

Abstract
Asparagine-linked glycans (N-glycans) are displayed on the majority of proteins synthesized in the endoplasmic reticulum (ER). Removal of the outermost glucose residue recruits the lectin chaperone malectin possibly involved in a first triage of defective polypeptides. Removal of a second glucose promotes engagement of folding and quality control machineries built around the ER lectin chaperones calnexin (CNX) and calreticulin (CRT) and including oxidoreductases and peptidyl-prolyl isomerases. Deprivation of the last glucose residue dictates the release of N- glycosylated polypeptides from the lectin chaperones. Correctly folded proteins are authorized to leave the ER. Non-native polypeptides are recognized by the ER quality control key player UDP-glucose glycoprotein glucosyltransferase 1 (UGT1), re-glucosylated and re-addressed to the CNX/CRT chaperone binding cycle to provide additional opportunity for the protein to fold in the ER. Failure to attain the native structure determines the selection of the misfolded polypeptides for proteasome-mediated degradation.
Keywords: Endoplasmic reticulum, N-glycosylation, protein folding and quality control, calnexin, calreticulin, UDP-glucose glycoprotein glucosyltransferase 1
1. Introduction
The ER is the site for maturation of secretory and membrane proteins in eukaryotic cells, which represent about one third of the total proteome of the cell (1–3). Although several proteins have been reported to acquire spontaneously their folded structure in vitro, a large number of newly synthesized polypeptides in the cell requires assistance to reach the final and biologically active conformations (4–6). The lumen of the ER contains resident molecular chaperones and folding factors to optimize the folding efficiency (7). The folding state of a protein is evaluated by quality control mechanisms: properly folded proteins are secreted or targeted to their final intra- or extra-cellular destination, whereas misfolded proteins are recognized as aberrant products and targeted for the ER-associated degradation ((ERAD) (8, 9) and reviewed by Benyair et al., in this issue). Most of the proteins designated for the secretory pathway are subjected to co- or post-translational glycosylation of asparagine residues side chains ((N-glycosylation) (10, 11) and reviewed by Shrimal et al., in this issue). The rapid removal of terminal glucose and mannose residues from protein- bound oligosaccharides and the regulated addition of a specific glucose residue dictate the sequential schedule of events occurring during maturation and selection for degradation. Here we present the body of knowledge concerning the sugar- regulated mechanisms that govern protein folding and quality control in the ER lumen.
2. Protein folding in the ER
2.1 N-glycosylation plays a key role in protein folding
The majority of the client proteins entering the ER lumen exhibit -N-X-S/T- (asparagine-any amino acid but proline-serine/threonine) sequences within the polypeptide chain (11, 12). The asparagine residue of this consensus motif is rapidly modified through the covalent attachment of a pre-formed oligosaccharide composed of three residues of glucose, nine mannoses and two N-acetylglucosamines (Glc3Man9GlcNAc2) (Fig. 1). The transfer of the 14-subunits oligosaccharide is catalyzed by the oligosaccharyltransferase complex (OST) (Fig. 2, step 1) (13) and thoroughly described in the article by Shrimal and Gilmore in this issue. N- glycosylation increases the solubility of the newly synthesized polypeptides and processing of the protein-bound oligosaccharides creates the signal required for the recruitment of ER-resident lectin chaperones that regulate glycopolypeptide folding (14–17).
Figure 1. N-glycan composition and processing.

The pre-formed oligosaccharide is composed of three glucoses (dark grey circles), nine mannoses (light grey circles) and two N-acetylglucosamines (black squares). The oligosaccharide branches are shown with A, B and C. The oligosaccharide processing enzymes are listed in the figure and their action is shown. The type of glycosidic bond is shown in color.
Figure 2. Scheme of the CNX/CRT cycle.

Upon addition of the 14-subunits oligosaccharide catalyzed by the OST complex (step 1), the first glucose is removed by α-glucosidase I (step 2). Di-glucosylated polypeptides associate with malectin. α-glucosidase II cleaves the second glucose from the glycan (step 3), generating mono-glucosylated polypeptides, which interact with CNX and CRT. Binding and release from lectin chaperones could occur (step 4). The release from the ER lectins is determined by the second cleavage by α-glucosidase II, which removes the last glucose residue (step 5). Re-glucosylation by UGT1 dictates the re-association of the polypeptides with CNX or CRT (step 6). Correctly folded polypeptides are exported from the ER (step 7). Terminally misfolded proteins are further processed by mannosidases (step 8) and eventually retrotranslocated for proteasomal degradation (step 9). The substrate-binding and catalytic domains of UGT1 are shown with s and c, respectively.
2.2 N-glycan processing dictates the binding to the ER-resident lectin chaperones
2.2.1 α-glucosidase I generates di-glucosylated glycans
2.2.1.1 Organization of α-glucosidase I
The modification of protein-bound oligosaccharides by ER-resident glycanases dictates the fate of newly synthesized polypeptides. As soon as the glycan is added to nascent chains, the first glucose is trimmed by α-glucosidase I (18–20) (Fig. 2, step 2), a single pass transmembrane ER protein with the catalytic domain facing the ER lumen (21). The crystal structure of an eukaryotic α-glucosidase I (the Cwh41p S. cerevisiae ortholog) reveals a globular protein of two domains connected by a linker: the N-domain is largely comprised of a 13 strand super β-sandwich and additional helices, and the C-domain contains 12 helices in an (α/α6) toroid bundle with an additional subunit termed C′-domain. The catalytic residues are proposed to lie in the center of the (α/α6) barrel (22).
