

Abstract
CdGAP is a Rac1/Cdc42 specific GTPase activating protein that localizes to cell–matrix adhesions through an interaction with the adhesion scaffold α-parvin/actopaxin to regulate lamellipodia formation and cell spreading. Herein we demonstrate, using a combination of siRNA-mediated silencing and over expression, that cdGAP negatively regulates directed and random migration by controlling adhesion maturation and dynamics through the regulation of both adhesion assembly and disassembly. Interestingly, cdGAP was also localized to adhesions formed in three-dimensional matrix environments and cdGAP depletion promoted cancer cell migration and invasion through 3D matrices. These findings highlight the importance of GAP proteins in the regulation of Rho family GTPases and the co-ordination of the cell migration machinery.
Keywords: Cancer, 3D matrix adhesions, Rho GTPase activity, focal adhesion, GTPase activating protein, GAP
Introduction
The ability of cells to migrate individually or as collective sheets is essential for organism development, wound repair, and immune surveillance, as well as pathologies including cancer metastasis [Bucar and Stamenkovic., 2008;Cahalan and Gutman., 2006;Sonneman and Bement., 2011;Weijer et al., 2009]. In order to efficiently translocate, cells must interact with and respond to their local microenvironment, which is rich in extracellular matrix (ECM) proteins [Chan et al., 2007;Lock et al., 2008]. The integrin family of heterodimeric transmembrane proteins are the primary and best-studied mediators of cell-ECM interactions [Huttenlocher and Horwitz, 2011]. The small GTPases Rac1, Cdc42 and RhoA are activated by integrin signaling in response to cell adhesion to the ECM [Arthur et al., 2002;Chan et al., 2007;Fukata et al., 2003;Hall, 2005;Hotchin and Hall, 1995]. These Rho family GTPases control cell migration through regulating cell polarization, lamellipodial extension, and force generation, as well as by coordinating the formation and dynamics of discrete cell-ECM adhesion structures collectively referred to as adhesion plaques or contacts [Abercrombie and Dunn, 1975;Beningo et al., 2001;DeMali and Burridge, 2003;Horwitz and Webb, 2003;Rottner et al., 1999]. Adhesion contacts provide a physical link between the ECM and the intracellular cytoskeleton to enable force transmission required for cell motility [Gardel et al., 2010]. They also serve as signaling nodes to relay bidirectional signals between cells and their environment to regulate migration, as well as cell morphology, growth and apoptosis [Huttenlocher and Horwitz, 2011].
Distinct groups of adhesion contacts formed by fibroblasts on 2D ECM substrates have been classified based primarily on their size, spatiotemporal localization and molecular content [Geiger and Bershadsky, 2001;Huttenlocher and Horwitz, 2011;Kirchner et al., 2003;Zamir and Geiger, 2001]. The formation of small peripheral focal complexes at the leading edge of protruding lamellae is dependent on the activation of Rac1 and Cdc42 [Nobes and Hall, 1995]. Upon the activation of RhoA or application of acto-myosin-driven contractile forces, focal complexes either rapidly disassemble or further mature into larger, more centrally located focal adhesions [Bershadsky et al., 2003;Hotchin and Hall, 1995;Ren et al., 1999;Ridley and Hall, 1992, Riveline et al., 2001]. In contrast to 2D systems, distinct populations of 3D matrix adhesions are less well defined. However, their formation and dynamics are also dependent on the spatially regulated activity of the Rho GTPases [Deakin and Turner, 2011;Geiger and Yamada, 2011;Harunaga and Yamada, 2011]. Indeed, a coordinated cycle of Rho GTPase-driven adhesion contact assembly, maturation, and disassembly is essential for efficient cell migration in both 2D and 3D microenvironments [Deakin and Turner, 2011;Doyle et al., 2012;Webb et al., 2002].
The activity of the Rho GTPases is regulated by guanine nucleotide exchange factors (GEFs), GTPase activating proteins (GAPs), and Rho guanine nucleotide dissociation inhibitors (GDIs) [Jaffe and Hall, 2005]. GEFs activate Rho family proteins, while GAPs and Rho GDIs act to inhibit their activity either by catalyzing GTP hydrolysis or preventing their localization to the cell membrane, respectively. In contrast to the extensive analysis of leading edge or adhesion localized GEFs, the role of GAP family proteins in controlling Rho GTPase activity associated with cell migration and adhesion dynamics is less well understood [Garrett et al., 2007;Lee et al., 2010;Nayal et al., 2006;Ten Klooster et al., 2006;Yu et al., 2009;Zhao et al., 2000].
CdGAP is a Rac1 and Cdc42 specific GAP [LaLonde et al., 2006;Lamarche-vane and Hall, 1998;Tcherkezian et al.,2006;Aoki et al., 2004;Kurokowa et al., 2004] that localizes to adhesion contacts via interaction with the scaffold protein α-parvin (actopaxin/CHILKBP) to regulate cell spreading and chemotaxis [LaLonde et al., 2006]. Recent studies have highlighted the importance of cdGAP in both normal human development [Southgate et al., 2011], and in the pathophysiology of cancer [He et al., 2011]. For example, Adams-Oliver disease is a rare developmental disorder characterized by vascular malformations, skin defects, and truncated limb development. In this condition, mutations in cdGAP result in enhanced Cdc42 activity and abnormally heightened migration in patient-derived fibroblasts [Southgate et al., 2011]. Importantly, the mechanism by which cdGAP controls cell migration is not well characterized. Herein, we show that cdGAP controls cell migration in 2D environments by influencing the type and dynamics of adhesion contacts. We further demonstrate that cdGAP regulates 3D ECM cancer cell invasive migration and identify cdGAP as the first 3D matrix adhesion-localized GAP.
Results
CdGAP regulates directed and random cell migration
Depletion of cdGAP in HeLa cells using siRNA was previously shown to enhance cell spreading [LaLonde et al., 2006]. To evaluate the role of cdGAP in cell migration, its expression was efficiently suppressed in the osteosarcoma-derived U2OS cell line using two independent siRNAs (Figure 1A). Scrape wound assays of confluent monolayers demonstrated that cells depleted of cdGAP exhibited enhanced closure rates and increased migration velocity at the wound edge compared to control RNAi-treated cells (Figure 1,B and C). The velocity of randomly migrating cells plated at low density was also increased upon cdGAP depletion (Figure 1,D and E). Highly motile U2OS cells have a crescent morphology akin to keratocytes during cell migration [Clarke et al., 2004;Khyrul et al., 2004]. Consistent with this, cdGAP siRNA-treated cells rapidly extended and retracted lamellipodia and exhibited an exaggerated crescent morphology as compared to control cells (see Movie S1, supplementary material). In contrast, over expression of GFP-cdGAP (long isoform) [Tcherkezian et al, 2005] (Figure 2A) significantly inhibited random cell migration velocity (Figure 2, B and C) as well as the formation of lamellipodia (see Movie S2, supplementary material), consistent with previous studies in which the over expressed short isoform of cdGAP inhibited lamellipodia formation during cell spreading [He et al., 2011;LaLonde et al., 2006;Tcherkezian et al., 2005;Tcherkezian et al., 2006]. Collectively, these data identify cdGAP as a regulator of migration velocity and thus as an important component of the cell migration machinery.
