

Abstract
Objective
Osteoporosis is a skeletal disorder characterized by low bone mass and increased bone fragility associated with aging, menopause, smoking, obesity, or diabetes. Persistent inflammation has been identified as an instigating factor in progressive bone loss. In addition to the role of fibrin in coagulation, inordinate fibrin deposition within a tissue matrix results in increased local inflammation. Given that fibrin accumulation is a hallmark of osteoporosis-related co-morbidities, we undertook this study to test the hypothesis that persistent fibrin deposition causes inflammatory osteoporosis.
Methods
Multiple imaging modalities, bone integrity metrics, and histologic analyses were employed to evaluate skeletal derangements in relation to fibrin deposition, circulating fibrinogen levels, and systemic markers of inflammation in mice that were plasminogen deficient and in plasminogen-deficient mice that were concomitantly either fibrinogen deficient or carrying a mutant form of fibrinogen lacking the αMβ2 binding motif.
Results
Mice generated with a genetic deficit in the key fibrinolytic protease, plasmin, uniformly developed severe osteoporosis. Furthermore, the development of osteoporosis was fibrin(ogen) dependent, and the derangements in the bone remodeling unit were mechanistically tied to fibrin(ogen)-mediated activation of osteoclasts via activation of the leukocyte integrin receptor αMβ2 on monocytes and secondary stimulation of osteoblasts by RANKL. Notably, the genetic elimination of fibrin(ogen) or the expression of a mutant form of fibrinogen retaining clotting function but lacking the αMβ2 binding motif prevented the degenerative skeletal phenotypes, resulting in normal local and systemic cytokine levels.
Conclusion
Taken together, these data reveal for the first time that fibrin promotes inflammation-driven systemic osteoporosis, which suggests a novel association between hemostasis, inflammation, and bone biology.
Bone integrity is maintained by “bone remodeling units” consisting of osteoclasts that resorb preexisting bone tissue and osteoblasts that produce new bone. The interactions between these cell types are dynamic and normally tightly regulated to ensure balance between osteolysis and osteogenesis, thereby maintaining tissue homeostasis. The typical consequence of “uncoupling” of the bone remodeling unit is a relative increase in osteoclast-mediated bone resorption over osteoblast-dependent bone formation (1–3). Over time, net bone loss results in osteoporosis, a skeletal disorder characterized by low bone mass and increased bone fragility. Although the etiology of osteoporosis is multifactorial, it is most commonly seen in association with aging, menopause, smoking, diabetes, and/or obesity (4–6). While the initiating endocrine or metabolic dysfunction(s) that uncouples the bone remodeling unit likely differs among these associated conditions, 2 common pathogenetic features are chronic inflammation within the bone microenvironment and osteoclast activation (3,7–11). However, the fundamental mechanisms that promote local and chronic inflammatory processes in bone are poorly understood. As a result, treatments designed to reduce the impact of proinflammatory factors remain elusive.
Coagulation factors that are traditionally associated with the control of fibrin deposition, maintenance of vascular integrity, and tissue repair are increasingly understood to be central to the local control of inflammatory processes in vivo (12). Recent work has established that extravascular fibrin accumulation is a potent stimulator of inflammatory responses through multiple mechanisms (13), including interactions with the leukocyte integrin receptor, αMβ2 (14) (CD11b/CD18, Mac-1 [15]). Inordinate or persistent fibrin deposition within a tissue matrix results in increased local inflammation, which may contribute to a wide spectrum of inflammatory disease (13). Eliminating fibrin or limiting the ability of fibrin to support inflammatory cell function through αMβ2 decreases inflammation and overall disease severity in arthritis, neuroinflammatory disease, inflammatory bowel disease, Duchenne’s muscular dystrophy, and glomerulonephritis (14,16–19).
Impaired fibrinolysis, or the inability to clear provisional fibrin matrices efficiently, has recently been linked to multiple conditions associated with osteoporosis and increased fracture risk. Diabetes, exposure to environmental toxins (such as tobacco smoke), obesity, and normal aging are all associated with decreased fibrinolysis and increased fibrin accumulation (20–25). Because elevated fibrinogen levels are seen in some chronic conditions strongly associated with osteoporosis, and because fibrin promotes local inflammation in vivo, we hypothesized that inordinate fibrin deposition within bone tissue is a plausible fundamental determinant of inflammation-mediated osteoporosis. In the present study, as an initial approach to explore the contribution of persistent fibrin deposition to bone homeostasis, we used plasminogen-deficient mice with profoundly defective fibrinolysis and consequent chronic fibrin deposition in multiple organ systems. Multiple imaging modalities, bone integrity metrics, and end point histologic analyses were employed to evaluate skeletal derangements in relation to fibrin deposition, circulating fibrinogen levels, and systemic markers of inflammation. Finally, we directly tested the concept that diminished bone integrity is driven by fibrin and, more specifically, fibrin-mediated engagement of αMβ2-expressing inflammatory cells by evaluating osteoporosis in plasminogen-deficient mice that were concomitantly either fibrinogen deficient or carrying a mutant form of fibrinogen retaining clotting function but lacking the αMβ2 binding motif.
MATERIALS AND METHODS
Animals
Wild-type mice, plasminogen-deficient (PLG−/−) mice (26), fibrinogen-deficient (FBG−/−) mice (27), mice deficient in both plasminogen and fibrinogen (PLG−/− FBG−/− mice) (28), and mice deficient in plasminogen and the αMβ2 binding site of fibrinogen (PLG−/− FBGg390–396A mice) (14) were killed at ages 5, 10, and 20 weeks by CO2 inhalation. The left femur and spine were fixed in 10% formalin for 24 hours, examined with micro–computed tomography (micro-CT), demineralized in 10% EDTA for 7 days, and subsequently processed in paraffin. The right femur was stored in phosphate buffered saline at −80°C for biomechanical analysis. Mice were housed at 22–24°C with a 12-hour light/dark cycle with standard mouse chow/water provided ad libitum. All animal procedures were approved by the Institutional Animal Care and Use Committee.
Radiographic analysis
Wild-type, PLG−/−, FBG−/−, and PLG−/− FBG−/− mice ages 3–20 weeks were radiographed once a week. Images were obtained for 8 seconds at 35 kV with a Faxitron LX-60 X-Ray System with mice prone in hip abduction and in the lateral position natural to the mouse. Individual measurements of the axial length were determined using ImageJ software (National Institutes of Health). The kyphotic index of the spine was calculated from the lateral position with a straight line drawn between the caudal margins of the last cervical vertebra to the caudal margin of L6 and a line drawn perpendicular to the dorsal edge of the vertebra at the point of greatest curvature. For vertebral body compression, the ratio of the length of the anterior vertebral body to the length of the posterior vertebral body as seen on lateral radiographs was determined.
Histologic analysis
Routine Safranin O and tartrate-resistant acid phosphatase (TRAP) staining (Sigma-Aldrich) was performed. Osteoclasts per bone surface area were quantified using Metamorph software (Molecular Devices). Immunofluorescence staining for fibrin was performed using a previously described rabbit antifibrin(ogen) antisera (diluted 1:1,000) in conjunction with an Alexa Fluor 647–conjugated secondary antibody (A212244; Life Technologies) and DAPI counterstain (D3571; Invitrogen) according to the protocol of the manufacturer (Life Technologies).