2.2.1.2 α-glucosidase I generates the ligand for malectin
For a long time, the di-glucosylated form of protein-bound oligosaccharides generated by the action of α-glucosidase I was considered an extremely short-lived trimming intermediate lacking biological significance. The discovery of malectin, a membrane-bound ER-resident lectin specifically binding di-glucosylated glycans, changed this view (23). Malectin is induced under conditions of ER stress (24) and is proposed to preferentially associate with off-pathway non-native conformers of well- studied glycoproteins like influenza hemagglutinin (HA) and null Hong Kong (NHK), a folding-defective variant of the secretory protein α-1-antitrypsin (α1AT). The putative capacity of malectin to detect terminally misfolded proteins so early after their expression in the ER lumen and to distinguish them from non-native intermediates of folding programs is a peculiar property that merits further investigation and may require the formation of a functional complex with the oligosaccharyl transferase complex subunit ribophorin I (24–26). This conclusion is reinforced by the observation that the overexpression of ribophorin I enhances malectin association with misfolded NHK (26).
2.2.2 α-glucosidase II induces the entry of glycoproteins into the CNX/CRT cycle
2.2.2.1 Organization of α-glucosidase II
α-Glucosidase II is a heterodimeric protein, with two non-covalently bound subunits. GIIα is the catalytic subunit and GIIβ contains an ER retention sequence in mammals and S. pombe but not in S. cerevisiae (27). GIIα activity requires the presence of GIIβ as shown in S. pombe and mammalian cells, as well as in cell free assays (28, 29). GIIβ contains a mannose-6-phosphate receptor homology (MRH) domain, which was proposed to regulate GIIα activity by interacting with the mannoses on the B and C branches of the nascent protein glycan (29).
2.2.2.2 Regulation of α-glucosidase II trimming for entry in the CNX/CRT cycle
α-Glucosidase II trims the second and the third glucose of the N-linked glycan (Fig. 2, step 3 and 5). The two trimming events play opposing roles. After the first trimming step, maturing substrates associate with the lectin chaperones CNX and CRT, which recognize mono-glucosylated glycans. The second trimming step releases the protein from the lectin chaperones due to the reduced affinity of these chaperones to glycans lacking terminal glucose residues (Fig. 2, step 5) (30–32). A series of de- glucosylations by α-glucosidase II (step 5) and re-glucosylations by a glucosyltransferase, the UGT1 (step 6) can occur (32, 33). Re-glucosylation by UGT1, which will be detailed in Section 3, directs the rebinding of CNX or CRT to the mono- glucosylated substrate, hence the name CNX/CRT binding cycle.
Despite having one catalytic site, the rates of the first and the second trimming events by the α-glucosidase II are different as the first cleavage occurs faster than the second cleavage (34, 35). Unlike the rapid trimming by α-glucosidase I, trimming by α-glucosidase II appears to be more regulated. Work in ER-derived microsomes and mammalian cells showed that more than one glycan on client proteins is required for recognition by α-glucosidase II (20).
The activity of α-glucosidase II appears to be influenced by the number of mannoses on a glycan because reduced activity of α-glucosidase II in rat liver microsomes was observed towards substrates possessing a reduced number of mannose residues (36). This was more recently supported by the observation that trimming of α1,2-bonded mannose residues in S. pombe resulted in a prolonged accumulation of mono-glucosylated proteins. The glycans with highest de- glucosylation rates harbored nine mannoses and the de-glucosylation rate decreased with glycans containing seven, six and five mannoses (37). On the contrary, another study using well defined synthetic glycopeptides as substrates in a cell free assay concluded that the lower number of mannose residues did not change α-glucosidase II trimming kinetics, as the trimming by α-glucosidase II of Glc1Man8 was identical or greater than that of Glc1Man9 (35). Mannose trimming has been proposed to act as a timer that allows proteins either to exit to the Golgi or to be targeted for degradation (11, 38). Reducing the α-glucosidase II activity with increasing mannose trimming could prevent premature release of the protein from the CNX/CRT cycle and thus provide additional time for the proteins to fold by prolonging the binding to these chaperones before they are targeted for trafficking to the Golgi or deemed misfolded and targeted for degradation.
2.2.3: The roles of CNX and CRT
2.2.3.1 Structure, mechanism of binding and similarities between CNX and CRT
CNX is a type I membrane protein. CRT is its soluble paralogue possessing 39% sequence homology (39). CNX and CRT share similar structural properties: they both have an N-terminal globular domain that contains the lectin binding site and a long arm domain. In CNX, the globular domain is a β-sandwich of concave and convex β-sheets and the arm domain, shaped in an overall hairpin like structure, is termed the P-domain. The latter protrudes 140 Å away from the globular domain and has two different proline rich motifs named 1 and 2, of four copies each (40). The four copies of motif 1 on one strand interact in a head to tail fashion with the four copies of motif 2 on the other strand of the hairpin (40). In CRT, the P-domain is similar but shorter than calnexin and has three instead of four tandem repeats (41). The tip of the P- domain interacts with the oxidoreductase ERp57 as will be detailed later in Section 2.3.1.2 (42). CNX co-crystallized with glucose has an extensive hydrogen-bonding pattern of the glucose with residues in the globular domain.
