

Abstract
In this study, we have used techniques from cell biology, biochemistry, and genetics to investigate the role of the tyrosine phosphatase Shp2 in tumor cells of MMTV-PyMT mouse mammary glands. Genetic ablation or pharmacological inhibition of Shp2 induces senescence, as determined by the activation of senescence-associated β-gal (SA-β-gal), cyclin-dependent kinase inhibitor 1B (p27), p53, and histone 3 trimethylated lysine 9 (H3K9me3). Senescence induction leads to the inhibition of self-renewal of tumor cells and blockage of tumor formation and growth. A signaling cascade was identified that acts downstream of Shp2 to counter senescence: Src, focal adhesion kinase, and Map kinase inhibit senescence by activating the expression of S-phase kinase-associated protein 2 (Skp2), Aurora kinase A (Aurka), and the Notch ligand Delta-like 1 (Dll1), which block p27 and p53. Remarkably, the expression of Shp2 and of selected target genes predicts human breast cancer outcome. We conclude that therapies, which rely on senescence induction by inhibiting Shp2 or controlling its target gene products, may be useful in blocking breast cancer.
Keywords: Kaplan–Meier analysis, pro-senescence therapy, PTPN11, relapse-free survival, Shp2-dependent gene signature
See also: M Serrano (June 2015)
Introduction
Breast cancer is a heterogeneous disease that can be divided into different therapeutic groups based on the status of the estrogen receptor (ER) and HER2 (van't Veer et al, 2002; Hynes & Lane, 2005; Weigelt & Reis-Filho, 2009). Recent genomic and proteomic technologies have led to the characterization of various mutations and gene amplifications in breast cancers (Cancer Genome Atlas Network, 2012; Stephens et al, 2012). Among the frequently altered genes are the tumor suppressor genes TP53, RB1, and the cyclin-dependent kinase inhibitor 1B (CDKN1B/p27). Alterations in tyrosine kinases and tyrosine phosphatases have also been commonly identified, such as amplifications of the receptor tyrosine kinases EGFR, FGFR, and MET, and mutations in the tyrosine phosphatases PTPN22 and PTPR, which highlight the importance of tyrosine phosphorylation-dependent oncogenic signaling in breast cancer (Cancer Genome Atlas Network, 2012; Stephens et al, 2012). Indeed, deregulation of several tyrosine phosphatases has been previously described in human breast cancer (Julien et al, 2011). The protein tyrosine phosphatase 1B gene is overexpressed in a large subset of breast cancers (72%), particularly in association with HER2 overexpression (Wiener et al, 1994). The protein tyrosine phosphatase Shp2 (the PTPN11 gene product) is upregulated in 70% of invasive breast cancers (Zhou et al, 2008), and mutations of SHP2 are frequently found in leukemia (reviewed in Chan et al, 2008). Shp2 is a major regulator of growth factor, cytokine, and integrin signaling (Neel et al, 2003). Shp2 plays essential roles in early embryogenesis and in the development and maintenance of various organs such as the nervous system, the kidney, and the intestine (reviewed in Grossmann et al, 2010, 2009; Willecke et al, 2011; Heuberger et al, 2014). Recent studies have linked Shp2 to breast cancer: A study on human breast cancer cell lines that overexpress the EGFR and HER2 showed that Shp2 mediated mitogenic and survival signals in cancer cells and promoted epithelial–mesenchymal transition (EMT), while inhibition of Shp2 leads to mesenchymal–epithelial transition (Zhou & Agazie, 2008). In a further study, shRNA interference with Shp2 inhibited tumor growth and cancer stem cells in xenograft models of human HER2+ and triple-negative breast cancer cells by controlling c-Myc and zinc finger E-box binding homeobox 1 (ZEB1) factor. A Shp2-dependent gene signature was associated with invasive behavior and poor prognosis in a large set of human primary breast cancers (Aceto et al, 2012). Shp2 has also been shown to promote cell motility in triple-negative breast cancer cells (Sausgruber et al, 2014).
Cellular senescence is an irreversible growth arrest originally identified in cell culture, but is also observed in normal and pathological processes in vivo including embryonic development, aging, and cancer (Schmitt, 2003; Collado et al, 2007; Banito & Lowe, 2013). Senescence occurs during the development of human and mouse tumors such as melanocytic naevi, colon adenomas, lymphomas, and prostate cancer (reviewed in Collado & Serrano, 2010; Rodier & Campisi, 2011). Senescence has been viewed as a physiological barrier against tumor initiation and progression: It can be triggered by the activation of oncogenes in a process called oncogene-induced senescence (Serrano et al, 1997) or by loss of tumor suppressor genes (Chen et al, 2005). Senescence prevails in many different precancerous lesions in humans and mice, but is reduced in malignant tumors. Inducing senescence by therapeutic regimens may be used as a strategy for cancer treatment (reviewed in Nardella et al, 2011; Schmitt, 2003). Characteristically, senescent cells increase in size, express senescence-associated β-galactosidase (SA-β-gal), display senescence-associated heterochromatin foci (SAHF), and may be cleared by the innate immune system (reviewed in Kuilman et al, 2010). The senescence program is activated by key effector systems such as p16INK4A-Rb and Arf-p53. Senescence is also controlled by genes including p15, p21, and p27 (Kuilman et al, 2010).
Recent evidence shows that senescence may play an inhibitory role in breast cancer. In HrasG12V-induced mouse mammary gland tumors, high Ras signaling initially induced senescent growth arrest by activating the senescence checkpoint genes p19, p53, p21, and p16. Ultimately, however, full transformation required the absence of wild-type p53 and p21 (Sarkisian et al, 2007). In MMTV-ErbB2 mouse mammary gland tumors, the genetic inhibition of IκB kinase α (IKKα) produced senescence and inhibited the self-renewal of tumor cells, but these effects were not seen in MMTV-v-Ha-ras, MMTV-PyMT, MMTV-c-myc, or MMTV-Wnt-1 lines (Cao et al, 2007). The T-BOX2 (TBX2)-encoding transcription factor gene has been identified as a senescence suppressor that inhibits the p19Arf checkpoint gene, and it is amplified in subsets of sporadic and familial human breast cancers (Jacobs et al, 2000). TWIST1 or TWIST2, either of which induces EMT, suppressed senescence by abrogating p16 and p21 checkpoint genes, cooperated with HER2 to transform mammary gland epithelial cells, and promoted invasion and metastasis (Ansieau et al, 2008). The poly ADP-ribose polymerase (PARP) inhibitor veliparib (ABT-888) induced senescence in breast cancer cells in combination with ionizing radiation and exerted therapeutic effects on xenografts (Efimova et al, 2010). Finally, based on recent genomic and proteomic analyses, several senescence activators such as TP53, RB1, and CDKN1B/p27 were frequently found to be mutated in breast cancer (Cancer Genome Atlas Network, 2012). Thus, senescence may represent a common mechanism to restrict breast cancer development and may be useful in therapeutic approaches.
Here, we used cell biological and biochemical experiments as well as mouse genetics to investigate the role of Shp2 in mammary gland tumors of MMTV-PyMT mice, a well-established model of breast cancer (Guy et al, 1992). PyMT is an oncogene that induces rapidly growing and highly metastatic mammary gland cancers by activating distinct signaling systems such as Ras/Mapk, Pi3k/Akt, and Plc-Pkc (Dilworth, 2002; Malanchi et al, 2012). Additional signaling molecules including ErbB3, integrin β1, and periostin are also involved in PyMT-induced tumorigenesis and metastasis (White et al, 2004; Cook et al, 2011; Malanchi et al, 2012). We found that Shp2 signaling is highly upregulated in PyMT tumors and is crucial for mammary gland tumorigenesis, primarily by suppression of senescence. As modulators of this effect, Shp2 and its downstream targets Src, Fak, and Mek1/Map kinase control the expression of Skp2, Aurka, and Dll1 and regulate p27- and p53-dependent senescence. Our data suggest that senescence induction by inhibiting Shp2 or controlling its downstream targets may be useful in therapeutic approaches to breast cancer.
Results
Ablation or inhibition of the tyrosine phosphatase Shp2 induces senescence of PyMT mammary gland tumor cells in culture and inhibits their self-renewal
We assessed the expression of the tyrosine phosphatase Shp2 in mouse mammary gland epithelia and in PyMT mammary gland tumors (Guy et al, 1992). We found strongly elevated levels of Shp2 protein in the tumors (Supplementary Fig S1A; compare normal tissue with tumors T1-3). We then undertook a functional analysis of the potential role of Shp2 in the PyMT tumor cells. Primary tumor cells from PyMT mammary glands were isolated from 12-week-old mice carrying in addition homozygous floxed Shp2 alleles (PyMT;Shp2fl/fl, see Materials and Methods) (Grossmann et al, 2009). The tumor cells were then infected in tissue culture with retroviruses that expressed either CreERT2-GFP or control GFP (see scheme in Fig1A). GFP+ cells purified by FACS were treated for 2 h with 4-hydroxytamoxifen (4OHT) to ablate Shp2. CD24+CD90+ cells were enriched by FACS (Supplementary Fig S1B) (Malanchi et al, 2012) and cultured in serum-free non-adherent conditions as mammospheres to measure the self-renewal capacity of the cells (Fig1A) (Ponti et al, 2005). Western blot analysis showed that Shp2 knockout (ko) cells exhibited an almost complete loss of Shp2 protein, suggesting high efficiency of Shp2 ablation (Fig1B).
Figure 1.

- Scheme of Shp2 ablation in cultures of PyMT;Shp2fl/fl tumor cells using retroviruses, which expressed CreERT2-GFP or control GFP.
- Western blot analysis of Shp2 in control and Shp2 ko cells.
- Immunofluorescence analysis of Ki67 on control and Shp2 ko cells at day 7. Scale bar, 50 μm.