Micro-CT
Femurs and L5 vertebrae were aligned with the scanning axis of a micro-CT apparatus (μCT40; Scanco) using the same standard tube size (diameter 12.3 mm). The regions of interest (metaphysis of the distal femur, mid-shaft of the femur, and vertebral body) were identified on a scout scan of each bone prior to image acquisition. Each region was scanned at 12-μm voxel size (isotropic), acquiring projections every 1° through 180°. X-ray source settings (55 keV and 145 μA) did not vary as each region of interest was scanned. After digital reconstruction using the manufacturer’s beam-hardening correction and calibration phantom (hydroxyapatite), a bone threshold value of 438.7 mg hydroxyapatite/cm3 was used to calculate the parameters of the skeletal architecture.
Biomechanics
Femurs were placed on the supports of a 3-point bending fixture with a span (L) of 8 mm, with the anterior side down and the medial side forward. Following a preload of 1N, the hydrated mid-shaft was monotonically loaded to failure at 3 mm/minute. Forces were recorded at 50 Hz from a 100N load cell (Honeywell), and whole bone strength in bending was the peak force (Pf) endured by the bone. Stiffness (δ) was determined as the slope of the initial linear portion of the force versus displacement curve. Using the moment of inertia (Iap) of the mid-shaft and the distance between the centroid and the bone surface in the anteroposterior direction (cap) determined by micro-CT, we estimated the modulus (δ×L3/Iap/48) and bending strength (Pf ×L × cap/Iap/4).
Plasma analysis
Mouse blood was collected in tubes containing 0.109M sodium citrate, and platelet-free plasma was prepared by centrifugation at 1,500g for 15 minutes and subsequent centrifugation of the supernatant at 13,000g for 15 minutes. Enzyme-linked immunosorbent assays for plasma soluble RANKL (sRANKL) (ab100749; Abcam), TRAP-5b (SB-TR103; Immunodiagnostic Systems), osteoprotegerin (OPG) (MOP00; R&D Systems), and fibrinogen (E90Fib; Immunology Consultants Laboratory) were performed according to the manufacturer’s protocol.
In vitro osteoclastogenesis assay
Splenocytes were isolated from the spleen of 8–10-week-old wild-type male mice using a cell strainer (70 mm; BD Biosciences). Cell suspensions were washed in α-minimum essential medium (Life Technologies)/10% fetal bovine serum and cultured at 5×105 cells/well in 24-well plates in media containing mouse RANKL (25 ng/ml) and mouse macrophage colony-stimulating factor (30 ng/ml). After 72 hours, fibrinogen (0.5 μM) was added with or without the γ-fibrinogen (377–395) peptide known to block leukocyte integrin receptor αMβ2 engagement of ligand (GL Biochem). Culture medium was changed every 2 days in all differentiation experiments. To quantify osteoclast formation, cells were fixed in acetone and stained for TRAP using an acid phosphatase kit (Sigma-Aldrich). TRAP-positive multinucleated cells containing ≥ 3 nuclei were considered osteoclasts. Osteoclast area was analyzed using Metamorph software.
Cytometric bead array immunoassay
Cytokine analyses were carried out using a Cytometric Bead Array kit (BD Biosciences) for simultaneous detection of 6 cytokines (interleukin-12 [IL-12] p70, tumor necrosis factor α[TNFα], interferon- γ [IFNγ], monocyte chemotactic protein 1 [MCP-1], IL-10, and IL-6) in plasma samples using a FACSCanto II flow cytometer according to the recommendations of the manufacturer (BD Biosciences).
Statistical analysis
Linear regression was performed for kyphotic index by animal age. The Mann-Whitney U test was used to compare micro-CT measurements, biomechanical data, and serum fibrinogen levels. Spearman’s correlation analysis was performed to determine the correlation between serum fibrinogen and IL-6 levels. P values less than 0.05 were considered significant. All statistical analyses were performed using GraphPad Prism software, version 6.
RESULTS
Plasminogen deficiency imposes early degeneration of axial and appendicular bone, resulting in skeletal fragility
Genetic elimination of the key fibrinolytic enzyme plasminogen in C57BL/6 inbred mice is known to result in reduced overall animal size/stature relative to their plasminogen-sufficient siblings, a phenotypic distinction that becomes increasingly obvious in the post-adolescent period (28). To explore the concept that persistent fibrin deposition is coupled to derangements in skeletal structure, we performed a comprehensive radiographic analysis of cohorts of control and PLG−/− mice at ages 5, 10, and 20 weeks. Because derangements in the key structural elements of the axial skeleton, vertebral bone, and intervertebral discs can result in overt kyphotic abnormalities (i.e., increased ventral curvature), we looked for genotype- and age-dependent manifestations of kyphosis by performing serial lateral radiographs (Figure 1A).
Figure 1.
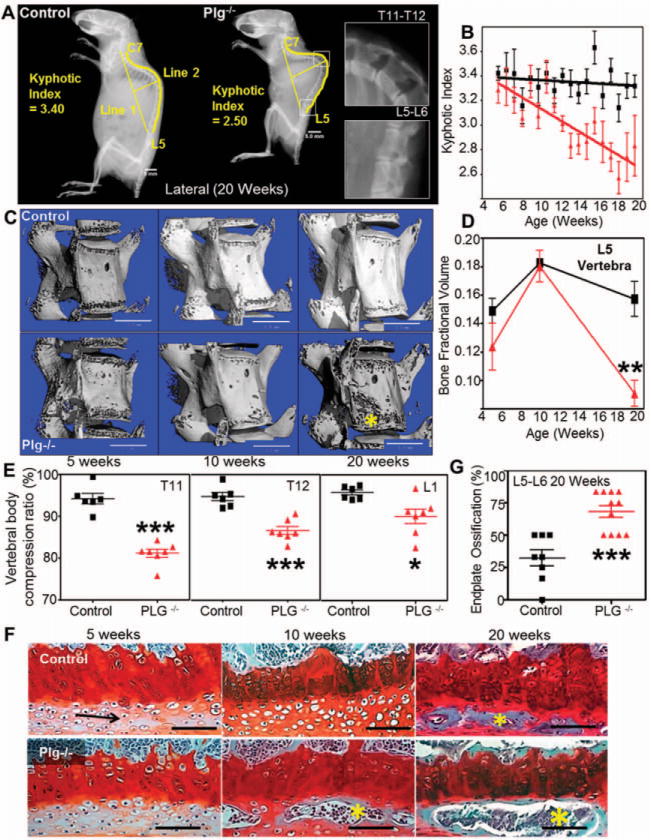
Plasminogen deficiency imposes early degeneration and biomechanical incompetency of the axial skeleton. A, Lateral radiographs of 20-week-old control and plasminogen-deficient (PLG−/−) mice show substantial kyphosis of the axial spine. Bars = 5 mm. B, Serial measurements of kyphotic indices indicate that following proper axial development through 5 weeks, PLG−/− mice (triangles) show progressive degeneration compared to control mice (squares) (P=0.0003 by two-way repeated-measures analysis of variance [ANOVA]). C, Three-dimensional renderings of micro–computed tomography (micro-CT) data show evidence of rapid bone loss in the L5 vertebrae (asterisk) of PLG−/− mice. Bars =1 mm. D, Shown are micro-CT–derived bone fraction volumes corresponding to images in C. Triangles represent PLG−/− mice; squares represent control mice. ** = P<0.01 versus control mice, by Student’s t-test. E, Vertebral body compression ratios of T11, T12, and L1 determined from lateral radiographs demonstrate biomechanical incompetence of the anterior cortex of the vertebral bodies at age 20 weeks in PLG−/− mice. *= P<0.05; ***= P<0.001 versus control mice, by two-way ANOVA with Bonferroni adjustment for multiple comparisons. F, Shown is premature ossification (asterisks) of the articular end plate of L5/L6 observed in PLG−/− mice at age 10 weeks and in both control and PLG−/− mice at age 20 weeks (Safranin O staining). Arrow indicates cartilage. Bars =100 μm. G, Shown is the percentage area of end plate ossification of L5/L6 determined by histologic examination at age 20 weeks (n = 8 wild-type mice; n = 11 PLG−/− mice). P<0.001 versus control mice, by Student’s t-test. Bars in B, D, E, and G show the mean ±SD. Symbols in E and G represent individual mice. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.38639/abstract.
