

Abstract
Early detection of primary immunodeficiency is recognized as important for avoiding infectious complications that compromise outcomes. In particular, severe combined immunodeficiency (SCID) is fatal in infancy unless affected infants can be diagnosed before the onset of devastating infections and provided with an immune system through allogenic hematopoietic cell transplantation, enzyme replacement, or gene therapy. A biomarker of normal T cell development, T cell receptor excision circles (TRECs), can be measured in DNA isolated from the dried blood spots routinely obtained for newborn screening; infants identified as lacking TRECs can thus receive confirmatory testing and prompt intervention. Early results of TREC testing of newborns in five states indicate that this addition to the newborn screening panel can be successfully integrated into state public health programs. A variety of cases with typical SCID genotypes and other T lymphocytopenic conditions have been detected in a timely manner and referred for appropriate early treatment.
Keywords: primary immunodeficiency, T cell receptor excision circle (TREC), severe combined immunodeficiency (SCID), lymphocytopenia, dried blood spot, newborn screening
Introduction
Population-based newborn screening began with a test for phenylketonuria (PKU) developed by Robert Guthrie in 19631 following the demonstration that a phenylalanine restricted diet instituted early in life can prevent serious neurodevelopmental impairment in children lacking phenylalanine hydroxylase. PKU was identified in neonates by finding elevated phenylalanine levels in drops of infant blood obtained by heelstick and applied to filter paper. Dried blood spot testing needed to be done in standardized laboratories, and infants with abnormal tests had to be contacted promptly and directed to metabolic disease specialists for dietary management. In the United States these tasks have been instituted, supported, and directed by each state’s public health program. Newborn screening efforts have grown as additional rare, but treatable conditions have been recognized that can be successfully identified by sensitive, specific, and inexpensive tests. Up to 50 or more metabolic disorders, hypothyroidism, and hemoglobinopathies have now been incorporated into dried blood spot testing of newborns in most states, and nursery-based tests to screen for deafness are also performed.2
Recently, severe combined immunodeficiency (SCID) has become the first genetic disorder of the immune system to be amenable to newborn screening. SCID and related conditions with low numbers of T lymphocytes can be identified by testing T cell receptor excision circles (TRECs), a DNA biomarker of normal T lymphocyte development, in dried blood spots routinely obtained for screening for other conditions. Coincidentally, the technology for SCID screening was developed at the same time as a new national oversight process was being rolled out to provide evidence-based assessments of new conditions proposed for addition to state newborn screening panels of tests. SCID became the first disease nominated to the national advisory committee that was reviewed and unanimously recommended for inclusion in the evidence-based uniform screening panel.3 SCID is also the first newborn screening test for which the primary analyte is DNA. This review summarizes the current case for SCID newborn screening, presenting evidence available before it was undertaken and now that early results are available from statewide pilot programs, as well as future challenges that remain to be addressed.
Evidence for adding SCID to the panel of newborn screening tests
The U.S. Advisory Committee on Heritable Disorders in Newborns and Children was established in part to work toward uniform, evidence-based newborn screening in what has traditionally been a patchwork of individual state programs.3 The Advisory Committee’s mandate is to solicit nominations of conditions to be added to newborn screening; consideration of each nominated condition includes an independent review of evidence by public health experts and input from knowledgeable physicians and stakeholders such as family advocacy groups. Upon weighing the evidence and arriving at a determination, the Committee reports to the Department of Health and Human Services (DHHS) Secretary. SCID was first nominated in 2008, and evidence was assembled and reported in 2009 and again in 2010, at which time it was considered strong enough to merit a unanimous favorable recommendation.4 Secretary of Health and Human Services Kathleen Sibelius accepted the recommendation and formally endorsed SCID screening in May 2010.
SCID is a collection of over 20 distinct genetic disorders characterized by profound defects in both cellular immunity and specific antibody production (Table 1). It is estimated to occur in 1 per 50,000 to 1 per 100,000 births, although true population incidence has been unknown prior to screening.4–10 All SCID infants have absent or extremely low production of T lymphocytes from the thymus, while some also have deficiencies of B cells, NK cells or both. Although there are individual exceptions, SCID genotypes have characteristic profiles of lymphocyte impairment, as shown in Table 1, which also gives rough estimates of relative frequency. The combined defects of T and B cells, plus absent NK cells in some forms of SCID, severely compromise an infant’s ability to resist infections, and the condition has thus been clinically defined since early descriptions by failure to thrive, thrush, Pneumocystis jiroveci pneumonia, and other bacterial, fungal, and viral infections.