- Quantification of the numbers of Ki67+ cells shown in (C). Error bars represent SEM (n = 3). **P < 0.01.
- Sphere formation by control and Shp2 ko cells at day 7. Scale bar, 200 μm.
- Quantification of the numbers of spheres formed by control and Shp2 ko cells shown in (E). Error bars represent SEM (n = 3). **P < 0.01.
- Growth kinetics of spheres formed by control and Shp2 ko cells by counting sphere cells at day 4, 7, and 12. Error bars represent SEM (n = 3).
- SA-β-gal staining on spheres formed by control and Shp2 ko cells at day 7. Scale bar, 100 μm.
- Quantification of the numbers of senescent cells in the spheres shown in (H). Error bars represent SEM (n = 3). **P < 0.01.
- Western blot analysis of H3K9me3, p27, p53 pS18 (Ser18-phosphorylated p53), total p53, Shp2, and α-tubulin in control and Shp2 ko cells.
- SA-β-gal staining on spheres formed by control cells and those treated by the Shp2 inhibitor GS493 at 15 μM at day 7. Scale bar, 100 μm.
- Quantification of the numbers of senescent cells shown in (K). Error bars represent SEM (n = 3). **P < 0.01.
- Quantification of the numbers of spheres formed by control cells and those treated by the Shp2 inhibitor GS493 at 15 μM. Error bars represent SEM (n = 3). **P < 0.01. Statistical significance was assessed by Student's unpaired t-test.
We then assessed the proliferative and self-renewal capacities of the tumor cells, using immunofluorescence for the Ki67 nuclear antigen and counting the numbers of spheres and sphere cells. A significant reduction in proliferation and self-renewal was observed in Shp2 ko cells at day 7, compared to controls (Fig1C and E; quantified in Fig1D and F). After 7 days, Shp2 ko spheres almost completely ceased expansion (Fig1G). We also examined senescence in spheres by determining the activity of senescence-associated β-gal, SA-β-gal (Dimri et al, 1995). Remarkably, Shp2 ko spheres at day 7 contained 10-fold higher numbers of SA-β-gal+ cells (Fig1H; quantified in Fig1I). Moreover, we observed strong increases in levels of histone 3 trimethylated lysine 9 (H3K9me3), p27, Ser 18-phosphorylated p53, and total p53, which are also characteristic of senescence (Fig1J) (Kuilman et al, 2010). In contrast, apoptosis was not induced by Shp2 ablation, as determined by cleaved caspase-3 staining (Supplementary Fig S1C). We then performed rescue experiments by overexpressing full-size human Shp2 prior to endogenous Shp2 ablation in PyMT mouse tumor cells (Supplementary Fig S1D). Indeed, overexpression of human Shp2 prevented senescence in Shp2 ko mouse cells, demonstrating the specificity of Shp2 ablation (Supplementary Fig S1E and F).
We also examined the effect of Shp2 inhibition on PyMT tumor cells using the Shp2 inhibitor GS493. GS493 is an improved analog of the Shp2 inhibitor phenylhydrazonopyrazolone sulfonate 1 (PHPS1) that was produced in our laboratory (Hellmuth et al, 2008). SA-β-gal+ cells were strongly induced upon GS493 treatment (Fig1K; quantified in Fig1L), while the numbers of spheres were reduced (Fig1M). Collectively, these data demonstrate that Shp2 deficiency induces senescence and inhibits the self-renewal of PyMT tumor cells, but does not induce apoptosis.
Shp2 activates the expression of Skp2, Aurka, and Dll1/Hey1 genes to suppress p27- and p53-dependent senescence in PyMT tumor cells
We performed gene expression profiling in PyMT tumor cells to examine genes that are controlled by Shp2. We compared PyMT;Shp2fl/fl tumor cells under the two conditions: where Shp2 had been ablated by retroviral CreERT2 activation (Shp2 ko cells), and where Shp2 had been inhibited by GS493 (Fig2A; see also above). The two types of interventions produced similar changes in gene expression (Supplementary Table S1; GEO accession number GSE50518): Overall, 428 genes were deregulated with a fold-change of 1.5 or above. These genes were annotated by gene ontology, revealing three classes that were overrepresented: (i) genes that control cell cycle progression (55 genes downregulated), (ii) those that control DNA replication (25 genes downregulated), and (iii) p53 target genes (seven genes upregulated) (Fig2A; Supplementary Tables S2, S3 and S4). The changes of expression of these classes of genes suggest a cell cycle arrest in Shp2-deficient cells.
Figure 2.

- Heatmaps for cell cycle, DNA replication, and p53 target genes defined by gene ontology terms in control, Shp2 ko, and Shp2-inhibited PyMT spheres at day 7.
- qRT–PCR analysis of mRNA levels of Skp2, Aurka, Dll1, and Hey1 in control, Shp2 ko, and Shp2-inhibited cells. Error bars represent SEM (n = 3).
- SA-β-gal staining on control, Shp2 ko, and Shp2 ko cells with overexpression of Skp2, Aurka, or N3IC cDNA. Scale bar, 100 μm.
- Quantification of the numbers of senescent cells shown in (C). Error bars represent SEM (n = 3). **P < 0.01; ***P < 0.001.
- Quantification of the numbers of spheres formed by cells shown in (C). Error bars represent SEM (n = 3). *P < 0.05; **P < 0.01; and ***P < 0.001. Statistical significance was assessed by Student's unpaired t-test.
- Scheme showing an overview of Shp2-dependent signaling systems and target genes, which are involved in senescence and cell cycle programs in PyMT tumor cells.
Upregulation of Skp2, amplification of Aurka, and translocation of Notch have been detected in human breast cancers (Signoretti et al, 2002; Anand et al, 2003; Robinson et al, 2011); these genes were also implicated in senescence inhibition of different cancers such as melanoma, prostate, or colon cancer (Huck et al, 2010; Lin et al, 2010; Kang et al, 2013). Interestingly, we found that Skp2, Aurka (encoding Aurora A kinase), Dll1 (encoding a Notch ligand), and the Notch target gene Hey1 were downregulated by Shp2 ablation or inhibition (Fig2B). We thus asked whether Skp2, Aurka, and Dll1 mediate the functions of Shp2 in senescence suppression of PyMT tumor cells. We treated the cells with small molecular inhibitors of the NEDD8-activating enzyme (NAE), which functions as an activator of the Skp2-Skp1-cullin1-Rbx1 complex, MLN4924, and of Aurora A, VX-680 (Harrington et al, 2004; Brownell et al, 2010). Both inhibitors induced senescence and reduced sphere formation in dosage-dependent manners (Supplementary Fig S2A, A′, B and B′). We then overexpressed Skp2 and Aurora A in PyMT tumor cells, followed by Shp2 ablation. Overexpression of both cDNAs reduced senescence and induced sphere formation (Fig2C, quantified in Fig2D and E). To test whether Dll1-mediated Notch signaling is also involved in Shp2-controlled senescence, we treated PyMT tumor cells with the Notch inhibitor DAPT (Micchelli et al, 2003). Senescence was indeed induced, and spheres were reduced (Supplementary Fig S2C and C′). Moreover, activation of Notch signaling through the expression of the cDNA of the Notch3 intracellular domain, N3IC, followed by Shp2 ablation, completely abrogated senescence and strongly increased sphere formation (Fig2C, quantified in Fig2D and E; summarized in the scheme of Fig2F).
p27 and p53 represent two important senescence effectors (Kuilman et al, 2010), and both were activated in Shp2 ko cells (see above). We therefore examined whether p27 and p53 might act downstream of Shp2 to execute senescence. We first knocked down p27 and p53 by retrovirus-based shRNAs, followed by ablation of Shp2 (Supplementary Fig S3A and B). Indeed, both p27 and p53 knockdowns reduced senescence and increased self-renewal (Fig3A; quantified in Fig3B and C). We then determined the protein levels of p27 and p53 in Shp2 ko and cDNA-rescued cells. Both proteins were elevated in Shp2 ko cells. p27 was reduced by overexpression of Skp2 but not Aurka or N3IC cDNA, and p53 was reduced by the overexpression of Aurka and N3IC but not Skp2 cDNA (Fig3D). We also measured p27 and p53 proteins in PyMT tumor cells, following treatment with pharmacological inhibitors. Inhibition of Skp2 by MLN4924 induced p27, and inhibition of Aurora A by VX-680 or inhibition of Notch by DAPT induced p53 (Fig3E). These data suggest that p27 acts downstream of Skp2 and p53 downstream of Aurora A and Notch. Moreover, the induction of senescence by MLN4924 was prevented by p27 knockdown, and senescence by VX-680 or DAPT was prevented by p53 knockdown (Fig3F–H). Overall, these data show that p27 and p53 indeed act downstream of Shp2 to control the senescence program and that their level and activity are modulated by Skp2, Aurka, and Dll1/Hey1 (summarized in the scheme of Fig2F).
Figure 3.