Although PLG−/− mice were generally indistinguishable from wild-type mice during their initial growth phase (<8 weeks), they subsequently developed severe progressive kyphosis (Figures 1A and B). To better elucidate the underlying cause of kyphosis, we evaluated the competency of the axial structural elements using multiple techniques. Assessment of vertebral bone structure by micro-CT (Figure 1C) (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38639/abstract) revealed advanced bone loss (bone fractional volume) in PLG−/− mice characterized by reduced trabecular thickness, but not by reduced trabecular number (Supplementary Table 1). The loss of vertebral structural rigidity in PLG−/− mice was associated with compression of the anterior vertebral elements that ultimately resulted in a wedge-shaped structure and kyphosis. Analysis of compression ratios at the apex of the kyphosis also showed that the observed bone loss in PLG−/− mice resulted in biomechanical failure of the vertebral bone elements (Figure 1E).
Additional axial deformity occurs upon failure of the intervertebral disc, as indicated by end plate ossification. While both PLG−/− and control mice showed signs of end plate ossification by age 20 weeks, end plate ossification developed prematurely and more extensively in the absence of plasminogen (Figures 1F and G). These results revealed an important and previously unknown role of this fibrinolytic protease in skeletal homeostasis. Furthermore, these studies establish that postnatal structural changes in the vertebral bodies and intervertebral discs in PLG−/− mice result in a biomechanically incompetent axial skeleton and development of severe kyphosis.
Plasminogen- and age-dependent differences in bone structure in young adulthood were not limited to the vertebral bodies. Bone fractional volume and cortical and trabecular thickness of the distal femur determined by serial micro-CT did not differ significantly during early development (5–10 weeks) (Figure 2A) (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38639/abstract). However, by age 20 weeks, PLG−/− mice showed rapid loss of cortical bone and trabecular thickness (Figures 2A–C) (Supplementary Table 1). While kyphosis is a hallmark of biomechanical incompetency in the axial spine, biomechanical incompetency of the appendicular skeleton manifests as fragility fractures. Therefore, in addition to resolving bone integrity through serial micro-CT analysis, we also tested appendicular fracture resistance in isolated femurs. Decreased trabecular and cortical thickness resulted in significant reductions in biomechanical properties (stiffness and modulus), which resulted in decreased resistance of the appendicular skeleton to fracture as measured by peak force to fracture and bending strength (Figures 2D and E) (Supplementary Table 1).
Figure 2.
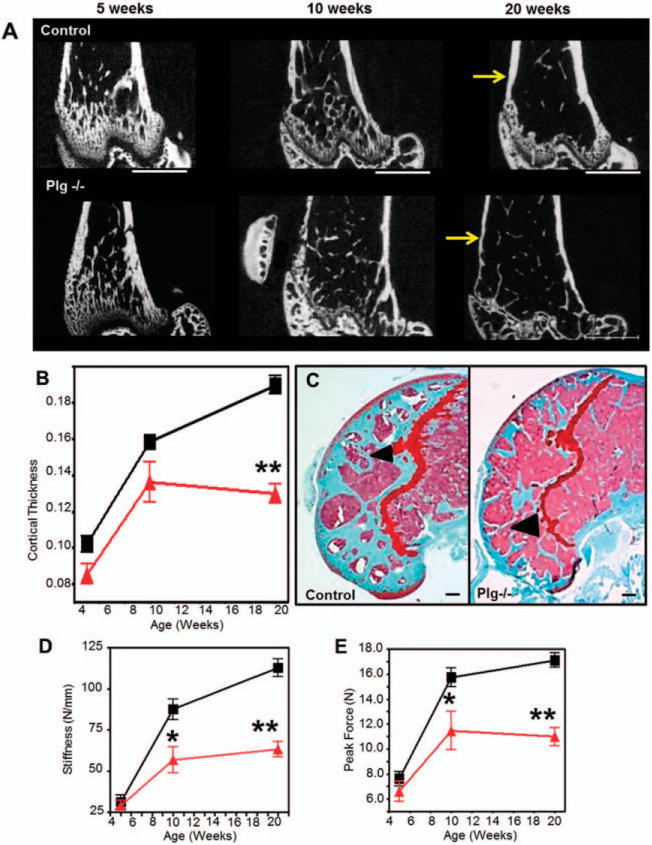
Plasminogen-deficient (PLG−/−) mice develop early degeneration and reduced fracture resistance of the appendicular skeleton. Triangles represent PLG−/− mice; squares represent control mice. A, Two-dimensional images rendered from micro–computed tomography (micro-CT) data reveal cortical thinning in PLG−/− mice compared to control mice (arrows) occurring at age 20 weeks despite relatively normal bone architecture in younger mice. Bars = 1 mm. B, Shown are tabulated cortical measurements (Student’s t-test was performed for each time point using data collected from separate mice at the time that they were killed). C, Safranin O staining of the distal femur at age 20 weeks demonstrates loss of trabecular bone in the distal femoral epiphysis in PLG−/− mice compared to control mice (arrowheads) (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38639/abstract). Bars = 200 μm. D and E, Biomechanical analyses of stiffness (D) and fracture resistance by peak force (E) indicate that observed progressive loss of bone results in significantly reduced biomechanical strength (means, standard deviations, and sample sizes, along with additional biomechanical and micro-CT data, are presented in Supplementary Tables 1 and 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38639/abstract). Bars show the mean ±SD (n = 8 wild-type mice; n=11 PLG−/− mice). *=P<0.05; **=P<0.01 versus control mice, by Student’s t-test. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.38639/abstract.
Skeletal degeneration in plasminogen-deficient mice is caused by a derangement in bone remodeling associated with a hyperinflammatory state
To determine if the loss of fibrinolytic potential correlated with a pathologic uncoupling of the bone remodeling unit, we compared plasminogen- and age-dependent changes in osteoclast activity by measuring circulating TRAP-5b, an enzyme highly expressed by bone-resorbing osteoclasts (29), and TRAP-5a, which is expressed by inflammatory macrophages and dendritic cells. Circulating TRAP-5b levels were similar in adolescent mice (i.e., age 5 weeks) with or without plasminogen, but TRAP-5b was significantly increased in PLG−/− mice compared to wild-type mice by age 8 weeks (Figure 3A), when significant differential bone loss ensued. By age 20 weeks, TRAP-5b levels normalized to levels seen in control mice. This pattern of circulating TRAP-5b suggested that osteoclastic bone resorption in PLG−/− mice is abnormally active immediately before trabecular and cortical bone loss becomes detectable. Reduction of TRAP-5 activity likely represents decreased osteoclastic activity as a result of reduced bone surface area available for degradation secondary to the degree of bone loss.
