

ABSTRACT
miR-122 is a liver-specific microRNA (miRNA) that binds to two sites (S1 and S2) on the 5′ untranslated region (UTR) of the hepatitis C virus (HCV) genome and promotes the viral life cycle. It positively affects viral RNA stability, translation, and replication, but the mechanism is not well understood. To unravel the roles of miR-122 binding at each site alone or in combination, we employed miR-122 binding site mutant viral RNAs, Hep3B cells (which lack detectable miR-122), and complementation with wild-type miR-122, an miR-122 with the matching mutation, or both. We found that miR-122 binding at either site alone increased replication equally, while binding at both sites had a cooperative effect. Xrn1 depletion rescued miR-122-unbound full-length RNA replication to detectable levels but not to miR-122-bound levels, confirming that miR-122 protects HCV RNA from Xrn1, a cytoplasmic 5′-to-3′ exoribonuclease, but also has additional functions. In cells depleted of Xrn1, replication levels of S1-bound HCV RNA were slightly higher than S2-bound RNA levels, suggesting that both sites contribute, but their contributions may be unequal when the need for protection from Xrn1 is reduced. miR-122 binding at S1 or S2 also increased translation equally, but the effect was abolished by Xrn1 knockdown, suggesting that the influence of miR-122 on HCV translation reflects protection from Xrn1 degradation. Our results show that occupation of each miR-122 binding site contributes equally and cooperatively to HCV replication but suggest somewhat unequal contributions of each site to Xrn1 protection and additional functions of miR-122.
IMPORTANCE The functions of miR-122 in the promotion of the HCV life cycle are not fully understood. Here, we show that binding of miR-122 to each of the two binding sites in the HCV 5′ UTR contributes equally to HCV replication and that binding to both sites can function cooperatively. This suggests that active Ago2–miR-122 complexes assemble at each site and can cooperatively promote the association and/or function of adjacent complexes, similar to what has been proposed for translation suppression by adjacent miRNA binding sites. We also confirm a role for miR-122 in protection from Xrn1 and provide evidence that miR-122 has additional functions in the HCV life cycle unrelated to Xrn1. Finally, we show that each binding site may contribute unequally to Xrn1 protection and other miR-122 functions.
INTRODUCTION
Hepatitis C virus (HCV) is a hepatotropic virus that infects an estimated 150 million humans worldwide, a significant portion of whom do not know their status due to the largely asymptomatic nature of the infection (1). The virus is transmitted by blood-to-blood contact, and humans are the only known reservoir. Chronic infection occurs in approximately 70% of cases and can lead to sequelae such as metabolic disease, steatosis, hepatocellular carcinoma, and decompensated liver disease late in infection (2).
One of the major determinants of the virus' hepatotropism is its requirement for the liver-specific, liver-abundant miR-122 microRNA (miRNA) (3, 4). miR-122 binds to two sites at the 5′ end of the virus' positive-sense RNA genome and has been shown to directly enhance viral RNA accumulation, since mutation of the miR-122 binding sites abolishes RNA accumulation, and the provision of exogenous miR-122 sequences that have compensatory mutations to restore binding also reinstates RNA accumulation (4–10). Argonaute-2, one of the key effector proteins in the microRNA pathway and a component of the RNA-induced silencing complex (RISC), binds in association with miR-122 and is required to increase HCV replication, while several other proteins in the microRNA pathway and RISC have been implicated in either the biogenesis or activity of miR-122 (5, 11–14). Although miR-122 uses canonical microRNA seed sequence binding and RISC components when interacting with the HCV genome, it also binds to HCV nucleotides outside the seed sequence, creating a double-stranded RNA-protein structure that overhangs the 5′ end of the viral genome, and also interacts with the “spacer” sequence located between miR-122 binding site 1 (S1) and S2 on the HCV 5′ untranslated region (UTR) (7, 11). We and others have ruled out any significant indirect influence of miR-122 on HCV in cell culture models; miR-122-mediated regulation of the cholesterol biosynthesis pathway had no significant effect on HCV RNA accumulation, and miR-122 binding mutant viral RNAs do not respond to wild-type (WT) miR-122 but will respond to mutant miRNAs the same as wild-type HCV responds to miR-122 (4, 6, 9).
Evidence suggests that there are multiple mechanisms involved in the increase in HCV RNA accumulation mediated by the interaction between miR-122 and the HCV 5′ UTR. miR-122 has been observed to modestly increase translation of the viral RNA, but this has recently been attributed to simply increased stability of the viral RNA in the assays, allowing more viral RNA to be translated (10, 11, 14, 15). In particular, miR-122 has been implicated in protecting the 5′ end of the viral RNA from degradation by Xrn1, the major cytoplasmic 5′-to-3′ exoribonuclease that normally functions to degrade decapped host mRNAs and other Flavivirus RNAs (16–19). One model posits that the interaction of miR-122 with S1 generates a 3′ overhang that shields the 5′ triphosphate genome terminus, but the influence of each miR-122 binding interaction in protection from Xrn1 remains unknown. Ultimately, though, the increase in translation and stability afforded by protection from Xrn1 appears insufficient to explain the increase in HCV RNA accumulation mediated by miR-122, and other mechanisms require exploration (10, 11, 18).
To better understand the roles of each miR-122 binding site on HCV replication and on protection from Xrn1, we analyzed the influence of Xrn1 knockdown and miR-122 binding to the individual binding sites on the 5′ end of the HCV genome. First, we verify that miR-122 protects the 5′ end of the viral genome from Xrn1 and are the first to show that knockdown of Xrn1 rescues transient replication of both subgenomic and full-length HCV RNA replication in the absence of miR-122, albeit incompletely. Thus, our results also support the finding that protection from Xrn1 is not the only function of miR-122 in the viral life cycle and that miR-122 must have additional functions (18). Next, we found that binding of miR-122 at each site contributes equally to replication promotion, while binding at both sites demonstrates a cooperative effect. Further, we demonstrate that in the absence of Xrn1, binding at S1 increased replication more than binding at S2, suggesting that S1 binding contributes more to an miR-122 function that is unrelated to Xrn1. Finally, we provide further evidence that the apparent function of miR-122 in promoting HCV translation is primarily through RNA protection from Xrn1 (18).
MATERIALS AND METHODS
Cell lines.
Huh7.5 (20) and Hep3B (21) cells were grown and maintained as described by Thibault et al. (4).
Plasmids and viral RNA.
Plasmids pSGR JFH-1 Fluc WT and pSGR JFH-1 Fluc GND contain bicistronic JFH-1-derived subgenomic replicon (SGR) cDNAs with a firefly luciferase reporter; the GDD-to-GND mutation renders the viral polymerase nonfunctional (22). pSGR JFH-1 S1:p3 Fluc WT, pSGR JFH-1 S2:p3 Fluc WT, and pSGR JFH-1 S1+S2:p3 Fluc WT contain C-to-G mutations at position 3 of miR-122 binding site 1, site 2, or sites 1 and 2, respectively, generated as described by Thibault et al. (4). Plasmids pJ6/JFH-1 FL Rluc WT and pJ6/JFH-1 FL Rluc GNN, also known as pJ6/JFH-1(p7Rluc2A) and pJ6/JFH-1(p7Rluc2A), were obtained from C. M. Rice. They contain monocistronic chimeric replicon cDNAs with J6-derived structural proteins and JFH-1-derived untranslated regions and nonstructural proteins, along with an in-frame Renilla luciferase reporter; the GDD-to-GNN mutation renders the polymerase nonfunctional (23). pJ6/JFH-1 FL S1:p3 Rluc WT, pJ6/JFH-1 FL S2:p3 Rluc WT, and pJ6/JFH-1 FL S1+S2:p3 Rluc WT contain C-to-G mutations as described for the concomitant S1:p3, S2:p3, and S1+S2:p3 subgenomic replicon (SGR) mutants. These were generated by replacing the 177-bp EcoRI-to-AgeI fragment from J6/JFH-1 with the fragment from the appropriate miR-122 binding site mutant SGR plasmid. Plasmids pT7 luciferase (containing firefly luciferase; Promega, Nepean, ON, Canada) and pRL-TK (containing Renilla luciferase; Promega) were used as the templates for production of mRNA. Viral RNA and mRNA were in vitro transcribed from these plasmids as described by Thibault et al. (4).
MicroRNAs, siRNAs, and miRNA antagonists.
miR-122, miR-122/p3, and miControl have been described previously (4). Small interfering RNAs (siRNAs) and their sequences are as follows (lowercase letters indicate DNA bases): siXrn1 (s29015; 5′-GAG AGU AUA UUG ACU AUG Att-3′), siXrn2 (s22412; 5′-GGA AAG UUG UGC AGU CGU Att-3′), siControl (5′-GAA GGU CAC UCA GCU AAU CAC ttc-3′). miRIDIAN microRNA hairpin inhibitors are human hsa-miR-122-5p and human hsa-miR-124-5p. All small RNAs were synthesized by Thermo Scientific Dharmacon (Lafayette, CO, USA).
Electroporation of Hep3B and Huh7.5 cells.
Electroporations were carried out as described by Thibault et al. (4). Both Hep3B and Huh7.5 cells were electroporated using the following conditions: 225 V, 950 μF, 4 mm, and ∞ Ω.
Transient HCV replication assays without knockdown.
On day 0, 6.0 × 106 cells in 400 μl Dulbecco's phosphate-buffered saline (PBS) were electroporated with 1 μg mRNA, 60 pmol miRNA, and 5 μg (Huh7.5) or 10 μg (Hep3B) in vitro-transcribed viral RNA. After electroporation, cells were resuspended in 4 ml cell culture medium. A total of 500 μl of cells was incubated in microcentrifuge tubes at 37°C for 2 h and then harvested for luciferase analysis. For all other luciferase analysis, 500 μl cells per well was plated in 6-well dishes and incubated at 37°C to be harvested at the indicated time points. A total of 2 ml cells was plated in a 4-mm tissue culture dish to be collected for RNA analysis 3 days postelectroporation.
Transient HCV replication assays with preknockdown.
Three days before day 0, 6.0 × 106 cells in 400 μl Dulbecco's PBS were electroporated with 60 pmol siRNA for preknockdown and were plated in 15-cm tissue culture dishes, two cuvettes per dish. siRNA-treated cells were incubated for 3 days at 37°C. On day 0, cells were collected from the dishes, prepared as described for electroporation (two cuvettes' worth from each dish), and then electroporated and plated as described above.