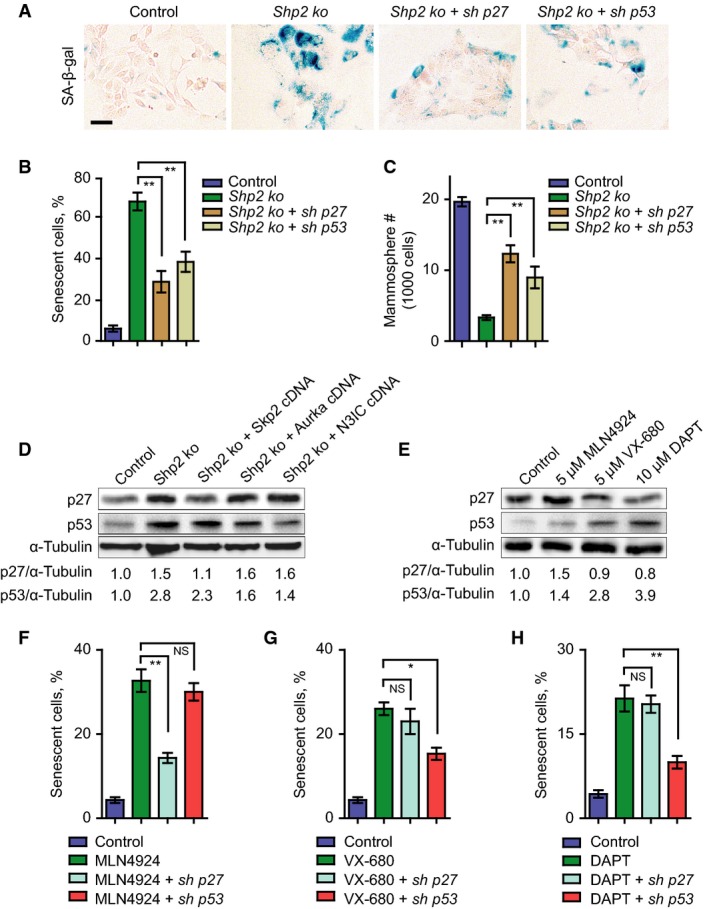
- SA-β-gal staining on control, Shp2 ko, and Shp2 ko cells with shRNA knockdown of p27 or p53. Scale bar, 50 μm.
- Quantification of the numbers of senescent cells shown in (A). Error bars represent SEM (n = 3). **P < 0.01.
- Quantification of the numbers of spheres formed by cells shown in (A). Error bars represent SEM (n = 3). **P < 0.01.
- Western blot analysis of p27, p53, and α-tubulin in control, Shp2 ko, and Shp2 ko cells with overexpression of Skp2, Aurka, or N3IC cDNAs. The numbers below indicate the relative ratios of band intensities between p27 and α-tubulin or between p53 and α-tubulin.
- Western blot analysis of p27, p53, and α-tubulin in PyMT cells treated with control (DMSO) or inhibitors against Skp2 (MLN4924), Aurora A (VX-680), or Notch (DAPT) at the indicated concentrations. The numbers below indicate the relative ratios of band intensities between p27 and α-tubulin or between p53 and α-tubulin.
- Quantification of the numbers of senescent cells in PyMT cells treated with control, MLN4924, MLN4924 plus p27 knockdown or MLN4924 plus p53 knockdown. Error bars represent SEM (n = 3). **P < 0.01; NS, not significant.
- Quantification of the numbers of senescent cells in PyMT cells treated with control, VX-680, VX-680 plus p27 knockdown, or VX-680 plus p53 knockdown. Error bars represent SEM (n = 3). *P < 0.05; NS, not significant.
- Quantification of the numbers of senescent cells in PyMT cells treated with control, DAPT, DAPT plus p27 knockdown, or DAPT plus p53 knockdown. Error bars represent SEM (n = 3). **P < 0.01; NS, not significant. Statistical significance was assessed by Student's unpaired t-test.
Shp2 activates Src, Fak, and Mek1 signaling to suppress senescence in PyMT tumor cells
We used pharmacological inhibitors to examine various signaling systems that had been shown to act downstream of Shp2 (Neel et al, 2003; Grossmann et al, 2009, 2010) in PyMT mammary gland tumor cells. Inhibition of Shp2 (by GS493), Src (by PP2), Fak (by TAE226), or Mek1 (by U0126), but not Met, Pi3k, or mTor, induced senescence in PyMT cells and reduced self-renewal in dosage-dependent manners (Fig4A, quantified in Fig4B and C; dosage dependency is shown in Supplementary Fig S4A and B; data not shown). We also detected crosstalk between Shp2 and downstream molecules in PyMT tumor cells at the level of posttranslational modifications; for instance, Shp2 inhibition increased Tyr 527 phosphorylation, which represents an inhibitory phosphorylation site of Src, and decreased Tyr 416 phosphorylation, which is an activating phosphorylation site of Src (Fig4D) (Grossmann et al, 2009). Moreover, phosphorylation of Tyr 925 of Fak, which is an activating phosphorylation site (Grossmann et al, 2009), and Erk1/2 activity were reduced by Shp2 inhibition (Fig4D). The effects of the pharmacological inhibitors of Shp2, Fak, Src, and Mek1 were also examined on the regulation of the newly identified genes Skp2, Aurka, Dll1, and Hey1, which control senescence and the cell cycle. The Src but not the Mek1 inhibitor reduced the expression of Skp2 and Aurka, the Mek1 but not the Src inhibitor reduced Dll1 and Hey1, and the Fak inhibitor reduced all four genes, as did the Shp2 inhibitor (Fig4E, see scheme in Fig4F). Apparently, Skp2 and Aurka expression are controlled by Src, and Dll1 and Hey1 expression are regulated by Mek1, while Fak controls the expression of all four genes (Fig4F). We have thus identified the network of expression of crucial genes downstream of Shp2 that controls senescence and the cell cycle in PyMT tumor cells (summarized in the scheme of Fig2F).
Figure 4.
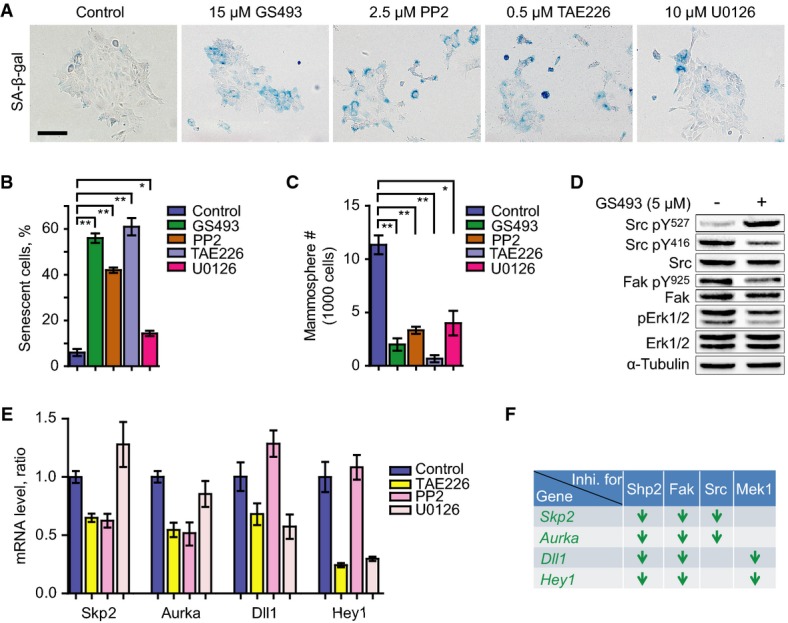
- SA-β-gal staining on PyMT tumor cells treated with control, Shp2 inhibitor GS493 (15 μM), Src inhibitor PP2 (2.5 μM), Fak inhibitor TAE226 (0.5 μM), or Mek1 inhibitor U0126 (20 μM). Scale bar, 100 μm.
- Quantification of the numbers of senescent cells in the presence or absence of the inhibitors, as shown in (A). Error bars represent SEM (n = 3). *P < 0.05; **P < 0.01.
- Quantification of the numbers of spheres in the presence or absence of the inhibitors, as in (A). Error bars represent SEM (n = 3). *P < 0.05; **P < 0.01.
- Western blot analysis of pY527, pY416 and total Src, pY925 and total Fak, pT202/pT204 and total Erk1/2, and α-tubulin in PyMT tumor cells with or without treatment with 5 μM GS493.
- qRT–PCR analysis of mRNA levels of Skp2, Aurka, Dll1, and Hey1 in control cells and those treated with the indicated inhibitors. Error bars represent SEM (n = 3).
- Summary of the deregulated genes by the chemical inhibition of Shp2, Src, Fak, and Mek1 signaling. Arrows represent downregulation of genes. Statistical significance was assessed by Student's unpaired t-test.
Shp2-regulated gene expression programs predict the survival of breast cancer patients
We performed retrospective Kaplan–Meier survival analyses on publically available data from 3,935 human patients with invasive breast cancers (Gyorffy & Schafer, 2009), based on the expression of Shp2 and Shp2-regulated genes. We found that higher mRNA levels of Shp2 significantly correlated with shorter survival of patients (Fig5A). When the overall expression of all Shp2-deregulated genes was considered, the findings were not predictive of patient outcome (Supplementary Fig S5A and B). However, the expression of Shp2 target genes that belong to the three overrepresented classes listed above (cell cycle progression, DNA replication, and p53 targets) was highly predictive of relapse-free survival times (Fig5B–D). Moreover, the expressions of the single target genes Skp2 or Aurka were also strong prognostic factors that predict patient survival (Fig5E and F). Our data thus show that the expression pattern of Shp2 and of selected Shp2 target genes is highly predictive for human breast cancer outcome.
Figure 5.

- A Kaplan–Meier survival analysis of human patients (n = 3,935) with invasive breast cancers divided into two groups based on the gene expression level of Shp2 (see Materials and Methods).
- B–D Kaplan–Meier survival analyses of the human patients (n = 3,935) divided into two groups based on the expression level of the genes of each overrepresented class determined in Fig2A: cell cycle genes, DNA replication genes, or p53 target genes.
- E, F Kaplan–Meier survival analyses of human patients (n = 3,935) divided into two groups based on the gene expression level of Skp2 or Aurka. HR, hazard ratio.