Figure 3.
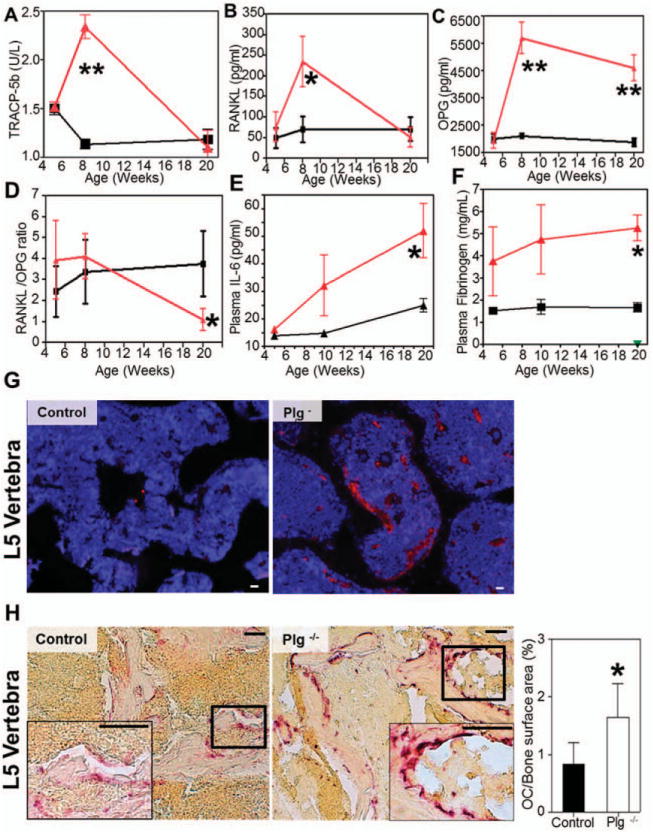
Exuberant osteoclast (OC) activity is associated with increased circulating levels of osteoprotegerin (OPG), interleukin-6 (IL-6), and fibrinogen and with accumulation of fibrin(ogen) in bone. Triangles represent plasminogen-deficient (PLG−/−) mice; squares represent control mice. A–D, Plasma levels of tartrate-resistant acid phosphatase 5b (TRAP-5b) (A), soluble RANKL (sRANKL) (B), and OPG (C) and the resulting sRANKL:OPG ratios (D) were determined by enzyme-linked immunosorbent assay (ELISA). Increased osteoclast activity was observed without a change in RANKL:OPG ratio. E and F, Plasma IL-6 levels determined by flow cytometry (E) and fibrinogen levels determined by ELISA (F) indicate a progressive increase in inflammation and acute-phase reactants at ages 5, 10, and 20 weeks (measurements for all data were collected from separate mice at the time that they were killed; Student’s t-test was performed at each time point). Bars show the mean ±SD (n=4 at each time point). *=P<0.05; ** = P<0.01 versus control mice. G, Immunolocalization of fibrinogen demonstrates abundant fibrin within the L5 vertebrae of 20-week-old PLG−/− mice (red signal), but not in control mice. Bars =100 μm. H, Left, The number of osteoclasts per unit bone surface in L5 vertebrae at age 8 weeks (time point of maximal TRAP-5b concentration) was determined by TRAP staining. Representative images from each group are shown. Insets are higher-magnification views of boxed areas. Original magnification ×200; ×400 in insets. Bars =500 μm. Right, Quantification of osteoclasts/bone surface area illustrates increased numbers of osteoclasts in PLG−/− mice (n=4) compared to control mice (n = 4) at age 8 weeks. Bars show the mean ±SD. *=P<0.01 versus control mice.
The primary mechanism by which osteoclasto-genesis occurs is through activation of RANK by RANKL on the surface of osteoclast precursors (30), a process controlled by OPG binding to RANKL as a decoy receptor. Therefore, as a complementary extension of this study, we serially evaluated plasma sRANKL and OPG levels in cohorts of control and PLG−/− mice. Plasma levels of sRANKL in PLG−/− mice were higher than those in wild-type mice only at age 8 weeks (Figure 3B). OPG levels were also significantly increased by age 8 weeks in PLG−/− mice compared to wild-type mice and remained elevated at 20 weeks (Figure 3C). Thus, contrary to 1 prior report (31), plasmin was not required for proteolytic processing of OPG; conversely, loss of plasmin(ogen) resulted in increased circulating OPG levels in parallel with the evident bone loss phenotype. Increased OPG has been reported in chronic inflammatory disease (32). The observation that RANKL:OPG ratios were comparable in PLG−/− and wild-type mice at age 8 weeks (coincident with maximum TRAP-5b levels) implicates an exogenous mechanism of osteoclast activation.
Chronic inflammation within the bone microenvironment and stimulation of osteoclast activation are prominent features of high-turnover osteoporosis (3,7–11,33–35). To better define the inflammatory consequences of plasminogen deficiency and activation of osteoclasts, detailed profiles of cytokines associated with inflammatory osteoporosis were developed in both PLG−/− and control mice. Unexpectedly, we found no significant changes in multiple proinflammatory cytokines including TNFα, IFNγ, MCP-1, or IL-12p70, or in the antiinflammatory cytokine IL-10, at either 5 or 20 weeks of age (Table 1). One notable exception to this pattern was an age-dependent increase in IL-6 levels in PLG−/− mice (Figure 3E and Table 1). IL-6 is a key cytokine affecting osteoclast function (3) as well as a modulator of fibrinogen biosynthesis (36). Indeed, PLG−/− mice also exhibited evidence of an acute-phase response in the form of a significant increase in circulating fibrinogen (Figure 3F) that strongly correlated with increased IL-6 levels (r=0.608).
Table 1.
Inflammation profile*
| 5 weeks
|
10 weeks
|
20 weeks
|
||||||
|---|---|---|---|---|---|---|---|---|
| WT mice (n = 5) |
PLG−/− mice (n = 4) |
WT mice (n = 5) |
PLG−/− mice (n = 4) |
WT mice (n = 5) |
PLG−/− mice (n = 7) |
PLG−/− FBG−/− mice (n = 4) |
PLG−/−FBGg390–396A mice (n = 5) |
|
| IL-12p70 | 87.15 ± 46.35 | 59.51 ± 21.71 | 71.45 ± 24.46 | 65.98 ± 31.84 | 69.76 ± 23.73 | 54.12 ± 15.00 | 73.83 ± 23.37 | 55.29 ± 32.48 |
| TNFα | 26.51 ± 7.37 | 26.9 ± 12.56 | 22.13 ± 7.22 | 30.91 ± 2.31 | 25.28 ± 6.97 | 29.08 ± 13.78 | 27.05 ± 7.64 | 24.92 ± 9.28 |
| IFNγ | 11.82 ± 8.45 | 7.81 ± 2.35 | 7.40 ± 2.41 | 8.05 ± 3.30 | 8.72 ± 2.85 | 8.66 ± 5.99 | 9.43 ± 2.96 | 5.75 ± 0.71 |
| MCP-1 | 55.41 ± 0.70 | 56.76 ± 2.86 | 55.95 ± 1.33 | 56.35 ± 1.57 | 70.88 ± 12.32 | 63.66 ± 9.99 | 76.05 ± 6.46 | 58.47 ±2.52 |
| IL-10 | 55.99 ± 2.13 | 55.56 ± 2.31 | 62.35 ± 14.08 | 56.61 ± 7.25 | 58.28 ± 2.19 | 67.16 ± 30.23 | 60.85 ± 12.85 | 55.13 ± 3.59 |
| IL-6 | 21.39 ± 6.9 | 27.07 ± 18.05 | 19.21 ± 6.23 | 41.07 ± 22.81 | 24.91 ± 5.5 | 46.56 ± 24.42† | 24.03 ± 7.306 | 34.74 ± 19.16 |
Values are the mean ±SD pg/ml. PLG−/− = plasminogen-deficient; FBG−/− = fibrinogen-deficient; FBGg390–396A = deficient in the αMβ2 binding site of fibrinogen; IL-12p70 =interleukin-12 p70; TNFα =tumor necrosis factor α; IFNγ = interferon-γ; MCP-1 = monocyte chemotactic protein 1.