In addition to structural similarities, CNX and CRT have similar lectin binding specificities; as shown by in-vitro binding studies as they both bind to the glucose as well as the three mannoses on branch A of the glycan with similar affinities in a calcium dependent manner (43). The oligosaccharide-binding sites of CNX and CRT are similar but not identical (44). Four out of the six residues that mediate binding in CNX are conserved in CRT.
The crystal structure of CRT with a glycan further confirmed previous observations that CRT binds to the glucose and the three-mannose residues of the glycan, which is mediated by extensive hydrogen bonding between the glucose ring and residues in the sugar-binding site of CRT. The rest of the tetra-saccharide was also involved in hydrogen bonding but to a lesser extent than the glucose (45). More in depth analysis of the mechanism of binding to CRT proposed that mono- glucosylated glycans are responsible for recruiting substrates to bind to CRT, while the proline-rich domain stabilizes the interaction by locking the substrate in a closed conformation (46). This might explain why the oxidation of substrates is arrested or delayed when chaperone binding is chemically trapped (32, 47, 48). These results are suggestive of global folding events occurring after the substrate is released from the lectin chaperones (49).
CNX and CRT are involved in several processes such as calcium homeostasis, immunity, cancer and apoptosis (50, 51). Here, we will only describe their roles as chaperones in the ER. Early studies proposed a “lectin only” model to describe their chaperone function (52). The studies relied on cell lines deficient in either α-glucosidase I or α-glucosidase II, or employed glucosidase inhibitors such as castanospermine and deoxynojirimycin-derivatives (31, 32, 53). These studies showed that inhibition of glucose trimming prevented substrate binding to these lectin chaperones supporting the requirement of mono-glucosylated glycans for association. It was also proposed that these lectins bind to glycoproteins independently of their conformation (54) and do not bind non-glycosylated proteins (55). However, later work proposed that they could act in a similar fashion to classical chaperones by binding to peptide regions of the substrates or to non-glycosylated proteins (56–58).
2.2.3.2 Salient differences in the roles of CNX and CRT
Despite the similarities between CNX and CRT, striking differences have been attributed to their roles or binding specificities. The location and the number of glycans recognized by each of the lectins could be quite distinct (59–61). Furthermore, even though CNX and CRT share some of the same substrates, some proteins are exclusively clients. One explanation for these differences is assigned to the different topologies. It was observed that CRT, when fused to the membrane segment of CNX, acquired the substrate specificity of CNX (62). Different peptide-binding specificities could also contribute to the different substrate selection by the two ER lectins (43).
Consistent with different substrate specificity and/or with specific functions in the ER lumen is the fact that CNX clients seem to associate with BiP rather than with CRT upon CNX deletion (63) and that CNX and CRT knockouts (KO), that in cultured cells are well tolerated, have different phenotypes in mice. CRT KO is embryonic lethal due to defective heart development (64), whereas CNX KO mice are viable and show motor disorders, associated with a dramatic loss of large myelinated nerve fibers (65). Finally, the depletion of either of the lectins may have opposing effects on folding. Depletion of CRT enhances the maturation of a subset of viral and cellular proteins with a minor decrease in folding efficiency. To the contrary, CNX depletion prevents the maturation of a substrate like HA, while its absence does not affect the maturation of other proteins (61). These differences highlight the unique roles of each of these lectins despite their similarities.
2.3 CNX and CRT engage enzymes required for proper protein folding
Specialized ER-resident enzymes catalyze the formation of native intra- and inter-molecular disulfide bonds (Fig. 3A) and the isomerization of peptidyl-prolyl bonds (Fig. 3B) of folding polypeptides. These are rate-limiting steps that are mediated by members of the protein disulfide isomerase (PDI) family and of the peptidyl-prolyl isomerase (PPI) family, respectively. Recent reports showed that at least one member from each family (i.e., the oxidoreductase ERp57 (42, 66) and the PPI CypB (67)) might form functional complexes with CNX and CRT. In this scenario, CNX/CRT-ERp57 complexes would promote the oxidative folding of nascent polypeptides, whereas CNX/CRT-CypB complexes would promote isomerization of peptidyl-prolyl bonds to attain the native polypeptide structure (67–70).
Figure 3. Disulfide and peptidyl-prolyl bonds.
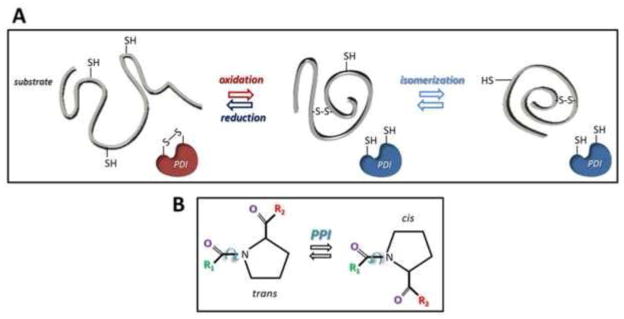
Mechanisms of disulfide bond oxidation, reduction and isomerization catalyzed by PDI proteins (upper panel). Mechanism of peptidyl-prolyl cis-trans isomerization catalyzed by PPI proteins (lower panel).