Figure 1. CdGAP silencing accelerates the speed of wound healing and random migration.

(A) U2OS cells were transfected with two separate siRNAs targeted against human cdGAP and knockdown efficiency was assessed by western blotting using an antibody specific for human cdGAP. (B) CdGAP siRNA-treated cells closed a wound more quickly then control siRNA-treated cells. U2OS cells treated with control or cdGAP siRNA were plated at high density, scrape wounded, and imaged over 16 hours. (C) Tracking of individual cells at the wound edge confirmed that cells treated with cdGAP siRNA migrated at a higher velocity than control siRNA-treated cells. A minimum of 40 cells were tracked in each wound per siRNA condition from each of three independent experiments and compared using Student’s t-test, *, p < 0.05 for each cdGAP siRNA when compared to the control. (D) Representative tracks generated from control and cdGAP siRNA-treated U2OS cells plated at low density migrating randomly on 10µg/ml collagen for 16 hours demonstrating the longer distance traveled by cdGAP silenced cells. (E) cdGAP siRNA-treated cells migrate at a higher velocity than control siRNA-treated cells. For random migration velocity calculations, a minimum of 30 cells from each of three independent experiments were tracked. Velocity measurements from the control siRNA-treatment and each cdGAP siRNA-treatment were statistically compared using Student’s t-test, *, p < 0.05 for both siRNAs. For Figure 1, all graphs have error bars representing +/− the standard error of the mean.
Figure 2. CdGAP over expression slows random cell migration.
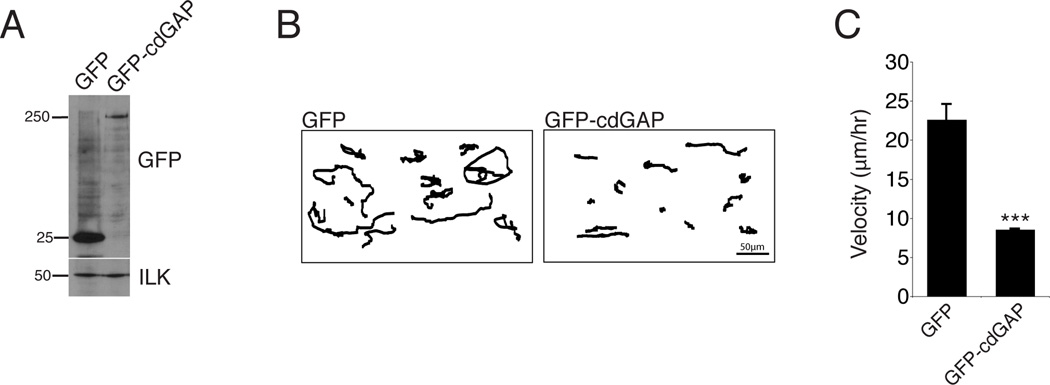
(A) U2OS cells were transfected with GFP or GFP-cdGAP. (B) Representative tracks of transfected U2OS cells plated at low density migrating on 10µg/ml collagen for 16 hours, with GFP-cdGAP transfected cells migrating a shorter distance than GFP control cells. (C) GFP expressing cells migrate at a higher velocity than GFP-cdGAP expressing cells. A minimum of 30 cells from three independent experiments were analyzed per condition and GFP versus GFP-cdGAP expressing cells compared with Student’s t-test,***, p < 0.001. Velocity graph has error bars representing +/− the standard error of the mean.
CdGAP controls the formation of small adhesion contacts
Previous studies demonstrated that a short isoform of cdGAP localizes to adhesions, suggesting a role for cdGAP in modulating adhesion localized, or proximal Rac1- and Cdc42-mediated signaling [LaLonde et al., 2006]. We confirmed that endogenous human cdGAP localizes with paxillin at adhesions in U2OS cells (Figure 3, A and B). Interestingly, RNAi depletion of cdGAP resulted in an increase in the number of small adhesions localized to the leading edge (Figure 3, C and D) reminiscent of Rac1- and Cdc42-driven focal complexes [Ren et al., 1999;Ridley and Hall, 1992] and a corresponding decrease in the average adhesion size per cell (Figure 3E). Representative masks of individual cells also demonstrate the enhanced crescent morphology of cdGAP RNAi-treated cells (Figure 3F) as was observed in the random migration movies (see Movie S1, supplementary material). Expression of active V12Rac1 or V12Cdc42 in U2OS cells (Figure 5), or inhibition of myosin II activity with blebbistatin (see Figure S1, supplementary material) produced a strikingly similar cell shape and adhesion morphology to cdGAP depletion. In contrast to cdGAP depletion, GFP-cdGAP over expression (see Figure 2A) resulted in a reduction in the number of cells with small leading edge localized adhesion contacts (Figure 4C) and a corresponding increase in the average adhesion size (Figure 4D). In addition, cells expressing adhesion localized GFP-cdGAP (Figure 4, A and B) spread less and were more angular than GFP control cells (Figure 4, A and E). The reduced cell area, large adhesion size, and angular morphology characteristic of U2OS cells over expressing GFP-cdGAP was eliminated by expression of either dominant active V12Rac1 or V12Cdc42 (Figure 5). U2OS cells co-expressing GFP-cdGAP and myc-V12Rac1 or myc-V12Cdc42 (Figure 5A) had small adhesion contacts, were well spread, and exhibited a crescent morphology, similar to cdGAP siRNA-treated cells (Figure 5B). Expressing wild-type versions of Rac1 or Cdc42 in U2OS cells did not alter the phenotype of either control or GFP-cdGAP expressing cells (unpublished observation). In contrast, treatment with the myosin II inhibitor blebbistatin also prevented the GFP-cdGAP over expression phenotype, inducing the formation of small adhesions and a crescent morphology (see Figure S1, supplementary material). Together, these data support a role for cdGAP in suppressing lamellipodia extension by inhibiting the activation of both Rac1 and Cdc42 to promote the growth and maturation of adhesion contacts.
Figure 3. CdGAP depletion promotes the formation of small adhesion contacts at the leading edge and decreases average adhesion area.

(A) U2OS cells were spread, fixed and stained for endogenous cdGAP two hours post plating on 10µg/ml collagen. Endogenous cdGAP co-localizes to adhesions that are positive for paxillin staining. Inset shows a zoom of adhesions positive for cdGAP and paxillin staining used to generate the line profile in (B). (B) A line profile through adhesions demonstrates that cdGAP and paxillin fluorescence intensity peaks in adhesions and diminishes in the surrounding cytoplasm. (C) Control and cdGAP siRNA-treated U2OS cells were spread on 10µg/ml collagen coated coverslips and stained for actin and paxillin. Merged images are overlays of the paxillin and actin channels. Inset shows enlarged area from each white box; small adhesions are frequently observed in the leading edge of migrating U2OS cells treated with cdGAP siRNA. (D) CdGAP depletion increases the percentage of cells with small, leading edge or peripheral adhesions. Cells were scored as positive if > 20% of their leading edge or periphery contained small paxillin-positive adhesions. A minimum of 80 cells from three separate experiments were scored per condition and control versus siRNA-treated cells compared with Student’s t-test, ***, p < 0.001 for both siRNAs. (E) Average adhesion size decreases in cdGAP siRNA-treated cells, a minimum of 30 cells from three independent experiments were analyzed and control-treated cells compared to each siRNA-treatment with Student’s t-test, ***, p < 0.001 for both siRNAs. (F) Masks of cdGAP siRNA-treated cells were made to display representative cell morphology. CdGAP siRNA-treatment promoted the formation of a crescent morphology in U2OS cells. For Figure 3, all error bars represent +/− the standard error of the mean.