Table 1.
Molecular causes of severe combined immunodeficiency
| Lymphocyte profile
|
|||||
|---|---|---|---|---|---|
| Gene defect | Defective protein, function, features | % SCIDa cases | Tb | B | NK |
| IL2RG (X-linked) | Common γ-chain (γc) of receptors for IL-2, -4, -7, -9, -15, and -21 | 45–50% (only males) | − | + | − |
| ADA | Adenosine deaminase enzyme | 16% | − | + | − |
| IL7R | α-Chain of IL-7 receptor | 9% | − | + | + |
| JAK3 | Janus kinase 3, activated by γc | 6% | − | + | − |
| RAG1, RAG2 | Recombinase activating genes required for T and B cell antigen receptor gene rearrangement | 5% | − | − | + |
| DCLRE1C (Artemis) | Part of T and B cell antigen receptor gene rearrangement complex, also required for DNA repair | < 5% | − | − | + |
| TCRD, TCRE, TCRZ | CD3 δ, ε, and ζ chains of the T cell receptor complex, required for T cell development | Rare | −/low | + | + |
| CD45 | Protein tyrosine phosphatase receptor (PTPRC), required for T and B cell activation by antigen | Rare | −/low | + | +/low |
| LCK | Lymphocyte tyrosine kinase p56lck, required for T cell development and activation | Rare | −/low | + | + |
| PNP | Purine nucleoside phosphorylase enzyme, deficiency also causes neurological impairment | Rare | Low | Low | +/low |
| LIG4 | DNA ligase IV required for antigen receptor gene rejoining | Rare | − | + | + |
| DNAPKCS | DNA protein kinase catalytic subunit, required for T and B cell antigen receptor rearrangement, and DNA repair | Rare | − | − | + |
| NHEJ1 (Cernunnos) | Nonhomologous end joining of DNA; deficiency also causes microcepahaly and radiation sensitivity | Rare | − | − | + |
| AK2 | Adenylate kinase 2; deficiency causes reticular dysgenesis with granulocytopenia, lymphocytopenia, and deafness | Rare | − | − | − |
| FOXN1 | Forkhead box N1, required for thymus and hair follicle development (ortholog of nude mouse) | Rare | −/low | + | + |
| STAT5a | Signal transducer and activator of transcription 5, phosphorylated after cytokine receptor engagement; deficiency also causes growth hormone–resistant growth failure | Rare | −/low | + | − |
| CORO1A | Coronin-1A, protein mediating lymphocyte migration and T cell emigration from the thymus | Rare | −/low | + | + |
| Currently unknown | Unknown defects, including SCID and congenital anomalies; SCID with multiple bowel atresias | ~10% | −/low | +/− | +/− |
The rationale for SCID newborn screening, outlined in Table 2, derives from our knowledge that SCID is potentially treatable, but is not recognized effectively prior to onset of devastating infections. Although treatment modalities have improved in the last four decades, SCID morbidity and mortality remain high. Affected infants do not survive unless provided with functional immunity, but this can be achieved by hematopoietic cell transplantation (HCT) from a healthy donor,5 by enzyme replacement in cases of adenosine deaminase (ADA) deficiency,6 and (although still experimental) by gene therapy for X-linked and ADA deficient SCID.6,7 Infants with SCID are healthy at birth, initially protected by transplacentally derived maternal IgG antibodies; but persistent, severe, and opportunistic infections typically develop by age four to seven months. Repeated observations have shown superior outcomes in SCID infants diagnosed at a young age, particularly those fortunate enough to have an affected relative to alert health providers of the diagnosis.5,8,11,12
Table 2.