2.3.1 The PDI family: formation of native disulfide bonds
The mammalian PDI family comprises about 20 members that can be either catalytically active or inactive. They all have in common one or more thioredoxin-like structural domains and are localized primarily in the ER (Table 1) (71–73).
Table 1.
PDI and PPI family members
| Protein | Domains | Active Sites | Activities | References |
|---|---|---|---|---|
| PDI | abb′xa′ | CGHC, CGHC | O, R, I, C | 70–78, 81, 83, 86–89 |
| ERp57 | abb′xa′ | CGHC, CGHC | O, R, I | 41–42, 64–66, 70–73, 76–77, 81–85, 90–98 |
| ERp72 | aoabb′xa′ | CGHC, CGHC, CGHC | O, R, I | 69–73 |
| P5 | aa′b | CGHC, CGHC | O, R, I | 70–73 |
| ERp44 | abb′ | CRFS | Ero1 binding | 71–73 |
| ERdj5 | Ja″baoaa′ | CSHC, CPPC, CHPC, CGPC | R | 70–73, 78–79 |
| PDIp | abb′xa′ | CGHC, CTHC | O, R, I, C | 70–73 |
| PDILT | abb′xa′ | SKQS, SKKC | Ero1 binding | 70–73 |
| PDIr | baoaa′ | CSMC, CGHC, CPHC | O, R, I | 70–73, 78–79 |
| ERp46 | aoaa′ | CGHC, CGHC, CGHC | ? | 70–73 |
| ERp18 | a | CGAC | O, VKOR binding | 70–73 |
| hAGR2 | a | CPHS | ? | 72–73 |
| hAGR3 | a | CQYS | ? | 72–73 |
| TMX1 | at | CPAC | R, VKOR binding | 72–73, 78–79 |
| TMX2 | ta | SNDC | ? | 71–73 |
| TMX3 | abt | CGHC | O | 71–73 |
| TMX4 | at | CPSC | O, R, VKOR binding | 71–73, 78–79 |
| TXNDC15 | at | CRFS | ? | 72–73 |
| ERp29 | b | - | ? | 70–73, 89 |
| ERp27 | bb′ | - | ERp57 binding | 71–73 |
| N33/Tusc3 | CSVC | OST complex subunit | 13–80 | |
| IAP/MagT1 | CVVC | OST complex subunit | 13–80 | |
| ERp90 | - | ERFAD interactor | 151 |
| Protein | Inhibitors | References |
|---|---|---|
| FKBP13 | FK506 | 70, 101–104 |
| FKBP19 | FK506 | 70, 101–104 |
| FKBP22 | FK506 | 70, 101–104 |
| FKBP23 | FK506 | 70, 101–104 |
| FKBP60 | FK506 | 70, 101–104 |
| FKBP65 | FK506 | 70, 101–104 |
| CypB | Cyclosporine A | 67–70, 101–106 |
| CypC | Cyclosporine A | 102 |
List of the PDI family members (upper table).
a, active thioredoxin-like domain; b, inactive thioredoxin-like domain; x, linker domain; J, J- domain; t, transmembrane domain. The aminoacidic composition of the PDI active sites is shown in the third column. O, oxidation; R, reduction; I, isomerization; C, chaperone activity. List of the ER-resident PPI members (lower table).
The thiol-reactive members of the PDI family contain at least one catalytic thioredoxin-like domain termed a domain. Each a domain conventionally consists of a Cys-Xxx-Xxx-Cys (CXXC) motif (74). In some cases, the CXXC motif shows cysteine to serine replacement (Table 1). Additionally, PDIs might possess one or more non- catalytic b domains. These are structurally similar to the a domains and may engage substrates, but do not contain any active cysteine residue (72, 75). ERp27 and ERp29 contain only a b domain and are catalytically inactive (Table 1). Furthermore, a linker domain, termed x domain, stabilizes the protein structure and has been identified in several PDI family members (such as in PDI and ERp57, Table 1) (76, 77). The most common organization of the functional domains is abb′xa′, where the prime symbol stands for the relative position within the protein sequence (Table 1).
Depending on the luminal redox environment, thiol-reactive PDIs can hypothetically catalyze the oxidation, the reduction or the isomerization of disulfide bonds (Fig. 3A) (78). The sequence of the catalytic site, but also the surrounding amino acids in the 3D structure may help in predicting if the given enzyme acts as an oxidase (i.e., it favors the formation of covalent bonds between substrate cysteine residues), as a reductase or as an isomerase. For example, while nearly all oxidases possess a histidine residue within the active site sequence, the presence of a proline residue (as in ERdj5, in TMX1 and in TMX4 (Table 1)) strongly hints at a reductive activity of the enzyme (78, 79). In this review, we will focus on ERp57 role in protein folding because of the specific engagement of this PDI family member in functional complexes with the ER lectins CNX and CRT. However, as a short note, the relevant function of the OST-associated oxidoreductases N33/Tusc3 and IAP/MagT1 in insuring efficient nascent protein glycosylation by delaying formation of substrate disulfide bonds should also be mentioned. This activity seems to be particularly important during brain development as dysfunction of these PDI family members is linked to mental retardation (13, 80).
