

Abstract
During development, progenitor cells with binary potential give rise to daughter cells that have distinct functions. Heritable epigenetic mechanisms then lock in gene expression programs that define lineage identity. Cd4 regulation in helper and cytotoxic T cells exemplifies this process, with enhancer- and silencer-regulated establishment of epigenetic memories for stable gene expression and repression, respectively. Using a genetic screen, we identified the DNA methylation machinery as essential for maintaining Cd4 silencing in the cytotoxic lineage. Further, we found a requirement for the proximal enhancer in mediating removal of Cd4 DNA methylation marks, allowing for stable expression in T helper cells. These findings suggest that stage-specific methylation and demethylation events in Cd4 regulate its heritable expression in response to the distinct signals that dictate lineage choice during T cell development.
INTRODUCTION
During metazoan development, a series of asymmetric cell divisions results in cells with a vast number of distinct phenotypes that are maintained throughout life. With rare exceptions, for example receptor gene segment rearrangements in B and T cells, the genome sequence remains unchanged as cells adopt new identities. Stable lineage commitment requires establishment of heritable patterns of gene expression or repression without alteration of DNA sequences, via epigenetic modifications. Despite a rapidly growing body of work that describes putative epigenetic regulation, physiological models in which epigenetic modulation can be functionally dissected and tested in fully differentiated cells are rare.
One of the rare examples in which heritable gene expression has been studied in depth is T cell lineage choice1. CD4+ helper and CD8+ cytotoxic T cells develop from common progenitors, based on the specificity of their T cell antigen receptors (TCRs) for peptide-major histocompatibility complex (MHC) class II or class I molecules, respectively. The CD4 and CD8 co-receptors are critical to the development and function of these lineages, as they facilitate TCR binding to MHCII (CD4) and MHCI (CD8). CD4 and CD8 expression defines distinct stages of thymocyte development, during which ordered Tcr gene rearrangements occur and serve as developmental checkpoints. Early CD4−CD8− double-negative (DN) progenitors transition through four distinct stages before up-regulating CD4 and CD8 to enter the CD4+CD8+ double-positive (DP) stage of development. DP cells then test their randomly rearranged TCRs for MHCI and II specificity. MHCI-specific cells stably down regulate CD4 to enter into the cytotoxic lineage, while MHCII-specific cells lose CD8 expression and maintain CD4 expression during helper lineage differentiation.
The regulation of Cd4 expression during T cell development is an ideal setting for studying epigenetic regulation, as Cd4 exhibits heritable active and silenced states that can be maintained independently of the initiating genomic elements1. Elements required for this regulation have been identified in a series of in vivo genetic studies and in vitro T cell culture assays2–6. These include a 434 bp cis-acting silencer (S4), located in the first intron of the Cd4 locus, and a 430 bp cis-acting proximal enhancer (E4P), located 13 kb upstream of the transcriptional start site (TSS). S4 is essential for Cd4 repression at two different stages of T cell development. First, germline S4 deletion leads to ectopic CD4 expression in DN cells, indicating that it is required for reversible silencing before the DP stage of development. Second, S4 is required for silencing Cd4 in mature CD8+ cytotoxic cells, since germline S4 deletion results in ectopic CD4 expression in cytotoxic lineage cells. However, Cre-mediated conditional S4 deletion in mature CD8+ lineage cells following their thymic egress does not affect CD4 expression even after multiple cell divisions5. Similarly, in mature cytotoxic cells, Cre-mediated deletion of genes encoding members of the RUNX protein complex that binds S4 to initiate Cd4 silencing3 fails to activate Cd4 gene expression (Egawa and Littman, unpublished). This failure to activate Cd4 expression within cytotoxic cells is not due to the loss of Cd4 gene expression potential because germline S4 deletion results in robust CD4 expression in CD8+ cells and E4P-Cd4 promoter reporter constructs exhibit strong activity upon delivery into mature CD8+ cells (J.R.H, D.R.L, unpublished). Thus, S4 initiates Cd4 silencing in developing cytotoxic cells, but is completely dispensable for the maintenance of that silenced state.
The proximal enhancer initiates an analogous epigenetically active Cd4 expression state in CD4+ helper cells7. Germline E4P deletion abrogated CD4 upregulation at the DN4 to DP transition during T cell development. However, a reduced number of MHCII-specific thymocytes were positively selected in Cd4E4PΔ/E4PΔ mice, and these cells displayed moderate, but unstable, CD4 expression. Thus, in vitro or in vivo proliferation of Cd4E4PΔ/E4PΔ helper cells resulted in the gradual loss of CD4. In contrast, Cre-mediated deletion of a loxP-flanked E4P in mature helper cells did not affect CD4 expression, even after multiple cell divisions in vitro and in vivo. Thus, E4P is required for the initiation of stable high expression of CD4, but is also dispensable for its maintenance.
The finding that established Cd4 silencing can be disassociated from the presence of S4 suggests the existence of a set of genes that epigenetically maintain silencing independently of S4. As T cells undergo multiple rounds of cell division after activation, these genes would need to both suppress CD4 re-expression (since CD8+ cells possess the capacity to express CD4) and actively pass the silenced state from parental to daughter cells independently of S4. To identify these putative trans–acting factors, we performed genetic screens using pooled retroviral shRNA libraries targeting the entire mouse genome. From these screens, DNA methyltransferase I (Dnmt1) was identified as a key factor in Cd4 silencing. Subsequent locus-wide methylation analyses revealed Cd4 hyper-methylation in DN, DP and CD8+ cells compared to CD4+ cells. We further found that in CD4+ and CD8+ T cells the Cd4 DNA methylation patterns are dependent on E4P and S4, respectively. E4P-dependent demethylation of the locus during the DP to CD4+CD8− thymocyte transition was achieved in the absence of cell division, consistent with the engagement of an active enzymatic process rather than passive demethylation. These results provide the first description of the epigenetic molecular machinery essential for the heritable regulation of Cd4 expression. Furthermore, they indicate that Cd4 gene regulation in mature T lymphocytes provides a unique opportunity to dissect the epigenetic mechanisms involved in establishing and maintaining gene expression or heritable silencing.
RESULTS
Unbiased screen for regulators of heritable Cd4 silencing
The factors that mediate bona fide epigenetic silencing of gene expression during T cell development remain poorly characterized. To identify trans-acting factors critical for maintaining Cd4 silencing in cytotoxic T cells (CD4−CD8+), we performed an unbiased, genome-wide retroviral shRNA screen (Supplementary Fig. 1a). We first activated spleen and lymph node CD8+ cells from Cd4S4-L/+Ubc-Cre-ER mice with anti-CD3 and anti-CD28 in the presence of OH-tamoxifen, to delete S4. We hypothesized that S4 deletion would allow for maximal sensitivity and ensure the identification of only true epigenetic modifiers by eliminating the possibility of S4 activity in mature CD8+ cells. After 18 h of activation, cells were transduced with pools of a retroviral shRNA library and expanded for 9 d with interleukin 2 (IL-2). On day 5, CD4+ cytotoxic cells were enriched using anti-CD4 magnetic beads. On day 10, a small percentage (~0.5%) of pooled shRNA virus-infected cells expressed CD4 in addition to CD8, but there was no expression in mock-infected cells (Fig. 1a). These CD4+CD8+ cells were sorted based on cell surface marker expression, and the shRNAs integrated into their genome were PCR-amplified, cloned and sequenced. Interestingly, 83% of the shRNAs isolated from infected CD4+CD8+ cells were specific for Dnmt1 (two different shRNA clones targeting Dnmt1 were independently identified), indicating that DNA methylation may be important for the maintenance of Cd4 silencing in fully differentiated cytotoxic T cells.
Figure 1. DNA methylation enzymes are essential for silencing CD4 expression in cytotoxic T cells.

Cd4S4-L; Ubc-Cre-ER cytotoxic cells were cultured in vitro and S4 was deleted by adding 400 nM OH-tamoxifen. (a–c) Flow cytometry of CD4 and CD8 expression. (a) Mock or pooled shRNA virus-infected cells (encoding a puromycin resistance gene) were enriched by MACS with anti-CD4 magnetic beads and cultured 4–5 more days in the presence of puromycin (2.5 μg/ml) before analysis. Cd4S4Δ/S4Δ CD4+8+ cells (germline S4 deletion) are shown as a staining control. Representative of 2 independent experiments. (b) Cytotoxic CD4−8+ cells from Dnmt1chip/chip mice were cultured in vitro for 5–6 days after control-ires-gfp or Dnmt1 shRNA-ires-gfp retroviral infection. Staining is on gated GFP+ cells. Representative of 3 independent experiments. (c) 1~1.5 × 106 peripheral CD4−8+ cells from control (Dnmt3aL/L; Dnmt3bL/+; Dnmt1L/+) or Dnmtsreduced (Dnmt3aL/L; Dnmt3bL/+; Dnmt1L/chip; Cd4-Cre+/−) animals were adoptively transferred into Rag2-deficient mice. TCRβ+CD8+ cells from the periphery were analyzed 16 days after transfer. Representative of 3 independent experiments.