Rationale for newborn screening for SCID
| Importance of early identification |
| Establish diagnosis and institute immediate lifesaving treatment |
| Avoid inefficient, costly, dangerous “diagnostic Odyssey” |
| Provide families with genetic diagnosis and advice on reproductive risks |
| Learn incidence and true spectrum of SCID |
| Educate providers and public about SCID |
| Permit multicenter collaborative trials to determine optimal treatments |
| Barriers to early diagnosis without screening |
| SCID is rare |
| Infections are common in all infants, not just those with SCID |
| Over 80% of cases are sporadic, with no family history |
| Family history can be missed, or nonspecific |
| SCID infants are protected by maternal IgG for their first months of life |
| Because both a gene defect and environmental exposure are required for overt disease, presentation is variable |
The first suggestion that screening infants for low lymphocytes could identify SCID in time for lifesaving treatment was by Buckley et al. in 1997.8 Further retrospective analysis of cases treated by Buckley at Duke,11 and recently in England,12 have underlined the better survival of SCID infants diagnosed before developing infections. However, although these reports showed a clear benefit for early diagnosis, they were limited to the potentially biased population of SCID infants admitted to specialized immunodeficiency transplant centers. A family-based survey by the Immune Deficiency Foundation and Chan et al. found even more striking differences due to the higher mortality of SCID infants not recognized at birth; half of the deceased infants in this study were either not diagnosed pre-mortem or were too ill to be transferred to a center for specialized treatment.13 Furthermore, confirmation that >80% of SCID infants were the first known to be affected in their family indicated that family history taking alone would not be sufficient to lead to identification of most SCID cases.
Mathematical modeling by two independent methods has also shown that a sensitive, specific, and economical newborn screening test for SCID would be likely to be cost effective.14,15
Another important development was the institution, beginning in 2006, of live attenuated vaccination of young infants against rotavirus infection, an important cause of infant diarrhea and dehydration leading to hospitalization and mortality. Infants receive two to three doses of rotavirus vaccine starting as early as six weeks to two months of age. Infants affected with SCID whose diagnosis had not been recognized and who unintentionally received the vaccination have developed severe diarrheal disease proven to be caused by the vaccine strain of rotavirus.16,17 While the vaccine is specifically contraindicated in infants with immune compromise, there is no way to know whether a healthy-appearing infant at that age has SCID, other than by performing an immunological blood test. Newborn screening for SCID thus has become an important consideration to balance the public health benefit of protection from diarrhea against the harm of vaccine-strain rotavirus infection that occurs in rare infants lacking immunity.
The TREC test for SCID using newborn dried blood spots
A breakthrough for the actualization of population-based newborn screening for SCID was the development of a screening test that could be performed using the dried blood spot samples already collected by state screening laboratories for routine newborn screening for other conditions. Early proposed screening methods included absolute lymphocyte count (requiring a separate liquid blood sample),8,11 IL-7 immunoassay,10 bead-bound antibody-based detection of T cell proteins CD3, CD4, and the leukocyte marker CD5,18 and gene resequencing microarrays.19 However, to date the only assay with adequate sensitivity and specificity for dried blood spot use is the TREC assay, first published in 2005 by Chan and Puck.20 Late in maturation, 70% of thymocytes that will ultimately express αβ T cell receptors form a circular DNA TREC from the excised TCRδ gene that lies within the TCRα locus.21 TRECs are stable because they lack free DNA ends to be attacked by DNA digesting enzymes, but because they have no origin of replication they do not increase in number when cells divide. Thus the TREC copy number, which can be measured by performing a quantitative PCR reaction across the joint of the circular TREC DNA, is an indicator of newly formed thymic emigrant T cells.
The frequency of TREC-bearing T cells in peripheral blood diminishes as newly formed T cells are diluted by T cells that have undergone mitosis. Normal newborns have a high rate of new T cell production, resulting in TREC numbers at about 10% of their total T cell numbers; in contrast older children and adults have progressively lower ratios of TRECs to T cells, reflecting peripheral T cell expansion.21 Infants with SCID, sampled both at the time of their SCID diagnosis and upon recovery of neonatal dried blood spots obtained when they were in the nursery, have very low or undetectable TRECs (Fig. 1, Table 3).20,22 Even maternal T cells present in the circulation of an infant with SCID do not falsely raise the TREC count to the normal range because maternal cells have very few TRECs. Thus, a normal number of TRECs is an excellent biomarker for new autologous T cell production, provided the DNA is adequate for PCR (shown by amplification of a control, such as a segment of the β-actin gene).
Figure 1.
