

Abstract
Cellular senescence is a response to stress that disables cell proliferation and orchestrates an inflammatory process that eliminates damaged cells. The first pro-senescence drugs for cancer treatment are now a clinical reality, but still few targets have been identified whose inactivation results in cancer cell senescence. Current work published in this issue of The EMBO Journal makes an important contribution to this area by discovering that pharmacological inhibition of the tyrosine phosphatase SHP2 blocks mouse mammary cancer through the induction of senescence (Lan et al, 2015).
See also: L Lan et al (June 2015)
The concept of cellular senescence has changed dramatically during last years (Munoz-Espin & Serrano, 2014). Whereas apoptosis is often deemed as ‘cellular suicide’ (cell autonomous), cellular senescence could be considered as ‘assisted cellular suicide’ (non-cell autonomous). At the end, both processes have a common goal, which is the elimination of cells under stress (or signaled for dismissal during development). Senescent cells are extremely active in secreting proteins, which could be the basis of their characteristic high levels of lysosomal enzymatic activities (most prominently β-galactosidase, hence known as senescence-associated β-galactosidase or SAβG). These secreted proteins include growth factors, cytokines and chemokines, and matrix remodeling proteases; the secretome of senescent cells is collectively known as the senescence-associated secretory phenotype (or SASP). The final outcome of the SASP is the recruitment of inflammatory cells, followed by the phagocytic elimination of the senescent cells and, finally, tissue repair. Therefore, senescent cells, in contrast to apoptotic cells, link their dismissal to a tissue reparative process that they orchestrate.
Considering the above scenario, it is not surprising that cellular senescence is an active component of embryonic developmental processes (reviewed in Munoz-Espin & Serrano, 2014) and tissue repair in adults, as during skin wound healing (Demaria et al, 2014). Senescence has, however, a negative side when damage is chronic and the virtuous sequence of senescence–inflammation–clearance–regeneration is corrupted. Indeed, senescent cells accumulate in multiple chronic pathologies in association with constitutive inflammatory levels and aberrant fibrosis (Munoz-Espin & Serrano, 2014).
Tumors thrive within a paramount stressful environment, and cancer cells usually have high levels of endogenous damage. Accordingly, cancer cells have evolved strategies to disable the mechanisms that trigger apoptosis and senescence (Fig1). The search for cancer therapies has been heavily focused on drugs that reactivate the apoptotic machinery of cancer cells, but a new trend is emerging aimed to reactivate senescence in cancer cells. This year has witnessed a landmark event with the approval by the FDA of Palbociclib for clinical use against a subset (ER+, HER2−) of advanced breast cancers. Palbociclib inhibits CDK4 and CDK6, and in this regard, it can be considered an activator of the p16/RB tumor suppressor pathway (Dickson, 2014). Although more studies are necessary, current evidence indicates that the main mode of action of Palbociclib is the induction of cancer cell senescence. Another class of drugs that are showing promising results derives from Nutlin-3a, an inhibitor of MDM2, that stabilizes p53 and that also induces senescence in many cancer cells (Barone et al, 2014). P16/RB and p53 are considered two of the most important routes to activate senescence, but other pathways certainly exist, such as the CDK2 inhibitor p27 (Munoz-Espin & Serrano, 2014). A better understanding of the oncogenic mechanisms that disable senescence in cancer cells is necessary for the development of additional pro-senescence therapies.
Figure 1. Cancer cells depend on SHP2 to cancel senescence and SHP2 inhibitors can reactivate senescence.
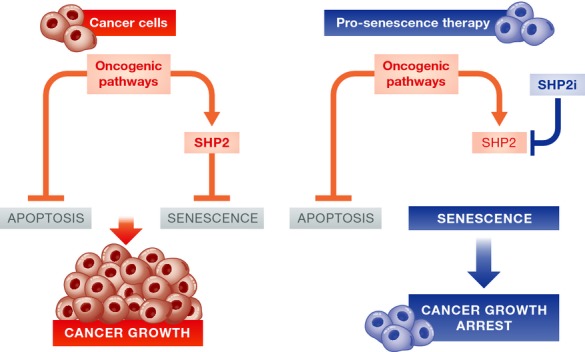
Left: Cancer cells use oncogenic pathways to silence the apoptotic and senescence programs. According to the current work (Lan et al, 2015), SHP2 is important for the silencing of the senescence program in breast cancer cells. Right: Pharmacological inhibition of SHP2 (SHP2i) reactivates the senescence program, cancer cells undergo senescence, and tumor growth is blocked.