Conditional ablation of Shp2 in mice delays the onset of PyMT-induced mammary gland tumors
To examine the role of endogenous Shp2 in PyMT mammary gland tumors in mice, we introduced a conditional mutation of Shp2 in the mammary gland of PyMT mice using MMTV-Cre (PyMT;MMTV-Cre;Shp2fl/fl, subsequently referred to as PyMT;coShp2 mutant) (Guy et al, 1992; Grossmann et al, 2009). To assess recombination, MMTV-Cre was used in combination with an EYFP reporter (MMTV-Cre;RosaEYFP−/fl) (Srinivas et al, 2001), where EYFP expression is generated after Cre-mediated deletion of a stop cassette. MMTV-Cre efficiently induced EYFP expression in the mammary gland epithelium (Supplementary Fig S6A). PCR analysis confirmed that MMTV-Cre recombined the floxed Shp2 allele in the mammary gland of PyMT mice (Supplementary Fig S6B). We found that conditional Shp2 mutation delayed mammary gland tumor formation induced by PyMT (Fig6A). PyMT;coShp2 mice presented with palpable tumors at 14 weeks (brown curve), whereas control PyMT mice that either lacked the MMTV-Cre allele (PyMT;Shp2fl/fl) or carried a heterozygous Shp2fl allele (PyMT;MMTV-cre;Shp2+/fl, also referred to as control PyMT mice) developed palpable tumors at 10 weeks (blue and green curves, respectively). A much stronger effect on tumor formation was observed at early stages, with a 75% reduction in hyperplastic nodules at 5–8 weeks (Fig6B; quantified in Fig6C).
Figure 6.
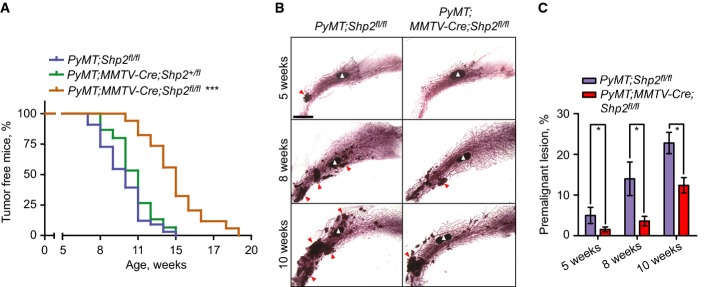
- Kaplan–Meier analyses of tumor formation for control (PyMT;Shp2fl/fl, blue, n = 33), heterozygous Shp2 mutant (PyMT;MMTV-Cre;Shp2+/fl, green, n = 15), and homozygous Shp2 mutant (PyMT;MMTV-Cre;Shp2fl/fl, also called PyMT;coShp2, orange, n = 34) mice. ***P < 0.001 (log-rank test).
- Mammary gland whole-mounts from control PyMT and PyMT;MMTV-Cre;Shp2fl/fl mice at indicated ages. The characteristic lymph node of the mammary gland fat pad is marked by white arrowheads; premalignant lesions are marked by red arrowheads. Scale bar, 4 mm.
- Quantification of premalignant lesions in mammary glands of control PyMT and PyMT;MMTV-Cre;Shp2fl/fl mice, as shown in (B). Error bars represent SEM (n = 6). *P < 0.05 (Student's unpaired t-test).
We analyzed EYFP expression at different stages of mammary gland tumor formation in control PyMT (PyMT;MMTV-Cre;Shp2+/fl) and PyMT;coShp2 mice that additionally carried the EYFP reporter. Remarkably, EYFP+ cells were gradually lost during tumor development in PyMT;coShp2 mice: At the hyperplasia stage, EYFP was broadly expressed both in areas of the mammary gland that displayed normal epithelial morphology and in hyperplasia (Fig7A, bottom left). At the adenoma stage, EYFP expression was only maintained in hyperplastic nodules, but not in adenomas (Fig7A, bottom middle; compare arrowheads and arrows). At the carcinoma stage, EYFP expression was completely absent (Fig7A, bottom right). In contrast, EYFP was widely expressed at all stages of mammary gland tumors in control PyMT;MMTV-Cre;Shp2+/fl mice (Fig7A, top panel). These data demonstrate that EYFP+ cells, in which Shp2 was recombined, were gradually out-competed by non-recombined PyMT tumor cells. Indeed, staining of late-stage mammary gland tumors of PyMT;coShp2 mice with anti-Cre antibody revealed absence of Cre (Fig7B, top right, see control without MMTV-Cre on top left). In contrast, Cre was maintained in tumors of control PyMT;MMTV-Cre;Shp2+/fl mice at the late stage (Fig7B, top middle). Metastases in the lungs were observable starting at 14–18 weeks in control PyMT and PyMT;coShp2 mice (Fig7B, bottom panel). Cre was produced in metastases of control PyMT;MMTV-Cre;Shp2+/fl mice, but was absent in PyMT;coShp2 mice (Fig7B, bottom middle and right, respectively; PyMT mice on the left). Furthermore, we measured mRNA and protein levels of Shp2 in late-stage mammary gland tumors. In control PyMT;MMTV-Cre;Shp2+/fl mice, we observed a 50% reduction in the levels of Shp2 mRNA and protein, while high levels were comparable between PyMT;coShp2 and PyMT;Shp2fl/fl mice (Fig7C and D; compare middle with right and left panels). These data confirm that late-stage tumors and metastases developed from cells that had escaped recombination, potentially because of initial mosaic patterns of MMTV-Cre expression. Escape from recombination has also been observed in other studies using MMTV-Cre and other Cre lines (Chmielowiec et al, 2007; Marcotte et al, 2012).
Figure 7.
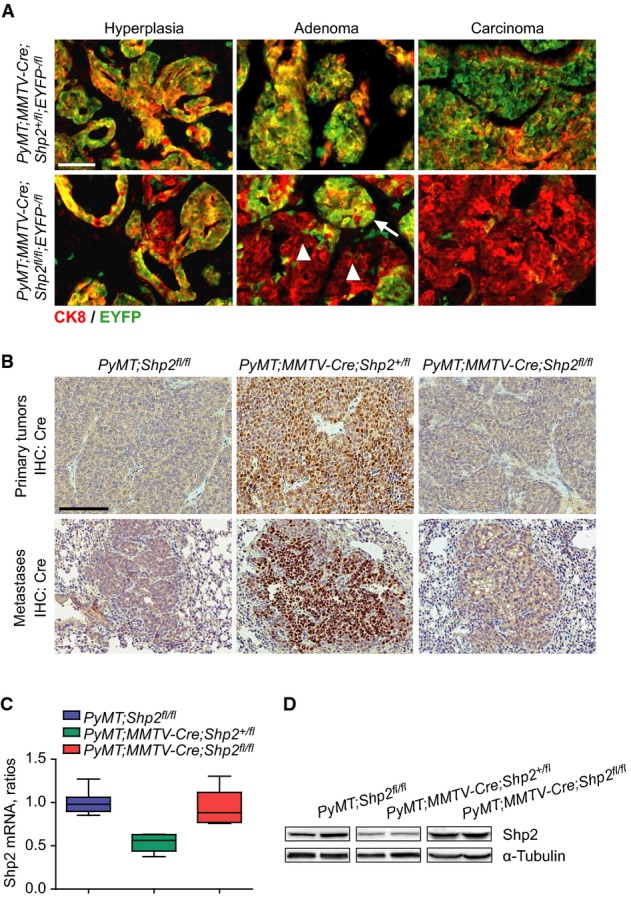
- Immunostaining of EYFP and CK8 on mammary gland sections from PyMT;MMTV-Cre;Shp2+/fl;EYFP−/fl and PyMT;MMTV-Cre;Shp2fl/fl;EYFP−/fl mice at hyperplasia, adenoma, and carcinoma stages. Scale bar, 100 μm. n = 8.
- Immunohistochemistry analysis of Cre recombinase on paraffin sections of primary tumors (top) and metastases (bottom) from PyMT;Shp2fl/fl, PyMT;MMTV-Cre;Shp2+/fl, and PyMT;MMTV-Cre;Shp2fl/fl mice. Scale bar, 100 μm. n = 8.
- qRT–PCR analyses of Shp2 mRNA levels in tumor tissues from PyMT;Shp2fl/fl (n = 8), PyMT;MMTV-Cre;Shp2+/fl (n = 6), and PyMT;MMTV-Cre;Shp2fl/fl (n = 6) mice. Error bars represent upper and lower quartiles.
- Western blot analysis of Shp2 protein levels in tumor tissues from PyMT;Shp2fl/fl, PyMT;MMTV-Cre;Shp2+/fl, and PyMT;MMTV-Cre;Shp2fl/fl mice.
Shp2 ablation induces senescence but not apoptosis of PyMT mammary gland tumors in mice
We also examined senescence in endogenous tumors of control PyMT;MMTV-Cre;Shp2+/fl and PyMT;coShp2 mice that additionally carried the EYFP reporter. We performed SA-β-gal staining of tumor sections: EYFP+ cells exhibited extensive SA-β-gal staining in tumors of PyMT;coShp2 mice, showing senescence in areas in which recombination had occurred and Shp2 was ablated (Fig8A, right, compare top and bottom; arrows on the serial sections mark EYFP+ and SA-β-gal+ areas). Conversely, senescent cells were virtually absent in areas where EYFP was negative and recombination was prevented (Fig8A, right, compare top and bottom; arrowheads on the serial sections mark EYFP-negative and SA-β-gal-negative areas). Sections of control PyMT tumors showed little SA-β-gal staining (Fig8A, left and middle). We then evaluated Ki67, p27, and p53 in tumor sections by immunofluorescence. Ki67 staining revealed high proliferation in hyperplasias and tumors derived from PyMT;coShp2 mice in areas where EYFP was absent, that is, where cells had escaped recombination (Fig8B, top right, marked by arrowheads). In contrast, Ki67 staining was absent in areas that were EYFP+ (Fig8B, top right, marked by arrows). In control mice (PyMT;MMTV-Cre;Shp2+/fl), EYFP and Ki67 staining co-localized extensively (Fig8B, top middle). Both p27 and p53 staining also co-localized with EYFP in tumors from PyMT;coShp2 mice (Fig8B, middle and bottom right, arrows), whereas p27 and p53 were not detected in EYFP-negative areas (Fig8B, middle and bottom right, arrowheads). These results demonstrate that conditional ablation of Shp2 in mammary gland tumors blocked cell proliferation and induced senescence. Further analyses of tissue sections from PyMT;coShp2;EYFP tumors at different stages (hyperplasia, adenoma, and carcinoma) clearly showed a gradual loss of EYFP+ cells and SA-β-gal+ cells and a steady gain of Ki67+ cells during tumor development, while the number of cells expressing the pan-epithelial marker keratin 8 (CK8) remained stable (Supplementary Fig S7A, top and bottom panel; quantified in Supplementary Fig S7B). Shp2 ablation did not induce apoptosis (Supplementary Fig S7A, middle panel). These findings show that ablation of Shp2 in PyMT mammary glands induces senescence in endogenous mouse tumors.