Figure 5. Dominant active V12Rac1 and V12Cdc42 block the cdGAP over expression phenotype.

(A) U2OS cells were co-transfected with either GFP or GFP-cdGAP and myc-tagged dominant active V12Rac1 or myc-tagged dominant active V12Cdc42 and the expression levels of each construct were evaluated by western blotting. (B) Cells expressing the indicated constructs were spread on 10µg/ml collagen coated coverslips and stained for paxillin and actin.
Figure 4. CdGAP over expression inhibits the formation of small adhesion contacts and increases adhesion area.

(A) U2OS cells transfected with GFP or GFP-cdGAP were spread on 10µg/ml collagen coated coverslips and stained for paxillin and actin. Inset shows the localization of GFP-cdGAP to adhesions that are positive for paxillin staining. The white bar in the merged inset for the GFP-cdGAP expressing cell marks adhesions through which the line profile in (B) was drawn. (B) A line profile through adhesions demonstrates that GFP-cdGAP and paxillin fluorescence intensity peaks in adhesions and diminishes in the surrounding cytoplasm. (C) The percentage of cells with small leading edge or peripheral adhesions decreased in GFP-cdGAP expressing cells. The percentage of small adhesions was quantified from a minimum of 60 transfected cells from each of three independent experiments per condition and GFP versus GFP-cdGAP expressing cells compared with Student’s t-test, *, p < 0.05. (D) Average adhesion size was measured in U2OS cells expressing GFP and GFP-cdGAP constructs from a total of 30 transfected cells per condition from three independent experiments and compared using Student’s t-test, ***, p < 0.001. (E) Masks of cells over expressing GFP or GFP-cdGAP demonstrate that GFP-cdGAP over expressing cells have a smaller spread area and an angular morphology. For Figure 4, all error bars represent +/− the standard error of the mean.
CdGAP regulates multiple aspects of focal adhesion dynamics
The manipulation of cdGAP by RNAi or over expression resulted in changes in adhesion morphology and size suggesting that cdGAP might be regulating adhesion dynamics during cell migration, as there is an inverse relationship between adhesion size and adhesion turnover [Lauffenburger and Horwitz, 1996;Nayal et al., 2006]. Furthermore, rapidly moving cells have accelerated adhesion dynamics [Bristow et al., 2009;Delorme-Walker et al., 2011;Gardel et al., 2010;Gupton and Waterman-Storer, 2006;Webb et al., 2004]. To assess the effect of cdGAP on adhesion dynamics, both control and cdGAP siRNA-treated cells expressing zyxin-GFP, as a marker for cell-ECM adhesions, (Figure 6A) were imaged using time-lapse microscopy (see Movie S3, supplementary material) and adhesion lifetime and dynamics measurements calculated [Deakin and Turner, 2011;Webb et al., 2004]. The overall adhesion lifetime of cdGAP siRNA-treated cells was significantly shortened as compared to control cells (Figure 6,B–D). In addition, cdGAP depletion enhanced both adhesion assembly and disassembly rates (Figure 6,E and F). In contrast, over expression of cdGAP in U2OS cells (Figure 7A) caused a significant increase in focal adhesion lifetime (Figure 7, B–D) by decreasing both the assembly and disassembly rates in comparison to control cells (Figure 7, E and F). Notably, many adhesions in cdGAP transfected cells exhibited lifetimes that were longer than the duration of the three-hour movies (see Movie S4, supplementary material). These highly stable adhesions were necessarily omitted from our analysis as adhesion lifetime or dynamics could not be accurately determined. Nevertheless, they serve as a further indication that the presence of cdGAP promotes adhesion stabilization.
Figure 6. CdGAP silencing accelerates adhesion dynamics.

(A) U2OS cells stably expressing zyxin-GFP were subjected to control or cdGAP siRNA-treatment and evaluated for cdGAP and zyxin-GFP expression by western blot. (B) Zyxin-GFP stable U2OS cells treated with control or cdGAP siRNA were imaged to quantify focal adhesion dynamics. White arrows indicate adhesions that are turning over during the time course and labels indicate the duration of each sequence in minutes. (C) The relative fluorescence intensity of representative individual adhesions with siRNA treatments plotted over time. (D) CdGAP siRNA shortens the average adhesion lifetime. (E) CdGAP siRNA accelerates adhesion assembly rates. (F) CdGAP siRNA enhances adhesion disassembly rates. Confocal image sequences were used to calculate the lifetime, adhesion assembly, and adhesion disassembly rates for each siRNA treatment. All lifetime and rate measurements were made from three independent experiments for a total of 50–70 adhesions, 4–7 cells per condition and compared with Student’s t-test, ***, p < 0.001 for assembly, disassembly, and lifetime calculations in comparison to control siRNA for both cdGAP siRNAs. For Figure 6 all error bars represent +/− the standard error of the mean.
Figure 7. CdGAP over expression slows adhesion dynamics.

(A) U2OS cells were transiently co-transfected with zyxin-GFP and either vector or myc-cdGAP and evaluated for cdGAP and zyxin-GFP expression by western blot. (B) Zyxin-GFP positive U2OS cells expressing myc-control vector or myc-cdGAP were imaged to quantify focal adhesion dynamics. White arrows indicate adhesions that are turning over during the time course and labels indicate the duration of each sequence in minutes. (C) The relative fluorescence intensity of representative individual adhesions plotted over time. (D) Myc-cdGAP over expression lengthens adhesion lifetime. (E) Over expression of myc-cdGAP inhibits adhesion assembly rates. (F) Over expression of myccdGAP inhibits adhesion disassembly rates. Confocal image sequences were used to calculate the lifetime, adhesion assembly, and adhesion disassembly rates for each condition. All lifetime and rate measurements were made from four independent experiments for a minimum of 50 adhesions from 6–10 cells per condition. Statistical comparisons were made between control and myc-cdGAP over expressing cells for the lifetime and adhesion dynamics measurements with a Student’s t-test,**, p < 0.005 for adhesion assembly rate, *** , p < 0.001 for lifetime and disassembly rate comparisons. For Figure 7, all error bars represent +/− the standard error of the mean.