The tyrosine phosphatase SHP2 (encoded by the PTPN11 gene) has been linked to cancer for a long time. This link is particularly compelling in the case of breast cancer because SHP2 levels are upregulated in up to 70% of ductal invasive breast carcinomas (Zhou et al, 2008). Previous studies have shown that SHP2 is critical for the expansion of breast cancer cells, and it appears responsible for their stem-like properties (Aceto et al, 2012). Inhibition of SHP2 prevented invasion, sphere formation, and the growth of tumor xenografts. Curiously, authors made note of the fact that these anti-tumoral effects of SHP2 inhibition were not accompanied by the induction of apoptosis, but other alternatives were not explored (Aceto et al, 2012). This sets the stage for the current study in The EMBO Journal where authors start by analyzing in further depth how SHP2 blocks breast cancer cells in vitro and they find that it induces all the hallmarks of senescence, including SAβG (Lan et al, 2015) (Fig1).
Remarkably, despite the wealth of information on the molecular activities of SHP2, we are still far from a detailed understanding of the signaling pathways controlled by SHP2. SHP2 possesses two SH2 domains for binding to tyrosine-phosphorylated residues characteristic of activated tyrosine kinase receptors. Therefore, SHP2 is at the apex of many signaling cascades and from that vantage point reinforces RAS/MAPK signaling, and inhibits JAK/STAT signaling. Furthermore, SHP2 is necessary to maintain high levels of key transcription factors, such as ZEB1 and cMYC, both implicated in self-renewal and stem-like properties (Aceto et al, 2012). However, the connection between SHP2 and senescence was unprecedented. To unravel the mechanisms involved, the authors of the current study in The EMBO Journal performed microarray gene expression analyses comparing mouse mammary cancer cells with or without genetic ablation of Shp2 (Lan et al, 2015). In this manner, the authors identified and validated (with gain- and loss-of-function experiments) a total of three SHP2 effectors that contribute to cancel senescence in cancer cells. These three effectors are the following ones: (1) upregulation of the E3 ubiquitin ligase SKP2 and, thereby, reduction in the levels of the cell cycle inhibitor p27; (2) upregulation of the mitotic kinase AURKA, which in turn contributes to inhibit p53; and (3) upregulation of the Notch receptor ligand DLL1, which also contributes to inhibit p53. Together, the increased levels of p27 and p53 provide a compelling explanation for the robust induction of senescence that occurs upon SHP2 inhibition.
An important bonus of the current study is the fact that all the above findings are recapitulated in vivo in a well-established model of mouse mammary cancer driven by the polyoma middle-t (PyMT) oncogene. In this model, the authors find that Shp2-null mammary glands undergo PyMT-driven hyperplasias to the same extent as the Shp2-wt controls. However, only Shp2-wt hyperplasias progress to form adenomas and carcinomas, whereas Shp2-null hyperplasias undergo senescence (Lan et al, 2015). Importantly, RNA expression analyses confirmed lower levels of Skp2, Aurka, and Dll1 in the senescent Shp2-null lesions
Phosphatases, from the early days of molecular oncology, were considered ‘undruggable’, and this, in turn, discouraged pharmaceutical companies from investing in the development of phosphatase inhibitors. Fortunately, this perception has changed during the last years and there are now promising advances regarding the pharmacological inhibition of SHP2 (Butterworth et al, 2014). Previously, an independent team reported that a SHP2 inhibitor, named PHPS1, was able to delay the growth of carcinogen-induced mammary cancers in rats and also of PyMT-driven mammary cancers in mice (Li et al, 2014). The authors of the current work in The EMBO Journal have used an improved SHP2 inhibitor, named GS493, that was able to prevent de novo PyMT-driven mammary cancer formation and, remarkably, it was also able to arrest the growth of already formed PyMT cancers. Importantly, cancers treated with the SHP2 inhibitor presented high levels of SAβG, p27 and p53, together with decreased expression of Skp2, Aurka, and Dll1 (Lan et al, 2015). In conclusion, this new study convincingly demonstrates that the anti-cancer therapeutic activity of SHP2 inhibition is associated with the induction of senescence.
An aspect that is left for future studies concerns the secretory phenotype elicited by SHP2 inhibition in breast cancer cells. This issue is of even higher relevance in light of an independent study connecting SHP2 with the SASP of prostate tumor cells (Toso et al, 2014). Prostate-specific expression of oncogenic Kras or deletion of the tumor suppressor Pten result in early prostate tumor lesions that undergo senescence.
