

Summary
Understanding of microbial metal reduction is based almost solely on studies of Gram-negative organisms. In this study, we focus on Desulfotomaculum reducens MI-1, a Gram-positive metal reducer whose genome lacks genes with similarity to any characterized metal reductase. Using non-denaturing separations and mass spectrometry identification, in combination with a colorimetric screen for chelated Fe(III)-NTA reduction with NADH as electron donor, we have identified proteins from the D. reducens proteome not previously characterized as iron reductases. Their function was confirmed by heterologous expression in E. coli. Furthermore, we show that these proteins have the capability to reduce soluble Cr(VI) and U(VI) with NADH as electron donor. The proteins identified are NADH:flavin oxidoreductase (Dred_2421) and a protein complex composed of oxidoreductase FAD/NAD(P)-binding subunit (Dred_1685) and dihydroorotate dehydrogenase 1B (Dred_1686). Dred_2421 was identified in the soluble proteome and is predicted to be a cytoplasmic protein. Dred_1685 and Dred_1686 were identified in both the soluble as well as the insoluble protein fraction, suggesting a type of membrane-association, although PSORTb predicts both proteins are cytoplasmic. This study is the first functional proteomic analysis of D. reducens and one of the first analyses of metal and radionuclide reduction in an environmentally relevant Gram-positive bacterium.
Introduction
Microorganisms capable of dissimilatory iron reduction are of interest due to their integral ecological roles and applications for heavy metal and radionuclide bioremediation (Weber et al., 2006; Mohapatra et al., 2010; Bird et al., 2011). Over two decades of research in model Gram-negative bacteria (i.e. species of Geobacter and Shewanella) focusing on electron transfer to Fe(III) and U(VI) has shaped understanding of microbial metal reduction. Common to model Gram-negative metal respirers is the abundance of annotated multiheme c-type cytochromes (MHCs), many of which are membrane-bound and predicted to be involved in iron and/or uranium reduction (Wall and Krumholz, 2006; Shi et al., 2009; Sharma et al., 2010).
Becoming increasingly apparent, however, is the diversity and environmental prevalence of Gram-positive organisms capable of dissimilatory metal reduction. Specifically, numerous Clostridia species have been detected commonly in subsurface environments with heavy metal contamination, and several of these species have been demonstrated to use various metals (including Fe(III) and U(VI)) as electron acceptors (Petrie et al., 2003; Suzuki et al., 2003; Cardenas et al., 2010; Williamson et al., 2013; Newsome et al., 2014). Mechanisms of electron transfer to metals in these phylogenetically distinct organisms are far less elucidated. Recent studies in two thermophilic Firmicutes, Carboxydothermus ferrireducens and Thermincola potens, support the involvement of MHCs localized on the cell surface (Carlson et al., 2012; Gavrilov et al., 2012). These MHC-rich thermophiles may be exceptions, however, as MHCs are scarce across the genomes of sequenced mesophilic Firmicutes (Sharma et al., 2010).
The sulfate reducing bacterium (SRB) Desulfotomaculum reducens MI-1, isolated from heavy-metal contaminated sediment, serves as a useful and novel system for the study of Gram-positive dissimilatory metal reduction. D. reducens has been shown to use a variety of metals including U(VI), Fe(III), Cr(VI), Mn(IV) as electron acceptors while oxidizing lactate or butyrate (Tebo and Obraztsova, 1998). D. reducens also reduces metals when grown fermentatively with pyruvate, and some studies have focused on metal reduction during this growth condition (Junier et al., 2009; Dalla Vecchia et al., 2014). A recent study concluded that although D. reducens does not appear to gain energy directly from the reduction of Fe(III) during fermentative growth on pyruvate, the Fe(III) serves as an electron sink, relieving thermodynamic limitations of fermentation resulting from H2-buildup. Furthermore, the study suggested that direct contact was not required for the reduction of insoluble Fe(III), and riboflavin and small amounts of FMN (flavin mononucleotide) in spent media were identified as potential electron shuttles (Dalla Vecchia et al., 2014). However, no insights regarding enzymes involved in Fe(III) reduction were provided in that study. Another unique capability of D. reducens following growth with pyruvate is U(VI) and Fe(III)-citrate reduction in the sporulated state, relevant to particular environments where conditions may vary dramatically over time (Junier et al., 2009).
The genome of D. reducens has been sequenced and contains only one operon annotated as a c-type cytochrome, encoded by the two genes Dred_0700 and Dred_0701 (Junier et al., 2010). However, all evidence to date suggests that this cytochrome is not involved in metal reduction. A transcriptomic study comparing gene expression in D. reducens when grown fermentatively with pyruvate versus pyruvate and U(VI) did not find differential expression of this c-type cytochrome (Junier et al., 2011). Furthermore, qRT-PCR analysis targeting Dred_0700 and Dred_0701 found expression levels to be around two orders of magnitude lower during Fe(III) reduction as compared with pyruvate fermentation. This study also failed to detect any peptides corresponding to the c-type cytochrome under Fe(III) reduction or fermentative conditions (Dalla Vecchia et al., 2014). Studies in our lab support these findings. Isobaric tag for relative and absolute quantitation (iTRAQ) based proteomic analysis of duplicate cultures of D. reducens grown with Fe(III)-citrate, pyruvate, and sulfate identified over 22,000 unique peptides. None of the detected peptides correspond to either gene encoding the c-type cytochrome (unpublished data).
A member of the Peptococcaceae family, D. reducens is a close relative of other environmentally relevant metal and radionuclide reducing Firmicutes, namely Desulfosporosinus and Desulfitobacterium species (Suzuki et al., 2004; Kim et al., 2012). No metal-reducing proteins have yet been described in any of these three genera. Our major objective of this study was to identify proteins capable of iron reduction from the proteome of D. reducens. Not only were iron reductases identified and confirmed through heterologous expression, but these reductases were also shown to reduce soluble Cr(VI) (in the form of sodium dichromate) and U(VI) (in the form of uranyl acetate). To the best of our knowledge, this is the first report identifying and validating metal reductases from a Gram-positive organism through heterologous expression.
Results
To accomplish our objective, we optimized and applied an efficient and high-resolution, non-denaturing protein separation workflow that allows for the purification of functional proteins and protein complexes. Resulting protein fractions were screened for iron reduction activity using a colorimetric assay for Fe(II), based on the reagent ferrozine, where the reduction of Fe(III)-NTA with NADH as electron donor was monitored. Subsequent LC-MS/MS analysis was performed, leading to the identification of proteins/protein complexes capable of iron reduction from the proteome of D. reducens. An overview of our implemented workflow is summarized in Figure 1. Proteins/protein complexes identified by this technique were then selected for functional validation by heterologous expression and biochemical characterization.
Figure 1. Workflow implemented to identify iron reduction candidate proteins in D. reducens:
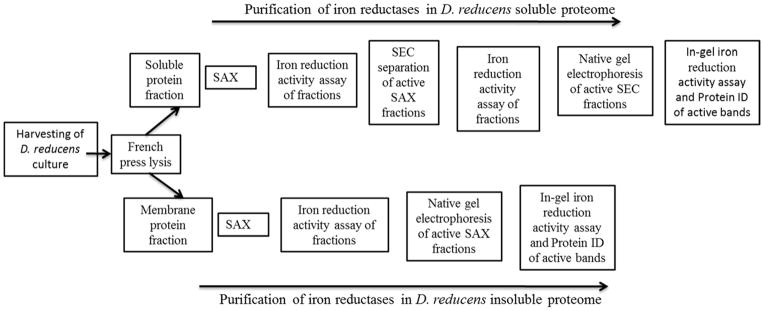
A series of non-denaturing protein separation steps, with a screen for iron reduction activity following each step, was implemented in order to identify iron reductases from the proteome of D. reducens. SAX= strong anion exchange chromatography. SEC= size exclusion chromatography.