P<0.05 versus wild-type (WT) mice.
Plasminogen-deficient mice accumulate fibrin within bone
To better understand the underlying mechanism(s) by which the loss of plasminogen results in a progressive failure of the biomechanical integrity of the skeletal system, we sought to determine which plasmin target(s) were responsible for these dramatic postnatal changes. In particular, we explored the hypothesis that alterations in bone structure were specifically due to spontaneous fibrin accumulation/persistence within bone. Consistent with this working hypothesis, immuno-fluorescence staining revealed significantly more deposition of fibrin within both the appendicular skeleton (data not shown) and the axial skeleton (Figure 3G) of 20-week-old PLG−/− mice compared to controls. In addition, to determine if deposition of fibrin was associated with increased osteoclastogenesis, we quantified the number of TRAP-positive osteoclasts along bone surfaces. Compared to controls, PLG−/− mice had significantly more osteoclasts per unit bone surface at the time of maximum osteoclast activity (peak TRAP-5b levels) (Figure 3H).
Fibrin(ogen) drives inflammation and bone loss in plasminogen-deficient mice, in part through the leukocyte integrin receptor αMβ2
To determine how increased fibrin(ogen) deposition causes acquired inflammatory osteoporosis in plasminogen-deficient mice, we examined the skeletal phenotype of mice in which plasminogen deficiency was combined with either 1) fibrinogen deficiency (PLG FBG−/−mice) or 2) a mutant form of fibrinogen (FBGg390–396A) that restores normal fibrinogen levels and coagulation activity but that lacks the αMβ2 binding motif and therefore cannot engage the leukocyte integrin receptor αMβ2 (PLG−/− FBGg390–396A mice) (14). Degenerative changes of the axial skeleton seen in PLG−/− mice were completely prevented in PLG−/− FBG−/− mice (Figure 4A). The kyphotic indices in PLG−/− FBG−/− mice from ages 2 weeks to 20 weeks were not significantly different from those in wild-type mice (Figure 4B).
Figure 4.
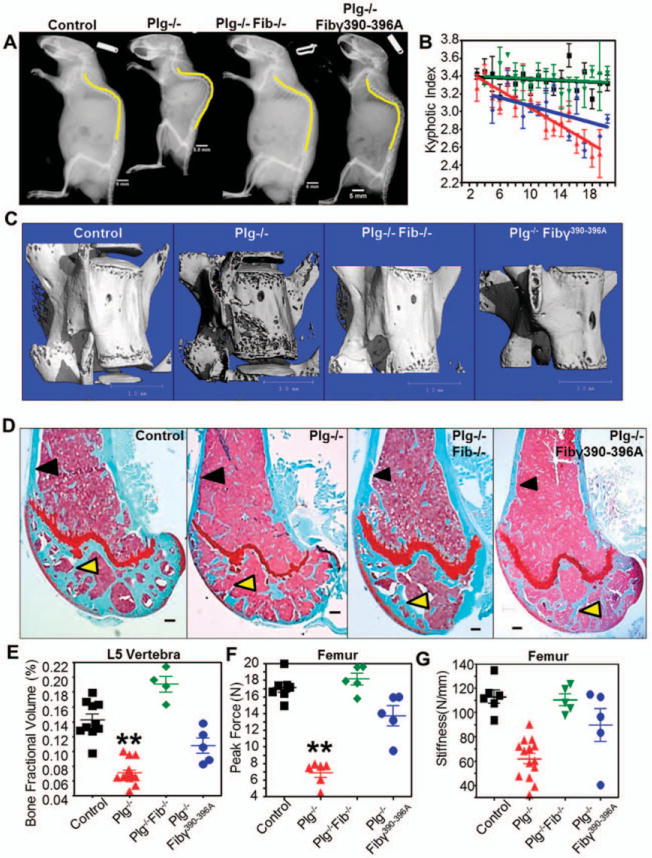
Targeting fibrin prevents bone loss in plasminogen-deficient (PLG−/−) mice. A, Lateral radiographs at age 20 weeks demonstrate that the degree of kyphosis seen in PLG−/− mice is reduced in mice deficient in both plasminogen and fibrinogen (PLG−/−FBG−/− mice [or Plg−/−Fib−/− mice]) as well as in plasminogen-deficient mice expressing a mutant fibrinogen unable to bind αMβ2 (PLG−/−FBGg390–396A mice [or Plg−/−Fib7390–396A mice]). Bars = 5 mm. B, By age 20 weeks there is complete prevention of kyphosis in PLG−/−FBG−/− mice (green symbols) and partial prevention in PLG−/−FBGg390–396A mice (blue symbols). Wild-type (WT) mice are represented by black symbols; PLG−/− mice are represented by red symbols. Bars show the mean ± SD. C, Three-dimensional reconstruction of micro-computed tomography data from L5 vertebrae in 20-week-old mice shows decreased bone loss in PLG−/−FBG−/− and PLG−/−FBGg390–396A mice compared to PLG−/− mice. Bars = 1 mm. D, Similar reductions in cortical bone loss (black arrows) and trabecular bone loss (yellow arrows) are observed in Safranin O-stained sections of distal femurs from 20-week-old PLG−/−FBG−/− and PLG−/−FBGg390–396A mice. Bars = 200 /μm. E-G, These changes in bone architecture are reflected by enhanced bone fraction volume (E) and biomechanical properties contributing to fracture resistance (peak force [F] and stiffness [G]). Symbols represent individual mice (n = 10 WT mice; n = 8 PLG−/− mice; n = 5 PLG−/−FBG−/− mice; n = 5 PLG−/−FBGg390–396A mice). Bars show the mean ± SD. ** = P < 0.01 versus control, by one-way analysis of variance. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.38639/abstract.