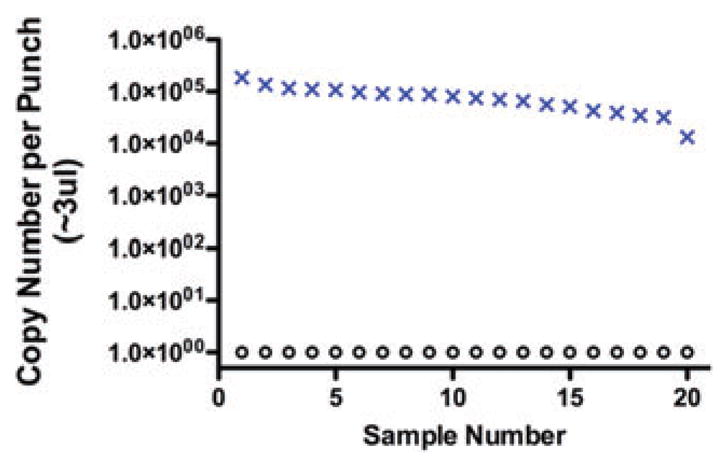
Copy number detected in actual nursery dried blood samples, recovered from state screening laboratories and tested by quantitative PCR of TRECs (o) and a control genomic DNA segment from the β-actin gene (×). Copy number is normalized to the amount of DNA isolated from a 3 mm punch from a dried blood filter, which is equivalent to about 3 μL of blood (J. Puck, unpublished data).
Table 3.
Summary of California TREC screening experience in the first year
|
Since all non-SCID screening is done first in regional labs, with samples then forwarded to a central lab for TREC testing, most newborns were two weeks old when the TREC result was available. When a SCID-specific repeat heelstick was needed, older age usually resulted in a normal TREC value on redraw.
Only one SCID case had a positive family history leading to testing at birth.
Signs of Omenn syndrome in the first weeks of life had led to the diagnosis just before the TREC test was reported.
Initiation of SCID screening programs
Although the TREC test in dried blood spots was a promising biomarker for insufficient T cell production, and retrospective analysis indicated that typical SCID cases would have been found by TREC screening had it been done in the past (Fig. 1), actual prospective tests in the field were required to establish clinical validity. The TREC test was first scaled up and adapted to a statewide newborn screening format by Baker et al. in Wisconsin.23 Other states, including Massachusetts, devised their own adaptations of this assay.24,25 Each state tailored its TREC screening according to individual program structure and requirements. A clinical study of TREC screening was also undertaken in selected hospitals on the Navajo Indian Reservation, because Navajo Native Americans are known to have a high rate of SCID (about 1 in 2,000 births) due to a founder mutation in the DCLRE1C (Artemis) gene.26 The Centers for Disease Control and Prevention developed standards to be offered as unknowns for quality control and calibration purposes. Consideration of the performance of TREC newborn screening as well as available clinical data described above led to the recommendations for universal adoption of SCID newborn screening.
The Wisconsin program, which began in 2008, was recently described.25 In the first year, in which 70,000 infants were screened, several were found with low T cell numbers, and one had a combined lymphocyte and granulocyte disorder, RAC2 deficiency, treated successfully by bone marrow transplantation.
Larger numbers of births and experience from additional state newborn screening programs would be needed to demonstrate clinical utility of SCID newborn screening. California, with over 500,000 births per year and a very diverse population, began its development program of statewide TREC screening in August 2010; a bill has now been passed and signed into law to make SCID a permanent addition to the California newborn screening panel. This state’s SCID screening algorithm is shown in Figure 2. A TREC quantitative PCR similar to those of Chan and Puck and the Wisconsin program is followed by a β-actin gene PCR control only if the TREC number is inadequate. If both TREC and control DNA copy numbers are below cutoff values after two separate DNA extractions, the sample is considered a “DNA amplification failure” (DAF), and a second heel stick is requested from the baby. Preterm and ill infants in intensive care units have modified DAF cutoff values. All samples with undetectable or below cutoff TRECs and adequate control PCR are considered positive. Infants with a positive screening test result or two DAF samples require second tier testing with T cell enumeration.
Figure 2.

California algorithm for newborn screening and follow-up for SCID and related conditions. TREC and β-actin gene PCR adapted by PerkinElmer for the California Genetic Disease Screening Program. Values expressed as copies per 1 μL of blood (J. Puck, J. Church, and F. Lorey, unpublished data).
An important feature of SCID screening in California is that a liquid blood sample following the positive screening result is an integral part of the program. Area service center staff work with providers and families so that blood is rapidly obtained at newborn blood drawing stations throughout the state that have been established for metabolic disease follow-up, and all samples are sent to a single central laboratory (Quest Nichols Institute, San Juan Capistrano, CA) for a complete blood count, differential count, and a specified flow cytometry panel of lymphocyte subset markers including naive and memory T cell markers. All results are interpreted by two designated immunodeficiency consultants for the program. In this centralized system infants receive a definitive diagnosis and are referred to a center of excellence for further management.