The D. reducens proteome under sulfate-reducing conditions confers an Fe(III) reducing phenotype
Initial proteomic separations were attempted with D. reducens cells grown with Fe(III)-citrate as electron acceptor and lactate as electron donor. However, attempts to extract active proteins from these cells were unsuccessful due to interference with Fe-precipitates. Therefore, cell culture conditions were modified to growth with sulfate as electron acceptor. Before protein separations were performed, cell suspension experiments were carried out to confirm Fe(III) reduction capability under these experimental culture conditions. Washed D. reducens cells grown with 28 mM sulfate and 20 mM lactate were shown to reduce Fe(III)-NTA immediately (Figure 2), suggesting that the sulfate-grown D. reducens proteome is capable of Fe(III) reduction. Reduction was dependent on lactate, although controls without lactate displayed a small amount of reduction. A likely explanation for Fe(III)-NTA reduction by live D. reducens cells lacking added electron donor is utilization of stored electrons. D. reducens was recently predicted to contain a type of capacitor that stores reducing equivalents for later reduction of Fe(III) (Dalla Vecchia et al., 2014).
Figure 2. Fe(III) reduction by D. reducens cell suspensions grown on sulfate reduction.

Washed cell suspensions of D. reducens grown with 28 mM sulfate and 20 mM lactate were tested for iron reduction capability and shown to express the proteome necessary for immediate dissimilatory reduction of Fe(III)-NTA. Cells killed by boiling and an abiotic sulfide control determined by the concentration of sulfide measured following cell washing (0.25 mM) demonstrated lack of iron reduction capability. Cell concentration was 3x109 cells/mL, equal to a protein concentration of ~0.9 mg/mL of protein assuming 60% protein per cell dry mass. Error bars display standard error duplicate reactions.
Recovery of iron reduction activity in extracted proteins
Following protein extraction, the soluble and insoluble proteomes of D. reducens were analyzed through implementation of the workflow outlined in Figure 1. Iron reduction activity obtained from the total soluble and insoluble fractions was quantified and is reported as nmol Fe(II) formed/minute and specific activity (nmol Fe(II) formed/mg protein/minute) (Table 1). The specific activity in the insoluble fraction is nearly two times that of the soluble fraction (22.42 versus 12.64 nmol Fe(II) formed/mg protein/minute).
Table 1.
Recovery of iron reduction activity in protein fractions:
| Sample | Protein Concentration (mg/mL) | Iron Reduction Activity (nmol Fe(II) formed/minute) | Specific Activity (nmol Fe(II) formed/mg protein/minute) |
|---|---|---|---|
| Soluble (S) fraction | 5.45 | 2.07 | 12.64 |
| Insoluble (IS) fraction | 1.21 | 0.81 | 22.42 |
| S Peak 1: SAX 16′ | <0.125 | 0.79 | >210.82 |
| S Peak 1: SAX 16′ SEC 9.5′ | <0.125 | 0.50 | >132.14 |
| S Peak 2: SAX 31–32′ | <0.125 | 0.19 | >50.09 |
| S Peak 2: SAX 31–32′ SEC 10.5′ | <0.125 | 0.16 | >43.94 |
| IS Peak 1: SAX 31′ | <0.125 | 0.08 | >22.16 |
Specific iron reduction activity is calculated for protein fractions using the iron reduction activity assay. Nearly twice the specific iron reduction activity was recovered in the total insoluble fraction versus the soluble fraction. Specific activities described as “>” values were limited in precision due to below detect protein concentrations as determined by Bradford assay. SAX= anion exchange chromatography. SEC= size exclusion chromatography.
Soluble protein fraction
The soluble proteome of D. reducens was separated using a series of three non-denaturing separation steps: strong anion exchange chromatography (SAX), size exclusion chromatography (SEC), and native gel electrophoresis. Iron reduction activity was screened for following each subsequent separation. Following SAX separation of the soluble protein fraction, peaks of iron reduction activity were recovered at 13′, 16′, 31–32′, and 47–48′, depicted by the gray line plotted in Figure 3a. These fractions were selected for high-resolution SEC separation, and iron reduction activity was retained following separation of both SAX 16′ and 31–32′ fractions, identified as Peak 1 and 2 respectively in Figure 3a, but not from the other fractions. The SEC-separated 16′ SAX fraction produced a peak of iron reduction activity in the 9.5′ SEC fraction, and the third dimension of separation (native gel electrophoresis followed by the in-gel activity assay) led to the identification of an iron reductase band (~280 kDa) visualized as a pink band due to the formation of the ferrozine-Fe(II) complex (Figure 3b). After analysis of in-gel digests of the active gel-band by LC-MS/MS and based on detection of at least two unique peptides, four proteins were identified including NADH:flavin oxidoreductase (Dred_2421), oligoendopeptidase F (Dred_2457), acetyl-CoA acetyltransferase (Dred_1784), and sulfate adenylyltransferase (Dred_0635) (Supplementary Table 1a).
Figure 3. Identification of iron reduction activity in the soluble proteome of D. reducens.

Protein concentration (determined by absorbance at 280nm and presented as mAU) is represented by the black chromatogram, while the gray line presents an overlay of iron reduction activity (μmol Fe(II) formed/minute). 3.a. SAX separation of the soluble protein fraction led to the recovery of two dominant iron reduction peaks that are maintained through two subsequent dimensions of separation. 3.b. SEC separation of Peak 1 (SAX 16′ fraction) led to the recovery of iron reduction activity in fraction 9.5′. Further separation with native gel electrophoresis recovered an active iron reductase band (visualized as a pink band at ~280 kDa, designated by gray arrow). 3.c. SEC separation of Peak 2 (SAX 31–32′ fraction) led to the recovery of iron reduction activity in fraction 10.5′. Further separation with native gel electrophoresis recovered an active iron reductase band (visualized as a pink band at ~244 kDa, designated by gray arrow). SAX= strong anion exchange chromatography. SEC= size exclusion chromatography.
The SAX 31–32′ fraction (Peak 2 in Figure 3a) was further separated with SEC and produced a peak of activity in the 10.5′ SEC fraction. Native gel electrophoresis followed by the in-gel activity assay indicated an iron reductase band at ~244 kDa (Figure 3c), again visualized as a pink band. LC-MS/MS identified four proteins in the excised band by at least two unique peptides including oxidoreductase FAD/NAD(P)-binding subunit (Dred_1685), dihydroorotate dehydrogenase 1B (Dred_1686), 4Fe-4S ferredoxin (Dred_0137), and pyruvate flavodoxin/ferredoxin oxidoreductase domain-containing protein (Dred_0047) (Supplementary Table 1b). Specific iron reduction activity of active fractions identified following separation of the soluble proteome is reported in Table 1. Due to protein concentrations below the detection limit of the Bradford assay, specific activities could only be approximated in separated fractions.
Insoluble protein fraction
The insoluble proteome of D. reducens, which was extracted in the presence of the detergent n-Dodecyl β-D-maltoside (DDM), was separated using two non-denaturing separation steps, including SAX and native gel electrophoresis. Previous experiments with all three phases of separation led to a loss of Fe(III) reduction activity in the insoluble fraction, and for this reason the SEC step was excluded from this workflow. Following SAX, a dominant activity peak was recovered at 31′ (Figure 4). Further separation of the 31′ fraction with native gel electrophoresis led to the identification of an iron reductase band at ~244 kDa (Figure 4). The subsequent LC-MS/MS analysis of the gel band identified five proteins based on at least two unique peptides, including again the proteins Dred_1685 and Dred_1686. The other proteins identified were ATP synthase F1 subunit alpha (Dred_3152), GntR family transcriptional regulator (Dred_0095), and adenylylsulfate reductase subunit alpha (Dred_0637) (Supplementary Table 1c). Specific iron reduction activity of the active fraction identified following separation of the insoluble proteome is reported in Table 1. Because protein concentration was below the detection limit of the Bradford assay, specific activity could only be approximated in this fraction.
Figure 4. Identification of iron reduction activity in the insoluble proteome of D. reducens.