HCV translation assays with preknockdown.
Cells were electroporated for preknockdown as described above. Three days later, cells were prepared as described above and electroporated again with 5 μg viral RNA, 1 μg mRNA, and 60 pmol miRNA and recovered in 4 ml culture medium. A total of 500 μl of cells was incubated at 37°C in microcentrifuge tubes for 3 h before being collected for luciferase analysis; 500 μl cells per well was plated in 6-well dishes for the 6-, 9-, and 12-h luciferase samples. A total of 2 ml of cells per well was plated in 6-well dishes for total RNA collection at 3 h postelectroporation.
Luciferase assays.
Each sample was harvested for luciferase by first being washed with Dulbecco's PBS and then being scraped into 100 μl 1× passive lysis buffer (Promega). Samples were assayed using the dual-luciferase assay kit, luciferase assay kit, or Renilla luciferase assay kit (Promega) according to the manufacturer's protocol and using the GloMax luminometer (Promega) with a 2-s delay and 10-s reading.
Western blotting.
Cell lysates were collected with 1× SDS lysis buffer 3 days post-first electroporation and 3 days post-second electroporation (6 days post-first electroporation). Proteins were separated on a 7.5% SDS-PAGE gel and transferred to Hybond-C nitrocellulose membrane (GE Healthcare; Fisher Scientific, Ottawa, ON, Canada) via wet transfer. The blot was probed with primary rabbit polyclonal anti-Xrn1, kindly provided by J. Lykke-Andersen (24), or affinity-purified rabbit polyclonal anti-Xrn2 (Bethyl Laboratories, Montgomery, TX), mouse monoclonal anti-β-actin (Abcam; Cambridge, MA, USA), and secondary IRDye-conjugated goat anti-rabbit and goat anti-mouse antibodies (Mandel Scientific; Guelph, ON, Canada) and then imaged with the Li-Cor Odyssey Classic (Mandel Scientific). Band density was quantified using Image Studio v3.1.
Data analysis.
All experiment results are shown as averages from at least three independent experiments, with error bars indicating standard errors of the mean (SEM). Statistical analyses were performed using GraphPad Prism 6.0.4; statistical significance was determined by the tests indicated in each figure legend (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001; NS, not significant [P > 0.05]).
RESULTS
Xrn1 knockdown affects miR-122-independent subgenomic HCV replication more than miR-122-dependent subgenomic replication.
Xrn1, a host cytoplasmic 5′-to-3′ RNA exonuclease, has been shown to target the 5′ end of HCV RNA for degradation, and miR-122 has been implicated to have a role in protecting the viral RNA from Xrn1 (18). In spite of this relationship, in a previous report, Xrn1 knockdown did not restore any detectible replication of an HCV genome to which miR-122 binding had been abolished; this finding was deemed evidence that protection from Xrn1 was not the only function for miR-122 during the HCV life cycle (18). We decided that our previously characterized system of miR-122-independent replication of HCV RNA would be ideal to further test whether knockdown of Xrn1 can restore or improve HCV replication in the absence of miR-122 binding and verify whether miR-122 plays a key role in limiting the impact of Xrn1 on HCV RNA (4). We have previously shown that subgenomic JFH-1 HCV RNA (SGR WT, a bicistronic construct that expresses a firefly luciferase reporter gene) can replicate at low levels without miR-122 binding to the 5′ UTR and that HCV RNA accumulation correlates with luciferase expression levels (4). In addition, we showed that that Hep3B cells can be used to study miR-122-independent HCV replication, because they do not express miR-122 detectable by reverse transcription-quantitative PCR (qRT-PCR) or by functional assays (4). In this study, we have investigated the impact of Xrn1 knockdown on miR-122-independent HCV replication in Hep3B cells using wild-type HCV genomes and in Huh7.5 cells using HCV genomes that have point mutations in the miR-122 seed binding sites (S1+S2:p3 UTR) that abolish miR-122 binding (as depicted in Fig. 1A) (4). We hypothesized that if miR-122 protects the 5′ end of the viral RNA from degradation by Xrn1, then removal of restriction by Xrn1 through knockdown should enhance miR-122-independent HCV replication. If protection from Xrn1 is the primary role for miR-122, then Xrn1 knockdown should restore miR-122-independent replication to levels similar to miR-122-dependent replication.
FIG 1.

Xrn1 knockdown increases miR-122-independent replication of subgenomic JFH-1 HCV RNA more than it increases miR-122-dependent replication. (A) Cartoon of experimental systems. Top, depiction of subgenomic JFH-1 HCV RNA (SGR), which contains a firefly luciferase reporter gene expressed from the HCV IRES and the viral nonstructural proteins expressed from an EMCV IRES; bottom, depiction of miR-122 binding sites on the 5′ UTR of HCV RNA. Hep3B cells lack expression of detectable miR-122 but can be supplemented with either synthetic miR-122 (left; binding at both sites, to achieve miR-122-dependent replication) or miControl (right; no binding, to achieve miR-122-independent replication) as shown. (B) Hep3B cells were electroporated with siXrn1 or siControl for preknockdown, and 3 days later (day 0) cells were electroporated again with the indicated siRNA, wild-type SGR HCV RNA, and miR-122 (miR-122 dependent) or miControl (miR-122 independent). Replication was measured by evaluating luciferase production at the indicated time points post-second electroporation. (C) The effect of Xrn1 knockdown on miR-122-dependent and miR-122-independent subgenomic HCV RNA replication in Hep3B cells was determined by measuring the fold increase in luciferase expression with siXrn1 treatment over luciferase expression with no knockdown (siControl), using luciferase data from panel B at the indicated time points. Significance was determined by paired parametric t test. (D) The effectiveness of siXrn1 at reducing Xrn1 protein levels in Hep3B cells was determined by Western blotting with antibodies against Xrn1 and β-actin. A representative blot is shown depicting Xrn1 protein levels 3 days post-first electroporation (at the time of second electroporation, which includes viral and miRNAs) in the first two lanes, and 6 days post-first electroporation or 3 days post-second electroporation. Percent knockdown ± standard deviation relative to the siControl-treated cells was determined by infrared fluorescence quantification on blots from three or more independent experiments. (E) Cartoon of experimental systems. Top, depiction of subgenomic JFH-1 HCV RNA (SGR); bottom, depiction of miR-122 binding at the 5′ UTR of HCV RNA. Huh7.5 cells endogenously express miR-122, and all wild-type replication is miR-122 dependent (left), so to study miR-122-independent replication, miR-122 binding was abolished by mutating both miR-122 binding sites (right). (F) Huh7.5 cells were treated as described in the legend to panel B, but no miRNA was added since Huh7.5 cells already express endogenous miR-122. “Dependent” samples were electroporated with wild-type SGR RNA, while “independent” samples were electroporated with the miR-122 binding site mutant SGR S1+S2:p3 RNA. (G) The fold increase in miR-122-dependent and -independent HCV replication induced by Xrn1 knockdown in Huh7.5 cells was determined as described in the legend to panel C. Significance was determined by unpaired parametric t test. (H) Xrn1 protein knockdown efficiency was determined by Western blot analysis of protein from three independent experiments as described in the legend to panel D, and a representative blot is shown. (I) The effect of siXrn1, viral RNAs, and microRNAs on cell survival was evaluated by WST-1 3 days postelectroporation from samples in panels B and F, and cell numbers are normalized to siControl-miControl or siControl-SGR S1+S2:p3 samples. (J) Transfection efficiency in the experiments in panels B and F was evaluated 2 h postelectroporation by measuring Renilla luciferase expression from a coelectroporated mRNA. Samples are normalized to siControl-miControl or siControl-SGR S1+S2:p3 samples. (K) Untreated Hep3B and Huh7.5 cell lysates were Western blotted to show steady-state levels of Xrn1 protein in each cell type.
To test this, we used siRNAs to knock down Xrn1 in Hep3B cells, and after waiting for 3 days to allow knockdown to occur, we electroporated the cells again with subgenomic viral RNA, transfection control Renilla luciferase reporter mRNA, and siRNA and miRNA as indicated. Our results (Fig. 1B) show that while knockdown of Xrn1 increased miR-122-independent replication levels by up to 35-fold (Fig. 1C; compare also Fig. 1B, siControl [Independent] to siXrn1 [Independent]), it did not reach the same levels as miR-122-dependent replication (compare siXrn1 [Independent] to siControl [Dependent]). We determined that the siXrn1 double knockdown reduced Xrn1 protein levels by 81% in Hep3B cells (Fig. 1D). We ensured that knockdown of Xrn1 did not affect cell growth during the course of the experiment (Fig. 1I) and that transfection efficiencies were similar for all samples by assessing Renilla luciferase expression 2 h post-second electroporation from a coelectroporated control mRNA (Fig. 1J). Thus, Xrn1 knockdown can augment miR-122-independent replication of HCV RNA but cannot restore it to miR-122-dependent levels, which suggests that protection from Xrn1 is not the only function for miR-122 in promoting HCV replication. Similar to the findings of Li et al., we observed that Xrn1 knockdown also enhanced levels of miR-122-dependent HCV RNA replication, suggesting that protection from Xrn1 is not complete (18).
If miR-122 functions to shield the viral RNA from Xrn1, then knockdown of Xrn1 would have a greater impact on HCV replication without miR-122 binding than with miR-122 binding. Conversely, if miR-122 did not play a role in shielding the viral RNA from Xrn1, then knockdown of Xrn1 would have the same impact on replication with and without miR-122 binding. Therefore, we compared the effect of Xrn1 knockdown on miR-122-bound and miR-122-unbound replication (Fig. 1C). We confirmed Li et al.'s observations that knockdown of Xrn1 had a positive impact on viral replication when miR-122 was bound, increasing replication 5-fold by 3 days post-second electroporation (18). However, we also demonstrated that knockdown of Xrn1 had a greater impact on miR-122-independent SGR WT RNA replication and increased it by an average of 35-fold on day 2 and 26-fold on day 3, considerably more than the impact of knockdown on miR-122-bound replication. Ultimately, this supports a model where miR-122 binding plays a role in protecting the HCV genome from Xrn1, but the protection is incomplete.