DNA methylation machinery maintains Cd4 silencing
To validate the results of our screen and to determine if DNA methylation enzymes are important for Cd4 silencing, we interfered with DNA methyltransferase activity by using shRNA knockdown and mice with targeted mutations in Dnmt genes. Cytotoxic T cells activated by CD3 and CD28 crosslinking and transduced with the Dnmt1 shRNAs identified in the screen exhibited increased surface CD4 expression compared to cells transduced with a control vector (Supplementary Fig. 1b and data not shown). To rule out off-target shRNA effects, we also tested CD4 expression after genetic manipulation of DNA methyltransferase activity. Since Dnmt1 deletion leads to lymphocyte death after a limited number of cell divisions8, we analyzed the maintenance of Cd4 silencing using a hypomorphic mutation in the Dnmt1 locus9, Dnmt1Chip, in which Dnmt1 expression is reduced to ~10% of wild-type. In mice homozygous for the hypomorphic mutation (Dnmt1Chip/Chip) or hemizygous for the mutation (Dnmt1L/ChipCd4-Cre ), 1–2 % of cytotoxic T cells upregulated CD4 expression following in vitro activation and expansion (Fig. 1b and data not shown). Further reduction of Dnmt1 expression in these cells using Dnmt1 shRNA led to CD4 expression on ~10% of the cytotoxic cells (Fig. 1b). If this result reflects a requirement for Dnmt1-directed maintenance methylation in Cd4 silencing, then CD4 expression may be expected to increase progressively with successive cell divisions due to passive DNA demethylation. To examine this, we transduced CD8+ T cells with control or Dnmt1 shRNAs, then labeled the cells with the fluorescent dye e670 and quantified CD4 expression through multiple rounds of cell division, as assessed by dye-dilution. Similarly to CFSE, e670 binds covalently to cellular proteins during staining and is distributed evenly between daughter cells upon cell division. Consistent with a role for Dnmt1-mediated maintenance methylation in Cd4 silencing, we found increased CD4 expression with increased numbers of cell divisions (Supplementary Fig. 1c).
To better assess the role of the DNA maintenance methylation machinery in Cd4 silencing, we monitored CD4 expression in long-term, in vivo cell proliferation assays. First, we adoptively transferred Dnmt1 hypomorphic cytotoxic T cells into T cell-deficient lymphopenic hosts. After 2–3 weeks, 10–20% of transferred cells expressed CD4 in addition to CD8 (data not shown). Since Dnmt1 deficiency can compromise proliferation, we attempted to reduce methylation content further in Dnmt1 hypomorphic mice by removing alternative methylation machinery, namely Dnmt3a and Dnmt3b. The de novo methyltransferases Dnmt3a and Dnmt3b have also been implicated in DNA maintenance methylation10,11 and their deletion does not significantly affect T cell proliferation (not shown). Compared to Dnmt3a deletion on a Dnmt1 hypomorphic background, Dnmt3b deletion had a relatively weak effect on Cd4 gene expression in cytotoxic T cells (data not shown). Intriguingly, eliminating Dnmt3a in Dnmt1 hypomorphic mice resulted in robust CD4 expression (>75%) after in vivo expansion (Fig. 1c). These data clearly demonstrate that DNA methylation machinery is critical for ensuring stable Cd4 silencing in cytotoxic T cells.
S4-dependent, cytotoxic lineage DNA hyper-methylation
The requirement for DNA methyltransferases in Cd4 silencing implied that there would be differences in 5′-methyl-cytosine (5mC) modifications of CpG dinucleotides within the locus in helper and cytotoxic T cells, and that the differences could be dependent on S4. To test this hypothesis, we isolated genomic DNA from peripheral wild-type CD4+ helper and CD8+ cytotoxic, and Cd4S4Δ/S4Δ CD4+CD8+ cytotoxic T cells, and enriched for the Cd4 locus flanked on each side by ~75 kb, by CATCH-Seq; this method uses BAC clone templates to generate probes for target capture hybridization-based locus enrichment, followed by bisulfite sequencing12. CATCH-Seq resulted in >30x sequencing coverage for 97.6% of target CpGs per sample, on average, and was highly reproducible between biological replicates (Supplementary Fig. 2). We identified a strongly differentially methylated region (DMR) from ~ +3.2 kb to −0.7 kb relative to the Cd4 TSS (Fig. 2a; referred to hereafter as the TSS-proximal DMR). This DMR was hyper-methylated in CD8+ cells compared to CD4+ cells (Fig. 2b and Supplementary Fig. 3; 21/55 CpGs in this region exhibited >40% methylation in CD8+ cells and >2x higher methylation in CD8+ versus CD4+ cells). Importantly, this TSS-proximal DMR straddles S4 and overlaps with the Cd4 promoter as well as a recently identified “maturation” enhancer (adjacent to S4) required to initiate stable CD4 expression in mature helper lineage cells (13 and P.D.I. and D.R.L., unpublished results), indicating that the DMR is likely to control functional cis-acting elements. We confirmed the existence of the DMR at a subset of CpGs by amplicon sequencing and methylation-sensitive restriction enzyme digests (Supplementary Figs. 4a–c). Furthermore, the cytotoxic lineage Cd4 locus hyper-methylation was silencer-dependent, as methylation patterns in Cd4S4Δ/S4Δ CD4+CD8+ peripheral T cells closely resembled those of wild-type CD4+ cells (Fig. 2b and Supplementary Fig. 4c).
Figure 2. Silencer-dependent DMR in the first intron of Cd4.

Biological replicates of naïve (Thy1.2+CD44loCD62L+CD25−) WT CD4+, WT CD8+ and Cd4S4Δ/S4Δ CD4+CD8+ cells were isolated from LNs, and their genomic DNA was subjected to CATCH-seq (BAC-mediated enrichment, bisulfite treatment and Illumina sequencing). (a) Percent CpG methylation was calculated at CpGs exhibiting >30x coverage, within ~75 kb of the Cd4 TSS (Chr6:124749635-124906460; mm9), and variance at each CpG across all samples was calculated and graphed on the y-axis versus genomic position on the x-axis. 98–98.5% of targeted CpGs were captured in each sample at >30x coverage; median CpG coverage exceeded 300x. Genes, S4 and E4P are indicated below the graph. (b) A heatmap depicts percent CpG methylation in WT CD4+, WT CD8+ and Cd4S4ΔS4Δ CD4+8+ cells for CpGs from +6200 to −669 relative to the Cd4 TSS (Chr #6:124832027-124838896; mm9). A red line underlines CpGs in the S4 silencer (indicated by the gaps), and a black arrow indicates the Cd4 TSS. (c) CFSE-labeled WT CD4+, WT CD8+ and Cd4S4Δ/S4Δ CD4+CD8+ cells were stimulated in vitro with anti-CD3, anti-CD28 and IL-2, and those cells that had undergone at least 6 divisions after 5 days were sorted for locus enrichment and high-throughput bisulfite sequencing (as above). A heatmap depicts CpG methylation percentage from +6200 to −669 relative to the Cd4 TSS (Chr6:124832027-124838896; mm9). Replicates were derived from two independent experiments.
To ascertain if the hyper-methylation pattern observed in CD8+ T cells was stable, we also examined DNA methylation after multiple cell divisions. Wild-type CD4+, wild-type CD8+ and Cd4S4Δ/S4Δ CD4+CD8+ cells that had undergone >5 divisions following in vitro stimulation, as assessed by CFSE dilution, maintained DNA methylation patterns similar to those of their ex vivo precursors (Fig. 2c and Supplementary Fig. 4d). These methylation patterns were also conserved after 20 days and >10 cell divisions of expansion in lymphopenic hosts (Supplementary Fig. 4e). Thus, consistent with a direct role for DNA methylation in regulating the heritable silencing of Cd4, S4 dictates Cd4 locus hyper-methylation in cytotoxic T cells, which in turn is stable through multiple cell divisions.
Cd4 locus DNA is hyper-methylated in T cell progenitors
The finding of heritable Cd4 DNA hyper-methylation in CD8+ T cells and hypo-methylation in CD4+ T cells suggested that the locus undergoes selective de novo methylation as DP cells differentiate towards the cytotoxic lineage. To determine if this was indeed the case, we analyzed DNA methylation by CATCH-Seq, as well as amplicon sequencing. Unexpectedly, we found that the Cd4 locus was already hyper-methylated both at the DN3 and DP stages (Fig. 3a, Supplementary Fig. 4f–g). Most of the methylated CpGs were retained in CD8 single-positive thymocytes (CD8SP; the “SP” suffix will be used to denote maturing helper and cytotoxic cells from the thymus), while a substantial number converted to the unmethylated state in CD4SP cells (Fig. 3a). Germline S4 deletion did not affect DNA methylation in DP cells (Fig. 3b, Supplementary Fig. 4h), suggesting that while silencer-mediated activity is required to sustain DNA methylation as cells transition from DP precursors to the CD8 lineage, it is not required to maintain locus methylation in DP cells. Overall these data indicate that the Cd4 locus becomes hyper-methylated early in T cell development, and is then selectively demethylated during specification of the CD4+ T-helper lineage.
Figure 3. Cd4 hyper-methylation in immature thymocytes.

(a) CATCH-seq was performed on genomic DNA from sorted populations of WT thymocytes: DN3 (Thy1.2+Lin−CD25+CD44−) (n=1), DP (TCRβloCD24+CD69−CD4+CD8+), CD4SP (TCRβhiCD24loCD69loCD4+CD8–−) and CD8SP
(TCRβhiCD24loCD69loCD4−CD8+); the SP suffix denotes thymus-derived helper and cytotoxic T cells. A heatmap depicts CpG methylation percentage from +6200bp to −669bp relative to the Cd4 TSS (Chr #6:124832027-124838896; mm9); Biological replicates are noted. (b) BAC-enrichment and bisulfite, high-throughput sequencing was performed on DP cells sorted from Cd4S4Δ/S4Δ mice. Data are displayed as in (a). Replicates were derived from two independent experiments.
DMR methylation does not grossly alter chromatin structure
We next considered how hyper-methylation of the Cd4 locus could impact silencing. As DNA methylation can regulate nucleosome stability and positioning in some contexts14,15, we evaluated the possibility that Cd4 locus DNA methylation could impact nucleosomes and, hence, chromatin compaction. Thus, we used micrococcal nuclease digestion and CATCH-seq to identify nucleosomes from diverse samples, including DN3, DP, CD4+, CD8+, Cd4E4PΔ/E4PΔ DP, Cd4E4PΔ/E4PΔ CD4+, Cd4S4Δ/S4Δ DP, and Cd4S4Δ/S4Δ CD8+. We found that the loss of nucleosomes at ~ +3.5 kb relative to the TSS, immediately downstream of the TSS-proximal DMR, correlated strongly with CD4 expression (Supplementary Fig. 5). However, DNA methylation content did not correlate well with differential nucleosome positioning, indicating that this is unlikely to be a critical function of Cd4 locus 5mC marks in facilitating silencing.