However, the two types of senescence appear different in the sense that oncogenic-Kras senescence is much stronger than Pten-loss senescence in protecting from the emergence of prostate carcinomas. Interestingly, the SASPs associated with the two types of senescence share a number of common factors but differ in their levels of immune suppressive cytokines, such as CXCL1, CXCL2, GM-CSF, M-CSF, IL-10, and IL-13, and thereby in their ability to suppress anti-tumoral immune responses. Remarkably, molecular analyses revealed that SHP2 is a critical factor that distinguishes both types of SASPs. In the case of oncogenic Kras prostates, SHP2 is active, and this causes low STAT3 signaling and reduced levels of immune suppressive cytokines in the SASP (favoring a potent anti-tumor immune response). In contrast, in Pten-deficient prostates, SHP2 levels are reduced, STAT3 is active, and the SASP is rich in immune suppressive cytokines (favoring escape from immune surveillance). In support of this model, inhibition of STAT3 or its upstream regulator JAK2 converts the immune suppressive SASP of Pten-deficient prostates into an immune stimulatory SASP similar to the one of oncogenic Kras prostates (Toso et al, 2014). These findings could be relevant for the possible therapeutic use of SHP2 inhibitors in breast cancer. The senescence response elicited by SHP2 inhibition would be expected to upregulate STAT3 and to generate an immune suppressive SASP (similar to the one in Pten-deficient prostates). If this were the case, the combination of pharmacological inhibitors targeting SHP2 and STAT3 (or JAK2) could be particularly efficient.
In summary, the current study (Lan et al, 2015) provides a timely target for pro-senescence cancer therapy. The recently approved CDK4 and CDK6 inhibitor Palbociclib has demonstrated that pro-senescence therapies are effective against breast cancer, and inhibitors of SHP2 could well join the ranks in the near future.
References
- Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G, Confalonieri S, Quarto M, Hu G, Balwierz PJ, Pachkov M, Elledge SJ, van Nimwegen E, Stadler MB, Bentires-Alj M. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat Med. 2012;18:529–537. doi: 10.1038/nm.2645. [DOI] [PubMed] [Google Scholar]
- Barone G, Tweddle DA, Shohet JM, Chesler L, Moreno L, Pearson AD, Van Maerken T. MDM2-p53 interaction in paediatric solid tumours: preclinical rationale, biomarkers and resistance. Curr Drug Targets. 2014;15:114–123. doi: 10.2174/13894501113149990194. [DOI] [PubMed] [Google Scholar]
- Butterworth S, Overduin M, Barr AJ. Targeting protein tyrosine phosphatase SHP2 for therapeutic intervention. Future Med Chem. 2014;6:1423–1437. doi: 10.4155/fmc.14.88. [DOI] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME, Hoeijmakers JH, de Bruin A, Hara E, Campisi J. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson MA. Molecular pathways: CDK4 inhibitors for cancer therapy. Clin Cancer Res. 2014;20:3379–3383. doi: 10.1158/1078-0432.CCR-13-1551. [DOI] [PubMed] [Google Scholar]
- Lan L, Holland JD, Qi J, Grosskopf S, Vogel R, Gyorffy B, Wulf-Goldenberg A, Birchmeier W. Shp2 signaling suppresses senescence in PyMT-induced mammary gland cancer in mice. EMBO J. 2015;34:1493–1508. doi: 10.15252/embj.201489004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Kang Y, Wei L, Liu W, Tian Y, Chen B, Lin X, Li Y, Feng GS, Lu Z. Tyrosine phosphatase Shp2 mediates the estrogen biological action in breast cancer via interaction with the estrogen extranuclear receptor. PLoS ONE. 2014;9:e102847. doi: 10.1371/journal.pone.0102847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, Pinton S, Zhang J, Kalathur M, Civenni G, Jarrossay D, Montani E, Marini C, Garcia-Escudero R, Scanziani E, Grassi F, Pandolfi PP, Catapano CV, Alimonti A. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9:75–89. doi: 10.1016/j.celrep.2014.08.044. [DOI] [PubMed] [Google Scholar]
- Zhou X, Coad J, Ducatman B, Agazie YM. SHP2 is up-regulated in breast cancer cells and in infiltrating ductal carcinoma of the breast, implying its involvement in breast oncogenesis. Histopathology. 2008;53:389–402. doi: 10.1111/j.1365-2559.2008.03103.x. [DOI] [PubMed] [Google Scholar]