Figure 8.

Shp2 ablation induces senescence in mammary gland tumors in PyMT mice
- Immunohistochemistry analysis of EYFP (top) and SA-β-gal staining (bottom) on cryosections of mammary gland tumors from control PyMT;Shp2fl/fl;EYFP−/fl, PyMT;MMTV-Cre;Shp2+/fl;EYFP−/fl, and PyMT;MMTV-Cre;Shp2fl/fl;EYFP−/fl mice. Note that SA-β-gal activity was present in the EYFP-positive (Shp2-negative) areas (right, arrows) and was absent in the EYFP-negative (Shp2-positive) areas (right, arrowhead) of mammary gland tumors of PyMT;MMTV-Cre;Shp2fl/fl;EYFP−/fl mice. Consecutive sections of EYFP and SA-β-gal staining for each group are shown. Scale bar: 100 μm. n = 5.
- Immunofluorescence analysis of Ki67, EYFP and CK8 (pan-epithelial marker) (top panel), p27 and EYFP (middle panel), and p53 and EYFP (bottom panel) on cryosections of mammary gland tumors from control PyMT;Shp2fl/fl;EYFP−/fl, PyMT;MMTV-Cre;Shp2+/fl;EYFP−/fl, and PyMT;MMTV-Cre;Shp2fl/fl;EYFP−/fl mice. DAPI was used for nuclear counterstain. Ki67 was absent in EYFP-positive cells (top right, arrows) and was present in EYFP-negative cells (top right, arrowheads); p27 was present in EYFP-positive cells (middle right, arrows) and was absent in EYFP-negative cells (middle right, arrowhead); and p53 was present in EYFP-positive cells (bottom right, arrows) and was absent in EYFP-negative cells (bottom right, arrowhead). Scale bar: top panel, 100 μm; middle and bottom panel, 50 μm. n = 5.
- qRT–PCR analysis of mRNA levels of Shp2, Skp2, Aurka, Dll1, and Hey1 in EYFP+ and EYFP− cells isolated by FACS from PyMT;MMTV-Cre;Shp2fl/fl;EYFP−/fl tumors. Error bars represent SEM (n = 5).
- Kaplan–Meier analysis of tumor formation in control (blue, n = 8) and GS493-pretreated (red, n = 8) PyMT;MMTV-Cre;Shp2fl/fl mice. The bar indicates the duration of treatment.
- Tumor growth curves for control (blue, n = 5) and GS493-treated (red, n = 5) PyMT mice. Note that after day 24, all mice in the control group were sacrificed due to the tumor size. Error bars represent SEM (n = 5).
We also examined the expression of Shp2 target genes in the in vivo settings. Primary tumor cells were freshly isolated from PyMT;coShp2;EYFP mice; EYFP+ and EYFP− tumor cells were then separated by FACS and collected for the analysis of gene expression. Shp2 expression was reduced in EYFP+ cells that had undergone efficient recombination (Fig8C). Importantly, the mRNA levels of Skp2, Aurka, Dll1, and Hey1 were also downregulated in EYFP+ cells that had lost Shp2 (Fig8C). Taken together, these data on endogenous tumors confirm that Shp2 activates Skp2, Aurka, and Dll1/Hey1 in senescence suppression.
The Shp2 inhibitor GS493 prevents tumor formation at early stages and inhibits tumor growth at late stages in PyMT mice
We examined whether GS493 could block tumor formation in PyMT;coShp2 mice. The compound was injected i.p. for 3 weeks, starting 7 weeks after birth, that is, before tumors became palpable and visible. Remarkably, GS493 completely blocked tumor formation in PyMT;coShp2 mice (Fig8D, Supplementary Fig S8A, red curves, compare with blue curves of untreated mice; the treatment period is marked by black bar). Treatment with the inhibitor in PyMT;coShp2 animals was well tolerated, as seen by the lack of effects on body weight (Supplementary Fig S8B; the increase in weight at later stages is due to tumors that developed in untreated but not in treated animals).
We also examined whether the Shp2 inhibitor GS493 could arrest tumor growth or regress tumors in tumor-bearing PyMT mice. The compound was administered i.p., starting when tumors were already palpable, and the kinetics of tumor growth was assessed during treatment. A significant inhibitory effect of GS493 on the growth of already existing tumors was seen after 10 days of treatment, and remarkably, tumor growth was halted at 3 weeks of treatment and in the following period (Fig8E, red curve, compared with blue curve of untreated mice). We carried out SA-β-gal staining as well as immunohistochemistry of p27 and p53 on tumor sections of the treated mice. Indeed, tumors from GS493-treated mice contained 10-fold more SA-β-gal+ cells, compared with tumors from untreated mice (Supplementary Fig S8C, top panel; quantified in Supplementary Fig S8D). Accordingly, the numbers of p27- and p53-positive cells were increased by sixfold and 12-fold, respectively (Supplementary Fig S8C, middle and bottom panel; quantified in Supplementary Fig S8D). Gene expression analysis revealed downregulation of mRNA levels of the Shp2 target genes Skp2, Aurka, Dll1, and Hey1 in tumors from GS493-treated mice (Supplementary Fig S8E). Taken together, these data demonstrate that the Shp2 inhibitor GS493 inhibits tumor growth by inducing senescence in PyMT mice.
Discussion
In this study, we have used techniques from cell biology, biochemistry, and genetics to investigate the role of the tyrosine phosphatase Shp2 in MMTV-PyMT mouse mammary gland tumor cells. Shp2 expression is highly upregulated in PyMT tumor cells, compared with normal mammary gland epithelial cells. Genetic ablation or pharmacological blockage of Shp2 inhibited cell proliferation and self-renewal of PyMT tumor cells. Remarkably, following Shp2 ablation and inhibition, we observed strong induction of senescence, as evidenced by characteristic markers: high levels of senescence-associated β-gal (SA-β-gal), p27, phosphorylated serine 18 of p53 (p53-pSer18), and histone 3 lysine trimethylation (H3K9me3). In contrast, blocking Shp2 did not lead to apoptosis. We also evaluated the role of known downstream signaling systems and identified target genes of Shp2 that are essential in senescence regulation: Shp2 controls Src, Fak, and Mek1 to activate the expression of the genes Skp2, Aurka, and Dll1/Hey1, which in turn block p27- and p53-mediated senescence (see scheme in Fig2F). Senescence also occurs in human breast cancers during therapeutic interventions: for instance, PARP inhibition induces senescence in irradiated breast cancers and xenograft tumors (Efimova et al, 2010). Recently, global genomic and proteomic analyses have revealed frequent amplification of the senescence suppressors CDK4 and CDK6 and inactivation of the senescence inducers p53, Rb1, and p27 (Cancer Genome Atlas Network, 2012). Overall, these data suggest that inhibition of the senescence program may be a common event during the development of breast cancer.
We used gene profiling and functional validation to identify effectors of Shp2 signaling that mediate senescence suppression in PyMT tumor cells (scheme in Fig2F). An essential downstream effector of Shp2 is Skp2, which is an F-box/E3-ubiquitin ligase in complex with Skp1-cullin1-Rbx1 (SCF), the importance of which we have confirmed through treatment with a small molecular inhibitor of cullin1 neddylation and overexpression of Skp2 in the context of Shp2 ablation. The SCF-Skp2 complex targets cyclin-dependent kinase inhibitors for degradation such as p27 and thus promotes cell cycle progression (reviewed in Nakayama & Nakayama, 2005). A senescence-suppressive function of Skp2 has been identified in several mouse and human tumors such as lymphoma and prostate cancer (Lin et al, 2010), but so far not in breast cancer. Of particular relevance are the facts that Skp2 is highly expressed in ER- and HER2-negative breast cancers (Signoretti et al, 2002), and that expression correlates with poor prognosis (Traub et al, 2006). A further effector of Shp2 is Aurora A (scheme in Fig2F), which is an important cell cycle regulator that controls mitosis entry and the assembly of the mitotic spindle (Vader & Lens, 2008). We have confirmed the importance of Aurora A through treatment with a new small molecular inhibitor and the overexpression of Aurora A in the context of Shp2 ablation. Aurora A plays a role in senescence inhibition in melanoma and colon cancer (Huck et al, 2010), but a similar function in breast cancer has not been addressed. However, amplification of Aurka has been reported in a subset of breast cancers (Anand et al, 2003). Finally, a downstream effector of Shp2 is Dll1/Notch3 (scheme in Fig2F), whose relevance in senescence we have confirmed in PyMT tumor cells through treatment with a specific Notch inhibitor and the overexpression of the Notch3 intracellular domain in the context of Shp2 ablation. A constitutive activation of Notch3 signaling induces mammary gland tumors in mice (Guo et al, 2011), and Notch3 expression is associated with poor survival of breast cancer patients (Reedijk et al, 2005). Note that N3IC fully rescued Shp2 ko-induced senescence. This may not mean that Notch signaling solely mediates Shp2 function, since Notch inhibition only partially phenocopied Shp2 deficiency. Moreover, N3IC is a constitutively active form of Notch3, and constitutive activation of Notch may lead to excessive signaling.