Cd4 hypo-methylation correlates with stable CD4 expression
Cd4 hypo-methylation in helper lineage cells led us to hypothesize that the removal of 5mC marks is critical for heritable, high-level CD4 expression. To determine if this may be the case, we assessed if 5mC content was dependent on the Cd4 proximal enhancer. E4P-deficient CD4+ helper T cells exhibited unstable CD4 expression upon cell division7. Interestingly, in naive Cd4E4PΔ/E4PΔ CD4+ cells there was Cd4 TSS-proximal DMR hyper-methylation, with 5mC content approaching that observed in mature wild-type CD8+ cells (Fig. 4a) and correlating with unstable CD4 expression. Further, when we stimulated naïve Cd4E4PΔ/E4PΔ CD4+ cells to proliferate in vitro and analyzed Cd4 methylation patterns locus-wide in CD4+ and CD4− cells after >5 cell divisions, those cells that lost CD4 expression exhibited more TSS-proximal DMR methylation (Fig. 4b). Thus, in helper T cells, Cd4 locus hypo-methylation correlated with more stable maintenance of CD4 expression (maintaining CD4 expression after > 5 cell divisions), while hyper-methylation correlated with loss of CD4 expression. In Cd4E4PΔ/E4PΔ thymic DP cells, which lack CD4 expression7, the Cd4 methylation pattern was most similar to that in wild-type DN3 cells, with hyper-methylation ±1 kb from the location of E4P (Supplementary Fig. 6). Taken together, these results suggest that E4P contributes to stable CD4 expression in the helper lineage by facilitating heritable Cd4 locus de-methylation.
Figure 4. E4P is required for Cd4 locus hypo-methylation in the T-helper lineage.
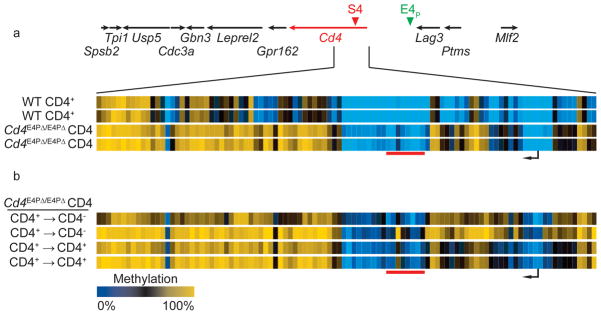
(a) CATCH-seq was performed on naïve CD4+ cells (Thy1.2+CD25−CD44loCD62L+CD4+) from Cd4E4PΔ/E4PΔ; mice. A heatmap depicts percentage CpG methylation in biological replicates from +6200bp to −669bp relative to the Cd4 TSS (Chr #6:124832027-124838896; mm9). Data from WT CD4 cells in Figure 2b are shown for comparison. (b) CFSE-labeled naïve CD4+ Cd4E4PΔ/E4PΔ cells were stimulated in vitro with anti-CD3, anti-CD28 and IL-2 for 5 days. CD4+ (CD4+ → CD4+) and CD4− (CD4+ → CD4−) that had undergone at least 6 divisions were sorted for CATCH-seq. Data are displayed as in (a). Results from 2 independent experiments are displayed.
Reduced DNMT1 rescues E4P-KO helper cell CD4 expression
To determine if there may be a causal link between Cd4 locus hypo-methylation and heritable CD4 expression, we interfered with DNMT1 expression in E4P- deficient helper cells. We sorted naïve CD4+ cells from Dnmt1L/LCd4-Cre+, Dnmt1L/ChipCd4-Cre+ and Dnmt1L/LCd4-Cre− mice, all on a Cd4E4PΔ/E4PΔ background, and analyzed CD4 expression at various time points after activation.
We measured CD4 protein expression, as it correlates with mRNA expression in Cd4E4PΔ/E4PΔ T cells7. At 72 h, 76.2% of Dnmt1-sufficient cells maintained CD4 expression (Fig. 5a). In contrast, Dnmt1-knockout and Dnmt1-hypomorphic cells exhibited higher proportions of CD4+ cells (94.9% and 87.6%, respectively; Fig. 5a). CD4+ Dnmt1-hypomorphic Cd4E4PΔ/E4PΔ cells also exhibited ~30% higher CD4 mean fluorescence intensity (MFI) compared to Dnmt1+/+ Cd4E4PΔ/E4PΔ CD4+ cells at 72h, indicating higher CD4 expression in the cells that remained CD4+ (Fig. 5b). The increased proportion of CD4+ cells (20–25% higher) and increased MFI (40–47% higher) in Dnmt1 hypomorphic versus Dnmt1-sufficient cells was also observed at 96 h and 120 h (Supplementary Fig. 7a–b). Importantly, these results were not due to differences in proliferative capacity because the fraction of CD4+ cells was consistently higher in Dnmt1-deficient cells compared to wild-type across each cell generation as measured by CFSE dilution (Fig. 5c, Supplementary Fig. 7c). Transduction of Cd4E4PΔ/E4PΔ CD4+ T cells with retroviruses encoding Dnmt1 shRNAs led to a similar result (Supplementary Fig. 7d–e). The results are thus consistent with E4P-dependent demethylation of the Cd4 TSS-proximal DMR being critical for establishing heritable high CD4 expression.
Figure 5. Reduction in Dnmt1 activity rescues CD4 expression in Cd4E4PΔ/E4PΔ helper T cells.

CFSE-loaded naïve CD4+ T cells from the indicated strains of mice were stimulated in vitro for 72 h with anti-CD3 and anti-CD28 antibodies. (a) Top, post-sort, pre-stimulation CD4 and CD8 expression; bottom, CD4 expression and CFSE dilution after 72 h. Percentages of CD4+ cells are indicated. (b) Graph of CD4 MFI after 72 h activation of Cd4E4PΔ/E4PΔDnmt1L/ChipCd4-cre+ (red) and Cd4E4PΔ/E4PΔDnmt1L/LCd4-Cre− CD4+ T cells (blue) (from CD4+ gates in (a)). Dnmt1 knockout cells were not included as they exhibited growth defects (i.e. lower forward scatter), precluding MFI comparison (data not shown). (c) Percent CD4+ cells from each cell division of samples in (a), as tracked using dilution of CFSE fluorescence. Cells were from mice of the genotypes Cd4E4PΔ/E4PΔDnmt1L/LCd4-Cre− (blue); Cd4E4PΔ/E4PΔDnmt1L/ChipCd4-cre+ (red); and Cd4E4PΔ/E4PΔDnmt1L/LCd4-Cre+ CD4+ (black). Results are representative of at least 4 independent experiments.
DMR demethylation occurs during helper lineage commitment
We next asked when DNA demethylation occurs within helper lineage cells. We examined Cd4 TSS-proximal DMR methylation by amplicon bisulfite sequencing at different stages after positive selection and during helper lineage differentiation using a Zbtb7bGFP reporter mouse16. Zbtb7b (which encodes ThPOK) is required for CD4-lineage commitment, and GFP is expressed from the Zbtb7bGFP allele specifically in MHCII-selected cells16–18. We sorted positively selected CD69+HSA+CD4+CD8lo cells prior to lineage commitment (GFP−; includes both MHCI- and MHCII-specific cells), early in helper lineage commitment (GFPmid) and late in commitment (GFPhi), as well as mature CD4SP cells (CD4+CD8−TCRβhiHSAloCD69loGFPhi) (Supplementary Fig. 8a–c). Using a representative TSS-proximal DMR amplicon (Fig. 6a), we found almost complete methylation in DP (Supplementary fig. 4g), CD4+CD8lo GFP− and GFPmid cells (Fig. 6b). DNA methylation began to decrease in GFPhi CD4+CD8lo cells, late in commitment, and was nearly absent in CD4SP cells, consistent with our earlier results (Fig. 6b). Thus, Cd4 locus differential methylation is initiated by the removal of DNA methylation marks late in helper lineage commitment, around the time of maximal Zbtb7b induction, and is completed at the mature CD4SP stage.
Figure 6. Cd4 locus demethylation occurs late in helper lineage differentiation and is largely independent of Zbtb7b.

(a) A Cd4 intron 1 amplicon from the indicated sorted cells in (b–d) was subjected to bisulfite sequencing: (b) GFP−, GFPmid or GFPhi CD4+CD8lo (also: CD69+HSAhiTCRβ+) and GFP+ CD4SP (CD69−HSAloTCRb+) thymocytes from Zbtb7bGFP/+ mice, (c) GFPhi CD4+CD8lo and GFP+ CD8SP thymocytes from Zbtb7bGFP/GFP mice. (d) WT CD8SP thymocytes (CD69−HSAloTCRb+). Filled circles indicate methylated CpGs and empty circles indicate unmethylated CpGs. Biological replicates from independent experiments are shown for (b–d). (e) CATCH-seq was performed on GFP+ (MHCII selected) and GFP− (MHCI selected) T cells from the lymph nodes of Zbtb7bGFP/GFP mice (n=1). The heatmap depicts the percentage CpG methylation from +6200bp to −669bp relative to the Cd4 TSS (Chr #6:124832027-124838896; mm9). Data from WT CD4 cells in Figure 2b, are shown for comparison. (f) Zbtb7bGFP/GFP GFP+ and GFP− lymph node CD8+ cells were stimulated in vitro for 3d with anti-CD3 and anti-CD28, and then analyzed for CD4, CD8 and GFP expression; results are representative of 2 independent experiments.