CdGAP localizes to 3D matrix adhesions to regulate 3D cell motility and invasion
Cell migration studies performed on 2D tissue culture plastic, where cells contact the ECM only on their ventral surface, may not accurately reflect the in vivo environment in which cells are often completely surrounded by ECM on all sides and where the organization, rigidity and composition of the ECM can vary greatly from 2D surfaces [Cukierman et al., 2001;Cukierman et al., 2002;Yamada and Cukierman, 2007]. To evaluate a potential role for cdGAP in the regulation of cell migration in these 3D ECM environments, control and cdGAP siRNA-treated HT1080 human fibrosarcoma or U2OS cells were seeded onto 3D cell-derived matrices (CDMs) [Cukierman et al., 2001;Deakin and Turner, 2011]. Immmunofluorescence analysis revealed that endogenous cdGAP localized to 3D matrix adhesions at the ends of actin filaments in both HT1080 cells (Figure 8A) and U2OS cells (see Figure S2A, supplementary material). The enrichment of cdGAP in 3D adhesions was confirmed by co-localization analysis of co-transfected GFP-cdGAP with mRFP-paxillin in both the HT1080 (Figure 8A) and U2OS cells (see Figure S2B, supplementary material).
Figure 8. CdGAP localizes to 3D matrix adhesions and cdGAP siRNA enhances Matrigel invasion and migration velocity in 3D CDMs.

(A) Endogenous cdGAP and GFP-cdGAP both localize to 3D matrix adhesions. HT1080 cells were plated onto coverslips coated with 3D CDMs before being fixed and stained for endogenous cdGAP, actin and fibronectin. HT1080 cells co-transfected with GFP-cdGAP and mRFP-paxillin were fixed and stained for fibronectin. (B) HT1080 cells were treated with cdGAP siRNAs and knockdown efficiency evaluated by western blotting. (C) CdGAP siRNA-treatment enhanced cell migration velocity in 3D CDMs. Migration velocity was calculated for individual cells moving within the 3D matrix from three independent experiments with a minimum of 30 cells per experiment and siRNA treatment, with ***, p < 0.001 for comparisons between control and cdGAP silenced cells. (D) HT1080 cells treated with cdGAP siRNA invade more efficiently through Matrigel than control siRNA-treated cells. Relative invasion was calculated from three independent experiments and compared using Student’s t-test. *, p < 0.05 for comparisons between control and each cdGAP siRNA-treatment. For Figure 8, all error bars represent +/− the standard error of the mean.
Recent studies have demonstrated that while U2OS cells are capable of forming 3D adhesions, they do not migrate efficiently within 3D matrices [Kubow and Horwitz, 2011] (and our unpublished observations). Therefore, in order to assess the role of cdGAP in 3D migration, HT1080 cells that have been shown previously to migrate effectively through 3D matrices were utilized [Wolf et al., 2003;Wolf and Friedl, 2009]. CdGAP-depleted HT1080 cells (Figure 8B) migrated at a significantly higher velocity through 3D CDMs than control RNAi cells (Figure 8C). Cells depleted of cdGAP migrated primarily in a mesencyhmal mode of migration, displaying an elongated morphology similar to control treated cells. In addition to increasing cell migration through the 3D CDMs, depletion of cdGAP also increased the capacity of HT1080 cells to invade through Matrigel-coated transwell inserts (Figure 8D).
Discussion
Efficient cell migration requires that the organization and dynamics of cell-ECM interactions are tightly coordinated [Gardel et al., 2010;Gupton and Waterman-Storer, 2006]. In the current study, we have identified cdGAP as an important regulator of both adhesion size and adhesion dynamics, providing insight into how it controls migration velocity. Perturbing cdGAP function with siRNA-treatment enhanced the formation of small leading edge adhesion contacts (Figure 3) that were phenotypically similiar to those observed in U2OS cells over expressing dominant active (V12) forms of either Rac1 or Cdc42 (Figure 5), or of adhesions in cells with inhibited myosin II activity (see Figure S1, supplementary material) [Kuo et al., 2011;Nobes and Hall.,1995]. Furthermore, unlike other adhesion proteins such as FAK, Src, and Erk that have been shown to modulate adhesion turnover by selectively modulating either adhesion assembly or disassembly [Webb et al., 2004;Shober et al., 2008], the adhesions in cdGAP-depleted cells exhibited increases in both adhesion assembly and disassembly rates that resulted in shortened lifetimes (Figure 5). A similar enhancement of adhesion assembly and disassembly rates has been observed in cells over expressing the Rac1-GEF Asef2, or in cells expressing constitutively active PAK1 (T423E), a Rac1 effector [Bristow et al., 2009;Delorme-Walker et al., 2011]. Thus, cdGAP silencing and activated Rac1 produce comparable effects on both adhesion morphology and dynamics. Activated Cdc42 may also perform a similar function in adhesion dynamics, but to our knowledge its effects on adhesion assembly and disassembly rates have not been examined. Taken together with previous studies that established the specific activity of cdGAP for Rac1 and Cdc42 both in vitro and in a cellular context [LaLonde et al., 2006;Lamarche-vane and Hall, 1998;Tcherkezian et al.,2006;Aoki et al., 2004;Kurokowa et al., 2004], our data suggest that cdGAP depletion likely enhances Rac1 and Cdc42 activity levels to drive the formation of small adhesions and accelerate adhesion dynamics.
In contrast to siRNA treatment, cells over expressing cdGAP were poorly spread with angular morphologies and large adhesions (Figure 4,6), all of which are properties of cells with increased levels of active RhoA and concomitant myosin II activity [Cox et al., 2001]. Inhibition of myosin II activity with blebbistatin prevented the cell and adhesion morphology typically observed in GFP-cdGAP over expressing cells (see Figure S1, supplementary material), further indicating that cdGAP over expression may be enhancing RhoA mediated signaling pathways that can regulate the control of acto-myosin generated tension. Studies on Rho family activation have demonstrated that increasing Rac1 or Cdc42 activity decreases the level of active RhoA [Sander et al., 1999], and vice versa, and that over expression of other Rac1 GAPs such as ArhGAP22 and FilGAP may indirectly influence RhoA activity [Ohta et al., 2006;Sanz-Moreno et al., 2008;Wu et al., 2009]. We can speculate that cdGAP may therefore function in a similar manner to indirectly shift the reciprocal balance of signaling from Rac1 and Cdc42 to RhoA during adhesion formation and maturation in order to produce the large adhesions observed in cdGAP over expressing cells. Importantly, coexpression of V12Rac1 or V12Cdc42 in U2OS cells with GFP-cdGAP induced the formation of small adhesion contacts and blocked the poorly spread, angular morphology of cdGAP over expressing cells, demonstrating that cdGAP over expression targeted both Rac1 and Cdc42.