2.3.1.1 ERp57
ERp57 exhibits great similarity in domain composition (abb′xa′) and length (481 amino acids) with PDI (81). The two oxidoreductases have an amino acid identity of about 29%, showing a large degree of variation at the level of the b′ domain. In ERp57, the b′ domain lacks the hydrophobic substrate-binding pocket insuring the PDI chaperone-like activity (82–84) and contains, instead, the binding region for the CNX and CRT P-domains (85).
2.3.1.2 Interaction between ERp57 and CNX or CRT
Whereas an association of PDI with the P-domain of CRT has originally been reported (86, 87), subsequent studies showed that this interaction requires unphysiologic calcium concentration and should, therefore, not have a physiologic relevance (83, 88, 89). In contrast, a large set of data proved a direct involvement of ERp57 in the CNX/CRT cycle (81, 82, 90–92). The carbohydrate-dependent association between either CNX or CRT, newly synthesized glycoproteins and ERp57 (81) led to the discovery that the two lectins and the oxidoreductase work in combination to assist glycoproteins folding ((91, 93, 94) and Section 2.3.1.3).
The peculiarities of ERp57-CRT and ERp57-CNX interactions have been revealed by TROSY-NMR spectroscopy. These studies established that ERp57 associates with the P-domain of CRT, more precisely with a region of the ER lectin comprising the residues 189–288. The interaction is short-lived, has low affinity (Kd of 9 μM) (82) and is stabilized by the presence of folding substrates (42). Isothermal titration calorimetry (ITC) allowed the mapping of the minimal domain required for ERp57-CRT interaction to the distal part of the CRT P-domain (residues 221–256) (41). The crystal structure of the ERp57 bb′ domain revealed a set of positively charged amino acids in the external part of the ERp57 b′ domain, which represents the moiety that binds to the negatively charged region of the CRT P-domain (84). The CNX P-domain (residues 337–353) is the motif required for ERp57 binding (88), with an interaction affinity calculated in a Kd of 26 μM using ITC technique (78, 84).
Altogether, these studies demonstrated that ERp57-CNX/CRT help with the productive maturation of glycoproteins by creating a strategic environment for protein folding.
2.3.1.3 ERp57 role in the CNX/CRT cycle
CNX and CRT bind the cargo protein oligosaccharides and ERp57 engages the folding proteins in mixed disulfides that represent short lived intermediates of the oxidative folding process (93). The use of the trapping mutant version of ERp57 revealed that proteins containing small, disulfide-rich glycoproteins with a surprisingly low level of secondary structures are endogenous substrates of ERp57 (95).
At least in cultured cells, deletion of ERp57 is well-tolerated showing that most cellular glycoproteins can engage other folding assistants to eventually attain their native structure (96). For example, ERp72 can efficiently replace ERp57 to ensure efficient maturation of Semliki forest virus glycoproteins (97). In this context, the impaired maturation of influenza hemagglutinin or of proteins that require an extensive rearrangement of disulfides in cells lacking ERp57 (95, 97) is a paradigm for those cellular proteins that are strictly dependent on CNX, CRT and ERp57 and whose incapacity to attain the functional conformation causes the embryonic or the early mortality in the corresponding knockout mice (64, 65, 98).
2.3.2 PPI family: peptidyl-prolyl bonds isomerization
Peptide bonds connecting any amino acid with a proline residue can adopt both a cis or a trans conformation, with a weak preference for the trans conformation (99). The isomerization of misarranged peptidyl-prolyl bonds can be a rate-limiting step during protein folding (Fig. 3B) (96, 100) and is catalyzed by members of the PPI family, which comprises three sub-groups, parvulins, cyclophilins (Cyps) and FK506 binding proteins (FKBPs) (Table 1) (101, 102). The mammalian ER contains six members of the FKBPs and two members of the Cyps sub-groups (Table 1) (102–104).
2.3.2.1 Cyclophilin B
Cyclophilin B (CypB) is a protein of 216 residues that is inhibited by cyclosporine A (103). Its function as a molecular chaperone has been established in studies on rhodopsin maturation and export in D. melanogaster (105). Recently, it was shown that CypB and BiP enhance IgG maturation and secretion in B cells and that CypB inactivation by cyclosporine A delayed IgG synthesis (106). Moreover, CypB was found to assist the oxidoreductase ERp72 in the folding of IgG CH1, which was reported to have a high content in cis prolines (70).