Protein concentration (determined by absorbance at 280nm and presented as mAU), is represented by the black chromatogram, while the gray line presents an overlay of iron reduction activity (μmol Fe(II) formed/minute). 3.a. SAX separation of insoluble protein fraction led to the recovery of a dominant peak at 31′. Further separation with native gel electrophoresis recovered an active iron reductase band (visualized as a pink band at ~244 kDa, designated by gray arrow). SAX= strong anion exchange chromatography. SEC= size exclusion chromatography.
Heterologous expression and characterization of metal reductase capability
The workflow outlined in Figure 1 led to short lists of potential iron reductases from the proteome of D. reducens (Supplementary Table 1). In order to confirm iron reductase activity, targets were selected for heterologous expression and affinity purification. Following tests for iron reduction activity, further characterization was performed.
Confirmation of iron reduction activity
From peak 1 of the soluble fraction, Dred_2421 (NADH: flavin oxidoreductase) was selected as the primary target for heterologous expression based on the highest protein score and its annotation as the sole oxidoreductase from the list (Supplementary Table 1a). An SDS gel of heterologously expressed and purified Dred_2421 confirmed its predicted molecular weight of ~72 kDa (Supplementary Figure 1). The purified protein was yellow in color due to bound flavin, and both FMN and FAD (flavin adenine dinucleotide) were detected with reversed-phase high performance liquid chromatography (Supplementary Figure 2). Tests for iron reduction activity were performed, and Dred_2421 was confirmed as an iron reductase (Figure 5). Activity was found to be NADH-dependent, as Dred_2421 does not use NADPH as an electron donor. The specific activity was calculated (based on the first 6 minutes of the iron reduction activity assay following Fe(III)-NTA injection), and is displayed in Table 2. Based on recovery of the active band at ~280 kDa in the native gel (Figure 3b), we predict that Dred_2421 (~72 kDa) functions as a homotetramer.
Figure 5. Iron reduction by Dred_2421.

Following heterologous expression and purification of Dred_2421, iron reduction capability was confirmed using the iron reduction activity assay. 0.5 mM Fe(III) was added at 7 minutes. The dotted lines display standard deviation across triplicate reactions. Reactions contained 1 μM Dred_2421 and 0.2 mM NADH as electron donor.
Table 2.
Iron reduction activity in heterologously expressed proteins:
| Locus Tag | Peak identified from (corresponding to Table 1) | Specific Activity (nmol Fe(II) formed/mg protein/minute) |
|---|---|---|
| Dred_2421 | S Peak 1 | 361.95 |
| Dred_1685–1686 | S Peak 2, IS Peak 1 | 627.68 |
Specific iron reduction activity is calculated for heterologously expressed and purified proteins identified in fractions described in Table 1. Micromoles of Fe(II) formed per minute were calculated based on the first 6 minutes (following Fe(III) injection) of the reactions displayed in Figures 5 and 6.
Dred_1685 and Dred_1686 were two other primary targets for heterologous expression based on their predicted annotations involving oxidoreductase activity as well as their identification in protein lists from Peak 2 of the soluble fraction as well as in the insoluble fraction peak (Supplementary Table 1b and 1c). Matching charge and size evidence leading to these protein lists (31–32′ or 31′ SAX fraction from the soluble and insoluble fractions respectively and ~244 kDa in both in-gel activity assays) supports that the same iron reductase was active in peak 2 of the soluble fraction and the insoluble fraction peak (Figures 3 and 4). Both individual proteins were expressed successfully. Dred_1685 was brownish in color, consistent with its annotated 2 iron, 2 sulfur cluster binding activity (https://img.jgi.doe.gov/). Dred_1686 was yellow in color, suggesting the presence of bound flavin. The iron reduction activity assay was initially performed with purified Dred_1685 and Dred_1686 expressed separately, and no iron reduction activity for each individual protein was observed (Figure 6). However, upon mixing of the two purified proteins, iron reduction was observed, providing evidence that the two proteins form a complex. In order to confirm this prediction, Dred_1685 and Dred_1686 were co-expressed on a single plasmid with a His6-tag only on Dred_1685. Nickel affinity column purification followed by SDS-PAGE electrophoresis resulted in the identification of two distinct protein bands of appropriate size (29 kDa for Dred_1685 and 32 kDa for Dred_1686), confirming the formation of a complex and suggesting a 1:1 stoichiometry (Supplementary Figure 3). The iron reduction activity assay demonstrated iron reduction capability in this complex (Figure 6). The specific activity was calculated (based on the first 6 minutes of the iron reduction activity assay following Fe(III)-NTA injection), and is displayed in Table 2. NADPH was also used as an electron donor for Fe(III)-NTA reduction by the Dred_1685–1686 complex, but at a rate nearly 5 times slower over the first 6 minutes than that calculated with NADH as electron donor. Based on the predicted size of the active complex in native gels (~244 kDa), we predict the complex is a heterooctamer consisting of four molecules from each of the two proteins.
Figure 6. Iron reduction by Dred_1685–1686 complex.

Following heterologous expression and copurification of Dred_1685 and Dred_1686, iron reduction capability was confirmed using the iron reduction activity assay. 0.5mM Fe(III)-NTA was added at 7 minutes. The copurified complex is necessary for iron reduction, as Dred_1685 and Dred_1686 individually do not demonstrate iron reduction capability. The dotted black lines display the standard deviation across triplicate reactions. Reactions contained 1 μM of purified proteins and 0.2 mM NADH as electron donor.
In order to thoroughly investigate proteins identified in the fractions with iron reduction activity, other proteins whose annotations suggested potential involvement were heterologously expressed. This included pyruvate flavodoxin/ferredoxin oxidoreductase domain-containing protein (Dred_0047), which was identified in peak 2 of the soluble fraction. Following heterologous expression and purification, Dred_0047 was found to not be able to reduce iron. Dred_0137, a 4Fe-4S ferredoxin, was also selected for heterologous expression, but multiple mutations throughout the cloning process have prevented expression, meaning Fe(III) reduction capability could not be tested. Although Dred_0637 was identified in the insoluble fraction peak and has an annotation as an oxidoreductase (adenylylsulfate reductase subunit alpha), it was not selected as it had a low protein score and its predicted involvement in the sulfate reduction pathway suggests that its presence in the active gel band was incidental.
Discovery of Cr(VI) and U(VI) reductase capability
As D. reducens is capable of reducing contaminant heavy metals and radionuclides, specifically Cr(VI) and U(VI), the discovered Fe(III) reductases were tested for soluble Cr(VI) (in the form of sodium dichromate) and U(VI) (in the form of uranyl acetate) reduction capability. For both metals, colorimetric assays were employed and the disappearance of the +6 oxidation state was monitored. Both Dred_2421 and the Dred_1685–1686 complex were capable of Cr(VI) reduction with NADH as electron donor (Figure 7). As we found that NADH was capable of some abiotic Cr(VI) reduction, we added 1 mM NADH in two steps, first at time zero and then at 80 minutes, in order to best visualize enzymatic reduction. The individual proteins Dred_1685 and Dred_1686 resembled the no protein control, again providing evidence that Dred_1685 and Dred_1686 form a functional complex that is capable of metal reduction.
Figure 7. Cr(VI) reduction by Dred_2421 and Dred_1685–1686 complex.

The disappearance of Cr(VI) over time is monitored based on the diphenylcarbazide method. In order to reduce the effects of abiotic reduction by NADH, only 1mM NADH was added at time zero. After an additional 1mM NADH was added at 80 minutes, complete Cr(VI) reduction is observed for both Dred_2421 and the Dred_1685–1686 complex. Individual Dred_1685 and Dred_1686 proteins resembled the no protein control. Reactions contained 2 μM purified proteins.
Discovered Fe(III) reductases were also tested for the capability to reduce U(VI) with NADH as electron donor. Both Dred_2421 and the Dred_1685–1686 complex displayed U(VI) reductase activity (Figure 8). As expected, the single proteins Dred_1685 and Dred_1686 were not capable of U(VI) reduction. As U(IV) can be oxidized back to U(VI) by oxygen, we exposed our reactions to air following the experiment. Re-oxidation of the reactions following U(VI) reduction provides further support that U(VI) was indeed being reduced, rather than simply disappearing from solution due to sorption (Supplementary Figure 4).