Huh7-derived cells are the most commonly used cell lineage when studying HCV in vitro, and so we also verified our findings in Huh7.5 cells. Because Huh7.5 cells express miR-122, in order to test miR-122-unbound replication, we made use of a miR-122 binding site mutant subgenomic RNA, SGR S1+S2:p3, that has already been shown to be unresponsive to wild-type and endogenous miR-122 (4). SGR S1+S2:p3 contains a C-to-G mutation at position 3 in both miR-122 binding sites (S1 and S2) that abolishes miR-122 binding (depicted in Fig. 1E) and demonstrates miR-122-independent levels of replication similar to SGR WT in Hep3B cells. We again observed that knockdown of Xrn1 increased replication of both miR-122-dependent and miR-122-independent SGR (Fig. 1F and G), that Xrn1 knockdown did not restore miR-122-unbound replication to miR-122-bound levels (Fig. 1G, compare siXrn1 [Independent] and siControl [Dependent]), and that miR-122-independent replication increased to a much greater degree (10-fold) than miR-122-dependent replication (2-fold; Fig. 1G). We also observed that siXrn1 treatment reduced Xrn1 protein levels by 84% in Huh7.5 cells (Fig. 1H), Xrn1 knockdown did not affect Huh7.5 cell growth (Fig. 1I), transfection efficiency was similar in all samples (Fig. 1J), and before knockdown both Huh7.5 and Hep3B cells express similar levels of Xrn1 (Fig. 1K). Thus, we show here that Xrn1 depletion has a positive impact on HCV replication and that it has a greater effect on HCV replication when miR-122 is not bound to the viral 5′ UTR, suggesting a specific role for miR-122 in protecting the viral RNA from Xrn1.
Knockdown of Xrn2 has a similar effect on both miR-122-independent and miR-122-dependent subgenomic HCV replication.
Xrn2 is another major host 5′-to-3′ RNA exonuclease but is normally found in the nucleus (19). We hypothesized that since Xrn1 knockdown did not fully restore miR-122-independent replication to miR-122-dependent levels, miR-122 may also protect HCV RNA from Xrn2; in the course of our work, this was indeed reported by another group (25). Thus, we tested miR-122-dependent and miR-122-independent replication with and without Xrn2 knockdown as we had with Xrn1. In Hep3B cells, as expected, knockdown of Xrn2 led to increased replication of both miR-122-dependent and miR-122-independent SGR WT RNA (Fig. 2A), but the difference in the impact of Xrn2 knockdown on dependent (2-fold) and independent (4-fold) replication (Fig. 2B), although statistically significant on day 3, was not as great as had been observed for Xrn1 knockdown in Fig. 1. siRNA treatment resulted in an 89% reduction in Xrn2 protein levels by 3 days post-second electroporation (Fig. 2C), cell numbers were unaffected (Fig. 2G), and transfection efficiencies were similar among all samples (Fig. 2H). When tested in Huh7.5 cells, the results were similar. Knockdown of Xrn2 led to increased miR-122-dependent and miR-122-independent replication of SGR WT (Fig. 2D), but the difference in increase between dependent (1.4-fold) and independent (3-fold) replication, although significant (Fig. 2E), was again not as dramatic as it had been with Xrn1. siXrn2 treatment resulted in a 96% reduction in Xrn2 mRNA levels in Huh7.5 cells (Fig. 2F), which may be due to the apparent lower level of endogenous Xrn2 in these cells than in Hep3B cells (Fig. 2I). As with Hep3B cells, knockdown did not impact cell growth (Fig. 2G) or transfection efficiency (Fig. 2H). Because the effects of Xrn2 knockdown on miR-122-dependent and miR-122-independent replication were similar in both Hep3B and Huh7.5 cells, we conclude that while Xrn2 restricts HCV replication, the presence or absence of miR-122 binding has little effect, and thus protection from Xrn2 by miR-122 appears to be a relatively minor function of the miRNA.
FIG 2.

The impact of Xrn2 knockdown on miR-122-dependent and miR-122-independent subgenomic HCV replication. (A) Hep3B cells were electroporated with siXrn2 or siControl for preknockdown and 3 days later (day 0) were electroporated again with the indicated siRNAs, miRNAs, mRNAs, and wild-type HCV SGR RNAs as described for Fig. 1B. “Dependent” samples were electroporated with miR-122, while “independent” samples were electroporated with miControl. (B) The effect of Xrn2 knockdown on miR-122-dependent and -independent replication was evaluated as described in the legend to Fig. 1C. Significance was determined by unpaired parametric t test. (C) The effectiveness of siXrn2 at reducing Xrn2 protein levels in Hep3B cells was determined by Western blotting with antibodies against Xrn2 and β-actin. A representative blot is shown depicting Xrn2 protein levels 3 days post-first electroporation (at the time of second electroporation, which includes viral RNA and miRNAs) in the first two lanes and 6 days post-first electroporation or 3 days post-second electroporation. Percent knockdown ± standard deviation relative to the siControl-treated cells was determined by infrared band quantification on blots from three independent experiments. (D) Huh7.5 cells were treated with siXrn2 as described in the legend to Fig. 1F. “Dependent” samples were electroporated with wild-type SGR RNA (depicted above), while “independent” samples were electroporated with mutant SGR S1+S2:p3 viral RNA, which does not respond to miR-122, as depicted in Fig. 1E. (E) The effect of Xrn2 knockdown on miR-122-dependent and -independent replication was evaluated as described in the legend to Fig. 1G. Significance was determined by paired parametric t test. (F) The effectiveness of siXrn2 in reducing Xrn2 protein levels in Huh7.5 cells was determined as described in the legend to panel C, and a blot representative of three independent experiments is shown. (G) The effect of siXrn2, viral RNAs, and microRNAs on cell survival was evaluated by WST-1 3 days postelectroporation from samples in panels A and D, and cell numbers are normalized to siControl-miControl or siControl-SGR S1+S2:p3 samples. (H) Transfection efficiency in the experiments in panels A and D was evaluated 2 h postelectroporation by measuring Renilla luciferase expression from a coelectroporated mRNA. Samples are normalized to siControl-miControl or siControl-SGR S1+S2:p3 samples. (I) Untreated Hep3B and Huh7.5 cell lysates were analyzed by using Western blotting to show steady-state levels of Xrn2 protein in each cell type.
Knockdown of Xrn1 enhances full-length viral RNA replication and permits detectable replication of full-length RNA in the absence of miR-122 binding.
The JFH-1 subgenomic replicon has been valuable in allowing us to study HCV RNA replication in the absence of miR-122, but it lacks the structural proteins and coding regions of the virus and also contains an additional internal ribosome entry site (IRES) derived from the encephalomyocarditis virus (EMCV) that drives translation of the viral nonstructural proteins; so while it responds to miR-122, it may not authentically model its influence on full-length HCV RNA. Thus, we next set out to verify our findings of the effect of Xrn1 on replication of the full-length J6/JFH-1 monocistronic viral RNA (FL WT) with and without miR-122 binding, as was done with SGR RNA in Fig. 1. We and others have been unable to detect replication of this construct in the absence of miR-122 binding (4, 6, 8, 10, 25, 26). However, knockdown of Xrn1 in Hep3B cells, in addition to increasing levels of miR-122-dependent replication (Fig. 3A; compare siXrn1 [Dependent] with siControl [Dependent]), also allowed detectable miR-122-independent replication of FL WT RNA, as determined by luciferase expression levels above those produced from the polymerase-negative GNN mutant virus (Fig. 3A; compare siXrn1 [Independent] to GNN or siControl [Independent]). This had not been reported previously. Detectable miR-122-independent replication of FL RNA continues at low levels for 4 and 5 days postelectroporation (data not shown) but does not increase further, perhaps due to cell confluence. Thus, restriction by Xrn1 contributes to our inability to detect replication of FL HCV RNA in the absence of miR-122.
FIG 3.
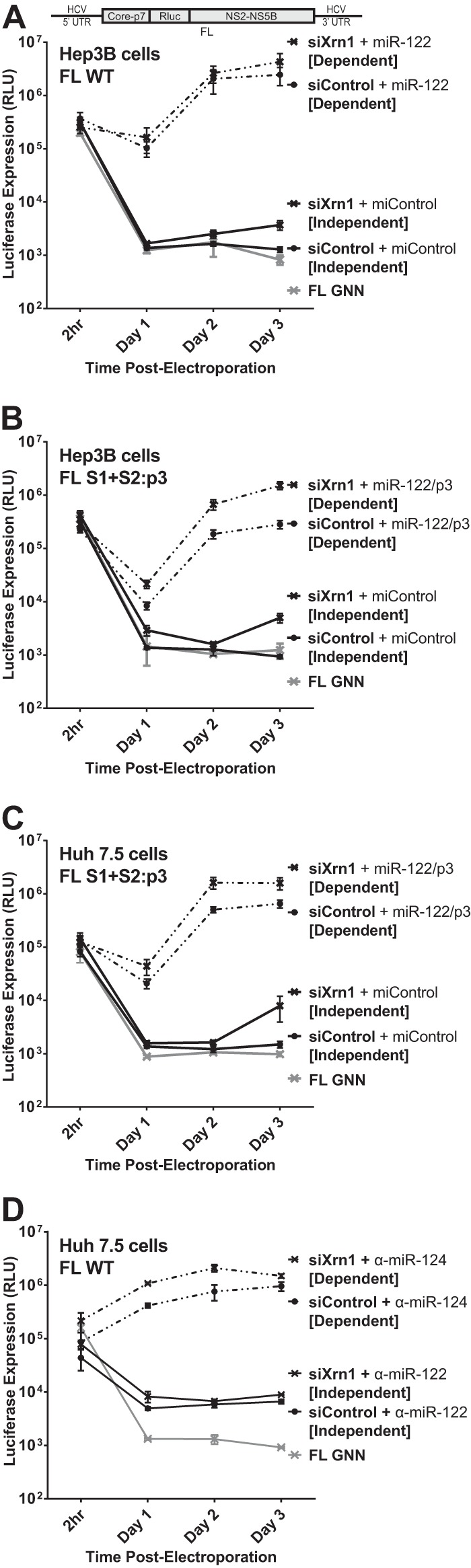
Knockdown of Xrn1 enhances full-length J6/JFH-1 viral RNA (FL WT) replication and permits detectable replication of full-length RNA in the absence of miR-122 binding. (A) Hep3B cells were electroporated with siXrn1 or siControl as described for Fig. 1B to knock down Xrn1. The effects of Xrn1 knockdown were assessed on replication of wild-type full-length J6/JFH-1 viral RNA “FL WT,” encoding an in-frame Renilla luciferase reporter (depiction above), miR-122-dependent replication was assessed in cells coelectroporated with miR-122, while miR-122-independent replication was assessed in cells coelectroporated with miControl. FL GNN is a full-length genome with a GDD-to-GNN polymerase-inactivating mutation to determine background Renilla luciferase expression in the absence of HCV RNA replication. (B) Hep3B cells were electroporated with siXrn1 or siControl to assess the influence of Xrn1 knockdown on HCV replication as described for Fig. 1B, but in this case miR-122-independent replication was evaluated using the miR-122 binding site mutant full-length J6/JFH-1 viral RNA FL S1+S2:p3, a full-length mutant with the p3 mutation in miR-122 binding sites S1 and S2 that does not respond to wild-type miR-122. miR-122-dependent replication was assessed in cells coelectroporated with miR-122/p3, an miR-122 mimic bearing a G-to-C mutation at position 3 that restores binding to the mutant genome. (C) Huh7.5 cells were electroporated with siXrn1 or siControl as described for Fig. 1F, and the effect of Xrn1 knockdown on miR-122-independent replication or miR-122-dependent replication of full-length J6/JFH-1 viral RNA was assessed in cells coelectroporated with FL S1+S2:p3 and miControl or miR-122/p3, respectively. (D) Huh7.5 cells were treated as described for panel C but were electroporated with wild-type full-length J6/JFH-1 viral RNA (FL WT). To establish miR-122-independent replication, the cells were treated with an miR-122 antagonist (α-miR-122) or a control miRNA antagonist targeting miR-124 (α-miR-124).