The large adhesions observed in cdGAP over expressing cells were exceptionally stable, whereas adhesions in cdGAP siRNA-treated cells were dynamic and rapidly turned over. Small adhesions similar to those we observed forming at the leading edge of cdGAP siRNA-treated cells exert high traction forces on the ECM relative to their size [Beningo et al., 2001]. Thus, it is interesting to speculate that cdGAP over expressing cells have static, hyper-mature adhesion contacts that slow cell migration by anchoring the cell body to the ECM, while siRNA-treated cells migrate more quickly as the result of an increase in the population of small adhesions that provide optimal traction forces for rapid cell migration. Importantly, we observed that endogenous cdGAP localized to both small adhesions at the leading edge as well as larger, more mature adhesions (Figure 3,4), and that actomyosin generated contractility was not necessary for GFP-cdGAP’s localization to small adhesion contacts (see Figure S1, supplementary material). This indicates that cdGAP is present in adhesions at all stages of adhesion maturation. The enhanced GAP activity of cdGAP observed in the later stages of cell spreading [LaLonde et al., 2006] suggests that during cell migration the normal function of cdGAP may be to stabilize adhesions and positively regulate their maturation as its GAP activity is stimulated in response to integrin signaling.
Cell migration models incorporating 3D matrix topography and rigidity offer a more physiologically relevant environment for evaluating the likely in-vivo function of proteins involved in coordinating cell migration, as compared to the more traditional 2D tissue culture plastic models [Even-Ram and Yamada, 2005;Pankov et al., 2005]. Indeed, adhesion morphology is different in 3D matrices [Cukierman et al., 2001] and some adhesion proteins have been found to play strikingly different roles in regulating the speed and mode of cell migration depending on the environmental context [Deakin and Turner., 2011;Even-Ram and Yamada, 2005;Pankov et al., 2005;Petrie et al., 2009]. For example, Hic-5 expression is not essential for 2D adhesion formation, but is necessary for the formation of 3D matrix adhesions and the mesenchymal phenotype in MDA-MB-231 breast cancer cells, as well as for their efficient migration and invasion [Deakin and Turner., 2011]. In contrast, our results demonstrate that cdGAP localizes to adhesions in both 2D and 3D environments and appears to play a similar role in inhibiting cell migration in both situations (Figure 7). Optimal mesenchymal migration in 3D requires efficient 3D matrix adhesion turnover, as well as the tight regulation of Rac1 and RhoA activity. Indeed, suboptimal or elevated global levels of Rac1 activity impair cell migration in 3D and the latter promotes the formation of numerous, non-productive small off-axis protrusions [Pankov et al.,2005], while high RhoA activity levels may promote switching to an amoeboid mode of motility [Deakin and Turner.,2011]. Importantly, cdGAP-depleted cells migrated more rapidly and did not form off-axis protrusions or switch their migratory phenotype (unpublished observation). This suggests that cdGAP, consistent with its restricted localization to adhesions, influences primarily the localized, as opposed to global levels of Rho GTPase activity.
While 3D CDMs closely approximate the 3D architecture and makeup of loosely packed connective tissue and the tumor stroma [Deakin and Turner.,2011], tumor cell dissemination also requires the invasion of cancer cells through densely packed basement membranes. In this regard, our studies revealed that cdGAP also plays an important role in suppressing invasion through Matrigel, a standard model for cancer cell basement membrane translocation (Figure 7D). In contrast, a recent study reported that cdGAP knockdown inhibited invasion in response to TGF-β[He et al., 2011], indicating that cdGAP’s function may be cancer cell type or context specific. Nonetheless, these studies point to an important and potentially complex role for cdGAP in regulating cancer cell invasion.
In summary, we have identified an important role for cdGAP in regulating cell migration and demonstrate that cdGAP inhibits cell motility through promoting adhesion contact maturation and stability. The study further highlights the importance of GAPs in addition to GEFs, in controlling the dynamics of the cell migration machinery in both 2D and 3D environments.
Materials and Methods
Antibodies and Reagents
Antibodies used in this study were cdGAP (Cell Signaling Technology, Beverly, MA), α-actinin (Sigma, St. Louis, MO) ILK, Rac1, paxillin (BD Bioscience, Bedford, MA) and paxillin clone H114 (Santa Cruz Biotechnology, Santa Cruz, CA). GFP and myc-tagged plasmids containing the murine long isoform of cdGAP were a gift from Nathalie Lamarche-Vane [Tcherkezian et al., 2005] (McGill University, Montreal, Canada). Plasmids containing myc-tagged V12Rac1 and myc-tagged V12Cdc42 were previously described [Brown et al., 2002].
Tissue Culture
U2OS, Human Foreskin Fibroblast (HFF), and HT1080 cells were obtained from the American Tissue Culture Collection (ATCC, Manassas, VA). HT1080 cells were maintained in Eagle’s Minimum Essential Media (EMEM) (ATCC) supplemented with 10 I.U./ml penicillin, 10µg/ml streptomycin, and 10% FBS (v/v) (Atlas Biologicals, Ft. Collins, CO). U2OS and HFF cells were maintained in Dulbeco’s Minimum Essential Media (DMEM) supplemented with 2mM L-glutamine, 10% FBS (v/v), 5µg/ml kanamycin, 10 I.U./ml penicillin, 10µg/ml streptomycin, and 1mM sodium pyruvate. Cells were maintained at 5% CO2 and 37°C. For blebbistatin experiments, cells were treated with DMSO vehicle control or 20µM blebbistatin (EMD Chemicals, San Diego, CA) for 30 minutes in complete DMEM before being fixed.
siRNA and GFP tagged protein transfection
HT1080 and U2OS cells were transfected with a non-specific control siRNA or two separate siRNAs to human cdGAP. U2OS cells were transfected using Oligofectamine (Invitrogen, Carlsbad, CA) as per the manufacturer’s instructions, while HT1080 cells were transfected with Lipofectamine 2000 (Invitrogen) as per the manufacturer’s instructions-siRNAs were used at a final concentration of 0.3 µM for both transfection reagents. The siRNA sequences directed against human cdGAP were as follows (Ambion, Grand Island, NY): cdGAP(1) 5’-GGACAGAUCUCUACAUAGA-3’, cdGAP(2) 5’-CCUCAGCGGAGAUCAGUAA-3’. The control siRNA sequence was 5’ACUCUAUCUGCACGCUGAC-3’. Transfection of tagged proteins into U2OS and HT1080 cells was performed with Fugene HD (Roche, Indianapolis, IN) according to the manufacturer’s instructions.
Immunofluoresence Microscopy
Cells plated on glass coverslips coated in either 2D or 3D ECM were fixed and permeabilized simultaneously using a mixture of 4% (w/v) paraformaldehyde (pH adjusted to 7.2) and 1% (v/v) Triton X-100 in phosphate buffered saline (PBS). Coverslips were washed in PBS containing 0.05% (v/v) Tween 20 (for all wash steps). Fixed coverslips were quenched in PBS containing 0.1M glycine, before being washed in PBS-Tween 20 and blocked for a minimum of one hour in PBS containing 3% (w/v) BSA. Glass coverslips were incubated with primary antibodies for one hour at room temperature, and following three washes in PBS-Tween 20, were incubated for one hour with Dylight-conjugated secondary antibodies (Jackson Immunoresearch Labs, West Grove, PA) in PBS with 3% (w/v) BSA. Filamentous-actin was visualized using rhodamine-phalloidin (Invitrogen).