Figure 8. U(VI) by Dred_2421 and Dred_1685–1686 complex.

The disappearance of U(VI) over time is monitored using a method dependent on the reagent Arsenazo-III. Individual Dred_1685 and Dred_1686 proteins resembled the no protein control. Reactions contained 10 μM purified protein.
Discussion
The field of microbial metal reduction is dominated by studies in Gram-negative bacteria whose genomes encode an abundance of multiheme c-type cytochromes. Our aim was to discover enzymes capable of iron reduction in D. reducens, an organism that encodes only a single c-type cytochrome in its genome and is phylogenetically distinct from the model metal reducers. This led to the identification of a protein and a protein complex that are not only capable of reducing Fe(III)-NTA but also Cr(VI) and U(VI) with NADH as electron donor. Proteomic studies combined with protein biochemistry, as seen here, are essential in order to validate existing genome annotations and/or discover new functional annotations in uncharacterized organisms such as D. reducens, rather than relying on automated curation. Our separation methods were designed with the goal of maintaining the D. reducens proteome at a functional level. While it is possible that complexes or interacting complexes necessary for activity were broken up, the identification of two proteins that require the formation of a complex for iron reductase activity (Dred_1685 and Dred_1686) points towards the success of our methods. The non-denaturing separation workflow described allows rapid and high-resolution protein fractionation and can be employed for a diverse range of functional screens in a variety of organisms in the future.
Based on the lack of understanding of energy-producing processes in D. reducens, or the physiologically relevant electron donor for Fe(III) reduction, NADH was selected as an electron donor based on previous studies in model organisms (Magnuson et al., 2001; Elias et al., 2007). As such, the use of NADH as electron donor to screen for iron reduction activity leads to inherent selectivity towards NADH-dependent oxidoreductases, and thus it is uncertain whether the identified proteins have a physiologically relevant role in metal reduction. It is possible that these proteins are involved in the recently described thermodynamic relief mechanism when D. reducens is grown fermentatively with pyruvate, where Fe(III) reduction serves as an electron dump rather than an energy-deriving process (Dalla Vecchia et al., 2014). It is unclear at this time whether this is also the case for Fe(III) reduction by D. reducens with lactate as electron donor, although the original isolation paper reports use of Fe(III) by D. reducens as a true electron acceptor under these conditions (Tebo and Obraztsova, 1998). However, preliminary evidence suggests that D. reducens is not using its annotated Type 1 NADH dehydrogenase (Dred_2036–2046) in a classic respiration sense when grown on Fe(III)-citrate and lactate. Under this growth condition, global proteomic analysis detected only one of the 11 subunits of the NADH dehydrogenase operon (unpublished data). Additional studies are required in order to elucidate catabolic processes in D. reducens, and genetic inactivation studies are necessary in order to provide a link between the proteins identified in this study and an in vivo role in metal reduction. However, without a genetic system currently available, our study serves as an initial functional survey of a poorly characterized proteome and has identified Fe(III)-NTA, Cr(VI), and U(VI) reductase activity in a protein and protein complex distinct from described metal reductases.
One of the proteins identified in this study, Dred_2421, is a soluble protein classified as an Old Yellow Enzyme (OYE). The first class of flavin-dependent enzymes identified, this group has been studied for many years, but physiological roles remain elusive (Williams and Bruce, 2002). Previous reports have identified iron reduction capability in proteins annotated as flavin oxidoreductases, but these are described as assimilatory iron reductases and require the addition of exogenous flavin (Fontecave et al., 1994; Vadas et al., 1999; Mazoch et al., 2004). Dred_2421, however, contains tightly bound flavins (FMN and FAD, as shown in Supplementary Figure 2) and reduces Fe(III)-NTA without the addition of exogenous flavin. In fact, tests adding exogenous riboflavin did not increase rates of Fe(III)-NTA reduction by Dred_2421. Based on the annotation of Dred_2421 as an NADH:flavin oxidoreductase, as well as the predicted involvement of riboflavin and FMN in Fe(III) reduction in D. reducens when grown on pyruvate, Dred_2421 was tested for the ability to reduce riboflavin and FMN with NADH as electron donor (Dalla Vecchia et al., 2014). Neither flavin was reduced by Dred_2421, further supporting the role of Dred_2421 as a metal reductase.
Dred_2421 is predicted to be in its own operon, and in fact on the genome falls in the middle of a region encoding genes predicted to be involved in flagella-related processes. This presumable genetic rearrangement is not conserved in any other sequenced Desulfotomaculum species (https://img.jgi.doe.gov/). Interestingly, the twelve proteins with highest sequence similarity to Dred_2421 (60–80% identity across 99% query coverage) are all from species of Desulfosporosinus and Desulfitobacterium, both genera of Gram-positive metal reducers (http://blast.ncbi.nlm.nih.gov/). No studies have investigated pathways of Fe(III) reduction in either of these two genera. Close relatives of D. reducens, we predict that similar metal reductase activity would be found in these Desulfosporosinus and Desulfitobacterium NADH:flavin oxidoreductases. E. coli species encode an orthologous protein to Dred_2421 (36% identity across 98% query coverage), and the crystal structure has been solved (Hubbard et al., 2003, http://blast.ncbi.nlm.nih.gov/). This protein was heterologously expressed and purified by our group and found to lack Fe(III)-NTA reduction with both NADH and NADPH as electron donor (unpublished data). This finding supports the possibility that Dred_2421 has a specifically evolved physiological role as a metal reductase. Furthermore, a published transcriptomic study, with microarray data deposited at NCBI’s Gene Expression Omnibus, compares conditions of D. reducens grown with pyruvate versus pyruvate and U(VI) (Junier et al., 2011). Dred_2421 is increased in expression on U(VI) conditions by ~1.3 times during both mid and late exponential phase (http://www.ncbi.nlm.nih.gov/geo/).
Our studies also identified a complex capable of Fe(III), Cr(VI), and U(VI) reduction, composed of Dred_1685 and Dred_1686. This complex was recovered from both the soluble as well as the insoluble (presumably membrane) protein fraction. These genes are predicted to be involved in the fourth step of pyrimidine biosynthesis, oxidizing dihydroorotate to orotate with NAD+, and are in a predicted operon composed of Dred_1685–9. This operon is conserved across five sequenced species of Desulfotomaculum. One of these is the only other known Fe(III) reducing species in the genus, Desulfotomaculum hydrothermale, while tests of Fe(III) reduction aren’t reported in the literature for the other four species. This operon includes a lipoprotein signal peptidase, which is predicted to be localized to the cytoplasmic membrane according to PSORTb (Yu et al., 2010). This peptidase is missing in other sequenced Desulfotomaculum species including Desulfotomaculum acetoxidans, a species tested and unable to reduce Fe(III) based on a 1993 paper and confirmed in studies in our lab (Lovley et al., 1993).
Other studies have found dihydroorotate dehydrogenase 1B (the annotation for Dred_1686) and homologs of Dred_1685 (annotations vary) to form a complex that is required for functionality. For instance, in the Gram-positive bacterial model for these proteins (Lactococcus lactis), a homolog of Dred_1686 (51 % identity across 95% query coverage) and a homolog of Dred_1685 (34& identity across 93% query coverage) form a complex that is required for a functional enzyme (http://blast.ncbi.nlm.nih.gov/). However, in L. lactis this complex is predicted to be a heterotetramer, while our findings support the formation of a heterooctamer based on the molecular weight of the active complex in native gels (Nielsen et al., 1996). Furthermore, dihydroorotate dehydrogenase 1B in Gram-positive organisms like L. lactis and D. reducens are grouped into Type 1 dihydroorotate dehydrogenases, which are predicted to be localized to the cytosol. Type 2 dihydroorotate dehydrogenases, on the other hand, are associated with the inner membrane (Nørager et al., 2002). We recovered Dred_1685 and Dred_1686 in both the soluble and insoluble fractions. The insoluble protein fraction was thoroughly washed following separation from the soluble fraction, demonstrated by the lack of carryover of the most active soluble peak following SAX separation (peak 1 in Figure 3a) into the insoluble fraction. Therefore, we believe the localization of Dred_1685 and Dred_1686 to both the soluble and insoluble fraction is a true result and supports the claim that this complex is in some way associated with the membrane, in contrast to other Type 1 dihydroorotate dehydrogenases.