2.3.2.2 Interaction between CypB and CNX or CRT
Several groups identified CypB as a component of multiprotein complexes containing CNX and CRT within the ER lumen (Fig. 2) (67–70). Crystallography studies mapped the interaction site of CypB to the tip of the CNX and CRT P-domains. As such the association of ERp57 and of CypB to CNX and/or CRT is mutually exclusive (67). The association of CypB with CNX/CRT requires a positively charged lysine-rich motif of CypB. The CypB-CNX and the CypB-CRT binding affinities have a Kd of about 10 μM. This data supports an active role of CypB in the CNX/CRT cycle by promoting the correct orientation of peptidyl-prolyl bonds in nascent polypeptides emerging in the ER lumen.
2.4 Regulation of the chaperone function and sub-cellular localization of CNX and CRT
Post-translational modifications such as phosphorylation and palmitoylation have been described to play a major role in defining the functions of CNX, the only ER lectin displaying a long cytosolic tail. These modifications have been implicated in modulating its location depending on the functional demands for CNX (17, 107, 108).
Several phosphorylation sites have been identified for CNX on its cytosolic C- terminal tail. These sites are modified by kinases such as casein kinase II (CKII) and the mitogen activated protein kinase ERK1. Phosphorylation of CNX has been proposed to increase its association with ribosomes possibly facilitating/promoting CNX association with nascent chains (107). CNX phosphorylation is enhanced under conditions of ER stress that cause protein misfolding which prolongs interaction of CNX with maturing substrates such as α1AT thus delaying protein secretion (109).
Palmitoylation of CNX also plays a role in the regulation of its chaperone function and location, but data are somewhat controversial. In one study, palmitoylation was reported to target CNX to the ribosome-translocon complex to position it to better bind nascent chains entering into the ER lumen and ensure their proper maturation and secretion. Consistently, non-palmitoylated CNX shows reduced binding to substrates compared to palmitoylated CNX (110). In contrast, another study proposed that non-palmitoylated CNX plays a more prominent role in folding while palmitoylated CNX interacts with the calcium channel SERCA2b and is implicated in calcium regulation (108). This is demonstrated by a change in the localization of CNX. Palmitoylation caused enrichment of CNX at the mitochondrial associated membrane (MAMs), thus enhancing its role in calcium signaling while de- palmitoylation relocates CNX to the rough ER or the ER quality control compartment, thus enhancing its role in folding and quality control (108).
As some inconsistencies appear to exist concerning the role of palmitoylation in determining the function of CNX, further investigations are required to clarify the role of this modification. Post-translational modifications of CNX influence its localization and functions. Whether such mechanisms of regulation also occur for other ER resident proteins including CRT is an interesting issue that requires further investigation.
3. Role of UGT1 as a folding sensor
Release from the lectin chaperones CNX and CRT is followed by trimming of the innermost glucose residue by α-glucosidase II that prevents immediate re- association of the newly synthesized polypeptide to CNX and CRT. Here, a decision has to be made whether a protein is properly folded and should be targeted for anterograde trafficking or whether its forward transport should be prevented in order to maintain it longer in the folding environment or to select it for clearance from the ER lumen. Making such decisions requires the capacity to distinguish between folding intermediates to be retained in the folding environment (Fig. 2, step 6), native proteins to be released from the ER (step 7), and terminally misfolded proteins to be eventually destroyed (steps 8 and 9). Such virtues have been proposed to be possessed by UGT1. UGT1 has been described as a folding sensor because it can recognize structural imperfections such as exposed hydrophobic domains on maturing proteins, a function that is performed by its N-terminal domain stretching to about 80% of the protein (111, 112). Upon recognition of structural defects on cargo proteins, the UGT1 transfers a glucose residue onto the A branch of the glycans via its C-terminal catalytic domain (Fig. 2, step 6). This forces immature proteins to rebind to CNX and CRT for another round of folding attempts under the assistance of the associated enzymes ERp57 and CypB. An element of recognition by UGT1 on the glycan is the innermost GlcNAc residue (111). That other yet undetermined elements are also recognized by UGT1 cannot be excluded.
Cycles of de-glucosylation and re-glucosylation occur (30), indicative of a role of UGT1 in retaining intermediates of folding programs and folding-defective polypeptides in the ER to enhance folding efficiency and to hamper secretion of aberrant proteins (32, 33, 113, 114). UGT1 also contributes to the luminal retention of unassembled subunits of multimeric complexes, thus promoting efficient and complete assembly (115) and, at least for some polypeptides, is required for structural maturation necessary for dissociation from CNX and CRT (116).
Significantly, UGT1 deletion poorly affects the stringency of ER quality control, which is ensured from sequentially operating retention machineries relying on the intervention of luminal chaperones such as BiP (117). However, the sequential domain lysosomal protein prosaposin is an obligate substrate of UGT1 as it misfolds and is found in aggresome-like structures in its absence (118). Prosaposin is efficiently reglucosylated by UGT1 as demonstrated using a cell-based reglucosylation assay. A recent investigation also showed that UGT1 promotes solubility of substrates prone to aggregation in the ER, which further hints at a potential role of UGT1 in alleviating cellular damage caused by aggregation (119).
There is discrepancy in the literature on substrate features that elicit UGT1 activity. A study showed that UGT1 is highly affected by the amino acid sequence proximal to the glycan to be re-glucosylated (120), and that structural defects recognized by UGT1 are localized near the region containing the glycan (121). Another study suggests that UGT1 can recognize glycans that are more distant to the region containing the folding defect (122). To shed more light on this issue, structural analysis of the UGT1 substrate recognition domain may provide better understanding of the ability of UGT1 to differentiate between native and non-native substrates.