We have identified p27 and p53 as downstream mediators of Shp2-controlled senescence, as demonstrated by the fact that knockdown of both p27 and p53 in Shp2 ko cells abrogated senescence and restored proliferation (scheme of Fig2E). p27 and p53 represent major executors of senescence in cancer (Salama et al, 2014), and both p27 and p53 are frequently mutated in human breast cancers (Cancer Genome Atlas Network, 2012). We have also established a link between these senescence executors and Shp2 target genes: p27 is controlled by Skp2, and p53 is regulated by both Aurora A and Dll1/Notch3, since we observed that: (i) overexpression of Skp2 reduced p27, while inhibition induced p27, (ii) overexpression of Aurora A or N3IC reduced p53, while inhibition of both induced p53, (iii) senescence by inhibition of Skp2 is prevented by knockdown of p27, and (iv) senescence by inhibition of Aurora A or Notch is blocked by knockdown of p53. p27 is a target for ubiquitination and degradation by Skp2 (Nakayama & Nakayama, 2005), a mechanism which is likely to be exploited by PyMT tumor cells; however, more biochemical data are needed for confirmation. p53 is directly phosphorylated by Aurora A in a process, which disrupts p53 stability and DNA-binding activity (Vader & Lens, 2008). Whether this mechanism takes place in PyMT tumor cells remains to be investigated. How Notch signaling regulates p53 also needs to be studied by biochemical means.
We have taken efforts to dissect the signaling cascades that act downstream of Shp2 in PyMT tumor cells to regulate senescence. Shp2 activates the signaling molecules Src, Fak, and Mek1. These are crucial in the suppression of senescence in PyMT tumor cells, as we have shown by using specific pharmacological inhibitors (scheme in Fig2F). Our comparison of the gene expression profiles of cells treated with the different inhibitors revealed distinct Shp2 target genes for each signaling system: Src regulates both Skp2 and Aurka, and Mek1 regulates Dll1 and Hey1, but Fak regulates all these genes. Apparently, Src, Fak, and Mek1 regulate different subsets of gene expression programs in order to control senescence. Previous genetic studies in mice have revealed an important function of Src and Fak in PyMT tumors: Both are essential for PyMT-induced proliferation and transformation (Pylayeva et al, 2009; Marcotte et al, 2012). However, Src and Fak may have additional functions in PyMT tumors, since conditional ablation reduced the expression of cyclins A, B, and E (in case of Src) and induced apoptosis (in case of Fak) (Pylayeva et al, 2009; Marcotte et al, 2012). We did not observe these changes in the Shp2 ko and Shp2-inhibited PyMT tumor cells. Shp2 could also modulate Src, Fak, and Mek1 that act downstream of ErbB3 or integrin β1, which are also crucial for PyMT tumor development (White et al, 2004; Cook et al, 2011).
The functional role of Shp2 in breast cancer has recently been investigated by others (Zhou & Agazie, 2008; Aceto et al, 2012; Sausgruber et al, 2014). In human breast cancer, in breast cancer cell lines that overexpress HER2 (the lines BT474, SKBR3, and MCF10A-NeuNT), and in triple-negative breast cancer cells (the lines SUM159 and SUM1315), Shp2 activated the proliferation-controlling gene c-Myc and the EMT-inducer Zeb1. Moreover, Shp2 knockdown blocked tumor growth and invasion and suppressed tumor-initiating cells (Aceto et al, 2012). In the present study, we also observed a strong effect of Shp2 ablation on proliferation and self-renewal of PyMT mouse mammary gland tumor cells; however, we did not find an involvement of EMT. Instead, we observed that Shp2 ablation and inhibition induced senescence in PyMT tumor cells. The senescence response cannot be due to general imbalances in growth-promoting signals, because we found that inhibitors against Met, Pi3k, or mTor blocked proliferation but did not induce senescence. One explanation for the difference between human and mouse tumor cells could be that Shp2 regulates the distinct processes of proliferation, senescence, and EMT in a manner that depends on the oncogenes and their cellular contexts. Indeed, PyMT is an extremely potent oncogene, which affects a wide range of signaling systems including Ras/Mapk, Pi3k/Akt, and Plc-Pkc (Dilworth, 2002). Therefore, Shp2-mediated senescence suppression may be particularly critical for the survival of PyMT tumor cells. Of note, such functions of Shp2 may not be limited to the PyMT context, since PyMT mimics many potent human oncogenes such as Ras, Src, and multiple RTKs. We envision that the senescence-suppressive function of Shp2 may be important for human cancers that are addicted to strong oncogenes. Moreover, PyMT tumor cells appear not to undergo EMT (our data; Trimboli et al, 2008). In contrast, the cell lines used in the work of Aceto et al (2012) exhibited mesenchymal properties (Blick et al, 2008), which were further strengthened and maintained by Shp2.
We also performed retrospective Kaplan–Meier survival analyses on publically available data from 3,935 human patients with invasive breast cancers (Gyorffy & Schafer, 2009), based on the expression of Shp2-regulated genes. Remarkably, the expression patterns of Shp2 and genes of the three overrepresented classes (cell cycle progression, DNA replication and p53 targets) as well as the expression patterns of the single genes Skp2 or Aurka were highly predictive for human breast cancer outcome. Whether these genes or gene classes are prognostically relevant for molecular or therapeutic subgroups of breast cancer remains to be established. This can be investigated with larger public collections of gene expression data of human breast cancers (Mihaly et al, 2013). Interestingly, Shp2 has been recently shown as an independent prognostic factor in human breast cancer (Muenst et al, 2013). Gene expression tools such as Mammaprint® and Oncotype DxTM are now used in clinical settings worldwide, and their application has improved treatment strategies (Glas et al, 2006; Oakman et al, 2010). Recently, in a basal mammary gland cancer model in mice, our group identified a Wnt-Met 322 gene signature, which is predictive of poor survival of human patients with ER-negative, basal breast cancers (Holland et al, 2013). The present set of senescence-controlling genes may likewise be useful in predicting the outcome of human breast cancer.
Recently, pro-senescence therapy has been suggested as a promising strategy for cancer treatment (Nardella et al, 2011). The reactivation of p53 in mouse liver carcinomas and sarcomas, for example, resulted in tumor regression by inducing senescence (Nardella et al, 2011). Several compounds that enhance p53 activity are currently in phase I clinical trials (Nardella et al, 2011). Our study shows that inhibition of Shp2 by the small molecule GS493 blocks tumor growth by inducing senescence in PyMT mice, suggesting that a therapeutic strategy based on the induction of senescence by using small molecules targeting Shp2 may be useful in breast cancer. Although Shp2 plays an essential role in the development and maintenance of many tissues (Grossmann et al, 2010), it is commonly upregulated and highly activated in mouse and human cancers (this study; Chan et al, 2008; Grossmann et al, 2010). Inhibition of Shp2 at a safe dosage indeed blocked tumor formation without affecting normal tissues in adult PyMT mice. Establishing the therapeutic window for Shp2 inhibitors is therefore of particular importance. Moreover, pro-senescence therapies might also be developed by using new or existing compounds to modulate components that act downstream of Shp2, for example, by inhibiting Skp2 and Aurora A, which are frequently overexpressed in breast cancer (Signoretti et al, 2002; Nadler et al, 2008). The identification of these new targets may thus open the door to new therapeutic options in the treatment of breast cancer.
Materials and Methods
Cloning, virus production, and infection
To generate the pMSCV-CreERT2-IRES-GFP construct, a CreERT2 insert was prepared from the pMSCV-CreERT2-IRES-neo plasmid through digestion with EcoRI and then ligated to EcoRI-cut pMSCV-IRES-GFP. To generate cDNA-expressing vectors, cDNAs for mouse Skp2, mouse Aurka, human Shp2, and human Notch3 intracellular domain (N3IC) were amplified by PCR and cloned into the pMSCV-IRES-CFP construct. To generate shRNA-expressing vectors, DNA oligos for p27 or p53 shRNAs were ligated to the pSUPERIOR.retro.puro vector. To produce recombinant retroviruses, plasmids based on pMSCV or pSUPERIOR were introduced into Plat-E packaging cells by calcium phosphate transfection, and after 48 h, the supernatants were collected through 0.45-μm syringe filters. Supernatant were then added to cell cultures at 30–50% confluence, supplemented with 4 μg/ml polybrene. GFP+ or CFP+ cells were sorted by FACS, and puromycin-resistant cells were selected.
Isolation of tumor cells, primary culture, and fluorescence-activated cell sorting (FACS)
Mammary gland tumors dissected from female mice were chopped into pieces and then dissociated enzymatically at 37°C for 1 h in culture medium (DMEM/F12) containing 10 μg/ml insulin (Sigma), 500 ng/ml hydrocortisone (Sigma), 10 ng/ml EGF (Sigma), 20 ng/ml cholera toxin (Sigma), and 5% fetal bovine serum supplemented with 300 U/ml collagenase (Worthington) and 100 U/ml hyaluronidase (Worthington). Resulting organoids were collected and suspended in 0.25% trypsin-EDTA for 2 min. After collecting, the pellets were digested at 37°C for 1 h in culture medium supplemented with 2 mg/ml dispase (Roche Diagnostics) and 0.1 mg/ml DNase (Worthington). Dissociated cells were then depleted of red blood cells by suspending in 0.64% NH4Cl for 3 min and finally filtered through a 40-mm mesh. Single cells were stained with fluorochrome-conjugated antibodies and subjected to analysis or sorting using FACS Aria (BD Biosciences). Dead cells were excluded by gating out 7-AAD-positive cells. The following antibodies were used: PE-conjugated rat anti-mouse CD24 and APC-Cy7-conjugated anti-mouse CD90 (BD Pharmingen).