We also tested miR-122 binding site mutant full-length HCV RNA (FL S1+S2:p3) for the effect of Xrn1 on miR-122-dependent and independent replication to confirm the phenotype in both Hep3B and Huh7.5 cells (Fig. 3B and C, respectively). As with SGR S1+S2:p3, FL S1+S2:p3 has the C-to-G mutations at position 3 in both miR-122 binding sites (S1 and S2), depicted in Fig. 1E, but is otherwise identical to FL WT. FL S1+S2:p3 replicates to levels similar to those of FL WT when supplemented with miR-122/p3, a synthetic miR-122 that bears the G-to-C mutation at position 3 to complement the p3 mutation in the viral construct. Knockdown of Xrn1 in both Hep3B and Huh7.5 cells increased both miR-122-dependent and miR-122-independent replication of FL S1+S2:p3, and like we observed with FL WT in Hep3B cells, Xrn1 knockdown permitted detectible, but low-level, miR-122-independent replication of FL S1+S2:p3 above the background levels detected without Xrn1 knockdown (compare siControl [Independent] to GNN). As a further control, we also tested electroporation of Huh7.5 cells with FL WT viral RNA and mimicked miR-122-independent replication through use of an miR-122 antagonist (Fig. 3D, α-miR-122) and again observed a small increase in luciferase expression following Xrn1 knockdown. Collectively, our results demonstrate that Xrn1 knockdown can relieve Xrn1-mediated suppression of full-length HCV RNA, restoring replication to detectible levels without miR-122, but that in the context of full-length HCV, protection from Xrn1 degradation is not the most significant function for miR-122.
Contribution of miR-122 binding at sites S1 and S2 in HCV RNA accumulation.
In the interest of further evaluating the role of miR-122 binding in HCV replication, we examined the possibility that each miR-122 binding site has a different function or contributes to the effect of miR-122 differently. In order to evaluate this, we generated subgenomic and full-length constructs bearing the p3 mutation in either miR-122 binding site S1 or S2 (SGR S1:p3, SGR S2:p3, FL S1:p2, and FL S2:p3). Using these constructs, we can assess the impact of miR-122 binding to one or both miR-122 sites since the unmutated site would still be able to use wild-type miR-122, but the mutated site would require supplementation with the mutant miRNA, miR-122/p3; these conditions in Huh7.5 cells are depicted in Fig. 4A.
FIG 4.

The impact of miR-122 binding at site 1 compared to binding at site 2 with subgenomic and full-length replicons in Huh7.5 cells. (A) By mutating either S1 or S2 to the p3 mutation and supplementing with miR-122/p3 as shown, we can specifically analyze the influence of miR-122 binding to both sites (i), neither site (ii), or each site alone, as in panels iii and iv. (B) To assess the influence of miR-122 binding to each site, Huh7.5 cells were electroporated with subgenomic HCV RNA bearing the p3 C-to-G mutation in miR-122 binding site S1 (S1:p3) or in binding site S2 (S2:p3). To assess replication of all constructs when both sites are occupied, the S1 and S2:p3 RNAs were supplemented with miR-122/p3 as shown in panel A, wild-type HCV RNA replication was supplemented with miControl, and the S1+S2:p3 double-binding site mutant was supplemented with miR-122/p3 as shown in panel Ai. Replication in the absence of miR-122 binding was assessed using S1+S2:p3 HCV RNA replication with miControl, where neither binding site was occupied. For the determination of background luciferase expression in the absence of replication, SGR GND RNA bearing a GDD-to-GND polymerase-inactivating mutation was used. Replication was measured by firefly luciferase expression at the indicated time points. (C) The fold increase in HCV replication induced by miR-122 binding at 3 days postelectroporation over that observed using miR-122-unbound subgenomic viral RNA, S1+S2:p3 [S1+S2 Unbound], dotted line. Significance for relevant comparisons was determined by unpaired parametric t test. (D) Experiments similar to those presented in panel B were done to assess the influence of miR-122 binding to each site on full-length HCV RNA. Huh7.5 cells were electroporated with full-length HCV RNA bearing the p3 mutation in miR-122 binding site S1 or S2 and were supplemented with miRNAs and evaluated as described for panel B. Replication was measured by Renilla luciferase expression at the indicated time points; the FL GNN polymerase-inactivated mutant is a no-replication control. (E) The fold increase in HCV replication induced by miR-122 binding at site S1 or S2 was evaluated as described for panel C. Significance for relevant comparisons was determined by unpaired parametric t test.
In Huh7.5 cells without additional miRNA supplementation, subgenomic S1:p3 and S2:p3 RNAs replicate to an equal magnitude.
We first tested replication of subgenomic S1:p3 and S2:p3 mutants in Huh7.5 cells with both or only one binding site occupied (Fig. 4B). Because Huh7.5 cells express endogenous miR-122, this was achieved by supplementation with either miControl or miR-122/p3, as depicted in Fig. 4A for each construct. When both binding sites were occupied through supplementation with exogenous miR-122/p3, the S1:p3 and S2:p3 mutants replicated to similar levels as SGR WT and the S1+S2:p3 RNA supplemented with miR-122/p3, suggesting that the mutations did not impair replication in any way other than in their ability to bind miR-122 (Fig. 4B and C).
When only one binding site was occupied (by endogenous miR-122) (Fig. 5B, S2:p3 [S1 Bound] and S1:p3 [S2 Bound]), replication of each mutant was increased by the same degree over miR-122-independent (unbound) replication (S1+S2:p3 + miControl [Unbound]), as shown in Fig. 5C. From these results, we conclude that miR-122 binding at S1 or S2 impacts HCV SGR RNA replication equally.
FIG 5.

The impact of miR-122 binding at site 1 compared to binding at site 2 with full-length replicons in Hep3B cells. (A) Because Hep3B cells lack expression of detectable miR-122, we used them to test the influence of miR-122 binding to each site separately or together in both S1:p3 and S2:p3 mutant HCV RNAs through supplementation with the appropriate miRNAs, either miR-122 or miR-122/p3 (i through iv). (B) A time course of virus replication in Hep3B cells electroporated with full-length S1:p3 RNA (or the indicated control viral RNAs: FL WT + miR-122, S1+S2:p3 + miR-122/p3 or miControl, and FL GNN) and supplemented as indicated with miR-122, miR-122/p3, both, or miControl, to achieve miR-122 binding site combinations depicted in panel Aiii. Replication was measured by Renilla luciferase expression at the indicated time points. (C) The effect of binding at miR-122 site S1 or S2 on replication of FL S1:p3 HCV RNA is shown relative to luciferase expression from unbound full-length viral RNA, S1+S2:p3 [S1+S2 Unbound], dotted line, at 3 days postelectroporation. Significance for relevant comparisons was determined by unpaired parametric t test. (D) A time course of virus replication in Hep3B cells electroporated with full-length S2:p3 RNA (or the indicated control viral RNAs: FL WT + miR-122, S1+S2:p3 + miR-122/p3 or miControl, and FL GNN) and supplemented with miRNAs as indicated to achieve miR-122 binding site combinations shown in panel Aiv. Replication was measured by Renilla luciferase expression at the indicated time points. (E) The effect of binding at miR-122 site S1 or S2 on replication of FL S2:p3 HCV RNA is shown relative to luciferase expression from unbound full-length viral RNA, S1+S2:p3 [S1+S2 Unbound], dotted line, at 3 days postelectroporation. Significance for relevant comparisons was determined by unpaired parametric t test.
In Huh7.5 cells without additional miRNA supplementation, full-length S2:p3 demonstrates almost undetectable replication, while S1:p3 replicates to an intermediate level.
To analyze the influence of miR-122 binding to each individual binding site in the context of a full-length HCV genome, we first tested the full-length S1:p3 and S2:p3 mutants with both binding sites occupied (by supplementation with miR-122/p3) to ensure their fitness (Fig. 4D and E). Both RNAs replicated to similar levels as FL WT and FL S1+S2:p3 RNA when both miR-122-binding sites were occupied. However, we found that when only one site was occupied (by endogenous miR-122 present in Huh7.5 cells), the S2:p3 construct demonstrated only barely detectable replication, while the S1:p3 construct demonstrated an intermediate level of replication, similar to that observed using S1:p3 subgenomic constructs. Specifically, S2:p3 (having miR-122 binding to only site 1) replicated more poorly than S1:p3 (having miR-122 binding only to site 2). These data suggest that, contrary to our conclusions using SGR RNA, binding of miR-122 at each site contributes unequally to HCV RNA replication in full-length HCV RNA and that binding to S2 is more important for HCV augmentation. However, subsequent experiments in Hep3B cells, shown below, confirmed that the binding site mutant RNAs used in this experiment have a mutant phenotype independent from abolishing miR-122 binding.