The performance of the TREC test in Wisconsin, Massachusetts, and California has been excellent to date, with no missed SCID cases that have come to light. The California experience is summarized in Table 3. There have been very few DAF samples necessitating a second heelstick, 1 per 1,250 births. Only 50 infants required a liquid blood sample (1 per 10,000 births); of these, 40% proved to have true T lymphocytopenia, indicating that TRECs recovered from blood spots are, as predicted, an excellent T cell biomarker. All infants with T lymphocytopenia were under the care of a primary immunodeficiency expert by one month of age (J. Puck and F. Lorey, unpublished information). The data in Table 3 suggest an incidence of typical SCID of 1 per 70,000 births, within the range of 1 per 50,000–100,000 estimated before screening began. Tracking of cases by race and ethnicity has suggested a somewhat higher rate than expected of T lymphocytopenia among Hispanics, and the large and diverse population of California will make possible assessment of rates between different ancestral groups. Compared to SCID and Omenn syndrome with seven cases, about twice as many cases of non-SCID T lymphocytopenia have been found.
Conditions that may be detected by newborn screening
The states that perform SCID newborn screening have successfully identified a range of typical SCID and other T lymphocytopenic conditions that would not otherwise have been identified before the onset of serious infections (Tables 3). The first case of typical SCID due to JAK3 deficiency was reported from Massachusetts after screening 100,000 infants.27 In addition to typical SCID, SCID-related disorders are increasingly appreciated, as reported by the early Wisconsin experience.28 It is clear that more data will be needed to understand the full range of conditions detected. Not only the total incidence but also the relative incidence in different population subgroups remains to be defined. The conditions with low or absent TRECs fall into five categories (Table 4):28–31 (A) Typical SCID is defined by the Primary Immune Deficiency Treatment Consortium (PIDTC) as fewer than 300 autologous T cells/μL of peripheral blood (normal infants have 2,250 to 5,000/μL) and less than 10% of normal T cell proliferation to the mitogen PHA.32,33 (B) Leaky SCID, which has no maternal engraftment and T cells ranging from 300–1,500/μL, may have a later age of onset of clinical symptoms or may present in infancy as Omenn syndrome (defined by erythroderma rash, adenopathy, and oligoclonal, poorly functioning T cells, due to hypomorphic mutations in RAG1, RAG2 or other known SCID genes). (C) Variant SCID is defined as the absence of a known SCID gene defect and 300–1,500 autologous T cells/μL with impaired responses to mitogens. (D) Multisystem syndromes with T cell defects have a wide spectrum of T cell dysgenesis. Those with severely affected T cell production are expected to have positive TREC screens, while others with the same syndrome but more normal T cells will not be detected. In several instances infants with these syndromes have been found to have abnormally low TRECs (Table 4D). DiGeorge syndrome, usually associated with chromosome 22q.11 deletion; trisomy 21; and CHARGE syndrome (with ocular coloboma, heart defect, atresia of nasal choanae, retardation of growth and development, genitourinary abnormality, and ear abnormality) can all present with life-threatening infections in infancy due to T cell deficiency and have been identified by neonatal TREC screening.28–30 In addition, RAC2 deficiency, previously known only as a granulocyte disorder, was diagnosed following newborn screening with low TRECs and T lymphocytopenia,28 as was Jacobsen syndrome associated with terminal deletions of chromosome 11q24. Siblings affected with DOCK8 deficient hyper-IgE syndrome with lymphocytopenia had undetectable TRECs during childhood, though newborn samples have not yet been available.31 Finally, (E) secondary T cell defects are characterized by acquired conditions with increased T cell loss. These include congenital heart defects, neonatal leukemia, lymphocyte loss by extravasation or third spacing, lymphangiectasia and possibly congenital HIV infection. Some of the secondary defects, such as lymphocytopenia associated with extreme low birthweight, may resolve over time. We expect that previously unrecognized conditions will come to light with screening. Furthermore, we recognize that many primary immunodeficiency diseases will not be detectable through TREC screening, and it is important to educate providers to remain alert for signs of these conditions in patients who have been screened.
Table 4.
Conditions detected by low or absent TRECs
|
Observed to have low or absent TRECs upon newborn screening in one or more cases to date in U.S. pilot programs or published reports.
Observed to have low or absent TRECs in one or more cases after diagnosis; newborn samples not available.