As with Dred_2421, the Dred_1685–1686 complex was found to not have riboflavin or FMN reduction capability with NADH as electron donor, nor was Fe(III)-NTA reduction enhanced with addition of exogenous riboflavin. A small increase was seen in the expression of Dred_1685 and Dred_1686 on pyruvate versus pyruvate and U(VI), based on the available transcriptomic data (http://www.ncbi.nlm.nih.gov/geo/). Furthermore, the Dred_1685–1686 complex is capable of using NADPH as an electron donor for Fe(III)-NTA reduction, although at a slower rate. These findings, along with the predicted role for Dred_1685 and Dred_1686 in pyrimidine biosynthesis, calls into question whether this complex is physiologically relevant to metal reduction. However, because it was identified as the most active fraction from the insoluble (presumably membrane) protein pool, it should not be discounted. In fact, in the study where a fraction containing OmcB, a characterized in vivo soluble and insoluble iron reductase, was originally purified from the membrane of G. sulfurreducens, specific activity was reported as 17.1 nmol Fe(II) formed/mg protein/minute (Magnuson et al., 2000). The specific activity of the insoluble/membrane fraction where Dred_1685–1686 was identified is >22.16 nmol Fe(II) formed/mg protein/minute (See Table 1). This specific activity is grossly underestimated, as protein concentration was below detection but is clearly much lower than other fractions where concentration was also below detection (see Figures 3 and 4). The calculated specific activity of the Dred_1685–1686 complex following heterologous expression and purification is much higher, 627.68 nmol Fe(II) formed/mg protein/minute (Table 2).
In conclusion, our studies employing functional screens of fractions of the D. reducens proteome have uncovered metal and radionuclide reductases that are quite distinct from the multiheme c-type cytochromes described in Gram-negative iron reducers. Further studies combining proteomic and biochemical techniques are essential for better elucidation of key functional enzymes in the proteome of D. reducens and other poorly characterized organisms.
Experimental procedures
Culturing
Desulfotomaculum reducens MI-1 was obtained from ATCC and cultured anaerobically with an 80/20 N2/CO2 headspace at 30° C on Widdel Low Phosphate (WLP) media minus pyruvic acid (Junier et al., 2009). Twenty mM sodium lactate (Fisher Scientific, Pittsburgh, PA USA) was added as electron donor with 25 mM Fe(III)-citrate (Santa Cruz Biotechnology, Inc., Dallas, TX USA) or 28 mM sodium sulfate (Fisher Scientific) as electron acceptor.
Iron reduction activity assay
Iron reduction activity of crude extracts and protein fractions was determined and screened for using a 96-well plate ferrozine-based assay described previously (Elias et al., 2007), where the reduction of Fe(III)-NTA with NADH as electron donor was monitored at 562nm. The reaction mixture contained 160 μL of assay buffer (40 mM MgCl2 (Sigma-Aldrich, St. Louis, MO USA) 0.2 mM NADH (Thermo Fisher Scientific Acros Organics, Pittsburgh, PA USA) and 0.5 mM Ferrozine® Iron Reagent (J.T. Baker/Avantor Performance Materials, Inc., Center Valley, PA USA) in 100 mM HEPES (EMD Chemicals, Inc., San Diego, CA USA) (pH 7.0) with 10% (v/v) glycerol (Sigma-Aldrich) and 30 μL of sample. Ten μL of 10mM Fe(III)-NTA (FeCl3 X 6H2O (Fisher Scientific), Nitrilotriacetic acid (Sigma-Aldrich), NaHCO3 (Fisher Scientific)) was added to commence the reaction. Absorbance of the ferrozine-Fe(II) complex was measured every 60 seconds over a 20 minute reaction time in a Spectra MAX plus spectrophotometer (Molecular Devices LLC, Sunnyvale, CA USA). The assay was also implemented for confirmation of iron reduction activity in purified proteins following heterologous expression (Tecan Infinite 200 series microplate reader, Tecan Group Ltd, Männedorf, Switzerland), and an N2 headspace was used. Reactions were incubated in this N2 atmosphere for 7 minutes prior to Fe(III)-NTA injection.
Whole cell experiments
The capability for sulfate-grown D. reducens cells to reduce Fe(III) was tested. Late exponential phase D. reducens cells grown with sulfate and lactate were harvested anaerobically, washed 3 times with HEPES buffer (100 mM HEPES (EMD Chemicals), 40 mM MgCl2 (Sigma-Aldrich), 10% (v/v) glycerol (Sigma-Aldrich), pH 7.0) and resuspended in 3 mL of the buffer to a concentration of 3x109 cells/mL. Residual sulfide was quantified using the Cline Assay in order to take into account any potential abiotic Fe(III) reduction (Strocchi et al., 1992). Ten mM Fe(III)-NTA and 10 mM lactate was added to the cell suspension, and the accumulation of Fe(II) over time was monitored using the ferrozine assay (Lovley and Phillips, 1987).
Soluble and insoluble protein fraction preparation
The soluble and insoluble proteome was prepared with modifications to a previously described protocol (Magnuson et al., 2000). Late exponential phase cells were harvested anaerobically at 4°C, washed, and resuspended in 5 mL of Tris-HCl extraction buffer (50 mM Tris-HCl (J.T. Baker/Avantor), 2 mM MgCl2 (Sigma-Aldrich) in 10% (v/v) glycerol (Sigma-Aldrich), pH 7) with protease inhibitor (cOmplete, Mini Protease Inhibitor Cocktail Tablets, Roche Applied Science, Indianapolis, IN USA). Following disruption with a French pressure cell at 8000 psi, unlysed cells were removed through centrifugation for 20 minutes at 7000g. Total protein extract in the supernatant was ultracentrifuged in a tabletop ultracentrifuge (Beckman Coulter, Brea, CA USA) at 100,000g for 60 minutes, the soluble fraction was removed to a separate tube, and the insoluble pellet was washed three times with Tris-HCl buffer and resuspended in 2 mL of Tris-HCl buffer with 0.5% wt/wt n-Dodecyl-β-D-maltoside (DDM) (Thermo Fisher Scientific Acros Organics). The insoluble protein fraction was extracted (solubilized) by stirring anaerobically at 4°C overnight and separated from unextracted protein by ultracentrifugation at 100,000g. Protein concentrations were quantified using the Bradford Assay (Bradford, 1976) (Thermo Scientific ™ Pierce ™ Coomassie (Bradford) Protein Assay, Fisher Scientific).
Strong anion exchange chromatography (SAX) separation
The soluble and extracted insoluble proteins were fractionated separately using strong anion exchange (SAX). SAX was performed on an Agilent 1100 Binary Solvent HPLC (Agilent Technologies, Inc., Wilmington, DE USA) using a Mono Q HR 5/5 column (GE Healthcare Bio-sciences, Pittsburgh, PA USA). All buffers were vacuum filtered through a 0.22 μm Durapore GV membrane (EMD Millipore Corporation, Billerica, MA USA) and stored at 4°C until use. A linear gradient (0-2-2.5-42-42.5-47.5-48-58 minutes, 0-0-5-50-75-75-0-0 % B) was employed using Buffer A (20 mM bis-tris (Sigma-Aldrich)/10% (w/v) glycerol (Fisher Scientific) pH = 6.9) and Buffer B (20 mM bis-Tris/1M sodium chloride (Fisher Scientific)/10% (w/v) glycerol pH = 6.9). The flow rate was set to 0.5 mL/min and 100 μL injections were performed. All samples were filtered through a Costar Spin-X 0.22 μm cellulose acetate centrifuge tube filter (Corning Incorporated, Corning, NY USA) prior to injection. Column effluent was monitored by UV absorbance at 280nm. Manual fraction collection was performed at 1-minute intervals beginning at 2 minutes and ending at 49 minutes post-injection for a total of 48 fractions. Fractions were stored on ice until the assay for iron reduction activity, and active fractions were selected for subsequent separation.