Interestingly, partial prevention of the phenotype was also seen in PLG−/− FBGg390–396A mice (Figure 4A). Although the effect of the FBGg390–396A variant on kyphotic index only trended toward significance (Figure 4B), diminished degeneration of vertebral bone was observed using 3-dimensional micro-CT analyses in both PLG−/− FBG−/− and PLG−/− FBGg390–396A mice at age 20 weeks (when osteoporotic-like bone loss was pronounced in PLG−/− mice) (Figure 4C). Furthermore, quantitative analysis revealed that bone fractional volumes in PLG−/− FBG−/− mice were virtually identical to those in control mice. PLG−/− FBGg390–396A mice also showed a moderate but statistically significant improvement in this bone structure metric (Figures 4C and D) (see Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38639/abstract). Control studies of bone structure and integrity in both FBG−/− and FBGg390–396A mice with normal circulating plasminogen showed no differences compared to wild-type mice (data not shown).
To establish that the interplay between the plasminogen–fibrinogen axis and bone degeneration extended to other skeletal components, we performed a histologic examination of the distal femurs of control and plasminogen-deficient mice with and without fibrinogen or homozygous for the FBGg390–396A mutation. While PLG−/− mice displayed a qualitative reduction in femoral cortical thickness at age 20 weeks, the cortical thickness in PLG−/− FBG−/− mice was similar to that in wild-type mice (Figure 4D) (see Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38639/abstract). A similar statistically significant prevention of cortical thickness loss was observed in PLG−/− FBGg390–396A mice (Figure 4D). The apparent preservation of anatomic bone structure in PLG−/− mice lacking fibrinogen was confirmed by biomechanical analysis, with significant improvement in fracture resistance (i.e., peak force and stiffness) (Figures 4F and G) (Supplementary Table 2). Neither FBG−/− mice nor PLG−/− FBGg390–396A mice showed evidence of altered bone phenotypes in biomechanical analyses (data not shown).
To determine if fibrin(ogen) was directly capable of inducing cellular changes leading to osteoporosis, we explored the hypothesis that fibrin(ogen) is an exogenous, matrix-associated regulator of the bone remodeling unit. TRAP staining of splenic monocytes cocultured with fibrinogen showed a marked increase in osteoclastogenesis (Figure 5A). Furthermore, consistent with the previous data showing that the fusion of monocytes to form multinucleated osteoclasts is regulated by engagement of the leukocyte integrin receptor αMβ2 by fibrin-(ogen), osteoclastogenesis in vitro was strongly impeded by a synthetic decoy ligand for αMβ2 based on the fibrinogen -chain sequence (Figures 5B and C) (16).
Figure 5.
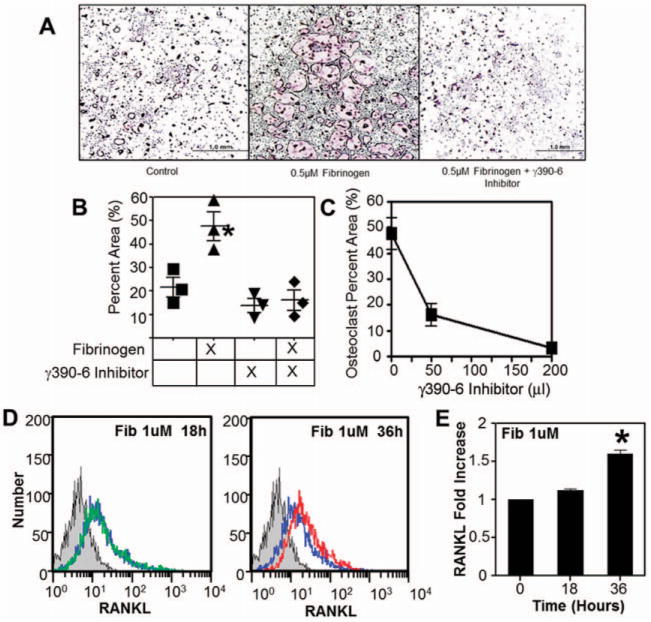
Fibrin(ogen) (Fib) stimulates osteoclastogenesis by engaging αMβ2 and promoting osteoblast RANKL expression through an alternative mechanism. A, Isolated splenic monocytes incubated with 0.5 μM fibrinogen show increased osteoclastogenesis as indicated by tartrate-resistant acid phosphatase (TRAP) staining of multinucleated cells. Bars =1 mm. B, Shown is the percent of TRAP-stained area. Symbols represent individual mice. *= P<0.01 versus control, by one-way analysis of variance (ANOVA) followed by Dunn’s multiple comparison test. C, The observed stimulatory behavior of fibrinogen is inhibited in a dose-dependent manner by addition of a γ390–396A inhibitor. D, Fibrin(ogen) stimulates a 2-fold increase in cell surface expression of RANKL by MC3T3 cells (an osteoblast precursor cell line). Blue line indicates normal RANKL production; green line indicates RANKL production in the presence of 1 μM fibrinogen for 18 hours; red line indicates RANKL production in the presence of 1 uM fibrinogen for 36 hours. E, Shown is the fold increase in cell surface expression of RANKL. *= P<0.01 versus 0 hours, by one-way ANOVA followed by Dunn’s multiple comparison test. Bars show the mean ±SD. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.38639/abstract.
Fibrin(ogen) not only supported osteoclastogenesis, but also appeared to stimulate osteoclast activity by increasing osteoblast production of RANKL. MC3T3 cells, an osteoblast precursor cell line, expressed 2-fold more surface-bound RANKL in the presence of fibrinogen compared to MC3T3 cell cultures without added fibrinogen (Figures 5D and E). However, here the fibrinogen-induced RANKL expression appeared to be independent of αMβ2, as addition of αMβ2 soluble decoy ligand had no effect on this response (data not shown).
Taken together, these findings demonstrate that hemostatic factors, particularly both plasminogen and fibrinogen, are fundamental determinants of bone remodeling and osteolysis. Furthermore, the mechanism by which plasminogen deficiency alters the bone remodeling unit in osteoporosis is coupled to fibrin, the fibrin αMβ2 binding motif, and chronic inflammation.
DISCUSSION
In accordance with previous work by Kanno et al (31), these studies establish that plasmin, a serine protease traditionally associated with thrombolysis and/or fibrinolysis, is essential for maintaining bone integrity and preventing osteoporosis. Specifically, we demonstrate that plasmin(ogen) deficiency results in significant loss of trabecular and cortical bone and spinal deformity (kyphosis) associated with impaired biomechanical strength due to high bone turnover. Furthermore, the present studies establish that persistent fibrin deposition is central to degenerative bone disease in that the simple elimination of fibrinogen prevents the osteoporotic phenotype of PLG−/− mice. A strong implication of these studies is that therapeutic interventions at the level of hemostatic factors may constitute an effective means of limiting some forms of degenerative bone disease.
Plasmin is capable of proteolysis of multiple bone-related proteins such as matrix glycoproteins (e.g., laminin and fibronectin), type I collagen, latent growth factors (e.g., transforming growth factor β1), and protease zymogens (matrix metalloproteinase 9, pro–urokinase plasminogen activator) (37–39). Thus, at least in principle, plasmin(ogen) may exert its effects on the bone remodeling unit through mechanisms other than fibrinolysis. A previous study suggested that plasmin(ogen) activates the proteolytic processing of OPG, a soluble inhibitor of the osteoclast activator RANKL (31). However, our finding that the level of circulating OPG is in fact elevated in mice lacking plasminogen is inconsistent with this possibility and implies that an alternative plasmin target is responsible for the osteoporotic phenotype. In the present study, we demonstrate that exuberant fibrin deposition within the local bone environment of PLG−/− mice is strongly linked to degenerative bone loss. In addition, we determined that fibrin(ogen) directly activates bone remodeling activity in osteoblasts and osteoclasts. Finally, we found that all examined aspects of the degenerative bone phenotype observed in PLG−/− mice are effectively prevented by removing fibrin(ogen). Taken together, these data indicate that the principal function of plasmin coupled to bone biology is fibrinolysis. While these studies do not formally exclude a fibrin-independent contribution of plasmin to bone homeostasis, any biologic importance of other plasmin substrates would appear to be limited or subtle.