Zbtb7b is partially dispensable for Cd4 demethylation
Our observation that DNA demethylation in the helper lineage occurs after Zbtb7b is maximally induced suggested that Zbtb7b may play a role in the removal of these methylation marks. To test this hypothesis, we first assessed DNA methylation by amplicon sequencing in MHCII-selected, Zbtb7bGFP/GFP CD4+CD8lo and CD8SP T cell subsets. In Zbtb7bGFP/GFP mice, MHCII selected cells continue to express the GFP reporter allele, but are redirected into the cytotoxic lineage due to Zbtb7b deficiency16. We found that, despite the absence of Zbtb7b, MHCII-selected cells began to lose DNA methylation at the CD4+CD8loGFPhi stage (Fig. 6c). While Zbtb7bGFP/GFP CD8SP GFP+ cells exhibited higher methylation than Zbtb7bGFP/+ GFP+ CD4SP cells, they were hypo-methylated compared to wild-type CD8SP cells (Fig. 6c,d). Thus Zbtb7b appears to be at least partially dispensable for intronic DMR demethylation in MHCII-selected cells. We confirmed this result by bisulfite CATCH-Seq across the Cd4 locus in mature peripheral MHCI (GFP−)- and MHCII (GFP+)- selected Zbtb7bGFP/GFP CD8+ cells (Fig. 6e). GFP+ cells exhibited TSS-proximal DMR hypo-methylation similar to wild-type CD4+ cells, while MHCI-selected GFP− cells exhibited hyper-methylation similar to wild-type CD8+ cells (Fig. 6e). Interestingly, when induced to proliferate in vitro, GFP+ Zbtb7bGFP/GFP CD8+ cells maintained high CD8 expression, but also initiated CD4 expression (Fig. 6f). Thus, MHCII selection can induce Cd4 locus DNA demethylating signals even in the absence of Zbtb7b expression, and the resulting hypo-methylation correlates with failure to silence Cd4 in redirected CD8+ cells. Moreover, Zbtb7b is not required for Cd4 expression.
Cell division-independent Cd4 locus demethylation
The removal of DNA methylation marks in the Cd4 locus late in lineage commitment raises the question of whether demethylation is passive, by way of cell division, or active, through biochemical removal of 5mC marks. Among the 21 differentially methylated CpGs of the TSS-proximal DMR, the median DP:CD4SP CpG methylation ratio is 4.3 (mean: 27.6). Thus, passive demethylation would require an average of at least 2 cell divisions during helper lineage differentiation to effect passive demethylation, and over 9 divisions to explain the demethylation observed at some residues. Previous reports have suggested that there is no cell division between the DP and CD4SP stages of development19, or that a minority (<20%) of CD4SP cells may divide after HSA down-regulation and within hours of thymic emigration20,21. Moreover, our amplicon analysis of CD4+CD8lo MHCII-selected cells revealed stochastic loss of DNA methylation consistent with biochemical removal or passive demethylation with some remethylation (for example, seemingly random loss of methylation at only a subset of CpGs on a given allele), rather than the mixture of all methylated and all demethylated alleles that would be indicative of purely passive demethylation resulting from cell division without maintenance methylation. To further rule out the possibility of proliferation-mediated passive demethylation, we used a CFSE labeling and thymic injection technique to determine if cells divide during differentiation into the helper lineage. We sorted CD69− DP cells from CD45.2 B2mΔ/ΔH2-Ab1Δ/Δ OT-II–TCR-transgenic mice, mixed these with CD45.1 CD69− DP cells, stained with CFSE and injected into the thymi of CD45.1 mice that expressed MHCI and MHCII. At four days after injection, thymi were isolated and CFSE intensity and lineage differentiation were assessed by flow cytometry (Supplementary Fig. 8d–f). Among both CD45.1+ and CD45.2+ injected cells, we could identify pre–selected cells (CD69− DP), positively selected (CD4+CD8loCD69+TCRbmed/hi) and mature CD4SP cells (CD4+CD8−CD69loTCRbhi). All three groups expressed similarly high CFSE labeling, indicating no difference in cell divisions after injection. Importantly, the lack of cell division was not due to loss of viability, as these cells were able to differentiate, modulating the expression of CD69, TCRβ, CD4 and CD8. Thus, without evidence of significant proliferation following positive selection, we conclude that DNA methylation is removed from the Cd4 locus during helper lineage commitment via an active biochemical process, independent of cell division.
Ten Eleven Translocation (TET) enzyme-mediated oxidation of 5mC to 5-hydroxymethyl-cytosine (5hmC) has recently been shown to contribute to active DNA demethylation pathways22. To determine if TET-mediated hydroxymethylation of 5mCs in the Cd4 locus could contribute to locus demethylation, we examined 5hmC content by T4-phage β-glucosyltransferase (T4-βGT) - mediated restriction enzyme protection-qPCR. We isolated genomic DNA from DP, CD4+ and MHCII-selected CD4+CD8lo cells (CD69+HSA+CD4+CD8loGFP+ cells from Zbtb7bGFP/+ mice) and incubated the DNA with or without T4-βGT in the presence of glucose-UTP, before digestion with a restriction enzyme sensitive to modified 5hmC, MspI. T4-βGT transfers glucose-UTP specifically to 5hmC residues, blocking MspI digestion of CCGG motifs. In DP and CD4+ cells, we found very little T4-βGT protection of a differentially methylated CCGG motif at +190 bp relative to the Cd4 TSS (Fig. 7a, <20%). However, we found significantly greater T4-βGT-mediated protection of the same motif in MHCII-selected CD4+CD8lo cells (~40%), consistent with TET-dependent 5hmC-mediated demethylation in differentiating helper lineage T cells. Importantly, this CpG lies near the middle of the TSS-proximal DMR, and its methylation status during T cell differentiation is representative of the other dynamically and differentially methylated CpGs in the TSS-proximal DMR. To confirm and expand on these results, we performed oxidative-bisulfite amplicon sequencing on a group of three representative CpGs located at +1407 to +1487 relative to the TSS. While both 5mC and 5hmC are read as cytosine after bisulfite treatment, 5hmC can be oxidized by KRuO4 to 5-formylcytosine, which is then read as thymine after bisulfite treatment23. Thus, bisulfite treatment, with or without oxidation, can be used to differentiate 5mC from 5hmC modifications.
Figure 7. Cd4 TSS-proximal hydroxymethylation in MHCII-selected CD4+CD8lo cells.

(a) Genomic DNA from DP, naïve CD4+ and MHCII-selected CD4+CD8lo cells (CD69+HSA+CD4+CD8loGFP+ cells from Zbtb7bGFP/+ mice) was incubated with Uridine Diphosphate Glucose (UDG), with or without β-glucosyltransferase (βGT), before digestion with MspI. T4-βGT transfers UDG specifically to 5hmC residues, blocking MspI digestion of CCGG motifs. MspI digestion (at Chr6:124838036; +191bp relative to the Cd4 TSS) was assessed by qPCR comparison to undigested DNA. As a loading control, values were normalized to an adjacent amplicon without a MspI site. The mean and SD of 3 (CD4+) or 4 (DP and CD4+CD8lo) independent biological samples analyzed in two independent experiments are shown; P values (unpaired student’s t-test) below 0.05 (*) and 0.01 (**) are noted. (b–c) Genomic DNA from DP and CD4+CD8lo cells was subjected to bisulfite amplicon sequencing (BS) or KRuO4-oxidation followed by bisulfite amplicon sequencing (OxBS). (b) Pie charts represent the mean percentage of the indicated cytosine modifications at three CpGs located from +1407 to +1487 relative to the Cd4 TSS (CpG 1 = Chr #6:124836820; CpG 2 = Chr #6:124836779; CpG 3 = Chr #6:124836740), in DP (n=2) and CD4+CD8lo cells (n=2); Data were combined from 2 independent experiments: 4–13 amplicons (BS) or 11–17 amplicons (OxBS) were analyzed for each sample. (c) The mean and SD of the indicated modifications at the 3 CpGs in (b) were graphed for DP and CD4+CD8lo cells (circles and triangles represent the measurements for individual DP and CD4+CD8lo CpGs, respectively). 5hmC levels were calculated by subtracting the percentage “C” after OxBS treatment (5mC only) from the percentage “C” after BS treatment (5mC + 5hmC). P values (paired student’s t-test) below 0.05 (*) and 0.01 (**) are noted.
In DP cells, we found that all three CpGs were highly methylated (>90% 5mC/5hmC by bisulfite treatment alone), with low amounts of 5hmC at two CpGs (<20% 5hmC at CpGs 1 and 2) and moderate amounts at a third (< 50% 5hmC at CpG 3) (Fig. 7b). While overall methylation content remained similar, we observed decreased 5mC and increased 5hmC in post-selection CD4+CD8lo cells compared to DP cells (Fig. 7b,c), in keeping with our previous results. It should be noted that CD4+CD8lo cells are predominantly (> 2:1) MHCII selected16. Taken together, these results are consistent with our finding that demethylation of the TSS-proximal DMR after selection on MHCII does not require cell division, and suggest that it may be achieved enzymatically via a hydroxymethylated intermediate.
DISCUSSION
The molecular mechanism for heritable epigenetic Cd4 silencing in cytotoxic T cells has remained elusive despite characterization of the key cis- and transacting factors required to establish the silenced state. We have shown here that DNA methyltransferases are required to maintain silencing in CD8-lineage T cells, and linked this requirement to Cd4 locus methylation. Conversely, our data indicate that stable CD4 expression in CD4-lineage T cells is regulated by proximal enhancer-dependent Cd4 locus DNA demethylation. Surprisingly, the Cd4 TSS-proximal lineage DMR was methylated early in development, in both DN and DP thymocytes, indicating that a critical function of the silencer is to antagonize demethylation in CD8-lineage cells. Finally, demethylation of the DMR in CD4-lineage thymocytes appears to involve an active enzymatic process that is likely mediated by TET-dependent oxidation of methylcytosines. Thus, heritable Cd4 silencing versus expression is directed by a DNA demethylation switch under the control of the silencer and proximal enhancer.
Our results indicate that Cd4 locus methylation is necessary, but not sufficient, for stable Cd4 silencing. Indeed, DP cells express similar amounts of CD4 to helper T cells, but exhibit Cd4 locus hyper-methylation comparable to cytotoxic lineage cells. This uncoupling of TSS-proximal DMR methylation from transcription implies that Cd4 locus methylation/demethylation is critical for establishing heritable Cd4 expression states rather than transcriptional activity. Still, the question remains: how do DP cells express CD4 in the face of TSS-proximal methylation? It could be due to developmental stage specific transacting factor expression. For example, Runx1 is down-regulated at the DN4 to DP transition3,24, which could contribute to reduced S4 activity, allowing CD4 expression in spite of DNA hyper-methylation. Alternatively, or in conjunction with low Runx levels, DP cells may express yet to be identified stage-specific Cd4 activators. Further, it should be noted that there are a handful of Cd4 locus CpGs that are hypo-methylated in DP cells and CD4+ cells relative to CD8+ and DN cells; hypo-methylation of these residues in DP cells could contribute to CD4 expression in DP cells, while their de novo methylation in CD8+ cells could contribute to silencing. Finally, it is possible that CD8 lineage cells express lineage specific factors, possibly including Runx3, that collaborate with 5mC modifications to impose a silenced state.