In Hep3B cells, binding of miR-122 to either S1 or S2 of full-length HCV RNA impacts replication to the same magnitude.
To further test the impact of miR-122 binding to each individual binding site in a full-length HCV RNA genome, we used Hep3B cells, which do not express endogenously active miR-122, to design experiments in which we could test the impact of miR-122 binding to either S1 or S2 (or both) in the context of the same mutant viral RNA. By electroporating either full-length viral RNAs (S1:p3 or S2:p3) alone (miControl) or with miR-122, miR-122/p3, or both, we could control miR-122 binding to either or both sites in the context of each mutant HCV genomic RNA. Specifically, coelectroporation of HCV S1:p3 RNA with control miRNA will achieve miR-122-unbound conditions as depicted throughout Fig. 5A, and supplementation with either miR-122, miR-122/p3, or both miRNAs will test the effects of binding to either S1, S2, or both, as shown in Fig. 5Aiii. These conditions can be mirrored using the S2:p3 mutant RNA and miRNAs, as depicted in Fig. 5Aiv, to verify the influence of miR-122 binding to each site in each viral RNA.
When we tested replication of the FL S1:p3 mutant viral RNA under the various miR-122 binding conditions outlined in Fig. 5Aiii (Fig. 5B), we observed that miR-122 binding to each site had similar impacts on HCV replication, which mirrors our observations using SGR mutants in Huh7.5 cells (Fig. 4B). When S1 was occupied by miR-122/p3, luciferase expression was increased by an average of 17-fold over unbound luciferase expression, while when S2 was occupied by miR-122, luciferase expression was increased 18-fold over unbound levels (Fig. 5C); the difference between these was not significant. Also as expected, replication of FL S1:p3 when both sites are miR-122 bound was similar to replication of FL S1+S2:p3 and FL WT with both sites bound, and replication of both FL S1:p3 and FL S1+S2:p3 with neither site bound was equivalent to that of the background (GNN).
We then tested replication of the FL S2:p3 mutant viral RNA under the different miR-122 binding conditions outlined in Fig. 5Aiv (Fig. 5D). Again, binding of miR-122 at S1 increased replication by the same amount (2.1-fold) as binding of miR-122/p3 at S2 (1.8-fold) over miR-122-unbound luciferase expression (Fig. 5E), although much lower in magnitude than with FL S1:p3 RNA, confirming that binding of miR-122 at each site contributes equally in magnitude to HCV RNA replication. Replication of this RNA was equivalent to that of S1+S2:p3 with both sites bound and similar to FL WT, and replication with neither site bound was equivalent to that of the background.
In Hep3B cells, FL S2:p3 RNA with either S1 or S2 occupied demonstrates significantly impaired replication in comparison to FL S1:p3 RNA.
By comparing results in Hep3B cells, the replication capacity of S1:p3 and S2:p3 when one site is bound differs greatly and clarifies the results we observed in Huh7.5 cells (Fig. 4). Data from Hep3B cells suggest that the full-length S2:p3 RNA has a defect in replication that is not observed in full-length S1:p3, nor in the SGR S1:p3 or S2:p3, and is observed only when one site (either S1 or S2) is bound by miR-122 (Fig. 5D and E). The phenotype of this mutant RNA suggests that binding to S2 is not solely responsible for the defect in replication we observed for FL S2:p3 viral RNA in Huh7.5 cells and explains why it replicated at much lower levels than the FL S1:p3 viral RNA (Fig. 4D and E). Interestingly, FL S2:p3 RNAs replicated to levels similar to those of wild-type RNA when both miR-122 binding sites were occupied (Fig. 4D and E and 5D and E). Thus, the impairment in replication was apparent only when one miR-122 binding site was unbound but could be masked or complemented when both miR-122 binding sites were occupied together. This suggests that full-length S2:p3 has an impairment other than simply miR-122 binding that is apparent only in the absence of miR-122 binding at one site, but we cannot exclude the possibility that full-length S1:p3 may also or instead exhibit an unnatural phenotype when a single miR-122 binding site is bound. However, our combined data derived from full-length RNAs tested in all possible states of miR-122 binding and SGR RNAs in Huh7.5 cells indicate that miR-122 binding to each site has an equivalent influence on HCV replication.
Binding of miR-122 to both sites at the same time increases HCV RNA replication cooperatively.
Our data suggest that the impact on HCV replication of miR-122 binding to both sites exceeds the combined effect of binding to each site alone and indicates cooperation between the effects of miR-122 binding at each site. Using data derived from experiments using subgenomic replicon RNA (Fig. 4B and C), in Fig. 6 we show the fold impact of miR-122 binding to each site to HCV replication (S1 bound/both sites unbound) and compare the calculated impact of binding to both sites if the sites do not act cooperatively (theoretical; the product of binding to each site individually) with the observed data when both sites are bound (experimental). Our data show that when both sites are bound, the influence on replication is an average of 3.3-fold higher than that of the products of binding at each site alone and thus cooperative. Experiments using full-length constructs in both Huh7.5 cells (Fig. 4D and E) and in Hep3B cells (Fig. 5) also suggest cooperation between miR-122 binding to each site, but since miR-122-unbound replication in full-length constructs was equivalent to the background, and likely below the limit of detection of our assay, then calculations similar to those presented in Fig. 6 are not possible. However, in all cases, the influence of miR-122 binding to both sites was significantly greater than the product of the effect of binding at both sites alone.
FIG 6.
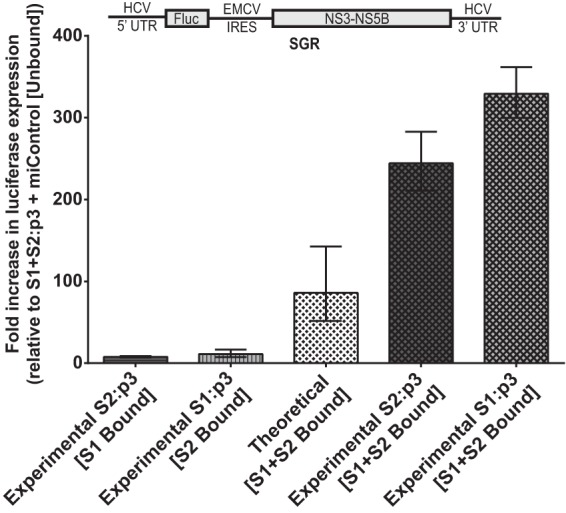
The effects of miR-122 binding at each site are cooperative rather than merely multiplicative. Subgenomic miR-122 binding site mutants from Fig. 4B and C were evaluated in Huh7.5 cells for the contribution of miR-122 binding at each site to the overall replication of the viral RNA on day 3. Geometric mean fold increase in luciferase expression of subgenomic S2:p3 and S1:p3 viral RNAs over S1+S2:p3 [Unbound] is shown for two independent experiments and is representative of three independent experiments. S2:p3 [S1 Bound] and S1:p3 [S2 Bound] were the indicated viral RNAs supplemented with miControl. Theoretical [S1+S2 Bound] was determined by multiplying the fold increase of [S1 Bound] by the fold-increase of [S2 Bound] to show the theoretical impact of binding at both sites, if their contribution was multiplicative. Experimental S2:p3 [S1+S2 Bound] and Experimental S1:p3 [S1+S2 Bound] were the indicated viral RNAs supplemented with miR-122/p3. Error bars are geometric standard deviations.
In Huh7.5 cells, saturation of binding at S1 requires more miR-122 than at S2.
Because many published experiments that attempted to characterize the function of miR-122 in the HCV life cycle have used Huh7-derived cells in which the endogenous levels of miR-122 in the cells are supplemented with synthetic miR-122 (5, 10–13, 15, 27, 28), we also examined the effect of miR-122 supplementation on each individual miR-122 binding site using subgenomic and full-length RNA with S1:p3 and S2:p3 mutations. Replication of SGR S2:p3 (Fig. 7A, S2:p3 + miControl [S1 Bound]) is increased 4.1-fold on day 3 by the addition of exogenous miR-122 (Fig. 7A, S2:p3 + miR-122 [S1 Bound], and C), indicating that endogenous levels of miR-122 in Huh7.5 cells are not sufficient to saturate binding of miR-122 to S1. However, the addition of miR-122 to SGR S1:p3 (Fig. 7B, compare S1:p3 + miControl [S2 Bound] to S1:p3 + miR-122 [S2 Bound], and C) does not affect replication, indicating that endogenous levels of miR-122 in Huh7.5 cells are sufficient to saturate binding of miR-122 to S2 in our hands.
FIG 7.

Increased miR-122 levels saturate S1 at a lower level of miR-122 than S2. (A) A time course of RNA replication in Huh7.5 cells electroporated with subgenomic S2:p3 RNA or the indicated control viral RNAs, supplemented with either miControl or with additional miR-122 to ensure that binding site S1 was maximally occupied. Replication was measured by firefly luciferase expression at the indicated time points. (B) A time course of RNA replication in Huh7.5 cells electroporated with subgenomic S1:p3 RNA or the indicated control viral RNAs, supplemented with either miControl or with additional miR-122 to ensure that binding site S2 was maximally occupied. Replication was measured as described for panel A. (C) The impact on HCV replication of exogenous miR-122 supplementation to the binding of either S1 or S2 from panels A and B was determined by comparing luciferase expression of the supplemented samples to that of subgenomic S1+S2:p3 [S1+S2 Unbound] (dotted line) on day 3 postelectroporation. (D) A time course of RNA replication in Huh7.5 cells electroporated with the indicated full-length viral RNAs and miRNAs as described for panel A. Replication was measured by Renilla luciferase expression at the indicated time points. (E) A time course of RNA replication in Huh7.5 cells electroporated with the indicated full-length viral RNAs and miRNAs as described for panel B. Replication was measured as described for panel D. (F) The impact on HCV replication of exogenous miR-122 supplementation to the binding at either S1 or S2 from panels D and E was determined by comparing luciferase expression to that of full-length S1+S2:p3 [S1+S2 Unbound] (dotted line) on day 3 postelectroporation.
When these comparisons are made in Huh7.5 cells using full-length S2:p3 (Fig. 7D) and S1:p3 (Fig. 7E), while the addition of miR-122 results in increased replication of both constructs, the increase is more drastic (8.8-fold) with S2:p3 (compared to a statistically insignificant change with miR-122 supplementation in S1:p3), suggesting again that binding at S1 is not saturated by endogenous miR-122 (Fig. 7F). Together, these data suggest that experiments involving supplementation of additional miR-122 in Huh7.5 cells may impact binding to S1 more than binding to S2.