Remaining challenges
Screening for SCID is a matter of fairness, access, and early awareness, designed to give all affected infants the advantages of early diagnosis and treatment that have previously been available primarily to families with means and second-born affected children. Parents in such families may have already lost an infant whose SCID diagnosis was delayed or unrecognized. However, in order for the case for newborn screening to be completely compelling, a seamless progression from prenatal education to screening to definitive diagnosis to optimal treatment must be established. This goal is challenging even for large states with centers of excellence in pediatric immunology and bone marrow transplantation; smaller states or those without such centers will have to consider regional collaborations and referrals of their SCID cases to treatment centers beyond their borders.
How best to perform HCT treatment in SCID to achieve maximum survival, minimal toxicity and B cell as well as T cell reconstitution remains controversial. In the absence of compelling data from well controlled multicenter trials, opinions remain divided as to whether cytoreductive or ablative versus no chemotherapy conditioning should be used, and what source of donor hematopoietic stem cells are best, among other questions.32–34 Although HCT provides reliable T cell immunity for SCID, B cell recovery remains problematic, and some transplants may not be durable over many years. Pre-transplantation conditioning does not guarantee that B cell function will develop; therefore, one must decide whether there is justification for using agents that compromise innate immunity and have intrinsic toxicities to gain B cell immune reconstitution. Pharmacokinetic studies of chemotherapy drugs commonly used in older children and adults have not been done in infants, and risks judged acceptable for patients with lethal malignancies may not be prudent for young, healthy infants with SCID detected by newborn screening. The risks of delaying treatment and exposing infants to infection pending a search for a matched unrelated donor must be balanced against the option of performing a T cell–depleted haploidentical HCT from a parent. Even the laboratory workup essential for very small infants with SCID is controversial, since blood tests may cause anemia, requiring transfusions. Centers that perform HCT for children with malignant disease cannot be assumed to have the specialized knowledge and experience required for optimal success with HCT for SCID. Fortunately a national rare disease network, the PIDTC has been funded by the National Institutes of Health to conduct prospective studies of SCID treatments and outcomes, and eventually multicenter clinical treatment trials will identify the best approaches for each individual SCID genotype.32,33
Convincing states to add a disease to their newborn screening panel requires a coalition of immunologists, geneticists, parent advocates, nonprofit agencies, public health officials, and politicians. In times of financial restraint, state officials are asking for proof that SCID screening is cost effective. While it is widely believed that early diagnosis of SCID and related disorders through screening will save lives and also lead to savings in medical care dollars, population-based proof of better outcomes and lower costs through screening remains to be shown. Such proof will take time to acquire as states accumulate data from screening and outcome tracking. Beyond TREC testing for newborns states will need to have programs to assure that expert diagnostic follow-up and treatment are available to infants with positive screening results.
Finally, current controversies surrounding the collection, testing, storage, and use of dried blood spots from infants by state newborn screening programs highlight the need for better outreach and education regarding the public health benefits of newborn screening. States need to be sure that their policies are clear and consistent,35 and better information needs to be transmitted to the public, perhaps by better linking of prenatal obstetrical care and counseling to postnatal newborn screening, particularly as DNA-based testing, started with TREC testing for SCID, becomes more widespread.36 Raising public awareness of rare disorders, including primary immunodeficiencies, is also an important activity in support of early diagnosis through newborn screening.37
SCID screening is justified because early identification is associated with higher survival rate and better outcome. Newborn screening for SCID is being adopted by an increasing number of state public health programs following its successful initiation in Wisconsin, followed by Massachusetts, California, New York, Louisiana, and Puerto Rico. Colorado, Connecticut, and Michigan as well as additional states are beginning to screen infants for SCID. Classic SCID, leaky SCID, and Omenn syndrome as well as additional known disorders with T lymphocytopenia have been found, as well as cases with low T cell production of unknown cause. For the population as a whole and for groups of distinct ancestry or ethnicity, newborn screening will make it possible for the first time to determine the true incidence and spectrum of SCID and related disorders.
Acknowledgments
Thanks to Dr. Fred Lorey and colleagues at the California Department of Public Health and Genetic Disease Laboratory, and to Perkin Elmer and Quest Nichols Institute for design and conduct of testing; Drs. Joseph Church, Mort Cowan, Christopher Dvorak, Nina Kapoor, David Lewis, Sean McGhee, Ted Moore, E. R. Stiehm, Chu Ri Shin, and Ken Weinberg for helpful discussions; and agencies supporting this work: The Jeffrey Modell Foundation, NIH NIAID for support of PIDTC U54 AI082973, NICHD for RO3 HD 060311, NIAID for RO1 AI078248, and NCRR UL1 RR024131 for the UCSF CTSI.