Our data strongly suggest that stable helper lineage CD4 expression depends on an E4P-directed active DNA demethylation process in MHCII-selected thymocytes: first, E4P is required for CD4-lineage Cd4 locus hypo-methylation; second, there is little to no cell division between the DP (hyper-methylated) and CD4SP (hypo-methylated) stages of development; third, we found 5hmC in MHCII-selected CD4+CD8lo cells at TSS-proximal DMR CpGs, which are demethylated during the DP to CD4SP transition, consistent with 5mC oxidation by TET enzymes. E4P becomes dispensable in mature cells7, in which a “maturation enhancer”, putatively localized adjacent to the silencer element in intron 1 (13 and PDI and DRL, unpublished), and within the TSS-proximal DMR, programs CD4 expression. Thus, it seems likely that the critical function of E4P is to effect TSS-proximal demethylation across the promoter and sequences flanking intronic regulatory elements, which then allows the maturation enhancer to direct CD4 expression. It is worth noting, however, that Tet enzymes can further oxidize 5hmC to 5-formylcytosine (5fC) and 5-carboxycytosine (5caC)25–27, which are read as “unmodified” cytosines by bisulfite sequencing23. Thus we cannot rule out that the unmethylated CpGs identified in differentiating CD4 lineage cells represent 5fC or 5caC. Further studies will be required to determine how demethylation and/or oxidation of cytosine residues are critical for establishing heritable CD4 expression.
Central questions raised by our study are how E4P directs demethylation in the CD4-lineage and how S4 maintains the minimally altered methylation pattern following the DP stage to CD8-lineage transition. While it is possible that Tet enzymes are recruited to E4P upon TCR signaling, we also noticed that the TSS-proximal DMR contains some 5hmC modifications in wild-type DP cells and that E4P is responsible for DP cell hypo-methylation of E4P proximal CpGs. Thus it is tempting to hypothesize that E4P recruits Tet enzymes in DP cells or earlier, to poise the locus for demethylation before positive selection and lineage commitment. Could S4 then inhibit the recruitment or activity of TET enzymes in the Cd4 locus in MHCI-selected cells? Runx3 expression is induced in MHCI-selected cells during the DP to CD8SP transition28, and the RUNX fusion proteins found in acute myeloid leukemia have been shown to recruit DNMT proteins to target genes, presumably through an indirect mechanism29,30. Thus it is possible that Runx3 could recruit DNMTs to the Cd4 locus to ensure maintenance methylation. It will be important to determine if and how TET enzymes are targeted to the Cd4 locus in an E4P-dependent manner and whether DNMT enzymes are recruited in an S4- and Runx3-dependent manner during the CD4+CD8lo to CD8+SP transition.
Cd4 is arguably the best-characterized locus in vertebrates for the study of heritability, but the mechanisms by which its heritable states are controlled have remained elusive for years. The findings that the DNA methylation machinery is critical for the establishment and maintenance of silencing and that de-methylation is critical for heritable expression represent a significant advance and offer new opportunities to dissect the signaling pathways involved in thymocyte lineage choice. These results also establish Cd4 as a unique model to understand how DNA demethylation is effected and regulated. Further investigation of how the Cd4 locus is controlled via DNA methylation can thus provide important insights into how fully differentiated somatic cells achieve heritable states of gene expression.
METHODS
Mice
Dnmt1L 31 and Dnmt1Chip 9 mice were a kind gift from R. Jaenisch. Dnmt3aL 32 and Dnmt3bL 33 were from the Mutant Mouse Regional Resource Center (MMRRC). Cd4E4PΔ 7, Cd4S4Δ, Cd4S4-L 5, and Zbtb7bGFP 16 were previously described. WT C57BL/6, Cd4-Cre 8, Ubc-CreER-T2 34, H2-AbI 35, B2m 36, Rag2, Ly5.1, Ly5.2 and Tg(TcraTcrb)425Cbn (OT-II TCR tg) mice were purchased from the Jackson Laboratory. All mice were maintained under specific pathogen-free conditions in the Skirball Institute Animal Facility. All experiments were performed in accordance with the protocol approved by the IACUC at the NYU School of Medicine.
shRNA screen
The mouse shRNAmir library37 was used to generate ten retroviral pools, each comprising ~6000 shRNA clones38. CD62L+CD25−CD8+ cells, isolated from spleens and lymph nodes of Cd4S4-L/+; Ubc-CreERT2 mice, were cultured with anti-CD3 (0.25 μg/ml) and anti-CD28 (1 μg/ml) in the presence of OH-tamoxifen (400 nM) for 16 h before transduced with the retroviral pools. After 7 days culture in the presence of IL-2 (100 U/ml), CD4-positive cells were enriched by CD4+ MACS column. After 3 days, double-positive cells (CD4+CD8+) were sorted and their genomic DNA isolated. To identify the candidate shRNAs, the shRNA region of the transduced virus was PCR amplified, cloned and sequenced. Individual shRNAs were either obtained from the Open Biosystems library or synthesized. Two shRNA clones targeting Dnmt1 were identified (5′-GTACACCTTTCATGATGTGAAA-3′ and 5′-TCCCGAAGATCAACTCACCAAA-3′).
Flow cytometry and sorting
Monoclonal antibodies were purchased from eBioscience or BD Bioscience. Clones used were: anti-CD4 (RM4-5), anti-CD8α (53–6.7), anti-TCRβ (H57-597), anti-HSA (M1/69), anti-CD69 (H1.2F3), anti-CD44 (IM7), anti-CD62L (MEL-14), anti-CD45.1 (A20), anti-CD45.2 (104), CD25 (PC61) and anti-Thy1.2 (53–2.1). CFSE and e670 were obtained from Molecular Probes. After staining with antibodies and DAPI (Molecular Probes), cells were analyzed with an LSRII flow cytometer (BD Biosciences) or sorted with an Aria II (BD Biosciences). Post sort sample purity was >98%. In some cases, anti-CD4, anti-PE, anti-B220 magnetic beads (Miltenyi) were used for enrichment and depletion on the Auto-MACS platform (Miltenyi) before sorting. Flow cytometry data was analyzed using Flowjo software (Tree Star).
Cell culture
Tissue culture plates were incubated overnight with polyclonal goat anti-hamster IgG (MP biomedicals), washed 3x with PBS, and purified CD4+CD8−CD25−CD62L+CD44lo or naïve CD4−CD8+CD25−CD62L+CD44lo T cells were added, along with anti-CD3 (145-2C11, 0.25 μg/ml) and anti-CD28 (37.5.1, 1 μg/ml) antibodies. At day 3, cultures were supplemented with 100 U/ml recombinant IL-2 (Peprotech). For Dnmt1 knockdown, the Dnmt1 shRNA sequences were inserted into a miR-30-based hairpin of the pMSCV-LMP vector (Open Biosystems) according to the manufacturer’s instruction. Retroviruses were packaged in PlatE cells39 by transient transfection using TransIT 293 (Mirus Bio). Cells were transduced by spin infection at 1,200 × g at 30 °C for 90 min in the presence of 10 μg/ml polybrene (Sigma).
Methylation analysis
Genomic DNA was isolated using Purelink genomic DNA isolation kits (Invitrogen). For locus-wide bisulfite sequencing, CATCH-seq was performed as described12, using BAC clone RP24-330J12 (BACPAC Resource Center, CHORI). For amplicon sequencing, bisulfite conversion was performed using the EpiTect Bisulfite Kit (Qiagen). Amplicons were prepared using Hot Start Ex-Taq Polymerase (TaKaRa) and the following primers: TSS-F: 5′-GGGGTATTTATTGTTTTGAGTAT-3′, TSS-R: 5′-TTTAATTTTTCAACTTCCCCAAC-3′, +1600-F: 5′-GGTTATTTGGAGTTTTTTTTTAG-3′, +1600-R: 5′-CTTCAATTCATAAACTTATTCCC-3′, and TA cloned using the Qiagen PCR cloning kit. Bisulfite analysis of Sanger sequenced clones was performed using QUMA40. Oxidative bisulfite treatment was performed as previously described41 and amplicons were analyzed as above using the following primers: +1400-F: 5′-AAGTGTTTAAAATGTGTTAATTATTG-3′, +1400-R: 5′-TTAAAAACAAAACTAAAAAAACCC-3′. T4-βGT-mediated 5mC- and 5hmC- sensitive restriction enzyme digest was performed using the EpiMARK 5-hmC and 5mC analysis kit according to the manufacturer’s instructions (New England Biolabs). Quantitative PCR was performed using HotStart-IT SYBR Green qPCR Master Mix (Affymetrix-USB) and a LightCycler 480 (Roche). Percent digestion was calculated using ΔΔCt.
Nucleosome analysis
Nucleosomes were prepared as previously described42. Briefly, 1.1 × 107 cells were lysed in digestion buffer (50 mM Tris-HCl, pH 7.5, 1 mM CaCl2, 0.2% Triton X-100) supplemented with protease and deacetylase inhibitors. Nuclei were then treated with 12 U Micrococcal Nuclease (Worthington Biochemical) in 135 μl digestion buffer for 5 min at 37 °C to produce >90% mono-nucleosomes. The reaction was quenched by adding EDTA to 25 mM EDTA and EGTA to 10 mM. Samples were spun for 5 min at 2500g and supernatants with solubilized mononucleosomes were reserved (digestion supernatants). Pelleted nuclei were then lysed twice for 1 h on ice in lysis buffer (150 μl of 1 mM Tris-HCl, pH 7.5, 0.25 mM EDTA; supplemented with Protease and deacetylase inhibitors), following gentle sonication. After removing nuclear debris by centrifugation (5 min at 11,000g), nuclear lysis supernatants were pooled with digestion supernatants. Mononucleosome fragments were then subjected to CATCH-seq.