Binding at S1 increases FL RNA replication more than binding at S2 when Xrn1 is knocked down in Hep3B cells.
We have confirmed that one of the functions of miR-122 binding at the HCV 5′ UTR is to protect the viral RNA from the effects of Xrn1. Further, we have established that protection from degradation by Xrn1 is not the only role for miR-122. Thus, we propose that by evaluating the impact on viral replication of miR-122 binding to each binding site in the context of Xrn1 knockdown, we can determine their individual contributions to the additional (Xrn1-independent) functions of miR-122. We compared the effect of binding at both sites, neither site, and either S1 or S2, in the context of Xrn1 knockdown on FL S1:p3 and FL S2:p3 replication (Fig. 8A and C, respectively). Despite our earlier observation that under normal cellular conditions S1 and S2 contribute to FL replication to an equal degree (Fig. 5; see also Fig. 8B and D), we observed that when Xrn1 was knocked down, binding at S1 had a slightly greater contribution than S2 (Fig. 8E). Specifically, when Xrn1 was knocked down in cells supporting FL S1:p3, binding at S1 increased replication 19-fold (±6.6) over unbound replication, while binding at S2 increased replication only by 13-fold (±4.6) by day 3 (Fig. 8E). In cells supporting FL S2:p3, the difference was greater: S1 increased replication 25-fold (±7.6) and S2 only 4-fold (±0.7) over unbound replication on day 3 (Fig. 8E). However, it is likely that the differences between these calculated fold increases are artificially magnified by our inability to detect unbound replication of this RNA. Overall, data from both S1:p3 and S2:p3 showed that S1 had a slightly greater influence on enhancing HCV replication when Xrn1 was knocked down. We also analyzed the impact of Xrn1 on replication of HCV RNA when miR-122 occupied each individual binding site by calculating the increase in replication when Xrn1 is knocked down compared to replication in the presence of Xrn1 (siXrn1/siControl) for binding at either site (Fig. 8F). As expected, Xrn1 knockdown increased replication when either miR-122 binding site was unoccupied, but we consistently observed that knockdown had a greater impact when binding site S2 was unoccupied, suggesting that Xrn1 may restrict replication more when S2 is unoccupied than when S1 is unoccupied, but these differences were not statistically significant. These findings suggest that binding to S1 has a slightly greater impact than S2 on the function(s) of miR-122 unrelated to protection from Xrn1 and that binding at S2 may have a greater role than S1 in protecting from Xrn1.
FIG 8.

The impact of miR-122 binding at site 1 compared to binding at site 2 with full-length HCV RNA when Xrn1 is knocked down. To assess the impact of miR-122 binding to site 1 or site 2 independent from that of protection from Xrn1, we tested the influence of miR-122 on replication of HCV in Xrn1-depleted cells. (A and B) We performed a time course of FL S1:p3 viral RNA replication in Hep3B cells with Xrn1 knockdown (A) or no knockdown (B). Replication of FL S1:p3 was assessed following supplementation with miR-122/p3 (S1 Bound), miR-122 (S2 Bound), miR-122/p3 and miR-122 (S1+S2 Bound), or miControl (S1+S2 Unbound) as depicted in Fig. 5Aiii, and replication was measured by evaluating reporter Renilla luciferase expression at the indicated time points. Results are an average from five independent experiments. (C and D) Xrn1 (C)- and siControl (D)-treated Hep3B cells were treated as described for panels A and B, respectively, but were electroporated with FL S2:p3 RNA, with miRNAs resulting in miR-122 binding site occupation as depicted in Fig. 5Aiv. Results are an average from five independent experiments. (E) In the context of Xrn1 knockdown, the fold increase in replication of FL S1:p3 RNA (light gray, left) and FL S2:p3 RNA (dark gray, right) when either S1 or S2 is bound was compared to unbound replication. Significance was determined using a ratio-paired parametric t test of relevant samples. (F) The effect of Xrn1 knockdown on FL S1:p3 or FL S2:p3 with either site 1 or site 2 unoccupied is shown by comparing the fold increase in replication with Xrn1 knockdown from panel A or C to replication with siControl (no knockdown) treatment from panel B or D, respectively. The dotted line indicates 1-fold or no increase in replication due to knockdown. Significance was tested using ratio-paired t tests of relevant samples.
miR-122 binding at either S1 or S2 does not impact FL S1:p3 RNA translation when Xrn1 is knocked down in Hep3B cells.
One of the challenges in studying the role of miR-122 in the HCV life cycle is the difficulty in separating effects on translation from effects on replication. Although we observed an impact of Xrn1 on replication, it may actually affect RNA translation, either directly or indirectly, and over the course of our 3-day experiments, this impacts replication. Thus, we chose to examine the effect of miR-122 binding to each site, both, or none on translation in the context of Xrn1 knockdown. To assess translation, we used FL S1:p3 GNN, which is incapable of replication, in Hep3B cells and added wild-type or mutant miR-122 to control binding to each site. When cells were treated with siControl (Fig. 9A), we observed that binding at both sites (S1+S2) increased translation over unbound translation up to 1.9-fold over the 12-hour time period, which is within the range of others' findings (5, 10, 11, 13, 15). We also observed that binding at either S1 or S2 had a lesser but still-discernible impact on translation and that there was no significant difference between the impact of S1 or S2 binding, suggesting that binding at each site plays an equivalent role in the effect of miR-122 on translation. However, when Xrn1 was knocked down (Fig. 9B), binding at either S1 or S2 alone did not increase translation at all, and binding at both sites had a much weaker impact on translation. This suggests that most or perhaps all of the apparent impact of miR-122 on translation is due to protection from Xrn1; we suggest that the remaining impact of binding at S1+S2 when Xrn1 is knocked down is due to minimal amounts of Xrn1 remaining in the cell (see Fig. 1D) rather than to a direct impact of miR-122 on translation independent of protection from Xrn1. Knockdown of Xrn1 increased overall viral RNA translation considerably (Fig. 9C) and also positively impacted control mRNA translation (data not shown). Particularly, knockdown of Xrn1 had a greater impact on S1+S2-unbound translation (4.8-fold) than it did on either S1-bound (2.9-fold), S2-bound (3.6-fold), or S1+S2-bound (3.6-fold) translation, providing further evidence that binding at these sites protects from Xrn1.
FIG 9.

Knockdown of Xrn1 increases FL S1:p3 RNA translation overall but reduces the contribution miR-122 binding makes to viral translation. Hep3B cells were electroporated with siXrn1 or siControl for preknockdown. Three days later, cells were electroporated with FL S1:p3 GNN (replication-incompetent S1 miR-122 binding mutant) viral RNA, transfection control firefly mRNA, and the indicated miRNAs as depicted in Fig. 5Aiii. Because the viral RNA is replication incompetent, measuring luciferase at the indicated 3, 6, 9, and 12 h post-second electroporation measures viral translation. Viral translation was normalized to transfection control mRNA firefly luciferase expression levels measured at 3 h post-second electroporation. The effect on translation of FL S1:p3 GNN viral RNA by miRNA binding at S1, S2, or both sites was compared to translation when neither site was bound in siControl-treated cells (A) or siXrn1-treated cells (B), where the dotted line represents 1-fold or no increase in translation. (C) Stimulation of translation by Xrn1 was determined by comparing normalized viral translation in siXrn1-treated cells to viral translation in siControl-treated cells. The dotted line indicates 1-fold or no stimulation of viral translation. Significance for all figures was determined using two-way analysis of variance (ANOVA), with Tukey's multiple-comparison test performed to compare differences of samples within a given time point. Only significant differences are indicated; all other comparisons were not significant.
DISCUSSION
We have shown that miR-122 has a specific function in shielding the 5′ untranslated region of the HCV RNA from the host RNA exonuclease Xrn1 (Fig. 1 and 3) and to a lesser extent from Xrn2 (Fig. 2). We made use of our miR-122-independent system of subgenomic HCV RNA replication (4) to verify that miR-122 was specifically protecting the viral RNA from restriction by Xrn1 and Xrn2, by calculating the difference in the effect between miR-122-dependent and miR-122-independent replication to determine how much miR-122 prevents the host restriction factor Xrn1 from inhibiting the virus' life cycle. We found that knockdown of both Xrn1 and Xrn2 did indeed affect miR-122-independent replication more strongly than miR-122-dependent replication (Fig. 1 and 2) but that this difference was much more drastic for Xrn1 knockdown than for Xrn2 knockdown. A role for miR-122 in protecting HCV RNA from Xrn2 was recently reported (25), but a conflicting publication also indicates no effect of Xrn2 on genotype chimera and luciferase reporter HCV RNA stability (29). Our data suggest that both exoribonucleases may contribute to restriction of HCV to differing degrees, and we cannot rule out differing mechanisms of restriction, but we chose to focus our efforts on determining protection from Xrn1, because it restricted miR-122-independent replication to a much greater degree in our assays.
We have also demonstrated that replication of full-length HCV RNA in the absence of miR-122 can be detected following knockdown of Xrn1, and we speculate that full-length HCV RNA likely replicates at undetectable levels in the absence of miR-122 binding (Fig. 3) and becomes detectible when we remove Xrn1 (Fig. 3 and 8). Because knockdown of Xrn1 does not allow miR-122-unsupplemented replication to reach miR-122-supplemented levels in either subgenomic or full-length RNA contexts, like Li et al. (18), we conclude that miR-122 has additional functions in viral replication above that of protecting the viral RNA from Xrn1, and this is further supported by the observed direct effect of miR-122 on HCV RNA synthesis (29). Our data also support the conclusion that the positive impact of miR-122 on HCV translation is due to protection of the viral RNA from Xrn1 (Fig. 9) (18).