Mammosphere culture
Single cells were plated in poly-HEMA-coated plates at a density of 5,000 cells/ml. Cells were grown in serum-free DMEM/F12 medium (Invitrogen), supplemented with B27 (Invitrogen), 20 ng/ml FGF, 20 ng/ml EGF, and 4 μg/ml heparin (Sigma-Aldrich) for 7–10 days. To passage, spheres were collected by gentle centrifugation, dissociated enzymatically and mechanically, and then filtered through a 40-μm sieve. The single cells were reseeded and cultured to form spheres.
SA-β-gal staining
SA-β-gal staining was performed as described previously (Dimri et al, 1995). Cryosections or cultured cells were fixed with 2% formaldehyde/0.2% glutaraldehyde in PBS for 15 min, washed with PBS containing 1 mM MgCl2, and stained for 3–4 h in 40 mM citric acid/sodium phosphate (pH = 5.5) containing 1 mM MgCl2, 1 mg/ml X-Gal, and 5 mM of each potassium ferricyanide and potassium ferrocyanide. Sections were counterstained with eosin and mounted using Permount (Thermo Scientific).
Immunostaining and immunoblotting
Tumor cells or tissues were fixed in 4% formaldehyde. Immunofluorescence and immunohistochemistry were performed on cultured cells, 7-μm paraffin-embedded sections, or 12-μm cyrosections. The following antibodies were used: rabbit anti-Cre (Novagen); rat anti-EYFP (Nacalai Tesque); rat anti-EYFP (Abcam); guinea pig anti-CK8 (Progen); rabbit anti-Ki67 (Thermo Scientific); mouse anti-p27 (BD Transduction Laboratories); rabbit anti-p53 (CM5, Novocastra), and anti-cleaved caspase-3 (Cell Signaling Technology). Secondary antibodies were conjugated with Cy2, Cy3, or Cy5 fluorochromes (Jackson ImmunoResearch Laboratories) or with peroxidase.
Total proteins were extracted by lysing tumor tissues, freshly isolated single cells, or cultured cells in RIPA buffer containing protease inhibitor cocktails (Roche), then separated by SDS–PAGE and transferred to PVDF membranes (Millipore). The following primary antibodies were used: mouse anti-Shp2 (BD Transduction Laboratories), rabbit anti-H3K9me3 (Abcam), rabbit anti-p53 (CM5, Novocastra), mouse anti-Src (Upstate), mouse anti-p27 (BD Transduction Laboratories), mouse anti-α-tubulin (Sigma-Aldrich), rabbit anti-p53-pS15, anti-Src pY527, anti-Src-pY416, anti-Fak pY925, anti-Fak, anti-pErk1/2, and anti-Erk1/2 (Cell Signaling Technology). For detection of antibodies, an ECL kit (PerkinElmer) was used. The intensity of bands was quantified using the ImageJ software.
Quantitative RT–PCR (qRT–PCR)
For qRT–PCR, total RNA from tumor tissues, freshly isolated single cells, or cultured cells were extracted using TRIzol, RNase-free DNase treatment (Invitrogen), and phenol/chloroform as specified by the manufacturer. First-strand cDNA was synthesized using 2 μg of total RNA and M-MLV reverse transcriptase (Promega) according to the manufacturer's instructions. qPCR was performed using a SYBR GreenER qPCR mix (Thermo Scientific), and relative mRNA expression was normalized to β-actin (primer sets are listed in Supplementary Table S5).
Microarray and bioinformatics analysis
For microarray and bioinformatics analyses, total RNA was isolated from mammospheres using TRIzol (Invitrogen) and the RNeasy kit (Qiagen). Total RNA (250 ng) was labeled and hybridized to Mouse Genome 430 2.0 chips (Affymetrix). Microarray data were analyzed in the R statistics using Bioconductor libraries. Normalization was performed using MAS5. ANOVA was first applied for class comparison to calculate the P-value for each gene. The P-values were then adjusted according to the Benjamini & Hochberg False Discovery Rate (FDR) method (Benjamini & Hochberg, 1995). Genes with the adjusted P-value, P(FDR) < 0.01, and fold-change > 1.5 were selected for showing significantly differential expression. Gene ontology annotations were performed using the DAVID tool. Data are deposited in GEO with the accession number GSE50518.
Kaplan–Meier survival analysis
Retrospective Kaplan–Meier relapse-free survival analyses of 3,935 human patients with invasive breast cancer, using an updated version of the previous database (Gyorffy & Schafer, 2009), were performed with the “survival” Bioconductor (www.bioconductor.org) package in R (www.r-project.org). Patients were divided into two groups according to the mean mRNA expression levels of downregulated genes, upregulated genes, overrepresented genes of each of three gene ontology classes, or single genes. Each percentile of expression between the lower and upper quartiles was computed, and the best performing threshold was used as cutoff for the Kaplan–Meier analyses. Differences in survival were assessed by computing Cox proportional-hazards regression.
Mouse strains, compound injection, and mammary gland whole-mount staining
The mouse strains MMTV-PyMT, MMTV-Cre;Shp2flox, and R26-EYFPflox and primers for genotyping were described previously (Guy et al, 1992; Wagner et al, 1997; Srinivas et al, 2001; Grossmann et al, 2009). MMTV-PyMT mice were kept on a pure FVB/N background and MMTV-Cre;Shp2flox, and Rosa26-EYFPflox mice were backcrossed with FVB/N mice for at least 6 generations. Animal experiments were performed according to the EU and national institutional regulations.
The modified Shp2 inhibitor of PHPS1 (Hellmuth et al, 2008), GS493, was administered in Cremophor (the control was Cremophor alone) i.p. into mice at 35 mg/kg body weight. In one therapy experiment with PyMT;coShp2 mice, GS493 was given from the 7th week on, five times per week for 3 weeks, and tumor formation was monitored by palpation for the following 3 months. In a second therapeutic experiment in PyMT mice with existing tumors, GS493 was given when tumors were palpable, and tumor growth was recorded by measuring the tumor volumes.
Mammary glands were dissected, fixed in Carnoy's fixative for 2 h, stained in carmine alum solution overnight, de-stained in 70% ethanol, washed in 100% ethanol, defatted in xylene overnight, and mounted using Permount (Thermo Scientific).
Acknowledgments
We thank Dr. Carmen Birchmeier, Dr. Clemens Schmitt, and Russ Hodge (MDC, Berlin) for helpful discussions and critical review of the manuscript, Dr. Hans-Peter Rahn (MDC, Berlin) for expertise in FACS, the Confocal Microscopy Core Facility at the MDC and Chris Eckert from the Screening Unit of the FMP, Berlin for advice, and Dr. Klaus Eckert and Dr. Susanne Flechsig (EPO, Berlin) for performing the therapy experiments in mice. This work was supported in part by a grant of the German Cancer Society/Deutsche Krebshilfe.