Size exclusion chromatography (SEC) separation
Fractions from the soluble protein fraction that retained activity after SAX were filtered through a Costar Spin-X 0.22 μm cellulose acetate centrifuge tube filter and concentrated using an Amicon Ultra 0.5 mL 10K MWCO Ultracel regenerated cellulose centrifugal filter (EMD Millipore) to a final volume of <100 μL. Concentrates were diluted to ~200 μL with SEC running buffer prior to injection. SEC fractionations were carried out using a Dionex UltiMate 3000 HPLC (Dionex Corporation, Sunnyvale, CA USA) outfitted with the fraction collection option and operated in isocratic mode. A high-resolution aqueous SEC Yarra SEC-2000 column (3 μm, 4.6 X 250mm) with column-appropriate guard cartridges (Phenomenex, Inc., Torrance, CA USA) was used for sample fractionation. Column performance was checked periodically by injecting Aqueous SEC 1 Standard (Phenomenex). SEC running buffer contained 20 mM Tris-base (PlusOne, GE Healthcare Bio-sciences, Pittsburgh, PA USA), 2 mM MgCl2 (Fisher Scientific), 150 mM NaCl (Fisher Scientific), and 10% (w/v) glycerol (Fisher Scientific) at pH = 6.8. Buffer was filtered as above and refrigerated prior to use. The maximum sample injection volume of 230 μL was used, the flow rate was set to 0.75 mL/min, and column effluent was monitored by dual wavelength UV absorbance at 230nm and 280nm. Fractions were collected in 0.5 mL 96-well microplates (Thermo Fisher Scientific Nunc A/S, Roskiide Denmark) at 15-second intervals beginning at 6 minutes and ending at 18 minutes and fractions were maintained at 4°C until the iron reduction activity assay. Active SEC fractions were selected for subsequent separation.
Native gel electrophoresis and in-gel activity assay
Active protein fractions selected following SAX or SEC separation were separated further with native gel electrophoresis using a discontinuous buffer system. For the native gel, the upper (cathode) buffer was 43 mM Tris-base (Fisher Scientific), 52 mM glycine (Fisher Scientific) adjusted to pH = 8.9 with hydrochloric acid (VWR International, West Chester, PA USA) in Milli-Q water (Millipore Corporation, Bedford, MA USA). The lower (anode) buffer was 120 mM Tris-base, 60 mM hydrochloric acid in Milli-Q water, pH = 8.1. The indicator running dye was bromophenol blue (Bio-Rad, Hercules, CA USA) saturated in water. The electrophoresis apparatus consisted of a Novex Mini-cell XCell Sure Lock PAGE unit (Life Technologies, Carlsbad, CA USA) connected to a PowerPac 300 power supply (Bio-Rad). Precast Tris-glycine gradient mini-gels, Novex 4–12% 1.5mm x 10 well or Novex 8–16% 1.0mm x 10 well were purchased from Life Technologies. Novex NativeMark Unstained Protein Standard (Life Technologies, Carlsbad, CA USA) was used as the molecular weight standard. Active fractions from SAX or SEC preparative runs (38 μL sample + 2 mL bromophenol blue solution) were loaded directly into the wells and 5 μL of molecular weight standard was used. Electrophoresis was performed at ambient temperature under constant voltage of 50V for 10 minutes, to allow the proteins to enter the gel and salts to dissipate, followed by separation at 125V for ~2 hours or until the dye front reached the bottom of the gel cassette. The developed gels were removed from the cassette, rinsed briefly in deionized water, and then an in-gel iron reduction activity assay was subsequently performed, designed by modifying an existing protocol (Gaspard et al., 1998). Gels were submerged in the iron reduction activity assay buffer (described above) with 0.5 mM Fe(III)-NTA for 40 minutes. Protein bands with enzymes/enzyme complexes capable of Fe(III)-NTA reduction stained pink and were excised and transferred to a 0.6 mL microcentrifuge tube (low retention polypropylene, Fisher Scientific). Gel-fixing solution (200mL) consisting of 50% methanol/7% glacial acetic acid (Fisher Scientific) in Milli-Q water was added to each gel slice followed by incubation at room temperature for a minimum of 45 minutes with occasional vortexing. The fixing solution was removed and gel storage solution (10% methanol/7% glacial acetic acid in Milli-Q water) was added to submerge the gel bands and then stored at 4°C until in-gel reduction/alkylation/digestion. To visualize banding patterns, gels were fixed as above, rinsed with deionized water and stained overnight in Invitrogen SYPRO Ruby protein gel stain (Life Technologies). Stained gels were destained following manufacturer’s recommended protocol and gel images were captured by a Typhoon 9400 Variable Mode Imager (GE Healthcare).
Protein identification by GeLC-MS/MS analysis
In-gel digestion (using modified trypsin from Promega (Madison, WI)) and tryptic peptide extraction were performed following a protocol from Shevchenko et al. 1996 and modified as described by Zhang et al. 2003 (Shevchenko et al., 1996; Zhang et al., 2003). All gel-extracted supernatants were combined and evaporated to dryness in a Speedvac SC110 (Thermo Savant, Milford, MA). Protein identification was carried out using nanoLC-MS/MS analysis with a Dionex UltiMate3000 system (Dionex, Sunnyvale, CA) and a hybrid triple quadrupole linear ion trap mass spectrometer, 4000 Q Trap from ABSciex (Framingham, MA). The gel-extracted peptides (5–10 μL) were injected onto a PepMap100 C18 trap column (5 μm, 100 Å, Dionex) at a flow rate of 20 μL/min for on-line desalting. They were then separated on a PepMap C18 RP nano column (3 μm, 75 μm x 15 cm, Dionex) and eluted in a 90-minute gradient of 5% to 40% acetonitrile in 0.1% formic acid at 300 nL/min. The 4000 Q Trap was equipped with Micro Ion Spray ion source II. MS data acquisition was performed using Analyst 1.4.2 software (Applied Biosystems) in the positive ion mode for information dependant acquisition (IDA) analysis. The nanospray voltage was 1.6 kV for all experiments in positive ion mode. Nitrogen was used as the curtain (value of 10) and collision gas (set to high) with heated interface on. The declustering potential was set at 50 eV and Gas1 was 20 (arbitrary unit). In IDA analysis, after each survey scan for m/z 400 to m/z 1550 and an enhanced resolution scan, the three highest intensity ions with multiple charge states were selected for tandem MS (MS/MS) with rolling collision energy applied for detected ions based on different charge states and m/z values. The exclusion time was set to 45 seconds.
MS/MS data generated from LC/ESI-based IDA analysis were submitted to Mascot 2.3 for database searching using an in-house licensed Mascot local server and the search was performed using the D. reducens MI-1 protein database (downloaded from NCBI on September 20th, 2012 with 3276 entries) with one missed cleavage site by trypsin allowed. The peptide tolerance was set to 1.5 Da and MS/MS tolerance was set to 0.6 Da. Carbobamidomethyl modification of cysteine and a methionine oxidation were set as variable modifications. Peptides with significant scores, defined with >95% identity based on Mascot probability analysis, were considered (Mascot server, Matrix Science).