Supplementary Material
Supplementary Figure 1. DNA methylation machinery is essential for CD4 silencing in cytotoxic T cells. (a) Scheme for the retroviral shRNA screen. (b) Histogram showing CD4 expression (MFI) in wild-type cytotoxic T cells infected with a Dnmt1 shRNA-GFP retrovirus (shaded area) or sham shRNA-GFP retrovirus (open area). Representative of 2 independent experiments. (c) Cytotoxic CD4−8+ cells from Dnmt1chip/chip animals were loaded with e670 and cultured in vitro for 4 days after infection with Dnmt1 shRNA retroviruses. Virus-infected cells were selected with puromycin. Gates define non-, low-, medium- and highly-cycled cells (i.e. e670 dilution), and the percentage of cells de-repressing CD4 in each gate is indicated. The red line shows the CD4 staining level used to determine de-repression. Representative of 2 independent experiments.
Supplementary Figure 2. Reproducibility and coverage of locus-wide bisulfite sequencing. Genomic DNA was prepared from biological replicates of the indicated samples, and then subjected to CATCH-seq (Cd4 TSS +/− ~75kb). (a) For each replicate, the fraction methylation at CpGs with at least 30x coverage is graphed. Linear regressions were performed (red lines) and R2 calculated. (b) The median CpG sequencing coverage for the indicated samples is graphed. Error bars represent the 5th and 95th percentiles for CpG sequencing coverage.
Supplementary Figure 3. Silencer dependent DMR in the first intron of Cd4. Naïve (Thy1.2+CD44loCD62L+CD25−) WT CD4+, WT CD8+ and Cd4S4Δ/S4Δ CD4+8+ cells were isolated from LNs. Their genomic DNA was either isolated immediately or after 5 days of in vitro expansion, using CFSE-labeling and dilution to identify and sort cells that had completed at least 5 divisions. Genomic DNA was then subjected to CATCH-seq. Percent CpG methylation was graphed on the UCSC genome browser for (a) Chr #6:124,746,000-124,909,000 and (b) Chr #6:124,814,000-124,855,000. UCSC genes are indicated below the graphs. The samples correspond to those Fig. 2. Replicates were derived from two experiments.
Supplementary Figure 4. Hyper-methylation of the Cd4 locus in the cytotoxic lineage and in immature T cell progenitors. To confirm locus-wide bisulfite sequencing, two amplicons were chosen for targeted bisulfite sequencing (a–b, d–h) and four amplicons were chosen for methylation-sensitive restriction enzyme digest analysis (c) (locus and CpG analysis map at top: S4: Silencer; black arrow: TSS; lollipops: CpGs; blue bars: bisulfite sequencing amplicons; purple arrows: HpaII sites with positions relative to TSS). (a–b) Naive WT CD4+ and CD8+ T cells were sorted, genomic DNA was prepared and bisulfite treated, and amplicons were cloned and sequenced. Filled circles indicate methylated CpGs and empty circles indicate unmethylated CpGs. Colored bars correspond to amplicons in map. The 5′ and 3′ amplicon methylation patterns are shown in (a) and (b), respectively. Data are from 3 mice. (c) HpaII digestion of genomic DNA from naive WT CD4+, WT CD8+ and Cd4S4Δ/S4Δ CD8+ T cells was assessed by qPCR at the indicated CpGs. HpaII digests only unmethylated-CCGG motifs; thus, percent-undigested DNA corresponds to percent methylation. All samples were normalized to an HpaII-insensitive loading control amplicon in the Cd4 locus. Graphs represent the average and standard deviation (n=2 for WT CD4+ and WT CD8+) or amount of undigested DNA (n=1 for Cd4S4Δ/S4Δ) are shown. Data are representative of at least two 2 experiments. Average and standard deviation of percent methylation from locus-wide bisulfite sequencing of biological replicates is presented in the graphs on the left for each CpG (n=2; samples correspond to those in Fig. 2). (d) CFSE-labeled naive WT CD4+ and CD8+ T cells were expanded for 5 days in vitro with anti-CD3, anti-CD28 and IL-2, and cells that had undergone at least 6 divisions were sorted and subjected to bisulfite sequencing of the 3′ amplicon (n = 1). (e) CFSE-labeled naive WT CD4+ and CD8+ T cells were injected into Rag1−/− mice, and 20 days later CFSE-negative cells were sorted (>10 divisions) and subjected to amplicon bisulfite sequencing of the 3′ amplicon (n = 1). (f) DN3 and WT DP T cells were sorted and the 5′ amplicon was sequenced as in (a). (g–h) Bisulfite analysis of the 3′ intronic amplicon from WT (g) and Cd4S4Δ/S4Δ (h) DP cells. (f–h) Data are from 1 (DN3), 2 (Cd4S4Δ/S4Δ DP) or 3 (DP) mice.
Supplementary Figure 5. Nucleosomal positioning correlates with CD4 expression, rather than DNA methylation. Nuclei from the indicated samples were isolated and treated with micrococcal nuclease, and mono-nucleosome fragments from the Cd4 locus ~ +/−75kb were analyzed by CATCH-seq (without bisulfite treatment). The upper density graphs show nucleosome occupancy (blue = high nucleosome density, white: naked DNA). The lower graphs show coordinate-specific CpG methylation (data from samples in figures 2–4; red = hyper-methylation, green = hypo-methylation). Tracks were graphed with the IGV browser platform (Chr #6:124,831,500-124,838,486 (mm9)). For clarity, yellow lines separate CD4 high- and low-expressing samples (above and below, respectively). The red arrowhead indicates a region in which nucleosome paucity is highly correlated with CD4 expression. Replicate samples are from two experiments.
Supplementary Figure 6. E4P controls proximal demethylation events early in T cell development. CATCH-seq was performed on sorted populations of WT DN3 (Thy1.2+Lin-CD25+CD44−), WT and Cd4E4PΔ/E4PΔ DP (TCRβloCD24+CD69−CD4+CD8+), WT naïve (Thy1.2+CD25−CD44loCD62L+) CD4+, WT CD8+, Cd4E4PΔ/E4PΔ CD4+, and Cd4S4Δ/S4Δ CD8+ T cells. The heatmap depicts percentage CpG methylation (in biological replicates, where indicated) from −9270bp to −15869bp relative to the Cd4 TSS (Chr6:124847307-124853906; mm9). The approximate location of the region in the Cd4 locus is indicated above (genes, S4 and E4P are noted), and the lone CpG within the proximal enhancer is indicated below the heat map. Replicates are from 2 experiments. 7 CpGs in this region experienced complete or partial demethylation at the DN3 to DP transition, and this hypo-methylated state was preserved in mature T cell lineages. E4P is responsible for de-methylation before the DN3 stage (note Cd4E4PΔ/E4PΔ DP hyper-methylation compared to DN3 cells), as well as at the DN3 to DP transition (note that DN3 vs. DP differentially methylated CpGs are all highly methylated in Cd4E4PΔ/E4PΔ DP cells).
Supplementary Figure 7. Reduced Dnmt1 activity rescues CD4 expression in Cd4E4PΔ/E4PΔ helper T cells. (a–c) Naive CD4+ T cells (Thy1.2+CD25−CD8−CD44loCD62L+) from Dnmt1-deficient and control mice, both with deletions of E4P, were CFSE labeled and stimulated in vitro with anti-CD3, anti-CD28 and IL-2. Analysis was performed at 96 h and 120 h, to determine (a) the percentage of CD4+ cells, (b) the MFI of the CD4+ cells, and (c) the percentage of CD4+ cells at each cell division as measured by CFSE dilution. Representative of at least 4 independent experiments. (d–e) CFSE stained Cd4E4PΔ/E4PΔ CD4+ T cells were stimulated for 24 h with anti-CD3 and anti-CD28, infected with retroviral vectors expressing Puro, RFP and either Dnmt1 shRNA in a mir30 context (red, shDnmt1) or an empty mir30 (blue, vector). Transduced cells were maintained with puromycin selection and analyzed by gating for RFP+ cells. (a) The percentage of CD4+ cells, as well as the CD4 MFI of CD4+ cells, was measured by flow cytometry at 72 h, 96 h and 120 h. (b) CFSE dilution was used to measure the percentage CD4+ cells in each generation at 72 h, 96 h and 120 h. Representative of at least 4 independent experiments.
Supplementary Figure 8: ThPOK expression and cell division during helper T lineage differentiation. (a) Helper and cytotoxic T cell differentiation can be traced by CD4, CD8 and GFP (from the Zbtb7bGFP allele) expression 16. Upon positive selection, DP T cells (A gate) up-regulate CD69 and TCRβ (not shown) and down-regulate CD8 expression, becoming CD4+CD8lo (B gate). MHCI-selected cells then up-regulate CD8 expression (D gate) before finally down-regulating HSA (not shown) and silencing CD4 expression (E gate). MHCII-selected Zbtb7bGFP/+ cells begin to express GFP at the CD4+CD8lo stage (green filled circles, B gate), before fully down regulating CD8 and HSA (C gate and not shown). In Zbtb7bGFP/GFP MHCII-selected cells, GFP expression is still induced at the CD4+CD8lo stage (green filled circles, B gate), but cells then up-regulate CD8 (D gate) before extinguishing CD4 expression to become GFP+ cytotoxic T cells (E gate). (b) To sort CD4+CD8lo cells at different stages of helper cell differentiation, we enriched Zbtb7bGFP/+ and Zbtb7bGFP/GFP thymocytes for TCRβ expression using MACS beads, before sorting HSA+CD69+CD4+CD8lo cells (middle panel) based on Zbtb7b expression: GFP− (MHCI- or early MHCII-selected), GFPmid (MHCII-selected, initiating commitment) and GFPhi (MHCII-selected, late commitment). (c) MHCII-selected CD4SP (Zbtb7bGFP/+) and CD8SP (Zbtb7bGFP/GFP) thymocytes were MACS enriched for TCRβ+ expression, before gating of HSA−TCRβhiCD69−GFP+ cells and sorting of CD4+CD8− and CD4−CD8+ cells, respectively. (b–c) Stainings are representative of at least 3 experiments. (d) Scheme for thymic injection to assess cell division during helper T cell development. (e–f) 106 Tg(TcraTcrb)425Cbn(OT-IItg)+ B2mΔ/ΔH2-AbI Δ/Δ CD45.2+ and 107 CD45.2− DP cells (HSA+CD69−CD4+CD8+) were CFSE-labeled and injected intra-thymically into CD45.2− recipients. Tg(TcraTcrb)425Cbn+ B2mΔ/ΔH2-AbI Δ/Δ were used to ensure a homogenous population of unselected DP cells that would differentiate into the helper lineage. After four days, mice were sacrificed and thymocytes were analyzed for phenotype and CFSE dilution. (e) Gating of host and donor-derived cells in recipient thymus at day 4 after injection. (f) CFSE levels in cells from gates indicated in (e). Note that injected DP thymocytes do not dilute CFSE following positive selection. Representative of at least 2 experiments.