Scrape Wound Assays
Following siRNA-treatment, U2OS or HT1080 cells were spread for three hours on 10µg/ml collagen-coated 6-well or 12-well plates and allowed to form a monolayer before wounding with a pipette tip. Wounded monolayers were washed in warmed media three times to remove detached cells and debris. All wound assays were performed in media containing serum. Cells were placed into an environment controlled chamber at 5% CO2 and 37°C, and phase contrast images were acquired on a Nikon Eclipse Ti scope using a 10X/0.30 PL FLUOR Nikon objective and equipped with a Hamamatsu Orca R2 camera (Hamamatsu City, Japan) and Nikon NS-Elements software. Three points along each wound edge were taken per each experiment, and cells at the wound edge were tracked in ImageJ using the manual tracking plugin (Fabrice Cordelieres, Institut Curie, Orsay, France). Average cell migration velocity was calculated from raw data generated with the manual tracking plugin using the chemotaxis plugin in ImageJ from Ibidi (Martinsried, Germany).
Random Migration Analysis
U2OS cells were plated at low density in either 6 or 12 well plates on 10µg/ml collagen for two hours before being imaged in serum containing DMEM for 16 hours under phase contrast on a Nikon Eclipse Ti scope under a 10X/0.30 PL FLUOR Nikon objective with environment chamber maintained at 5% CO2 and 37°C. Velocity values were calculated as described above for the wound healing analysis. To generate the track images in Figure 1D and 2B, the xy positions of randomly migrating cells generated using the manual tracking plugin in ImageJ were plotted in Microsoft Excel (Redmond, WA).
Adhesion Morphology and Size Analysis
To quantify the percentage of cells containing small, leading edge localized adhesion contacts, cells were spread for two hours in 10% FBS containing DMEM on 10µg/ml collagen-coated glass coverslips. Cells were processed for immunofluoresence and stained with paxillin to mark adhesions and rhodamine phalloidin to visualize actin. Cells with small paxillin positive adhesions that encompassed >20% of either their leading edge or cell periphery were scored as positive for small adhesion contacts. Adhesion size was measured in ImageJ on background-subtracted images of paxillin stained adhesions. Thresholded particles >0.1µm2 were measured using the analyze particles function in ImageJ to give an average adhesion area.
Live Cell Imaging and Focal Adhesion Dynamics Analysis
For adhesion dynamics analysis, U2OS cells were either co-transfected with myc-cdGAP and zyxin-GFP, or U2OS cells stably expressing zyxin-GFP were transfected with a control siRNA or siRNAs targeting cdGAP. SiRNA-treated or over expressing cells were re-plated onto collagen-coated 35mm MatTek glass bottom dishes (MatTek Corp, Ashland, MA) overnight before being imaged on a Leica SP5 confocal microscope using an HCX PL APO 63×/1.40–0.60 OIL λ BL objective (Leica, Bannockburn, IL) equipped with an environment chamber maintained at 37°C and 5% CO2. Time-lapse movies were compiled and background subtracted before being analyzed in ImageJ. Only individual adhesions that could be followed from the point at which they were initially visible all the way through until complete disassembly were quantified. Adhesion assembly and disassembly rates were calculated as previously described by measuring the slopes of semilog plots of the mean fluorescence intensity over time for each individual adhesion [Deakin and Turner, 2011;Webb et al., 2004]. Adhesion lifetimes were determined from adhesions used for the assembly and disassembly rate calculations.
Generation of 3D CDMs and 3D random migration analysis
Cell derived matrices (CDMs) were generated using Human Foreskin Fibroblast (HFF) cells as previously described [Deakin and Turner, 2011]. Briefly, HFF cells were plated on either 1% (v/v) gluteraldehyde fixed gelatin-coated (0.02% gelatin (v/v) in PBS) glass coverslips and 6 or 12 well plastic dishes. Dishes and coverslips were quenched with 1M glycine and washed with PBS before HFF cells were added and grown to confluence. Cells were then cultured for an additional 12–14 days. Cells were grown in DMEM with 10% (v/v) FBS supplemented with 50µg/ml ascorbic acid to promote the stabilization of the collagen matrix. Ascorbic acid containing media was exchanged every two days until matrices were denuded with extraction buffer containing 20 mM NH4OH and 0.5% (v/v) Triton X-100. HT1080 cells were seeded onto the matrix for 4–6 hours before beginning migration studies. Cells were imaged on a Nikon Eclipse Ti scope using a 10X/0.30 PL FLUOR Nikon objective with environment chamber maintained at 5% CO2 and 37°C. Cell migration velocity was calculated as described for the scrape wound and random migration studies.
Matrigel Invasion Assays
Control and cdGAP siRNA-treated HT1080 cells (1.5×10^4 total cells per well) were placed into the upper chamber of a Matrigel coated transwell insert with 0.8µm pores prepared in accordance with the manufacturer’s instructions (BD bioscience). The bottom chamber was filled with 10% (v/v) FBS-containing media. Cells were allowed to invade for 10–12 hours before the Matrigel and any remaining cells were scraped off the top of each membrane with a cotton swab. Membranes were then fixed with 4% paraformaldehyde, permeabilized, and stained for actin using rhodamine phalloidin. Widefield images (10X10) of the bottom of each transwell were taken using a Nikon Eclipse Ti scope under a 10X/0.30 PL FLUOR Nikon objective. Stitched widefield images were used to count the total number of invaded cells. The total numbers of invaded cells were normalized to control siRNA treatments and presented as relative invasion.
Statistics
All statistics were performed in Microsoft Excel using the standard deviation, standard error of the mean, and Student’s t-test functions. Statistical analysis was considered significant if p values were < 0.05, and error bars represent +/− standard error of the mean. For all asterisks *,p < 0.05, **,p < 0.005 , ***, p < 0.001. All data were compiled from a minimum of three independent experiments.
Supplementary Material
Acknowledgements
We thank members of the Turner lab for helpful discussions regarding the data and manuscript. We are grateful to Nathalie Lamarche-Vane, who provided the myc and GFP-tagged cdGAP constructs. We also thank Cell Signaling Technology for their collaboration in generating the antibody against human cdGAP that was used in this study. This work was supported by NIH RO1HL070244 and RO1GM047607 to CET and a Susan G. Komen for the Cure Postdoctoral Fellowship to NOD.
Footnotes
Conflict of interest: The authors declare that they have no conflict of interest.