Footnotes
Conflicts of interest
The author declares no conflicts of interest.
References
- 1.Guthrie R, Susi I. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. 1963;32:318–343. [PubMed] [Google Scholar]
- 2.Newborn Screening Status Report (updated 3/01/10) National Newborn Screening and Genetics Resource Center; [Accessed Sept. 26, 2011]. Available at: http://genes-r-us.uthscsa.edu/nbsdisorders.pdf. [Google Scholar]
- 3.Howell RR, Lloyd-Puryear MA. From developing guidelines to implementing legislation: actions of the US Advisory Committee on Heritable Disorders in Newborns and Children toward advancing and improving newborn screening. Semin Perinatol. 2010;34:121–124. doi: 10.1053/j.semperi.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Lipstein EA, Browning MF, Green NS, et al. Systematic evidence review of newborn screening and treatment of severe combined immunodeficiency. Pediatrics. 2010;125:1226–1235. doi: 10.1542/peds.2009-1567. [DOI] [PubMed] [Google Scholar]
- 5.Buckley RH. Transplantation of hematopoietic stem cells in human severe combined immunodeficiency: longterm outcomes. Immunol Res. 2011;49:25–43. doi: 10.1007/s12026-010-8191-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaspar HB, Aiuti A, Porta F, et al. How I treat ADA deficiency. Blood. 2009;114:3524–3532. doi: 10.1182/blood-2009-06-189209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fischer A, Hacein-Bey-Abina S, Cavazzana-Calvo M. Gene therapy for primary adaptive immune deficiencies. J Allergy Clin Immunol. 2011;127:1356–1359. doi: 10.1016/j.jaci.2011.04.030. [DOI] [PubMed] [Google Scholar]
- 8.Buckley RH, Schiff RI, Schiff SE, et al. Human severe combined immunodeficiency: genetic, phenotypic and functional diversity in one hundred eight infants. J Pediatr. 1997;130:378–387. doi: 10.1016/s0022-3476(97)70199-9. [DOI] [PubMed] [Google Scholar]
- 9.Lindegren ML, Kobrynski L, Rasmussen SA, et al. Applying public health strategies to primary immunodeficiency diseases: a potential approach to genetic disorders. MMWR Recomm Rep. 2004;53:1–29. [PubMed] [Google Scholar]
- 10.Puck JM Newborn Screening Working Group. SCID population-based newborn screening for severe combined immunodeficiency: steps toward implementation. J Allergy Clin Immunol. 2007;120:760–768. doi: 10.1016/j.jaci.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 11.Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood. 2002;99:872–878. doi: 10.1182/blood.v99.3.872. [DOI] [PubMed] [Google Scholar]
- 12.Brown L, Xu-Bayford J, Allwood Z, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood. 2011;117:3243–3246. doi: 10.1182/blood-2010-08-300384. [DOI] [PubMed] [Google Scholar]
- 13.Chan A, Scalchunes C, Boyle M, Puck JM. Early vs. delayed diagnosis of severe combined immunodeficiency: a family perspective survey. Clin Immunol. 2011;138:3–8. doi: 10.1016/j.clim.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGhee SA, Stiehm ER, McCabe ER. Potential costs and benefits of newborn screening for severe combined immunodeficiency. J Pediatr. 2005;147:603–608. doi: 10.1016/j.jpeds.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Chan K, Davis J, Pai SY, et al. A Markov model to analyze cost-effectiveness of screening for severe combined immunodeficiency (SCID) Mol Genet Metab. 2011 Jul 12; doi: 10.1016/j.ymgme.2011.07.007. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Werther RL, Crawford NW, Boniface K, et al. Rotavirus vaccine induced diarrhea in a child with severe combined immune deficiency. J Allergy Clin Immunol. 2009;124:600. doi: 10.1016/j.jaci.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Patel NC, Hertel PM, Estes MK, et al. Vaccine-acquired rotavirus in infants with severe combined immunodeficiency. N Engl J Med. 2010;362:314–319. doi: 10.1056/NEJMoa0904485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janik DK, Lindau-Shepard B, Comeau AM, Pass KA. A multiplex immunoassay using the Guthrie specimen to detect T-cell deficiencies including severe combined immunodeficiency disease. Clin Chem. 2010;56:1460–1465. doi: 10.1373/clinchem.2010.144329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lebet T, Chiles R, Hsu AP, et al. Mutations causing severe combined immunodeficiency: detection with a custom resequencing microarray. Genet Med. 2008;10:575–585. doi: 10.1097/gim.0b013e31818063bc. [DOI] [PubMed] [Google Scholar]
