. Viral subversion of apoptotic enzymes: escape from death row. Annu Rev Microbiol 62:171–192. doi: 10.1146/annurev.micro.62.081307.163009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, Williams BR, Sen GC. 2010. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J 29:1762–1773. doi: 10.1038/emboj.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chattopadhyay S, Yamashita M, Zhang Y, Sen GC. 2011. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J Virol 85:3708–3716. doi: 10.1128/JVI.02133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolokoltsova OA, Grant AM, Huang C, Smith JK, Poussard AL, Tian B, Brasier AR, Peters CJ, Tseng CT, de la Torre JC, Paessler S. 2014. RIG-I enhanced interferon independent apoptosis upon Junin virus infection. PLoS One 9:e99610. doi: 10.1371/journal.pone.0099610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolff S, Becker S, Groseth A. 2013. Cleavage of the Junin virus nucleoprotein serves a decoy function to inhibit the induction of apoptosis during infection. J Virol 87:224–233. doi: 10.1128/JVI.01929-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunz S, Edelmann KH, de la Torre J-C, Gorney R, Oldstone MBA. 2003. Mechanisms for lymphocytic choriomeningitis virus glycoprotein cleavage, transport, and incorporation into virions. Virology 314:168–178. doi: 10.1016/S0042-6822(03)00421-5. [DOI] [PubMed] [Google Scholar]
- 39.Pasquato A, Rochat C, Burri DJ, Pasqual G, de la Torre JC, Kunz S. 2012. Evaluation of the anti-arenaviral activity of the subtilisin kexin isozyme-1/site-1 protease inhibitor PF-429242. Virology 423:14–22. doi: 10.1016/j.virol.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchmeier MJ, Lewicki HA, Tomori O, Oldstone MB. 1981. Monoclonal antibodies to lymphocytic choriomeningitis and pichinde viruses: generation, characterization, and cross-reactivity with other arenaviruses. Virology 113:73–85. doi: 10.1016/0042-6822(81)90137-9. [DOI] [PubMed] [Google Scholar]
- 41.Lefrancois L, Lyles DS. 1982. The interaction of antibody with the major surface glycoprotein of vesicular stomatitis virus. II. Monoclonal antibodies of nonneutralizing and cross-reactive epitopes of Indiana and New Jersey serotypes. Virology 121:168–174. [DOI] [PubMed] [Google Scholar]
- 42.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 43.Pasqual G, Rojek JM, Masin M, Chatton JY, Kunz S. 2011. Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog 7:e1002232. doi: 10.1371/journal.ppat.1002232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peters K, Chattopadhyay S, Sen GC. 2008. IRF-3 activation by Sendai virus infection is required for cellular apoptosis and avoidance of persistence. J Virol 82:3500–3508. doi: 10.1128/JVI.02536-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y, Bose S. 2009. Activation of innate immune antiviral responses by Nod2. Nat Immunol 10:1073–1080. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benedict CA, Norris PS, Ware CF. 2002. To kill or be killed: viral evasion of apoptosis. Nat Immunol 3:1013–1018. doi: 10.1038/ni1102-1013. [DOI] [PubMed] [Google Scholar]
- 47.Oldstone MB, Buchmeier MJ. 1982. Restricted expression of viral glycoprotein in cells of persistently infected mice. Nature 300:360–362. doi: 10.1038/300360a0. [DOI] [PubMed] [Google Scholar]
- 48.White CL, Chattopadhyay S, Sen GC. 2011. Phosphatidylinositol 3-kinase signaling delays Sendai virus-induced apoptosis by preventing XIAP degradation. J Virol 85:5224–5227. doi: 10.1128/JVI.00053-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hale BG, Randall RE. 2007. PI3K signalling during influenza A virus infections. Biochem Soc Trans 35:186–187. doi: 10.1042/BST0350186. [DOI] [PubMed] [Google Scholar]
- 50.Linero FN, Scolaro LA. 2009. Participation of the phosphatidylinositol 3-kinase/Akt pathway in Junin virus replication in vitro. Virus Res 145:166–170. doi: 10.1016/j.virusres.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chattopadhyay S, Fensterl V, Zhang Y, Veleeparambil M, Yamashita M, Sen GC. 2013. Role of interferon regulatory factor 3-mediated apoptosis in the establishment and maintenance of persistent infection by Sendai virus. J Virol 87:16–24. doi: 10.1128/JVI.01853-12. [DOI] [PMC free article] [PubMed] [Google Scholar]