Acknowledgments
We thank R. Jaenisch (Whitehead Institute for Biomedical Research) for mouse strains; the University of Massachusetts Medical School RNAi Core Faciility for providing shRNAs; A. Cuesta and A. Chen for technical help; and members of the laboratory for discussion. Supported by the US National Institutes of Health (R00DK091508 to J.R.H., 5 T32 CA009161-36 to M.S, and GM033977 to M.R.G.), the Jane Coffin Childs Fund (J.R.H.), the Cancer Research Institute (M.S.and P.D.I.) and the Howard Hughes Medical Institute (M.R.G. and D.R.L.).
Footnotes
ACCESSION CODES
BioProject ID PRJNA282735
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
J.R.H. did the genetic screen and follow-up analyses. M.S. and K.D. did the Cd4 locus-wide methylation and MNase analyses with bioinformatics support from D.A. M.S. and C.G. performed amplicon bisulfite sequencing. M.S. performed E4P rescue experiments, proliferation assays in thymus and T4-βGT analysis. P.D.I. performed oxidative bisulfite analysis. S.G. and M.R.G. provided the murine shRNA retroviral pools. J.R.H., M.S., and D.R.L. designed the experiments and wrote the manuscript with input from the other authors.
References
- 1.Gialitakis M, Sellars M, Littman DR. The epigenetic landscape of lineage choice: lessons from the heritability of CD4 and CD8 expression. Current topics in microbiology and immunology. 2012;356:165–188. doi: 10.1007/82_2011_175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taniuchi I, Ellmeier W, Littman DR. The CD4/CD8 lineage choice: new insights into epigenetic regulation during T cell development. Adv Immunol. 2004;83:55–89. doi: 10.1016/S0065-2776(04)83002-5. [DOI] [PubMed] [Google Scholar]
- 3.Taniuchi I, et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–633. doi: 10.1016/s0092-8674(02)01111-x. [DOI] [PubMed] [Google Scholar]
- 4.Taniuchi I, Sunshine MJ, Festenstein R, Littman DR. Evidence for distinct CD4 silencer functions at different stages of thymocyte differentiation. Mol Cell. 2002;10:1083–1096. doi: 10.1016/s1097-2765(02)00735-9. [DOI] [PubMed] [Google Scholar]
- 5.Zou YR, et al. Epigenetic silencing of CD4 in T cells committed to the cytotoxic lineage. Nat Genet. 2001;29:332–336. doi: 10.1038/ng750. [DOI] [PubMed] [Google Scholar]
- 6.Sawada S, Scarborough JD, Killeen N, Littman DR. Cell. 1994;77:917–929. doi: 10.1016/0092-8674(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 7.Chong MM, et al. Epigenetic propagation of CD4 expression is established by the Cd4 proximal enhancer in helper T cells. Genes Dev. 2010;24:659–669. doi: 10.1101/gad.1901610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee PP, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. S1074-7613(01)00227-8 [pii] [DOI] [PubMed] [Google Scholar]
- 9.Tucker KL, et al. Germ-line passage is required for establishment of methylation and expression patterns of imprinted but not of nonimprinted genes. Genes Dev. 1996;10:1008–1020. doi: 10.1101/gad.10.8.1008. [DOI] [PubMed] [Google Scholar]
- 10.Feng J, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nature neuroscience. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeong M, et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nature genetics. 2014;46:17–23. doi: 10.1038/ng.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Day K, Song J, Absher D. Targeted sequencing of large genomic regions with CATCH-Seq. PLoS One. 2014;9:e111756. doi: 10.1371/journal.pone.0111756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henson DM, Chou C, Sakurai N, Egawa T. A silencer-proximal intronic region is required for sustained CD4 expression in postselection thymocytes. J Immunol. 2014;192:4620–4627. doi: 10.4049/jimmunol.1302374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collings CK, Waddell PJ, Anderson JN. Effects of DNA methylation on nucleosome stability. Nucleic Acids Res. 2013;41:2918–2931. doi: 10.1093/nar/gks893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jimenez-Useche I, et al. DNA methylation regulated nucleosome dynamics. Scientific reports. 2013;3:2121. doi: 10.1038/srep02121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egawa T, Littman DR. ThPOK acts late in specification of the helper T cell lineage and suppresses Runx-mediated commitment to the cytotoxic T cell lineage. Nat Immunol. 2008;9:1131–1139. doi: 10.1038/ni.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun G, et al. The zinc finger protein cKrox directs CD4 lineage differentiation during intrathymic T cell positive selection. Nature immunology. 2005;6:373–381. doi: 10.1038/ni1183. [DOI] [PubMed] [Google Scholar]
- 18.He X, et al. The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature. 2005;433:826–833. doi: 10.1038/nature03338. [DOI] [PubMed] [Google Scholar]
- 19.Egerton M, Scollay R, Shortman K. Kinetics of mature T-cell development in the thymus. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:2579–2582. doi: 10.1073/pnas.87.7.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Penit C, Vasseur F. Expansion of mature thymocyte subsets before emigration to the periphery. Journal of immunology. 1997;159:4848–4856. [PubMed] [Google Scholar]
- 21.Ernst B, Surh CD, Sprent J. Thymic selection and cell division. The Journal of experimental medicine. 1995;182:961–971. doi: 10.1084/jem.182.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schubeler D. Function and information content of DNA methylation. Nature. 2015;517:321–326. doi: 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- 23.Booth MJ, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 24.Sato T, et al. Dual functions of Runx proteins for reactivating CD8 and silencing CD4 at the commitment process into CD8 thymocytes. Immunity. 2005;22:317–328. doi: 10.1016/j.immuni.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 25.He YF, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfaffeneder T, et al. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angewandte Chemie. 2011;50:7008–7012. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- 28.Egawa T, Tillman RE, Naoe Y, Taniuchi I, Littman DR. The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. J Exp Med. 2007;204:1945–1957. doi: 10.1084/jem.20070133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu S, et al. Interplay of RUNX1/MTG8 and DNA methyltransferase 1 in acute myeloid leukemia. Cancer Res. 2005;65:1277–1284. doi: 10.1158/0008-5472.CAN-04-4532. [DOI] [PubMed] [Google Scholar]
- 30.Cheng CK, et al. Secreted-frizzled related protein 1 is a transcriptional repression target of the t(8;21) fusion protein in acute myeloid leukemia. Blood. 2011;118:6638–6648. doi: 10.1182/blood-2011-05-354712. [DOI] [PubMed] [Google Scholar]
- 31.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. 0092-8674(92)90611-F [pii] [DOI] [PubMed] [Google Scholar]
- 32.Nguyen S, Meletis K, Fu D, Jhaveri S, Jaenisch R. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev Dyn. 2007;236:1663–1676. doi: 10.1002/dvdy.21176. [DOI] [PubMed] [Google Scholar]
- 33.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 34.Ruzankina Y, et al. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell stem cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grusby MJ, Johnson RS, Papaioannou VE, Glimcher LH. Depletion of CD4+ T cells in major histocompatibility complex class II-deficient mice. Science. 1991;253:1417–1420. doi: 10.1126/science.1910207. [DOI] [PubMed] [Google Scholar]
- 36.Zijlstra M, et al. Beta 2-microglobulin deficient mice lack CD4-8+ cytolytic T cells. Nature. 1990;344:742–746. doi: 10.1038/344742a0. [DOI] [PubMed] [Google Scholar]
- 37.Silva JM, et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet. 2005;37:1281–1288. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- 38.Gobeil S, Zhu X, Doillon CJ, Green MR. A genome-wide shRNA screen identifies GAS1 as a novel melanoma metastasis suppressor gene. Genes Dev. 2008;22:2932–2940. doi: 10.1101/gad.1714608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene therapy. 2000;7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 40.Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic acids research. 2008;36:W170–175. doi: 10.1093/nar/gkn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Booth MJ, et al. Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nat Protoc. 2013;8:1841–1851. doi: 10.1038/nprot.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. DNA methylation machinery is essential for CD4 silencing in cytotoxic T cells. (a) Scheme for the retroviral shRNA screen. (b) Histogram showing CD4 expression (MFI) in wild-type cytotoxic T cells infected with a Dnmt1 shRNA-GFP retrovirus (shaded area) or sham shRNA-GFP retrovirus (open area). Representative of 2 independent experiments. (c) Cytotoxic CD4−8+ cells from Dnmt1chip/chip animals were loaded with e670 and cultured in vitro for 4 days after infection with Dnmt1 shRNA retroviruses. Virus-infected cells were selected with puromycin. Gates define non-, low-, medium- and highly-cycled cells (i.e. e670 dilution), and the percentage of cells de-repressing CD4 in each gate is indicated. The red line shows the CD4 staining level used to determine de-repression. Representative of 2 independent experiments.