Heterologous expression of predicted iron reductases
Single protein heterologous expression
Locus tag Dred_2421, Dred_1685 and Dred_1686 from D. reducens MI-1 were amplified from genomic DNA by PCR and cloned into NdeI/XhoI, BamHI/SalI and BamHI/XhoI restriction sites of pET28a, respectively. The plasmids were transformed into 10G Escherichia coli cells selecting for kanamycin resistance. Based on colony PCR results, colonies containing the desired genes were selected, and plasmid DNA was isolated and sequenced using T7 primers. Sequence-confirmed plasmids were then transformed and expressed in BL-21 Rosetta cells. Cells were cultured at 37°C in 2 liters of Luria Broth (LB) media (VWR) with 50 μg/mL kanamycin and 20μg/mL chloramphenicol. At an OD600 of 0.7, 200 μM IPTG was used to induce expression and cells were further incubated at 16°C for 24 hours. Cells were harvested at 8000 rpm for 5 minutes and the cell pellet was stored in 30 mL of lysis buffer (20 mM Tris pH 8.0, 5 mM imidozale, 500 mM NaCl, 2% (v/v) glycerol) at −80°C until use. Cells were lysed using a cell disruptor and lysate was centrifuged at 20,000 rpm for 30 minutes. The soluble fraction was loaded onto a column containing 2.4 ml of Ni-NTA agarose resin (Qiagen), as each target protein contained an N-terminus His6 tag. The column was first washed with wash buffer (20 mM Tris pH 8.0, 30 mM imidozale, 500 mM NaCl) and then a linear gradient of 50–250 mM imidozale in wash buffer was used to elute the proteins. Fractions containing pure target protein were collected and the buffer was changed to 20 mM Tris pH 8.0, 150 mM NaCl using either dialysis or a desalting column (Econo-Pac, Bio-Rad).
Duet plasmid copurification
To co-express the predicted complex, Dred_1685 and Dred_1686 were cloned into BamHI/SalI and BglII/XhoI restriction sites in pETDuet vector, respectively. The co-expressed plasmid was transformed into 10G Escherichia coli cells selecting for ampicilin resistance. Based on colony PCR results, colonies containing the desired genes were selected and the plasmids were obtained and sequenced. The obtained plasmids were then transformed into BL-21 Rosetta cells for protein expression. Cells harboring the expression plasmid were cultured at 37°C in LB media with 100 μg/mL ampicillin and 20 μg/mL chloramphenicol. Induction, expression, and protein purification were performed as described above. In this expression construct, only Dred_1685 contained an N-terminus His6 tag. Following affinity purification, the Dred_1685–1686 complex was separated from excess Dred_1685 using size exclusion chromatography.
Cr(VI) reduction assay
Cr(VI) reduction was tested in anaerobic serum vials (N2 headspace) with 2 mL reaction volumes containing 1 mM NADH (Thermo Fisher Scientific Acros Organics), 0.5 mM sodium dichromate (Alfa Aesar, Ward Hill, MA USA) and 1 μM in HEPES buffer. Following the 80 minutes timepoint, an additional 1 mM NADH was added in order to allow for complete reduction of the 0.5 mM Cr(VI) to presumably Cr(III). The disappearance of Cr(VI) was monitored at 540nm using the diphenylcarbazide method (Urone, 1955). At each timepoint, the concentration of Cr(VI) in a reaction subsample was measured using an anaerobic plate reader (Tecan) at 540 nm. The reaction included 20 μL sample, 13 μL diphenylcarbazide solution (6mM 1,5-diphenylcarbazide (Sigma-Aldrich) in acetone (Fisher-Scientific)), and 167 μL 0.12M H2SO4 (Fisher-Scientific).
U(VI) reduction assay
U(VI) reduction was tested in anaerobic serum vials (N2 headspace) with 2 mL reaction volumes containing 1 mM NADH (Thermo Fisher Scientific Acros Organics), 0.5 mM uranyl acetate, (J.T. Baker/Avantor Performance Materials) and 10 μM protein in HEPES buffer. The disappearance of U(VI) was monitored over time with the reagent Arsenazo (III) based on an updated Arsenazo protocol (Golmohammadi et al., 2012). At each timepoint, the concentration of U(VI) in a reaction subsample was measured using an anaerobic plate reader (Tecan) at 651nm. The reaction included 8 μL of sample, 8 μL DTPA solution (2.5% diethylenetriaminepenta-acetic acid (Sigma-Aldrich)), 4 μL 10% L-tartaric acid (Sigma-Aldrich), 4 μL Arsenazo Solution (3.2 mM Arsenazo-III (Sigma-Aldrich), 0.5 N NaOH (Sigma-Aldrich)), and 176 μL of dilute H2SO4 (Fisher-Scientific) (pH 2). U(VI) oxidation experiments were performed following reduction of U(VI) and anaerobic incubation overnight. Following bubbling with 150 mL ambient air and aerobic incubation for 5 hours, U(VI) concentration was measured.
Supplementary Material
Acknowledgments
This project was funded by the Department of Energy’s Office of Biological and Environmental Research within the Office of Science, project number DE-SC0006644. The Department of Energy’s (DOE) Office of Biological and Environmental Research (OBER) Pan-omics program provided partial support for PNNL staff in collaboration with research activities conducted at Cornell. Pacific Northwest National Laboratory (PNNL) is a multiprogram national laboratory operated by Battelle for the DOE under contract DE-AC05-76RL01830. O.D.N is supported by National Institutes of Health/National Institute of General Medical Sciences grant 5T32GM008500.
References
- Bird LJ, Bonnefoy V, Newman DK. Bioenergetic challenges of microbial iron metabolisms. Trends in microbiology. 2011;19:330–340. doi: 10.1016/j.tim.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Cardenas E, Wu WM, Leigh MB, Carley J, Carroll S, Gentry T, et al. Significant Association between Sulfate-Reducing Bacteria and Uranium-Reducing Microbial Communities as Revealed by a Combined Massively Parallel Sequencing-Indicator Species Approach. Applied and Environmental Microbiology. 2010;76:6778–6786. doi: 10.1128/AEM.01097-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson HK, Iavarone AT, Gorur A, Yeo BS, Tran R, Melnyk RA, et al. Surface multiheme c-type cytochromes from Thermincola potens and implications for respiratory metal reduction by Gram-positive bacteria. Proceedings of the National Academy of Sciences. 2012 doi: 10.1073/pnas.1112905109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla Vecchia E, Suvorova EI, Maillard J, Bernier-Latmani R. Fe(III) reduction during pyruvate fermentation by Desulfotomaculum reducens strain MI-1. Geobiology. 2014;12:48–61. doi: 10.1111/gbi.12067. [DOI] [PubMed] [Google Scholar]
- Elias DA, Yang F, Mottaz HM, Beliaev AS, Lipton MS. Enrichment of functional redox reactive proteins and identification by mass spectrometry results in several terminal Fe(III)-reducing candidate proteins in Shewanella oneidensis MR-1. Journal of microbiological methods. 2007;68:367–375. doi: 10.1016/j.mimet.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Fontecave M, Covès J, Pierre J-L. Ferric reductases or flavin reductases? Biometals. 1994;7 doi: 10.1007/BF00205187. [DOI] [PubMed] [Google Scholar]
- Gaspard S, Vazquez F, Holliger C. Localization and Solubilization of the Iron(III) Reductase of Geobacter sulfurreducens. Appl Envir Microbiol. 1998;64:3188–3194. doi: 10.1128/aem.64.9.3188-3194.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilov SN, Lloyd JR, Kostrikina NA, Slobodkin AI. Fe(III) Oxide Reduction by a Gram-positive Thermophile: Physiological Mechanisms for Dissimilatory Reduction of Poorly Crystalline Fe(III) Oxide by a Thermophilic Gram-positive Bacterium Carboxydothermus ferrireducens. Geomicrobiology Journal. 2012;29:804–819. [Google Scholar]