Earlier studies agree that UGT1 prefers substrates that are incompletely folded over native proteins; however, the data is conflicting about the severity of the structural imperfection that UGT1 prefers. Seminal UGT1 studies using purified UGT1 or microsomal membranes suggested that UGT1 recognizes severely misfolded substrates such as those that are urea denatured when compared to its native counterpart (123–126). Yet, later studies showed that UGT1 has better affinity for nearly native proteins, late folding intermediates or proteins that contain only minor structural perturbations (127–130). The latter conclusion would suggest that UGT1 would have better affinity for proteins on the pathway to fold, and might ignore proteins deemed severely misfolded. However, a recent cellular study found that UGT1 modifies terminally misfolded ERAD substrates such as NHK, T-cell receptor α-subunit, and an alpha-N-acetylgalactosaminidase (α-NAGAL) mutant more efficiently than on-pathway wild type substrates (114). Additional studies are required to scrutinize the specificity for UGT1 in its native environment. A more detailed description of the UGT1 specificity is presented by Ito et al., in this issue.
UGT2 is a human homologue of UGT1 that shares 55% identity with UGT1, especially in the C-terminus domain that contains the catalytic site. However, its function remains unclear. It was originally proposed that UGT2 lacks re-glucosylation activity (131) yet, a recent study showed that UGT2 has re-glucosylation activity and similar specificity to UGT1. This study used a cell free re-glucosylation assay (132). Further investigations are required to confirm the function of UGT2.
3.1 Proposed mechanisms of how substrates exit the CNX and CRT cycle
It is still not fully understood how the re-glucosylation/de-glucosylation cycles are terminated. Extensive mannose trimming results in exit of folding-defective proteins from the CNX/CRT cycle (Fig. 2, step 8). Trimming of the mannose to which the glucose is added in the A-branch would stop re-glucosylation (133, 134), and trimming of the mannose residues of the B and C branches may decrease the affinity of UGT1 for glycans (124). Moreover, removal of terminal α1,2-bonded mannose residues exposes an α1,6 mannose residue on the C-branch of the protein-bound oligosaccharide that recruits the MRH domain-containing lectins XTP3-B and OS-9 that target aberrant polypeptides for ERAD (11). The selection of glycosylated ERAD substrates is discussed more thoroughly in the article by Benyair et al., in this issue.
4. Export from the ER: Proposed models and roles of ERGIC-53, VIP36 and other lectins in glycoprotein sorting
After leaving the CNX/CRT binding cycle, proteins that have folded properly will be targeted for trafficking to the Golgi via the ER/Golgi intermediate compartment (ERGIC) (Fig. 2, step 7). Exit from the ER to the Golgi could occur either by bulk flow, which allows the cargo to be incorporated into COPII vesicles without any sort of selection process or by receptor mediated transport, which involves interaction of the proteins of the coat of the vesicles with membrane receptors that bind to the cargo (135).
In the bulk flow model, it is proposed that proteins leave the ER via the fluid or the membrane of forming vesicles without the intervention of receptors. In this transport model, cargo is not concentrated (136). The flow of cargo out of the ER is completely controlled by factors that retain immature or non-native proteins such as the lectin chaperones CNX and CRT. Native and properly assembled proteins that no longer bind chaperones or ER factors are free to exit in COPII vesicles by bulk flow.
In the receptor mediated transport model, receptor interaction with cargo involves glycans. ERGIC-53, VIPL and VIP36 are classified as lectin receptors due to their carbohydrate binding capability. These lectins share similar structural characteristics in their carbohydrate binding domain (137). Yet, the specificity of these lectin receptors is not identical because the composition of the glycan to which these lectin receptors bind can vary. While VIP36 and VIPL bind to high mannose type oligosaccharides having a de-glucosylated branch A (137, 138), ERGIC-53 has a lower binding affinity and broader specificity and can bind to glycans with both glucosylated and de-glucosylated branch A (137). Structural analysis suggested that the broad specificity of ERGIC-53 is due to a shallower sugar binding pocket in ERGIC- 53 and the outward orientation of the 3-OH of the terminal mannose residue, which allows a glucose residue to fit in the binding pocket of ERGIC-53 without steric hindrance (139). Although they have been shown to bind different substrates, ERGIC- 53 and VIP36 appear to share some substrates such as α1AT (138, 140). Substrates specific to ERGIC-53 include the hemophilia associated blood coagulation factors V and VIII, as well as cathepsin Z and C (141–143). VIP36 binds to α-amylase and clusterin (144, 145) and the more recently identified substrate guanylyl cyclase C (146). While some of the carbohydrate and protein signals that are utilized by selective-transport route have been identified (147, 148), what determines that a substrate follows the bulk flow route or the selective receptor mediated transport route is not fully understood.