We show that fibrin(ogen) is an exogenous activator of the bone remodeling unit, favoring bone resorption and leading to osteoporosis and thus providing a novel, direct link between hemostasis and bone biology. In addition to examining direct stimulation of the bone remodeling unit by fibrin(ogen), we also explored the hypothesis that fibrin(ogen) causes osteoporosis through an inflammatory process. We specifically focused our investigation on IL-6, as this cytokine is a primary promoter of fibrinogen expression during chronic disease and, reciprocally, persistent fibrin deposition promotes IL-6 expression by a multitude of cells (40,41). Substantial clinical and experimental evidence indicates that the inflammatory system is directly and indirectly linked to bone turnover. Failure to resolve local and systemic inflammation results in osteoporosis, and this appears to be due in part to osteoclast activity outpacing osteoblastic osteogenesis (3,7–11,33–35). Previous studies have identified certain proinflammatory cytokines as critical to both normal bone remodeling and the pathogenesis of perimenopausal and late-life osteoporosis (10). In particular, IL-6 promotes osteoclastogenesis and activation (3,42). We show that PLG−/− mice have markedly elevated IL-6 levels compared to wild-type mice, indicative of a chronic inflammatory state. Importantly, we found a direct correlation between IL-6 and circulating fibrinogen levels, with normal levels in PLG−/− FBG−/− mice. These data indicate that plasmin(ogen) deficiency results in a hyperinflammatory state linked to fibrin(ogen).
To explore whether osteoporosis in PLG−/− mice is secondary to fibrin(ogen) stimulation of inflammatory cells, we assessed whether inactivation of a key sequence of fibrin(ogen) important for leukocyte receptor recognition, but not fibrin polymer formation per se, resulted in reduced bone loss. Of particular interest was the element of the fibrin(ogen) γ-chain recognized by leukocyte integrin receptor αMβ2 that has been implicated in inflammatory arthritis and neuroinflammatory disease among other inflammatory diseases (14–19). Elimination of the γ-chain αMβ2 binding motif resulted in both a reduced inflammatory response and prevention of the bone phenotype in PLG−/− mice. However, the phenotypes seen with this fibrinogen variant were less pronounced than those seen with genetic ablation of circulating fibrin(ogen). Thus, while local stimulation of inflammatory cells through fibrinogen engagement of αMβ2 appears to be in part responsible for inflammatory osteoporosis observed in PLG−/− mice, other fibrin(ogen)-mediated mechanisms remain to be defined.
In support of the notion of multiple mechanisms linking fibrin(ogen) to inflammatory skeletal degeneration, we found that fibrin(ogen) stimulation of monocyte fusion into osteoclasts was dependent upon fibrinogen engagement of αMβ2, while fibrinogen stimulation of RANKL expression in osteoblasts was independent of this pathway. Since increased RANKL expression is known to uncouple the bone remodeling unit and promote osteoclastic bone resorption (2), it is plausible that the partial prevention of bone loss observed in PLG−/− FBGg390–396A mice is due to a reduction in bone resorption through the αMβ2 osteoclast-dependent pathway despite increased osteoblast RANKL expression. Further studies are necessary to determine if this process occurs in vivo. Nevertheless, the distinct phenotypic changes seen in PLG−/− FBGg390–396A mice suggest that pharmacologic interventions targeting fibrinogen (but not compromising hemostatic function) could be a potentially low-risk but effective adjunct therapy in limiting osteoporosis.
In conclusion, these studies reveal a fundamental new insight into the pathogenesis of osteoporosis by establishing that hemostatic factors are powerful positive or negative determinants of bone loss. These studies show that accumulation of fibrin(ogen) due to a plasmin(ogen) deficiency uncouples the bone remodeling unit and results in bone loss. As hypercoagulable and hyperinflammatory states are integral components of many of the chronic diseases associated with osteoporosis, these findings have the potential to shift the paradigm for osteoporosis therapy toward selected targets within the coagulation and inflammatory systems coupled to bone remodeling.
Supplementary Material
Acknowledgments
Supported by the Orthopaedic Research and Education Foundation, the Musculoskeletal Transplantation Foundation, and the Caitlin Lovejoy Fund.
Footnotes
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Schoenecker had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Cole, Ohba, Nyman, Hirotaka, Flick, Degen, Schoenecker.
Acquisition of data. Cole, Ohba, Nyman, Schoenecker.
Analysis and interpretation of data. Cole, Ohba, Nyman, Cates, Schoenecker.
References
- 1.Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest. 2005;115:3318–25. doi: 10.1172/JCI27071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Canalis E, Giustina A, Bilezikian JP. Mechanisms of anabolic therapies for osteoporosis. N Engl J Med. 2007;357:905–16. doi: 10.1056/NEJMra067395. [DOI] [PubMed] [Google Scholar]
- 3.Mundy GR. Osteoporosis and inflammation. Nutr Rev. 2007;65:S147–51. doi: 10.1111/j.1753-4887.2007.tb00353.x. [DOI] [PubMed] [Google Scholar]
- 4.Daniell HW. Osteoporosis and smoking. JAMA. 1972;221:509. [PubMed] [Google Scholar]
- 5.Hofbauer LC, Brueck CC, Singh SK, Dobnig H. Osteoporosis in patients with diabetes mellitus. J Bone Miner Res. 2007;22:1317–28. doi: 10.1359/jbmr.070510. [DOI] [PubMed] [Google Scholar]
- 6.Nielson CM, Srikanth P, Orwoll ES. Obesity and fracture in men and women: an epidemiologic perspective. J Bone Miner Res. 2012;27:1–10. doi: 10.1002/jbmr.1486. [DOI] [PubMed] [Google Scholar]
- 7.Arron JR, Choi Y. Bone versus immune system. Nature. 2000;408:535–6. doi: 10.1038/35046196. [DOI] [PubMed] [Google Scholar]
- 8.Ginaldi L, Di Benedetto MC, De Martinis M. Osteoporosis, inflammation and ageing. Immun Ageing. 2005;2:14. doi: 10.1186/1742-4933-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lacativa PG, Farias ML. Osteoporosis and inflammation. Arq Bras Endocrinol Metabol. 2010;54:123–32. doi: 10.1590/s0004-27302010000200007. [DOI] [PubMed] [Google Scholar]
- 10.Manolagas SC, Jilka RL. Bone marrow, cytokines, and bone remodeling: emerging insights into the pathophysiology of osteoporosis. N Engl J Med. 1995;332:305–11. doi: 10.1056/NEJM199502023320506. [DOI] [PubMed] [Google Scholar]
- 11.Tilg H, Moschen AR, Kaser A, Pines A, Dotan I. Gut, inflammation and osteoporosis: basic and clinical concepts. Gut. 2008;57:684–94. doi: 10.1136/gut.2006.117382. [DOI] [PubMed] [Google Scholar]