We also quantified the relative contribution of miR-122 binding at each of the two 5′ miR-122 binding sites to HCV replication, showing that each site contributes with equal magnitude to enhancing replication of the virus and that binding at both sites increases replication cooperatively (Fig. 4 to 6). This suggests that functional Ago2–miR-122 complexes bind to each site, and each complex enhances the effect of adjacent complexes, similar to the proposed cooperativity between adjacent miRNA binding sites (within 19 nucleotides) for miRNA suppression (30, 31). We speculate that binding of the Ago2–miR-122 complex at one site may enhance recruitment to the second sites through either aggregative properties of the complex or through changing RNA structure to make the adjacent site more accessible to Ago2–miR-122, or it could be that Ago2–miR-122 complexes in close proximity to each other have enhanced downstream activities. However, it appears that the cooperative effect is less potent than the effects of miR-122 binding to each site since we found that HCV replication augmentation induced by miR-122 binding to a single site ranged from 7- to 17-fold, while the cooperativity effect was only 2- to 5-fold greater than the predicted product of binding at both sites. Thus, while the cooperative effect is consistent and relevant to HCV replication, its impact is less than that of individual miRNA binding interactions.
Our data also indicate that each miRNA binding site requires a different amount of miR-122 to saturate the effects of binding. Providing greater amounts of miR-122 exogenously to Huh7.5 cells enhanced both subgenomic (Fig. 7A to C) and full-length (Fig. 7D to F) RNA replication if it could bind to S1, suggesting that in our hands, S1 binding is not saturated in Huh7.5 cells. In contrast, the exogenous addition of miR-122 for binding at S2 did not augment replication of subgenomic RNA (Fig. 7B and C) and increased full-length RNA replication only slightly (Fig. 7E and F), suggesting that S2 is already saturated in Huh7.5 cells. These data may reflect the enhanced binding strength of miR-122 to S2 proposed by Mortimer and Doudna (32). This also has implications for explorations of the functions of miR-122, where Huh7.5 cells were supplemented with additional miR-122; and it is possible that supplementation may disproportionately measure effects of miR-122 binding to S1 (5, 10, 11, 15, 27, 28).
We identified that full-length S2:p3 exhibits an impaired replication phenotype that is apparent only when at least one miR-122 binding site is unoccupied but that can be completely rescued by miR-122 binding at both sites (Fig. 4 and 5). The phenotype of this mutant was unidentifiable in Huh7.5 cells and initially led us to incorrectly conclude that S2 was more important for miR-122's effect on HCV replication, but experiments using Hep3B cells allowed us to assess the impact of miR-122 binding to each site in this RNA and identify the nature of the impairment. While we do not know the mechanism of impairment of this mutant, interestingly it is apparent only when there is a miR-122-off state at either binding site, and no defect is observed when both sites are bound. However, it is difficult to speculate on the biological significance of this particular mutant's phenotype since there is no experimental data describing the dynamics of miR-122 binding during the virus' life cycle or when, how often, and where in the cell HCV RNA is or is not bound by miR-122. Also, interestingly, the mutation at S2:p3 strangely has a more severely impaired phenotype than a virus mutant having the p3 mutation at both S2 and S1, since Xrn1 knockdown can neither compensate for the S2:p3 impairment (Fig. 8C) nor rescue miR-122-unbound replication of FL S2:p3 to detectable levels but can rescue replication of FL S1+S2:p3 (Fig. 3B and C). It is tempting to speculate that the phenotype of this mutant may suggest that there is a benefit to having matching seed sequences at both sites, but this cannot be the sole reason for the defect, since S1:p3, which also has no matching seed sequences, did not exhibit the same phenotype. Thus, the severely impaired phenotype of S2:p3 seems to be displayed only when the RNA has the p3 mutation at S2 alone.
Our conclusion that there is an equal contribution to HCV replication by miR-122 binding at each site is contrary to other published work (8, 10, 33). Experiments by the Lemon group and the Bukh group found that binding at S1 was overall more important than binding at S2, but we believe that significant differences between the viral mutants and experimental approaches used may explain the differences (8, 10, 33). First, data presented by the Lemon lab compared the replication of an S1:p6 mutant genome with that of an S2:p6 genome in Huh7.5 cells and found that S1:p6 grew less efficiently than S2:p6, concluding that S1 must be more important (8, 10). However, their experiment cannot exclude the possibility that either or both S1:p6 or S2:p6 A-to-U mutant RNAs have a mutant phenotype that contributed to their observations, similar to what we observed with S2:p3 in Huh7.5 cells (Fig. 4D and E) (8, 10). Second, the study by the Bukh lab also concluded that S1 was more important than S2; however, the results of this study may have been affected by the affinity of the mutant binding sites for miR-122. This group tested responsiveness of S2 binding by using an miR-122 antagonist to compete off binding to S2 in viruses in which S1 had been mutated. When the RNAs failed to respond significantly to the antagonist, the authors concluded that S2 must not be important for replication (33). However, structural data and in vitro analysis of miR-122 binding affinities suggest that the binding at S2 is stronger and less sensitive to antagonism than binding at S1 and may explain the lack of response of the S1 mutant to the antagonist (32). By using Hep3B cells, which are devoid of functional miR-122 but support HCV replication, we were able to identify the mutant phenotype in S2:p3 and design controlled experiments to test the impact of miR-122 binding to neither site, either site, or both sites in the context of individual mutant viral RNA constructs. Thus, by using Hep3B cells as a model, we were able to confirm equal contributions of each binding site to replication in two different full-length HCV RNAs.
We further evaluated the effect of miR-122 binding at either S1 or S2 in the context of Xrn1 knockdown to assess the impact of miR-122 on its other roles in full-length viral replication (Fig. 8). We found that binding at both sites contributed to HCV replication when the need for protection from Xrn1 is reduced (Xrn1 knockdown), but binding at S1 increased replication more than binding at S2 (Fig. 8E and F), which suggests that S1 has a greater role than S2 in a function of miR-122 that is unrelated to protection from Xrn1. Our translation assays also support that binding at both sites contributes to protection from Xrn1 (Fig. 9). It has been hypothesized that miR-122 binding to the HCV 5′ UTR creates a double-stranded RNA structure that protects the 5′ end of the viral RNA from degradation. Our data confirming a specific impact of Xrn1 knockdown on miR-122-unbound replication support this hypothesis (18, 34). An alternative hypothesis suggested that the overhang generated by miR-122 binding to S1 may specifically mask and protect the 5′ end. Since our data show a role for both S1 and S2 in protection from Xrn1, they do not support the hypothetical importance of the overhang but instead suggest that miR-122 binding and the concomitant protein/RNA structures that assemble at both sites mediate both protection from Xrn1 and other functions (7, 8, 11, 13).
Our observations that knockdown of Xrn1 did not eliminate the requirement for miR-122 on viral replication in any context tested (Fig. 1, 3, and 8) also support the conclusion that protection from Xrn1 is not sufficient to account for the effect of miR-122 on HCV RNA accumulation (10, 11, 35). Another possible function for miR-122 is protecting the viral RNA from an as-yet-unidentified pyrophosphatase that removes the 5′ triphosphate (34, 36). HCV requires a 5′ triphosphate for efficient replication, but Xrn1 is more effective at degrading RNA having monophosphate 5′ ends, such as those produced by decapping of mRNAs, so degradation of the HCV genome by Xrn1 may require enzymes to generate a 5′ monophosphate substrate on the 5′ end of the viral RNA (18, 19, 34, 37). Other possible functions of miR-122 in protecting the 5′ end of the viral RNA include shielding the 5′ triphosphate from innate immune sensors such as RIG-I, IFIT-1, or IFIT-5 (34, 36). Thus, Xrn1 may not be the only cellular gene that targets the 5′ terminus of the viral genome and may explain why Xrn1 knockdown alone is insufficient to restore replication to miR-122-bound levels (18, 19, 34). Given the RIG-I defect found in Huh7.5 cells but not Hep3B cells, it is unlikely that RIG-I signaling restricts HCV in the absence of miR-122 (38, 39). Alternatively, miR-122 may have an additional function unrelated to end protection, such as in viral life cycle stages following translation, like the switch directing the viral genomic RNA to different functions (translation, transcription, packaging) or aiding in initiation of replication (40). In vitro work suggests that miR-122 is not involved in elongation by the virus' RNA-dependent RNA polymerase, NS5B, but a role in initiation of replication has not been thoroughly examined, nor has a role in polymerase elongation been studied in vivo, and a recent publication suggests that miR-122 does increase RNA synthesis (9, 29, 41).
Assessment of the impact of miR-122 and Xrn1 on HCV translation suggested that the observed effect of miR-122 on translation is mostly or solely due to protection from Xrn1 (Fig. 9). Other researchers have also found that the addition of miR-122 results in increased HCV translation (5, 10–15, 35, 42), and when the effect is compared to that of miR-122 on replication, it is most likely that miR-122 has a separate, additional role in replication (10, 11, 35). More recently, researchers have been exploring whether the effect of miR-122 on viral translation is merely a stabilization of the viral RNA (8, 11, 18, 32, 43). In particular, Xrn1 has been implicated as the major host factor destabilizing and degrading the viral RNA (17, 18, 29, 32), although not all screens involving Xrn1 knockdown show that it has a negative or any effect on HCV (17, 44–46). Our evidence (Fig. 9) combined with that of others suggests that the main impact that miR-122 has on translation in our assays is due to protecting the viral RNA from degradation by Xrn1, leaving more copies of the RNA to be translated. However, since our assays use an HCV RNA that does not replicate, we cannot omit the possibility that it may not detect a putative role for miR-122 in regulating the switch between translation and replication.
Overall, our efforts add to the growing picture that the function of miR-122 in the hepatitis C virus life cycle is nuanced and complicated. We find that miR-122 binding protects the viral RNA from Xrn1 during replication and translation but also confirm that it must have additional functions in the HCV life cycle. We provide evidence to suggest that replication of full-length HCV RNA can occur in the absence of miR-122 binding since detectible replication can be rescued by relieving suppression by Xrn1. We further determine that binding of miR-122 at both S1 and S2 contribute equally to replication and that their effects are cooperative when both sites are bound. This suggests a model for the mechanism of activity of miR-122 that includes two independently active ribonucleoprotein complexes bound to the HCV 5′ UTR. In addition, we speculate that the complexes binding to S1 and S2 may have slightly different roles in the various functions of miR-122, since when we remove the need to protect the viral RNA from Xrn1 (by Xrn1 knockdown), S1 binding increases replication more than S2 binding does. Finally, we have validated the Hep3B cell culture system as a model that permits accurate analysis of virus life cycle processes in the presence and absence of miR-122 binding and characterization of the roles of the different binding sites in the same HCV RNA construct. Further characterization of the effects of specific binding site point mutations may also shed light on the various functions of miR-122. By exploring the functions of miR-122 on the hepatitis C virus life cycle in this system, we have provided insight into both others' results with respect to the roles of miR-122 binding to the HCV genome and future avenues of exploration to further understand the mechanisms of miR-122's interaction with hepatitis C virus.