References
- Abercrombie M, Dunn GA. Adhesions of fibroblasts to substratum during contact inhibition observed by interference reflection microscopy. Exp. Cell Res. 1975;92:57–62. doi: 10.1016/0014-4827(75)90636-9. [DOI] [PubMed] [Google Scholar]
- Allen WE, Zicha D, Ridley AJ, Jones GE. A role for Cdc42 in macrophage chemotaxis. J. Cell Biol. 1998;141:1147–1157. doi: 10.1083/jcb.141.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki K, Nakamura T, Matsuda M. Spatio-temporal Regulation of Rac1 and Cdc42 activity during nerve growth factor-induced neurite outgrowth in PC12 cells. J. Biol. Chem. 2004;279:713–719. doi: 10.1074/jbc.M306382200. [DOI] [PubMed] [Google Scholar]
- Arthur WT, Noren NK, Burridge K. Regulation of rho family GTPases by cell-cell and cell-matrix adhesion. Biol. Res. 2002;35:239–246. doi: 10.4067/s0716-97602002000200016. [DOI] [PubMed] [Google Scholar]
- Bacac M, Stavenkovic I. Metastatic Cancer Cell. Annu. Rev. Path. Mech. Dis. 2008;3:221–247. doi: 10.1146/annurev.pathmechdis.3.121806.151523. [DOI] [PubMed] [Google Scholar]
- Banyard J, Anand-Apte B, Symons M, Zetter BR. Motility and Invasion are differentially modulated by Rho family GTPases. Oncogene. 2000;19:580–591. doi: 10.1038/sj.onc.1203338. [DOI] [PubMed] [Google Scholar]
- Beningo KA, Dembo M, Kaverina I, Small JV, Wang YL. Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J. Cell Biol. 2001;153:881–888. doi: 10.1083/jcb.153.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershadsky AD, Balaban NQ, Geiger B. Adhesion-dependent cell mechanosensitivity. Annu. Rev. Cell Dev. Biol. 2003;19:677–695. doi: 10.1146/annurev.cellbio.19.111301.153011. [DOI] [PubMed] [Google Scholar]
- Bristow JM, Sellers MH, Majumdar D, Anderson B, Hu L, Webb DJ. The rho-family GEF Asef2 activates rac to modulate adhesion and actin dynamics and thereby regulate cell migration. J. Cell. Sci. 2009;122:4535–4546. doi: 10.1242/jcs.053728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, West KA, Turner CE. Paxillin-dependent Paxillin Kinase Linker and p21-Activated Kinase Localization to Focal Adhesions Involves a Multistep Activation Pathway. Mol. Bio. Cell. 2002;13:1550–1565. doi: 10.1091/mbc.02-02-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan MD, Gutman GA. The sense of place in the immune system. Nat. Immunol. 2006;7:329–332. doi: 10.1038/ni0406-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KT, Cortesio CL, Huttenlocher A. Integrins in cell migration. Methods Enzymol. 2007;426:47–67. doi: 10.1016/S0076-6879(07)26003-3. [DOI] [PubMed] [Google Scholar]
- Clarke DM, Brown MC, LaLonde DP, Turner CE. Phosphorylation of actopaxin regulates cell spreading and migration. J. Cell Biol. 2004;166:901–912. doi: 10.1083/jcb.200404024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox EA, Sastry SK, Huttenlocher A. Integrin-mediated adhesion regulates cell polarity and membrane protrusion through the rho family of GTPases. Mol. Biol. Cell. 2001;12:265–277. doi: 10.1091/mbc.12.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- Cukierman E, Pankov R, Yamada KM. Cell interactions with three-dimensional matrices. Curr. Opin. Cell Biol. 2002;14:633–639. doi: 10.1016/s0955-0674(02)00364-2. [DOI] [PubMed] [Google Scholar]
- Deakin NO, Turner CE. Distinct roles for paxillin and hic-5 in regulating breast cancer cell morphology, invasion, and metastasis. Mol. Biol. Cell. 2011;22:327–341. doi: 10.1091/mbc.e10-09-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme-Walker VD, Peterson JR, Chernoff J, Waterman CM, Danuser G, DerMardirossian C, Bokoch GM. Pak1 regulates focal adhesion strength, myosin IIA distribution, and actin dynamics to optimize cell migration. J. Cell Biol. 2011;193:1289–1303. doi: 10.1083/jcb.201010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMali KA, Burridge K. Coupling membrane protrusion and cell adhesion. J. Cell. Sci. 2003;116:2389–2397. doi: 10.1242/jcs.00605. [DOI] [PubMed] [Google Scholar]
- Doyle AD, Kutys ML, Conti MA, Matsumoto K, Adelstein RS, Yamada KM. Microenvironmental control of cell migration: Myosin IIA is required for efficient migration in fibrillar environments through control of cell adhesion dynamics. J. Cell. Sci. 2012 doi: 10.1242/jcs.098806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even-Ram S, Yamada KM. Cell migration in 3D matrix. Curr. Opin. Cell Biol. 2005;17:524–532. doi: 10.1016/j.ceb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Fidyk NJ, Cerione RA. Understanding the catalytic mechanism of GTPase-activating proteins: Demonstration of the importance of switch domain stabilization in the stimulation of GTP hydrolysis. Biochemistry. 2002;41:15644–15653. doi: 10.1021/bi026413p. [DOI] [PubMed] [Google Scholar]
- Fukata M, Nakagawa M, Kaibuchi K. Roles of rho-family GTPases in cell polarisation and directional migration. Curr. Opin. Cell Biol. 2003;15:590–597. doi: 10.1016/s0955-0674(03)00097-8. [DOI] [PubMed] [Google Scholar]
- Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM. Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 2010;26:315–333. doi: 10.1146/annurev.cellbio.011209.122036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett TA, Van Buul JD, Burridge K. VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Exp. Cell Res. 2007;313:3285–3297. doi: 10.1016/j.yexcr.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A. Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 2001;13:584–592. doi: 10.1016/s0955-0674(00)00255-6. [DOI] [PubMed] [Google Scholar]
- Geiger B, Yamada KM. Molecular architecture and function of matrix adhesions. Cold Spring Harb Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton SL, Waterman-Storer CM. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell. 2006;125:1361–1374. doi: 10.1016/j.cell.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005;33:891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- Harunaga JS, Yamada KM. Cell-matrix adhesions in 3D. Matrix Biol. 2011;30:363–368. doi: 10.1016/j.matbio.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Northey JJ, Primeau M, Machado RD, Trembath R, Siegel PM, Lamarche-Vane N. CdGAP is required for transforming growth factor beta- and Neu/ErbB-2-induced breast cancer cell motility and invasion. Oncogene. 2011;30:1032–1045. doi: 10.1038/onc.2010.477. [DOI] [PubMed] [Google Scholar]
- Horwitz R, Webb D. Cell migration. Curr. Biol. 2003;13:R756–R759. doi: 10.1016/j.cub.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Hotchin NA, Hall A. The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J. Cell Biol. 1995a;131:1857–1865. doi: 10.1083/jcb.131.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harb Perspect. Biol. 2011;3:a005074. doi: 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Kirchner J, Kam Z, Tzur G, Bershadsky AD, Geiger B. Live-cell monitoring of tyrosine phosphorylation in focal adhesions following microtubule disruption. J. Cell. Sci. 2003;116:975–986. doi: 10.1242/jcs.00284. [DOI] [PubMed] [Google Scholar]