Author contributions
LL, JDH and WB designed the research; LL, JQ and AW-G performed the research; LL, JQ and BG analyzed the data; SG contributed new reagents/analytic tools; RV contributed mouse breeding, and LL, JDH and WB wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Legends
Review Process File
Source Data for Figure 3
Source Data for Figure 4
References
- Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G, Confalonieri S, Quarto M, Hu G, Balwierz PJ, Pachkov M, Elledge SJ, van Nimwegen E, Stadler MB, Bentires-Alj M. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat Med. 2012;18:529–537. doi: 10.1038/nm.2645. [DOI] [PubMed] [Google Scholar]
- Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3:51–62. doi: 10.1016/s1535-6108(02)00235-0. [DOI] [PubMed] [Google Scholar]
- Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, Valsesia-Wittmann S, Caron de Fromentel C, Puisieux A. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14:79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Banito A, Lowe SW. A new development in senescence. Cell. 2013;155:977–978. doi: 10.1016/j.cell.2013.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Blick T, Widodo E, Hugo H, Waltham M, Lenburg ME, Neve RM, Thompson EW. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin Exp Metastasis. 2008;25:629–642. doi: 10.1007/s10585-008-9170-6. [DOI] [PubMed] [Google Scholar]
- Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt AL, Ma J, Loke HK, Lingaraj T, Wu D, Hamman KB, Spelman JJ, Cullis CA, Langston SP, Vyskocil S, Sells TB, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37:102–111. doi: 10.1016/j.molcel.2009.12.024. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Luo JL, Karin M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc Natl Acad Sci USA. 2007;104:15852–15857. doi: 10.1073/pnas.0706728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;27:179–192. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielowiec J, Borowiak M, Morkel M, Stradal T, Munz B, Werner S, Wehland J, Birchmeier C, Birchmeier W. c-Met is essential for wound healing in the skin. J Cell Biol. 2007;177:151–162. doi: 10.1083/jcb.200701086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook RS, Garrett JT, Sanchez V, Stanford JC, Young C, Chakrabarty A, Rinehart C, Zhang Y, Wu Y, Greenberger L, Horak ID, Arteaga CL. ErbB3 ablation impairs PI3K/Akt-dependent mammary tumorigenesis. Cancer Res. 2011;71:3941–3951. doi: 10.1158/0008-5472.CAN-10-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilworth SM. Polyoma virus middle T antigen and its role in identifying cancer-related molecules. Nat Rev Cancer. 2002;2:951–956. doi: 10.1038/nrc946. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocket M, Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimova EV, Mauceri HJ, Golden DW, Labay E, Bindokas VP, Darga TE, Chakraborty C, Barreto-Andrade JC, Crawley C, Sutton HG, Kron SJ, Weichselbaum RR. Poly(ADP-ribose) polymerase inhibitor induces accelerated senescence in irradiated breast cancer cells and tumors. Cancer Res. 2010;70:6277–6282. doi: 10.1158/0008-5472.CAN-09-4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glas AM, Floore A, Delahaye LJ, Witteveen AT, Pover RC, Bakx N, Lahti-Domenici JS, Bruinsma TJ, Warmoes MO, Bernards R, Wessels LF, Van't Veer LJ. Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genom. 2006;7:278. doi: 10.1186/1471-2164-7-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann KS, Wende H, Paul FE, Cheret C, Garratt AN, Zurborg S, Feinberg K, Besser D, Schulz H, Peles E, Selbach M, Birchmeier W, Birchmeier C. The tyrosine phosphatase Shp2 (PTPN11) directs Neuregulin-1/ErbB signaling throughout Schwann cell development. Proc Natl Acad Sci USA. 2009;106:16704–16709. doi: 10.1073/pnas.0904336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann KS, Rosario M, Birchmeier C, Birchmeier W. The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res. 2010;106:53–89. doi: 10.1016/S0065-230X(10)06002-1. [DOI] [PubMed] [Google Scholar]
- Guo S, Liu M, Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim Biophys Acta. 2011;1815:197–213. doi: 10.1016/j.bbcan.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Schafer R. Meta-analysis of gene expression profiles related to relapse-free survival in 1,079 breast cancer patients. Breast Cancer Res Treat. 2009;118:433–441. doi: 10.1007/s10549-008-0242-8. [DOI] [PubMed] [Google Scholar]
- Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–267. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- Hellmuth K, Grosskopf S, Lum CT, Wurtele M, Roder N, von Kries JP, Rosario M, Rademann J, Birchmeier W. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc Natl Acad Sci USA. 2008;105:7275–7280. doi: 10.1073/pnas.0710468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuberger J, Kosel F, Qi J, Grossmann KS, Rajewsky K, Birchmeier W. Shp2/MAPK signaling controls goblet/paneth cell fate decisions in the intestine. Proc Natl Acad Sci USA. 2014;111:3472–3477. doi: 10.1073/pnas.1309342111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland JD, Gyorffy B, Vogel R, Eckert K, Valenti G, Fang L, Lohneis P, Elezkurtaj S, Ziebold U, Birchmeier W. Combined Wnt/beta-catenin, Met, and CXCL12/CXCR4 signals characterize basal breast cancer and predict disease outcome. Cell Rep. 2013;5:1214–1227. doi: 10.1016/j.celrep.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Huck JJ, Zhang M, McDonald A, Bowman D, Hoar KM, Stringer B, Ecsedy J, Manfredi MG, Hyer ML. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol Cancer Res. 2010;8:373–384. doi: 10.1158/1541-7786.MCR-09-0300. [DOI] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, Keblusek P, Robanus-Maandag E, Kristel P, Lingbeek M, Nederlof PM, van Welsem T, van de Vijver MJ, Koh EY, Daley GQ, van Lohuizen M. Senescence bypass screen identifies TBX2, which represses Cdkn2a (p19(ARF)) and is amplified in a subset of human breast cancers. Nat Genet. 2000;26:291–299. doi: 10.1038/81583. [DOI] [PubMed] [Google Scholar]
- Julien SG, Dube N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- Kang S, Xie J, Miao J, Li R, Liao W, Luo R. A knockdown of Maml1 that results in melanoma cell senescence promotes an innate and adaptive immune response. Cancer Immunol Immunother. 2013;62:183–190. doi: 10.1007/s00262-012-1318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, Cordon-Cardo C, Teruya-Feldstein J, Pandolfi PP. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464:374–379. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, Huelsken J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2012;481:85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- Marcotte R, Smith HW, Sanguin-Gendreau V, McDonough RV, Muller WJ. Mammary epithelial-specific disruption of c-Src impairs cell cycle progression and tumorigenesis. Proc Natl Acad Sci USA. 2012;109:2808–2813. doi: 10.1073/pnas.1018861108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micchelli CA, Esler WP, Kimberly WT, Jack C, Berezovska O, Kornilova A, Hyman BT, Perrimon N, Wolfe MS. Gamma-secretase/presenilin inhibitors for Alzheimer's disease phenocopy Notch mutations in Drosophila. FASEB J. 2003;17:79–81. doi: 10.1096/fj.02-0394fje. [DOI] [PubMed] [Google Scholar]
- Mihaly Z, Kormos M, Lanczky A, Dank M, Budczies J, Szasz MA, Gyorffy B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res Treat. 2013;140:219–232. doi: 10.1007/s10549-013-2622-y. [DOI] [PubMed] [Google Scholar]
- Muenst S, Obermann EC, Gao F, Oertli D, Viehl CT, Weber WP, Fleming T, Gillanders WE, Soysal SD. Src homology phosphotyrosyl phosphatase-2 expression is an independent negative prognostic factor in human breast cancer. Histopathology. 2013;63:74–82. doi: 10.1111/his.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler Y, Camp RL, Schwartz C, Rimm DL, Kluger HM, Kluger Y. Expression of Aurora A (but not Aurora B) is predictive of survival in breast cancer. Clin Cancer Res. 2008;14:4455–4462. doi: 10.1158/1078-0432.CCR-07-5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama KI, Nakayama K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol. 2005;16:323–333. doi: 10.1016/j.semcdb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer. 2011;11:503–511. doi: 10.1038/nrc3057. [DOI] [PubMed] [Google Scholar]
- Neel BG, Gu H, Pao L. The “Shping” news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- Oakman C, Santarpia L, Di Leo A. Breast cancer assessment tools and optimizing adjuvant therapy. Nat Rev Clin Oncol. 2010;7:725–732. doi: 10.1038/nrclinonc.2010.170. [DOI] [PubMed] [Google Scholar]
- Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest. 2009;119:252–266. doi: 10.1172/JCI37160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, Lockwood G, Egan SE. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65:8530–8537. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- Robinson DR, Kalyana-Sundaram S, Wu YM, Shankar S, Cao X, Ateeq B, Asangani IA, Iyer M, Maher CA, Grasso CS, Lonigro RJ, Quist M, Siddiqui J, Mehra R, Jing X, Giordano TJ, Sabel MS, Kleer CG, Palanisamy N, Natrajan R, et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat Med. 2011;17:1646–1651. doi: 10.1038/nm.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114. doi: 10.1101/gad.235184.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- Sausgruber N, Coissieux MM, Britschgi A, Wyckoff J, Aceto N, Leroy C, Stadler MB, Voshol H, Bonenfant D, Bentires-Alj M. Tyrosine phosphatase SHP2 increases cell motility in triple-negative breast cancer through the activation of SRC-family kinases. Oncogene. 2014;34:2272–2278. doi: 10.1038/onc.2014.170. [DOI] [PubMed] [Google Scholar]
- Schmitt CA. Senescence, apoptosis and therapy–cutting the lifelines of cancer. Nat Rev Cancer. 2003;3:286–295. doi: 10.1038/nrc1044. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Signoretti S, Di Marcotullio L, Richardson A, Ramaswamy S, Isaac B, Rue M, Monti F, Loda M, Pagano M. Oncogenic role of the ubiquitin ligase subunit Skp2 in human breast cancer. J Clin Invest. 2002;110:633–641. doi: 10.1172/JCI15795. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub F, Mengel M, Luck HJ, Kreipe HH, von Wasielewski R. Prognostic impact of Skp2 and p27 in human breast cancer. Breast Cancer Res Treat. 2006;99:185–191. doi: 10.1007/s10549-006-9202-3. [DOI] [PubMed] [Google Scholar]
- Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, Tanner SM, Creasap N, Rosol TJ, Robinson ML, Eng C, Ostrowski MC, Leone G. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008;68:937–945. doi: 10.1158/0008-5472.CAN-07-2148. [DOI] [PubMed] [Google Scholar]
- Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786:60–72. doi: 10.1016/j.bbcan.2008.07.003. [DOI] [PubMed] [Google Scholar]
- van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, Li M, Furth PA, Hennighausen L. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B, Reis-Filho JS. Histological and molecular types of breast cancer: is there a unifying taxonomy? Nat Rev Clin Oncol. 2009;6:718–730. doi: 10.1038/nrclinonc.2009.166. [DOI] [PubMed] [Google Scholar]
- White DE, Kurpios NA, Zuo D, Hassell JA, Blaess S, Mueller U, Muller WJ. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 2004;6:159–170. doi: 10.1016/j.ccr.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Wiener JR, Kerns BJ, Harvey EL, Conaway MR, Iglehart JD, Berchuck A, Bast RC., Jr Overexpression of the protein tyrosine phosphatase PTP1B in human breast cancer: association with p185c-erbB-2 protein expression. J Natl Cancer Inst. 1994;86:372–378. doi: 10.1093/jnci/86.5.372. [DOI] [PubMed] [Google Scholar]
- Willecke R, Heuberger J, Grossmann K, Michos O, Schmidt-Ott K, Walentin K, Costantini F, Birchmeier W. The tyrosine phosphatase Shp2 acts downstream of GDNF/Ret in branching morphogenesis of the developing mouse kidney. Dev Biol. 2011;360:310–317. doi: 10.1016/j.ydbio.2011.09.029. [DOI] [PubMed] [Google Scholar]
- Zhou X, Coad J, Ducatman B, Agazie YM. SHP2 is up-regulated in breast cancer cells and in infiltrating ductal carcinoma of the breast, implying its involvement in breast oncogenesis. Histopathology. 2008;53:389–402. doi: 10.1111/j.1365-2559.2008.03103.x. [DOI] [PubMed] [Google Scholar]
- Zhou XD, Agazie YM. Inhibition of SHP2 leads to mesenchymal to epithelial transition in breast cancer cells. Cell Death Differ. 2008;15:988–996. doi: 10.1038/cdd.2008.54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Legends
Review Process File
Source Data for Figure 3
Source Data for Figure 4