- 20.Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2005;115:391–398. doi: 10.1016/j.jaci.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Douek DC, McFarland RD, Keiser PH, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–695. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- 22.Morinishi Y, Imai K, Nakagawa N, et al. Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal Guthrie cards. J Pediatr. 2009;155:829–833. doi: 10.1016/j.jpeds.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 23.Baker MW, Grossman WJ, Laessig RH, et al. Development of a routine newborn screening protocol for severe combined immunodeficiency. J Allergy Clin Immunol. 2009;124:522–527. doi: 10.1016/j.jaci.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 24.Gerstel-Thompson JL, Baptiste JC, Navas JS, et al. High-throughput multiplexed T-cell-receptor excision circle quantitative PCR assay with internal controls for detection of severe combined immunodeficiency in population-based newborn screening. Clin Chem. 2010;56:1466–1474. doi: 10.1373/clinchem.2010.144915. [DOI] [PubMed] [Google Scholar]
- 25.Chase NM, Verbsky JW, Routes JM. Newborn screening for T-cell deficiency. Curr Opin Allergy Clin Immunol. 2010;10:521–525. doi: 10.1097/ACI.0b013e32833fd6fe. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Moshous D, Zhou Y, et al. A founder mutation in Artemis, an SNM1-like protein, causes SCID in Athabascan-speaking native Americans. J Immunol. 2002;168:6323–6329. doi: 10.4049/jimmunol.168.12.6323. [DOI] [PubMed] [Google Scholar]
- 27.Hale JE, Bonilla FA, Pai SY, et al. Identification of an infant with severe combined immunodeficiency by newborn screening. J Allergy Clin Immunol. 2010;126:1073–1074. doi: 10.1016/j.jaci.2010.08.043. [DOI] [PubMed] [Google Scholar]
- 28.Routes JM, Verbsky J, Laessig RH, et al. Statewide newborn screening for severe T-cell lymphopenia. JAMA. 2009;302:2465–2470. doi: 10.1001/jama.2009.1806. [DOI] [PubMed] [Google Scholar]
- 29.McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome) Medicine. 2011;90:1–18. doi: 10.1097/MD.0b013e3182060469. [DOI] [PubMed] [Google Scholar]
- 30.Ram G, Chinen J. Infections and immunodeficiency in Down syndrome. Clin Exp Immunol. 2011;164:9–16. doi: 10.1111/j.1365-2249.2011.04335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dasouki M, Okonkwo KC, Ray A, et al. Deficient T cell receptor excision circles (TRECs) in autosomal recessive hyper IgE syndrome caused by DOCK8 mutation: implications for pathogenesis and potential detection by newborn screening. Clin Immunol. 2011;141:128–132. doi: 10.1016/j.clim.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Primary Immune Deficiency Treatment Consortium. [Accessed Sept. 30, 2011]; Available at: http://rarediseasesnetwork.epi.usf.edu/PIDTC/SCID/index.htm.
- 33.Griffith LM, Cowan MJ, Kohn DB, et al. Allogeneic hematopoietic cell transplantation for primary immune deficiency diseases: current status and critical needs. J Allergy Clin Immunol. 2008;122:1087–1096. doi: 10.1016/j.jaci.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buckley RH. B-cell function in severe combined immunodeficiency after stem cell or gene therapy: a review. J Allergy Clin Immunol. 2010;125:790–797. doi: 10.1016/j.jaci.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Therrell BL, Jr, Hannon WH, Bailey DB, Jr, et al. Committee report: considerations and recommendations for national guidance regarding the retention and use of residual dried blood spot specimens after newborn screening. Genet Med. 2011;13:621–624. doi: 10.1097/GIM.0b013e3182147639. [DOI] [PubMed] [Google Scholar]
- 36.Hiraki S, Green NS. Newborn screening for treatable genetic conditions: past, present and future. Obstet Gynecol Clin North Am. 2010;37:11–21. doi: 10.1016/j.ogc.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 37.Modell F, Puente D, Modell V. From genotype to phenotype. Further studies measuring the impact of a Physician Education and Public Awareness Campaign on early diagnosis and management of primary immunodeficiencies. Immunol Res. 2009;44:132–149. doi: 10.1007/s12026-008-8092-3. [DOI] [PubMed] [Google Scholar]