Supplementary Figure 2. Reproducibility and coverage of locus-wide bisulfite sequencing. Genomic DNA was prepared from biological replicates of the indicated samples, and then subjected to CATCH-seq (Cd4 TSS +/− ~75kb). (a) For each replicate, the fraction methylation at CpGs with at least 30x coverage is graphed. Linear regressions were performed (red lines) and R2 calculated. (b) The median CpG sequencing coverage for the indicated samples is graphed. Error bars represent the 5th and 95th percentiles for CpG sequencing coverage.
Supplementary Figure 3. Silencer dependent DMR in the first intron of Cd4. Naïve (Thy1.2+CD44loCD62L+CD25−) WT CD4+, WT CD8+ and Cd4S4Δ/S4Δ CD4+8+ cells were isolated from LNs. Their genomic DNA was either isolated immediately or after 5 days of in vitro expansion, using CFSE-labeling and dilution to identify and sort cells that had completed at least 5 divisions. Genomic DNA was then subjected to CATCH-seq. Percent CpG methylation was graphed on the UCSC genome browser for (a) Chr #6:124,746,000-124,909,000 and (b) Chr #6:124,814,000-124,855,000. UCSC genes are indicated below the graphs. The samples correspond to those Fig. 2. Replicates were derived from two experiments.
Supplementary Figure 4. Hyper-methylation of the Cd4 locus in the cytotoxic lineage and in immature T cell progenitors. To confirm locus-wide bisulfite sequencing, two amplicons were chosen for targeted bisulfite sequencing (a–b, d–h) and four amplicons were chosen for methylation-sensitive restriction enzyme digest analysis (c) (locus and CpG analysis map at top: S4: Silencer; black arrow: TSS; lollipops: CpGs; blue bars: bisulfite sequencing amplicons; purple arrows: HpaII sites with positions relative to TSS). (a–b) Naive WT CD4+ and CD8+ T cells were sorted, genomic DNA was prepared and bisulfite treated, and amplicons were cloned and sequenced. Filled circles indicate methylated CpGs and empty circles indicate unmethylated CpGs. Colored bars correspond to amplicons in map. The 5′ and 3′ amplicon methylation patterns are shown in (a) and (b), respectively. Data are from 3 mice. (c) HpaII digestion of genomic DNA from naive WT CD4+, WT CD8+ and Cd4S4Δ/S4Δ CD8+ T cells was assessed by qPCR at the indicated CpGs. HpaII digests only unmethylated-CCGG motifs; thus, percent-undigested DNA corresponds to percent methylation. All samples were normalized to an HpaII-insensitive loading control amplicon in the Cd4 locus. Graphs represent the average and standard deviation (n=2 for WT CD4+ and WT CD8+) or amount of undigested DNA (n=1 for Cd4S4Δ/S4Δ) are shown. Data are representative of at least two 2 experiments. Average and standard deviation of percent methylation from locus-wide bisulfite sequencing of biological replicates is presented in the graphs on the left for each CpG (n=2; samples correspond to those in Fig. 2). (d) CFSE-labeled naive WT CD4+ and CD8+ T cells were expanded for 5 days in vitro with anti-CD3, anti-CD28 and IL-2, and cells that had undergone at least 6 divisions were sorted and subjected to bisulfite sequencing of the 3′ amplicon (n = 1). (e) CFSE-labeled naive WT CD4+ and CD8+ T cells were injected into Rag1−/− mice, and 20 days later CFSE-negative cells were sorted (>10 divisions) and subjected to amplicon bisulfite sequencing of the 3′ amplicon (n = 1). (f) DN3 and WT DP T cells were sorted and the 5′ amplicon was sequenced as in (a). (g–h) Bisulfite analysis of the 3′ intronic amplicon from WT (g) and Cd4S4Δ/S4Δ (h) DP cells. (f–h) Data are from 1 (DN3), 2 (Cd4S4Δ/S4Δ DP) or 3 (DP) mice.
Supplementary Figure 5. Nucleosomal positioning correlates with CD4 expression, rather than DNA methylation. Nuclei from the indicated samples were isolated and treated with micrococcal nuclease, and mono-nucleosome fragments from the Cd4 locus ~ +/−75kb were analyzed by CATCH-seq (without bisulfite treatment). The upper density graphs show nucleosome occupancy (blue = high nucleosome density, white: naked DNA). The lower graphs show coordinate-specific CpG methylation (data from samples in figures 2–4; red = hyper-methylation, green = hypo-methylation). Tracks were graphed with the IGV browser platform (Chr #6:124,831,500-124,838,486 (mm9)). For clarity, yellow lines separate CD4 high- and low-expressing samples (above and below, respectively). The red arrowhead indicates a region in which nucleosome paucity is highly correlated with CD4 expression. Replicate samples are from two experiments.
Supplementary Figure 6. E4P controls proximal demethylation events early in T cell development. CATCH-seq was performed on sorted populations of WT DN3 (Thy1.2+Lin-CD25+CD44−), WT and Cd4E4PΔ/E4PΔ DP (TCRβloCD24+CD69−CD4+CD8+), WT naïve (Thy1.2+CD25−CD44loCD62L+) CD4+, WT CD8+, Cd4E4PΔ/E4PΔ CD4+, and Cd4S4Δ/S4Δ CD8+ T cells. The heatmap depicts percentage CpG methylation (in biological replicates, where indicated) from −9270bp to −15869bp relative to the Cd4 TSS (Chr6:124847307-124853906; mm9). The approximate location of the region in the Cd4 locus is indicated above (genes, S4 and E4P are noted), and the lone CpG within the proximal enhancer is indicated below the heat map. Replicates are from 2 experiments. 7 CpGs in this region experienced complete or partial demethylation at the DN3 to DP transition, and this hypo-methylated state was preserved in mature T cell lineages. E4P is responsible for de-methylation before the DN3 stage (note Cd4E4PΔ/E4PΔ DP hyper-methylation compared to DN3 cells), as well as at the DN3 to DP transition (note that DN3 vs. DP differentially methylated CpGs are all highly methylated in Cd4E4PΔ/E4PΔ DP cells).
Supplementary Figure 7. Reduced Dnmt1 activity rescues CD4 expression in Cd4E4PΔ/E4PΔ helper T cells. (a–c) Naive CD4+ T cells (Thy1.2+CD25−CD8−CD44loCD62L+) from Dnmt1-deficient and control mice, both with deletions of E4P, were CFSE labeled and stimulated in vitro with anti-CD3, anti-CD28 and IL-2. Analysis was performed at 96 h and 120 h, to determine (a) the percentage of CD4+ cells, (b) the MFI of the CD4+ cells, and (c) the percentage of CD4+ cells at each cell division as measured by CFSE dilution. Representative of at least 4 independent experiments. (d–e) CFSE stained Cd4E4PΔ/E4PΔ CD4+ T cells were stimulated for 24 h with anti-CD3 and anti-CD28, infected with retroviral vectors expressing Puro, RFP and either Dnmt1 shRNA in a mir30 context (red, shDnmt1) or an empty mir30 (blue, vector). Transduced cells were maintained with puromycin selection and analyzed by gating for RFP+ cells. (a) The percentage of CD4+ cells, as well as the CD4 MFI of CD4+ cells, was measured by flow cytometry at 72 h, 96 h and 120 h. (b) CFSE dilution was used to measure the percentage CD4+ cells in each generation at 72 h, 96 h and 120 h. Representative of at least 4 independent experiments.
Supplementary Figure 8: ThPOK expression and cell division during helper T lineage differentiation. (a) Helper and cytotoxic T cell differentiation can be traced by CD4, CD8 and GFP (from the Zbtb7bGFP allele) expression 16. Upon positive selection, DP T cells (A gate) up-regulate CD69 and TCRβ (not shown) and down-regulate CD8 expression, becoming CD4+CD8lo (B gate). MHCI-selected cells then up-regulate CD8 expression (D gate) before finally down-regulating HSA (not shown) and silencing CD4 expression (E gate). MHCII-selected Zbtb7bGFP/+ cells begin to express GFP at the CD4+CD8lo stage (green filled circles, B gate), before fully down regulating CD8 and HSA (C gate and not shown). In Zbtb7bGFP/GFP MHCII-selected cells, GFP expression is still induced at the CD4+CD8lo stage (green filled circles, B gate), but cells then up-regulate CD8 (D gate) before extinguishing CD4 expression to become GFP+ cytotoxic T cells (E gate). (b) To sort CD4+CD8lo cells at different stages of helper cell differentiation, we enriched Zbtb7bGFP/+ and Zbtb7bGFP/GFP thymocytes for TCRβ expression using MACS beads, before sorting HSA+CD69+CD4+CD8lo cells (middle panel) based on Zbtb7b expression: GFP− (MHCI- or early MHCII-selected), GFPmid (MHCII-selected, initiating commitment) and GFPhi (MHCII-selected, late commitment). (c) MHCII-selected CD4SP (Zbtb7bGFP/+) and CD8SP (Zbtb7bGFP/GFP) thymocytes were MACS enriched for TCRβ+ expression, before gating of HSA−TCRβhiCD69−GFP+ cells and sorting of CD4+CD8− and CD4−CD8+ cells, respectively. (b–c) Stainings are representative of at least 3 experiments. (d) Scheme for thymic injection to assess cell division during helper T cell development. (e–f) 106 Tg(TcraTcrb)425Cbn(OT-IItg)+ B2mΔ/ΔH2-AbI Δ/Δ CD45.2+ and 107 CD45.2− DP cells (HSA+CD69−CD4+CD8+) were CFSE-labeled and injected intra-thymically into CD45.2− recipients. Tg(TcraTcrb)425Cbn+ B2mΔ/ΔH2-AbI Δ/Δ were used to ensure a homogenous population of unselected DP cells that would differentiate into the helper lineage. After four days, mice were sacrificed and thymocytes were analyzed for phenotype and CFSE dilution. (e) Gating of host and donor-derived cells in recipient thymus at day 4 after injection. (f) CFSE levels in cells from gates indicated in (e). Note that injected DP thymocytes do not dilute CFSE following positive selection. Representative of at least 2 experiments.