ACKNOWLEDGMENTS
We thank Jens Lykke-Andersen for the kind gift of anti-Xrn1 antibody, Charlie Rice for pJ6/JFH-1(p7Rluc2A), and Takaji Wakita for pSGR-JFH-1-Luc.
P.A.T. acknowledges the National CIHR Research and Training Program in Hepatitis C (NCRTP-HepC) for Ph.D. funding, Y.A.-C. acknowledges the Saskatchewan Health Research Foundation for Postdoctoral Research Fellowship funding, and J.E.G. acknowledges NSERC for USRA funding. This work was also funded by CIHR (MOP-133458) and NSERC (342475-2012) grants to J.A.W.
REFERENCES
- 1.WHO. 2012. Hepatitis C. World Health Organization fact sheet no. 164. WHO, Geneva, Switzerland. [Google Scholar]
- 2.Strader DB, Wright T, Thomas DL, Seeff LB. 2004. Diagnosis, management, and treatment of hepatitis C. Hepatology 39:1147–1171. doi: 10.1002/hep.20119. [DOI] [PubMed] [Google Scholar]
- 3.Thibault PA, Wilson JA. 2014. Transient replication of hepatitis C virus subgenomic RNA in murine cell lines is enabled by miR-122 and varies with cell passage. PLoS One 9:e89971. doi: 10.1371/journal.pone.0089971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thibault PA, Huys A, Dhillon P, Wilson JA. 2013. MicroRNA-122-dependent and -independent replication of hepatitis C virus in Hep3B human hepatoma cells. Virology 436:179–190. doi: 10.1016/j.virol.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Huys A, Thibault PA, Wilson JA. 2013. Modulation of hepatitis C virus RNA accumulation and translation by DDX6 and miR-122 are mediated by separate mechanisms. PLoS One 8:e67437. doi: 10.1371/journal.pone.0067437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jopling CL, Schutz S, Sarnow P. 2008. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 4:77–85. doi: 10.1016/j.chom.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Machlin ES, Sarnow P, Sagan SM. 2011. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci U S A 108:3193–3198. doi: 10.1073/pnas.1012464108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimakami T, Yamane D, Welsch C, Hensley L, Jangra RK, Lemon SM. 2012. Base pairing between hepatitis C virus RNA and microRNA 122 3′ of its seed sequence is essential for genome stabilization and production of infectious virus. J Virol 86:7372–7383. doi: 10.1128/JVI.00513-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Norman KL, Sarnow P. 2010. Modulation of hepatitis C virus RNA abundance and the isoprenoid biosynthesis pathway by microRNA miR-122 involves distinct mechanisms. J Virol 84:666–670. doi: 10.1128/JVI.01156-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jangra RK, Yi M, Lemon SM. 2010. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J Virol 84:6615–6625. doi: 10.1128/JVI.00417-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, Lemon SM. 2012. Stabilization of hepatitis C virus RNA by an Ago2–miR-122 complex. Proc Natl Acad Sci U S A 109:941–946. doi: 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang C, Huys A, Thibault PA, Wilson JA. 2012. Requirements for human Dicer and TRBP in microRNA-122 regulation of HCV translation and RNA abundance. Virology 433:479–488. doi: 10.1016/j.virol.2012.08.039. [DOI] [PubMed] [Google Scholar]
- 13.Wilson JA, Zhang C, Huys A, Richardson CD. 2011. Human Ago2 is required for efficient microRNA 122 regulation of hepatitis C virus RNA accumulation and translation. J Virol 85:2342–2350. doi: 10.1128/JVI.02046-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts AP, Lewis AP, Jopling CL. 2011. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res 39:7716–7729. doi: 10.1093/nar/gkr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henke JI, Goergen D, Zheng J, Song Y, Schuttler CG, Fehr C, Junemann C, Niepmann M. 2008. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 27:3300–3310. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roby JA, Pijlman GP, Wilusz J, Khromykh AA. 2014. Noncoding subgenomic flavivirus RNA: multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses 6:404–427. doi: 10.3390/v6020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones DM, Domingues P, Targett-Adams P, McLauchlan J. 2010. Comparison of U2OS and Huh-7 cells for identifying host factors that affect hepatitis C virus RNA replication. J Gen Virol 91:2238–2248. doi: 10.1099/vir.0.022210-0. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Masaki T, Yamane D, McGivern DR, Lemon SM. 2013. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc Natl Acad Sci U S A 110:1881–1886. doi: 10.1073/pnas.1213515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagarajan VK, Jones CI, Newbury SF, Green PJ. 2013. XRN 5′ to 3′ exoribonucleases: structure, mechanisms and functions. Biochim Biophys Acta 1829:590–603. doi: 10.1016/j.bbagrm.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB. 1979. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 282:615–616. doi: 10.1038/282615a0. [DOI] [PubMed] [Google Scholar]
- 22.Kato T, Date T, Miyamoto M, Sugiyama M, Tanaka Y, Orito E, Ohno T, Sugihara K, Hasegawa I, Fujiwara K, Ito K, Ozasa A, Mizokami M, Wakita T. 2005. Detection of anti-hepatitis C virus effects of interferon and ribavirin by a sensitive replicon system. J Clin Microbiol 43:5679–5684. doi: 10.1128/JCM.43.11.5679-5684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol 81:8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lykke-Andersen J, Wagner E. 2005. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev 19:351–361. doi: 10.1101/gad.1282305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sedano CD, Sarnow P. 2014. Hepatitis C virus subverts liver-specific miR-122 to protect the viral genome from exoribonuclease Xrn2. Cell Host Microb 16:257–264. doi: 10.1016/j.chom.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 27.Jangra RK, Yi M, Lemon SM. 2010. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not IRES-directed translation. J Virol 84:6810–6824. doi: 10.1128/JVI.00397-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cox EM, Sagan SM, Mortimer SAW, Doudna JA, Sarnow P. 2013. Enhancement of hepatitis C viral RNA abundance by precursor miR-122 molecules. RNA 19:1825–1832. doi: 10.1261/rna.040865.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Yamane D, Lemon SM. 11 February 2015. Dissecting the roles of the 5′ exoribonucleases Xrn1 and Xrn2 in restricting hepatitis C virus replication. J Virol doi: 10.1128/JVI.03692-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rinck A, Preusse M, Laggerbauer B, Lickert H, Engelhardt S, Theis FJ. 2013. The human transcriptome is enriched for miRNA-binding sites located in cooperativity-permitting distance. RNA Biol 10:1125–1135. doi: 10.4161/rna.24955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mortimer SA, Doudna JA. 2013. Unconventional miR-122 binding stabilizes the HCV genome by forming a trimolecular RNA structure. Nucleic Acids Res 41:4230–4240. doi: 10.1093/nar/gkt075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y-P, Gottwein JM, Scheel TK, Jensen TB, Bukh J. 2011. MicroRNA-122 antagonism against hepatitis C virus genotypes 1-6 and reduced efficacy by host RNA insertion or mutations in the HCV 5′ UTR. Proc Natl Acad Sci U S A 108:4991–4996. doi: 10.1073/pnas.1016606108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Masaki T, Lemon SM. 2013. miR-122 and the hepatitis C RNA genome: more than just stability. RNA Biol 10:919–923. doi: 10.4161/rna.25137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts APE, Doidge R, Tarr AW, Jopling CL. 2014. The P body protein LSm1 contributes to stimulation of hepatitis C virus translation, but not replication, by microRNA-122. Nucleic Acids Res 42:1257–1269. doi: 10.1093/nar/gkt941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson JA, Sagan SM. 2014. Hepatitis C virus and human miR-122: insights from the bench to the clinic. Curr Opin Virol 7:11–18. doi: 10.1016/j.coviro.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Sastre A, Evans MJ. 2013. miR-122 is more than a shield for the hepatitis C virus genome. Proc Natl Acad Sci U S A 110:1571–1572. doi: 10.1073/pnas.1220841110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Z, Dou C, Jia Y, Li Q, Zheng X, Yao Y, Liu Q, Song T. 2015. RIG-I suppresses the migration and invasion of hepatocellular carcinoma cells by regulating MMP9. Int J Oncol 46:1710–1720. doi: 10.3892/ijo.2015.2853. [DOI] [PubMed] [Google Scholar]
- 39.Li K, Chen Z, Kato N, Gale M, Lemon SM. 2005. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-β production in hepatocytes. J Biol Chem 280:16739–16747. doi: 10.1074/jbc.M414139200. [DOI] [PubMed] [Google Scholar]
- 40.Diaz-Toledano R, Ariza-Mateos A, Birk A, Martinez-Garcia B, Gomez J. 2009. In vitro characterization of a miR-122-sensitive double-helical switch element in the 5′ region of hepatitis C virus RNA. Nucleic Acids Res 37:5498–5510. doi: 10.1093/nar/gkp553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villanueva RA, Jangra RK, Yi M, Pyles R, Bourne N, Lemon SM. 2010. miR-122 does not modulate the elongation phase of hepatitis C virus RNA synthesis in isolated replicase complexes. Antiviral Res 88:119–123. doi: 10.1016/j.antiviral.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niepmann M. 2009. Activation of hepatitis C virus translation by a liver-specific microRNA. Cell Cycle 8:1473–1477. doi: 10.4161/cc.8.10.8349. [DOI] [PubMed] [Google Scholar]
- 43.Conrad KD, Giering F, Erfurth C, Neumann A, Fehr C, Meister G, Niepmann M. 2013. microRNA-122 dependent binding of Ago2 protein to hepatitis C virus RNA is associated with enhanced RNA stability and translation stimulation. PLoS One 8:e56272. doi: 10.1371/journal.pone.0056272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheller N, Mina LB, Galao RP, Chari A, Gimenez-Barcons M, Noueiry A, Fischer U, Meyerhans A, Diez J. 2009. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc Natl Acad Sci U S A 106:13517–13522. doi: 10.1073/pnas.0906413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, Ikeda M, Kato N. 2011. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J Virol 85:6882–6892. doi: 10.1128/JVI.02418-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pager CT, Schutz S, Abraham TM, Luo G, Sarnow P. 2013. Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules. Virology 435:472–484. doi: 10.1016/j.virol.2012.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]