- Golmohammadi H, Rashidi A, Safdari SJ. Simple and rapid spectrophotometric method for determination of uranium (VI) in low grade uranium ores using arsenazo (III) Chemistry & Chemical Technology. 2012;6:245–249. [Google Scholar]
- Hubbard PA, Liang X, Schulz H, Kim JJP. The crystal structure and reaction mechanism of Escherichia coli 2,4-dienoyl-CoA reductase. The Journal of biological chemistry. 2003;278:37553–60. doi: 10.1074/jbc.M304642200. [DOI] [PubMed] [Google Scholar]
- Junier P, Frutschi M, Wigginton NS, Schofield EJ, Bargar JR, Bernier-Latmani R. Metal reduction by spores of Desulfotomaculum reducens. Environmental microbiology. 2009;11:3007–3017. doi: 10.1111/j.1462-2920.2009.02003.x. [DOI] [PubMed] [Google Scholar]
- Junier P, Junier T, Podell S, Sims DR, Detter JC, Lykidis A, et al. The genome of the Gram-positive metal- and sulfate-reducing bacterium Desulfotomaculum reducens strain MI-1. Environmental microbiology. 2010;12:2738–2754. doi: 10.1111/j.1462-2920.2010.02242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junier P, Vecchia ED, Bernier-Latmani R. The Response of Desulfotomaculum reducens MI-1 to U(VI) Exposure: A Transcriptomic Study. Geomicrobiology Journal. 2011;28:483–496. [Google Scholar]
- Kim SH, Harzman C, Davis JK, Hutcheson R, Broderick JB, Marsh TL, Tiedje JM. Genome sequence of Desulfitobacterium hafniense DCB-2, a Gram-positive anaerobe capable of dehalogenation and metal reduction. BMC Microbiology. 2012;12:21. doi: 10.1186/1471-2180-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovley DR, Phillips EJP. Rapid Assay for Microbially Reducible Ferric Iron in Aquatic Sediments. Applied and Environmental Microbiology. 1987;53:1536–1540. doi: 10.1128/aem.53.7.1536-1540.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovley DR, Roden EE, Phillips EJ, Woodward J. Enzymatic iron and uranium reduction by sulfate-reducing bacteria. Marine Geology. 1993;113:41–53. [Google Scholar]
- Magnuson TS, Hodges-Myerson AL, Lovley DR. Characterization of a membrane-bound NADH-dependent Fe3+ reductase from the dissimilatory Fe3+-reducing bacterium Geobacter sulfurreducens. FEMS Microbiology Letters. 2000;185:205–211. doi: 10.1111/j.1574-6968.2000.tb09063.x. [DOI] [PubMed] [Google Scholar]
- Magnuson TS, Isoyama N, Hodges-Myerson AL, Davidson G, Maroney MJ, Geesey GG, Lovley DR. Isolation, characterization and gene sequence analysis of a membrane-associated 89 kDa Fe(III) reducing cytochrome c from Geobacter sulfurreducens. Biochemical Journal. 2001;359:147–152. doi: 10.1042/0264-6021:3590147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazoch J, Tesařík R, Sedláček V, Kučera I, Turánek J. Isolation and biochemical characterization of two soluble iron(III) reductases from Paracoccus denitrificans. European Journal of Biochemistry. 2004;271:553–562. doi: 10.1046/j.1432-1033.2003.03957.x. [DOI] [PubMed] [Google Scholar]
- Mohapatra BR, Dinardo O, Gould WD, Koren DW. Biochemical and genomic facets on the dissimilatory reduction of radionuclides by microorganisms – A review. Minerals Engineering. 2010;23:591–599. [Google Scholar]
- Newsome L, Morris K, Lloyd JR. The biogeochemistry and bioremediation of uranium and other priority radionuclides. Chemical Geology. 2014;363:164–184. [Google Scholar]
- Nielsen FS, Andersen PS, Jensen KF. The B form of dihydroorotate dehydrogenase from Lactococcus lactis consists of two different subunits, encoded by the pyrDb and pyrK genes, and contains FMN, FAD, and [FeS] redox centers. The Journal of biological chemistry. 1996;271:29359–65. doi: 10.1074/jbc.271.46.29359. [DOI] [PubMed] [Google Scholar]
- Nørager S, Jensen KF, Björnberg O, Larsen S. E. coli dihydroorotate dehydrogenase reveals structural and functional distinctions between different classes of dihydroorotate dehydrogenases. Structure (London, England: 1993) 2002;10:1211–23. doi: 10.1016/s0969-2126(02)00831-6. [DOI] [PubMed] [Google Scholar]
- Petrie L, North NN, Dollhopf SL, Balkwill DL, Kostka JE. Enumeration and Characterization of Iron(III)-Reducing Microbial Communities from Acidic Subsurface Sediments Contaminated with Uranium(VI) Applied and Environmental Microbiology. 2003;69:7467–7479. doi: 10.1128/AEM.69.12.7467-7479.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Cavallaro G, Rosato A. A systematic investigation of multiheme c-type cytochromes in prokaryotes. JBIC Journal of Biological Inorganic Chemistry. 2010;15:559–571. doi: 10.1007/s00775-010-0623-4. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Analytical chemistry. 1996;68:850–8. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Shi L, Richardson DJ, Wang Z, Kerisit SN, Rosso KM, Zachara JM, Fredrickson JK. The roles of outer membrane cytochromes of Shewanella and Geobacter in extracellular electron transfer. Environmental Microbiology Reports. 2009;1:220–227. doi: 10.1111/j.1758-2229.2009.00035.x. [DOI] [PubMed] [Google Scholar]
- Strocchi A, Furne JK, Levitt MD. A modification of the methylene blue method to measure bacterial sulfide production in feces. Journal of Microbiological Methods. 1992;15:75–82. [Google Scholar]
- Suzuki Y, Kelly SD, Kemner KM, Banfield JF. Enzymatic U(VI) reduction by Desulfosporosinus species. Radiochimica Acta. 2004;92:11–16. [Google Scholar]
- Suzuki Y, Kelly SD, Kemner KM, Banfield JF. Microbial Populations Stimulated for Hexavalent Uranium Reduction in Uranium Mine Sediment. Applied and Environmental Microbiology. 2003;69:1337–1346. doi: 10.1128/AEM.69.3.1337-1346.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebo BM, Obraztsova AY. Sulfate-reducing bacterium grows with Cr(VI), U(VI), Mn(IV), and Fe(III) as electron acceptors. FEMS Microbiology Letters. 1998;162:193–198. [Google Scholar]
- Urone PF. Stability of Colorimetric Reagent for Chromium, s-Diphenylcarbazide, in Various Solvents. Analytical Chemistry. 1955;27:1354–1355. [Google Scholar]
- Vadas A, Monbouquette HG, Johnson E, Schröder I. Identification and characterization of a novel ferric reductase from the hyperthermophilic Archaeon Archaeoglobus fulgidus. The Journal of biological chemistry. 1999;274:36715–36721. doi: 10.1074/jbc.274.51.36715. [DOI] [PubMed] [Google Scholar]
- Wall JD, Krumholz LR. Uranium reduction. Annual review of microbiology. 2006;60:149–166. doi: 10.1146/annurev.micro.59.030804.121357. [DOI] [PubMed] [Google Scholar]
- Weber KA, Achenbach LA, Coates JD. Microorganisms pumping iron: anaerobic microbial iron oxidation and reduction. Nature reviews Microbiology. 2006;4:752–64. doi: 10.1038/nrmicro1490. [DOI] [PubMed] [Google Scholar]
- Williams RE, Bruce NC. “New uses for an Old Enzyme” - the Old Yellow Enzyme family of flavoenzymes. Microbiology. 2002;148:1607–1614. doi: 10.1099/00221287-148-6-1607. [DOI] [PubMed] [Google Scholar]
- Williamson AJ, Morris K, Shaw S, Byrne JM, Boothman C, Lloyd JR. Microbial reduction of Fe(III) under alkaline conditions relevant to geological disposal. Applied and environmental microbiology. 2013;79:3320–6. doi: 10.1128/AEM.03063-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics (Oxford, England) 2010;26:1608–15. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Van Pelt CK, Henion JD. Automated chip-based nanoelectrospray-mass spectrometry for rapid identification of proteins separated by two-dimensional gel electrophoresis. Electrophoresis. 2003;24:3620–32. doi: 10.1002/elps.200305585. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.