- Kubow KE, Horwitz AR. Reducing background fluorescence reveals adhesions in 3D matrices. Nat. Cell Biol. 2011;13:3–5. doi: 10.1038/ncb0111-3. author reply 5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo JC, Han X, Hsiao CT, Yates JR, 3rd, Waterman CM. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011;13:383–393. doi: 10.1038/ncb2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokowa K, Itoh R, Yoshizaki H, Nakamura YOT, Matsuda M. Coactivation of Rac1 and Ccdc42 at Lamellipodia and Membrane Ruffles induced by Epidermal Growth Factor. Mol. Bio. Cell. 2004;3:1003–1010. doi: 10.1091/mbc.E03-08-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhyl AKMW, Lalonde DP, Brown MC, Levinson H, Turner CE. The Integrin-linked Kinase Regulates Cell Morphology and Motility in a Rho-associated Kinase-dependent Manner. J. Cell. Biol. 2004;279:54131–54139. doi: 10.1074/jbc.M410051200. [DOI] [PubMed] [Google Scholar]
- LaLonde DP, Grubinger M, Lamarche-Vane N, Turner CE. CdGAP associates with actopaxin to regulate integrin-dependent changes in cell morphology and motility. Curr. Biol. 2006;16:1375–1385. doi: 10.1016/j.cub.2006.05.057. [DOI] [PubMed] [Google Scholar]
- Lamarche-vane N, Hall A. CdGAP a novel proline-rich GTPase-activating protein for Cdc42 and Rac. J. Biol. Chem. 1998;44:29172–29177. doi: 10.1074/jbc.273.44.29172. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: A physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Lee CS, Choi CK, Shin EY, Schwartz MA, Kim EG. Myosin II directly binds and inhibits dbl family guanine nucleotide exchange factors: A possible link to rho family GTPases. J. Cell Biol. 2010;190:663–674. doi: 10.1083/jcb.201003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock JG, Wehrle-Haller B, Stromblad S. Cell-matrix adhesion complexes: Master control machinery of cell migration. Semin. Cancer Biol. 2008;18:65–76. doi: 10.1016/j.semcancer.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Nayal A, Webb DJ, Brown CM, Schaefer EM, Vicente-Manzanares M, Horwitz AR. Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J. Cell Biol. 2006;173:587–589. doi: 10.1083/jcb.200509075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Ohta Y, Hartwig JH, Stossel TP. FilGAP, a rho- and ROCK-regulated GAP for rac binds filamin A to control actin remodelling. Nat. Cell Biol. 2006;8:803–814. doi: 10.1038/ncb1437. [DOI] [PubMed] [Google Scholar]
- Pankov R, Endo Y, Even-Ram S, Araki M, Clark K, Cukierman E, Matsumoto K, Yamada KM. A rac switch regulates random versus directionally persistent cell migration. J. Cell Biol. 2005;170:793–802. doi: 10.1083/jcb.200503152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrini MC, Sadou-Dubourgnoux A, Aoki K, Kunida K, Biondini M, Hatzoglou A, Poullet P, Formstecher E, Yeaman C, Matsuda M, et al. SH3BP1, an exocyst-associated RhoGAP, inactivates Rac1 at the front to drive cell motility. Mol. Cell. 2011;42:650–661. doi: 10.1016/j.molcel.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010;11:633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 2009;10:538–549. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein rho by cell adhesion and the cytoskeleton. EMBO. J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Riveline D, Zamir R, Balaban N, Schwarz U, Ishizaki T, Narumiya S, Kam Z, Geiger B, Bershadsky A. Focal Contacts as Mechanosensors: Externally Applied Local Mechanical Force Induces Growth of Focal Contacts by an mDia1-dependent and ROCK-independent Mechanism. J. Cell Biology. 2001;153:1175–1185. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottner K, Hall A, Small JV. Interplay between rac and rho in the control of substrate contact dynamics. Current Biology. 1999;9:640–649. doi: 10.1016/s0960-9822(99)80286-3. [DOI] [PubMed] [Google Scholar]
- Sander ES, ten klooster JP, van Delft S, van der Kammen RA, Collard JG. Reciprocal Balance between Both Gtpases determines cellular morphology and Migratory Behavior. J. Cell Biology. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135:510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- Shober M, Raghaan S, Nikolova M, Polak L, Posolli HA, Beggs HE, Reichardt LF, Fuchs E. ACF7 Regulates Cytoskeletal-Focal Adhesion Dynamics and Has ATPase activity. Cell. 2008;135:137–148. doi: 10.1016/j.cell.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonneman KJ, Bement WM. Wound Repair, toward understanding and integration of single-cell and multicellular wound responses. Annu Rev Cell Dev Biol. 2011;27:237–263. doi: 10.1146/annurev-cellbio-092910-154251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southgate L, Machado RD, Snape KM, Primeau M, Dafou D, Ruddy DM, Branney PA, Fisher M, Lee GJ, Simpson MA, et al. Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am. J. Hum. Genet. 2011;88:574–585. doi: 10.1016/j.ajhg.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherkezian J, Danek EI, Jenna S, Triki I, Lamarche-Vane N. Extracellular signal-regulated kinase 1 interacts with and phosphorylates CdGAP at an important regulatory site. Mol. Cell. Biol. 2005;25:6314–6329. doi: 10.1128/MCB.25.15.6314-6329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherkezian J, Triki I, Stenne R, Danek EI, Lamarche-Vane N. The human orthologue of CdGAP is a phosphoprotein and a GTPase-activating protein for Cdc42 and Rac1 but not RhoA. Biol. Cell. 2006;98:445–456. doi: 10.1042/BC20050101. [DOI] [PubMed] [Google Scholar]
- Ten Klooster JP, Evers EE, Janssen L, Machesky LM, Michiels F, Hordijk P, Collard JG. Interaction between Tiam1 and the Arp2/3 complex links activation of rac to actin polymerization. Biochem. J. 2006;397:39–45. doi: 10.1042/BJ20051957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Klooster JP, Jaffer ZM, Chernoff J, Hordijk PL. Targeting and activation of Rac1 are mediated by the exchange factor beta-pix. J. Cell Biol. 2006;172:759–769. doi: 10.1083/jcb.200509096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004;6:154–161. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- Webb DJ, Parsons JT, Horwitz AF. Adhesion assembly, disassembly and turnover in migrating cells -- over and over and over again. Nat. Cell Biol. 2002;4:E97–E100. doi: 10.1038/ncb0402-e97. [DOI] [PubMed] [Google Scholar]
- Weijer CJ. Collective cell migration in development. J. Cell Science. 2009;15:3215–3223. doi: 10.1242/jcs.036517. [DOI] [PubMed] [Google Scholar]
- Wolf K, Friedl P. Mapping proteolytic cancer cell-extracellular matrix interfaces. Clin. Exp. Metastasis. 2009;26:289–298. doi: 10.1007/s10585-008-9190-2. [DOI] [PubMed] [Google Scholar]
- Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Brocker EB, Friedl P. Compensation mechanism in tumor cell migration: Mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 2003;160:267–277. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, Hahn KM. A genetically encoded photoactivatable rac controls the motility of living cells. Nature. 2009;461:104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130:601–610. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Yu JA, Deakin NO, Turner CE. Paxillin-kinase-linker tyrosine phosphorylation regulates directional cell migration. Mol. Biol. Cell. 2009;20:4706–4719. doi: 10.1091/mbc.E09-07-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J. Cell. Sci. 2001;114:3583–3590. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
- Zhao ZS, Manser E, Loo TH, Lim L. Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol. Cell. Biol. 2000;20:6354–6363. doi: 10.1128/mcb.20.17.6354-6363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.