- 12.Stutz CM, O’Rear LD, O’Neill KR, Tamborski ME, Crosby CG, Devin CJ, et al. Coagulopathies in orthopaedics: links to inflammation and the potential of individualizing treatment strategies. J Orthop Trauma. 2013;27:236–41. doi: 10.1097/BOT.0b013e318269b782. [DOI] [PubMed] [Google Scholar]
- 13.Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol. 2012;34:43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- 14.Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, et al. Leukocyte engagement of fibrin(ogen) via the integrin receptor αMβ2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. 2004;113:1596–606. doi: 10.1172/JCI20741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Todd RF., III The continuing saga of complement receptor type 3 (CR3) J Clin Invest. 1996;98:1–2. doi: 10.1172/JCI118752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, et al. The fibrinderived γ377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med. 2007;204:571–82. doi: 10.1084/jem.20061931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. 2012;3:1227. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinbrecher KA, Horowitz NA, Blevins EA, Barney KA, Shaw MA, Harmel-Laws E, et al. Colitis-associated cancer is dependent on the interplay between the hemostatic and inflammatory systems and supported by integrin αMβ2 engagement of fibrinogen. Cancer Res. 2010;70:2634–43. doi: 10.1158/0008-5472.CAN-09-3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vidal B, Ardite E, Suelves M, Ruiz-Bonilla V, Janue A, Flick MJ, et al. Amelioration of Duchenne muscular dystrophy in mdx mice by elimination of matrix-associated fibrin-driven inflammation coupled to the αMβ2 leukocyte integrin receptor. Hum Mol Genet. 2012;21:1989–2004. doi: 10.1093/hmg/dds012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alzahrani SH, Ajjan RA. Coagulation and fibrinolysis in diabetes. Diab Vasc Dis Res. 2010;7:260–73. doi: 10.1177/1479164110383723. [DOI] [PubMed] [Google Scholar]
- 21.Billimoria JD, Pozner H, Metselaar B, Best FW, James DC. Effect of cigarette smoking on lipids, lipoproteins, blood coagulation, fibrinolysis and cellular components of human blood. Atherosclerosis. 1975;21:61–76. doi: 10.1016/0021-9150(75)90094-5. [DOI] [PubMed] [Google Scholar]
- 22.De Pergola G, Pannacciulli N. Coagulation and fibrinolysis abnormalities in obesity. J Endocrinol Invest. 2002;25:899–904. doi: 10.1007/BF03344054. [DOI] [PubMed] [Google Scholar]
- 23.Dunn EJ, Ariens RA. Fibrinogen and fibrin clot structure in diabetes. Herz. 2004;29:470–9. doi: 10.1007/s00059-004-2607-z. [DOI] [PubMed] [Google Scholar]
- 24.Paraskevas KI, Baker DM, Vrentzos GE, Mikhailidis DP. The role of fibrinogen and fibrinolysis in peripheral arterial disease. Thromb Res. 2008;122:1–12. doi: 10.1016/j.thromres.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 25.Undas A, Topor-Madry R, Tracz W, Pasowicz M. Effect of cigarette smoking on plasma fibrin clot permeability and susceptibility to lysis. Thromb Haemost. 2009;102:1289–91. doi: 10.1160/TH09-03-0187. [DOI] [PubMed] [Google Scholar]
- 26.Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, et al. Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med. 1996;2:287–92. doi: 10.1038/nm0396-287. [DOI] [PubMed] [Google Scholar]
- 27.Suh TT, Holmback K, Jensen NJ, Daugherty CC, Small K, Simon DI, et al. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–33. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]
- 28.Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell. 1996;87:709–19. doi: 10.1016/s0092-8674(00)81390-2. [DOI] [PubMed] [Google Scholar]
- 29.Halleen JM, Tiitinen SL, Ylipahkala H, Fagerlund KM, Vaananen HK. Tartrate-resistant acid phosphatase 5b (TRACP 5b) as a marker of bone resorption. Clin Lab. 2006;52:499–509. [PubMed] [Google Scholar]
- 30.Takahashi N, Udagawa N, Suda T. A new member of tumor necrosis factor ligand family, ODF/OPGL/TRANCE/RANKL, regulates osteoclast differentiation and function. Biochem Biophys Res Commun. 1999;256:449–55. doi: 10.1006/bbrc.1999.0252. [DOI] [PubMed] [Google Scholar]
- 31.Kanno Y, Ishisaki A, Kawashita E, Chosa N, Nakajima K, Nishihara T, et al. Plasminogen/plasmin modulates bone metabolism by regulating the osteoblast and osteoclast function. J Biol Chem. 2011;286:8952–60. doi: 10.1074/jbc.M110.152181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guzel S, Seven A, Kocaoglu A, Ilk B, Guzel EC, Saracoglu GV, et al. Osteoprotegerin, leptin and IL-6: association with silent myocardial ischemia in type 2 diabetes mellitus. Diab Vasc Dis Res. 2013;10:25–31. doi: 10.1177/1479164112440815. [DOI] [PubMed] [Google Scholar]
- 33.Armour KJ, Armour KE. Inflammation-induced osteoporosis: the IMO model. Methods Mol Med. 2003;80:353–60. doi: 10.1385/1-59259-366-6:353. [DOI] [PubMed] [Google Scholar]
- 34.Bai P, Sun Y, Jin J, Hou J, Li R, Zhang Q, et al. Disturbance of the OPG/RANK/RANKL pathway and systemic inflammation in COPD patients with emphysema and osteoporosis. Respir Res. 2011;12:157. doi: 10.1186/1465-9921-12-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breuil V, Ticchioni M, Testa J, Roux CH, Ferrari P, Breittmayer JP, et al. Immune changes in post-menopausal osteoporosis: the Immunos study. Osteoporos Int. 2010;21:805–14. doi: 10.1007/s00198-009-1018-7. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Fuentes NL, Fuller GM. Characterization of the IL-6 responsive elements in the γ fibrinogen gene promoter. J Biol Chem. 1995;270:24287–91. doi: 10.1074/jbc.270.41.24287. [DOI] [PubMed] [Google Scholar]
- 37.Toriseva M, Kahari VM. Proteinases in cutaneous wound healing. Cell Mol Life Sci. 2009;66:203–24. doi: 10.1007/s00018-008-8388-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jenkins G. The role of proteases in transforming growth factor-β activation. Int J Biochem Cell Biol. 2008;40:1068–78. doi: 10.1016/j.biocel.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 39.Roth D, Piekarek M, Paulsson M, Christ H, Bloch W, Krieg T, et al. Plasmin modulates vascular endothelial growth factor-A-mediated angiogenesis during wound repair. Am J Pathol. 2006;168:670–84. doi: 10.2353/ajpath.2006.050372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jensen T, Kierulf P, Sandset PM, Klingenberg O, Joo GB, Godal HC, et al. Fibrinogen and fibrin induce synthesis of proinflammatory cytokines from isolated peripheral blood mononuclear cells. Thromb Haemost. 2007;97:822–9. doi: 10.1160/th07-01-0039. [DOI] [PubMed] [Google Scholar]
- 41.Sitrin RG, Pan PM, Srikanth S, Todd RF., III Fibrinogen activates NF-κB transcription factors in mononuclear phagocytes. J Immunol. 1998;161:1462–70. [PubMed] [Google Scholar]
- 42.Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–37. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.