5. Concluding remarks
While a great wealth of information has been acquired on the role of glycans in maturation and quality control of proteins expressed in the ER of Eukarya, the recent discovery of additional lectins and quality control factors highlights both the complex principles governing protein production and the needs for further studies. Many questions remain to be answered and perhaps models to be re-visited regarding the mechanism of action of the known folding and quality control machineries. To understand how these respond to variation in the quantity and the quality of the cargo load is crucial for designing therapeutic strategies for diseases caused by production, accumulation or destruction of aberrant polypeptides or to intervene to contrast infection by pathogens that exploit the host cell protein factory during their infection cycle. For instance, one strategy has been the use of α-glucosidase I and II inhibitors to inhibit viral infectivity (149) based on the findings that viral glycoproteins are more dependent on CNX/CRT assistance for folding than cellular proteins (63). Inhibiting glucosidase trimming prevents the entry of viral proteins into the CNX/CRT cycle (150). This leads to misfolding of viral proteins and their eventual degradation and subsequent activation of the immune response. Derivatives of the iminosugars castanospermine and deoxynojirmycin have been widely used to inhibit the infectivity of a vast range of both DNA and RNA viruses. Some of these inhibitors have entered clinical trials (149). Optimization of the lectin chaperone pathway may also help to correct the folding of defective proteins associated with loss of function diseases (96) or increase the expression of recombinant proteins used as therapeutics. Future studies will shed more light on how exploiting the components of the quality control network can ameliorate disease phenotypes or provide more effective therapies.
Acknowledgments
MM is supported by Signora Alessandra, by the Foundation for Research on Neurodegenerative Diseases, the Swiss National Science Foundation and the Comel, Gabriele and Gelu Foundations. This work was also supported by the National Institutes of Health under award numbers GM086874 and GM094848 (to D.N.H.); and a Chemistry-Biology Interface program training grant (T32 GM08515 to A.T.).
References
- 1.Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:867. doi: 10.1126/science.189.4206.867-b. [DOI] [PubMed] [Google Scholar]
- 2.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–41. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 3.Lynes EM, Simmen T. Urban planning of the endoplasmic reticulum (ER): How diverse mechanisms segregate the many functions of the ER. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2011;1813(10):1893–905. doi: 10.1016/j.bbamcr.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anfinsen CB. Principles that Govern the Folding of Protein chains. Science. 1973;181:223–30. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 5.Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–9. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 6.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–81. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 7.Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutation research. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 8.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nature reviews Molecular cell biology. 2003;4:181–91. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 9.Romisch K. Endoplasmic reticulum-associated degradation. Annual review of cell and developmental biology. 2005;21:435–56. doi: 10.1146/annurev.cellbio.21.012704.133250. [DOI] [PubMed] [Google Scholar]
- 10.Molinari M. N-glycan structure dictates extension of protein folding or onset of disposal. Nat Chem Biol. 2007;3(6):313–20. doi: 10.1038/nchembio880. [DOI] [PubMed] [Google Scholar]
- 11.Aebi M, Bernasconi R, Clerc S, Molinari M. N-glycan structures: recognition and processing in the ER. Trends in biochemical sciences. 2010;35(2):74–82. doi: 10.1016/j.tibs.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Burda P, Aebi M. The dolichol pathway of N-linked glycosylation. Biochimica et biophysica acta. 1999;1426:239–57. doi: 10.1016/s0304-4165(98)00127-5. [DOI] [PubMed] [Google Scholar]
- 13.Mohorko E, Glockshuber R, Aebi M. Oligosaccharyltransferase: the central enzyme of N-linked protein glycosylation. Journal of inherited metabolic disease. 2011;34:869–78. doi: 10.1007/s10545-011-9337-1. [DOI] [PubMed] [Google Scholar]
- 14.Wormald MR, Dwek RA. Glycoproteins: glycan presentation and protein-fold stability. Structure. 1999;7:R155–60. doi: 10.1016/s0969-2126(99)80095-1. [DOI] [PubMed] [Google Scholar]
- 15.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–9. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 16.Braakman I, Hebert DN. Protein folding in the endoplasmic reticulum. Cold Spring Harbor perspectives in biology. 2013;5:a013201. doi: 10.1101/cshperspect.a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hebert DN, Lamriben L, Powers ET, Kelly JW. The intrinsic and extrinsic effects of N-linked glycans on glycoproteostasis. Nat Chem Biol. 2014;10(11):902–10. doi: 10.1038/nchembio.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–64. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 19.Parodi AJ. Protein glucosylation and its role in protein folding. Annu Rev Biochem. 2000;69:69–93. doi: 10.1146/annurev.biochem.69.1.69. [DOI] [PubMed] [Google Scholar]
- 20.Deprez P, Gautschi M, Helenius A. More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Molecular Cell. 2005;9(2):183–95. doi: 10.1016/j.molcel.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 21.Shailubhai K, Pukazhenthi BS, Saxena ES, Varma GM, Vijay IK. Glucosidase I, a transmembrane endoplasmic reticular glycoprotein with a luminal catalytic domain. Journal of Biological Chemistry. 1991;266(25):16587–93. [PubMed] [Google Scholar]
- 22.Barker MK, Rose DR. Specificity of Processing α-Glucosidase I Is Guided by the Substrate Conformation: CRYSTALLOGRAPHIC AND IN SILICO STUDIES. Journal of Biological Chemistry. 2013;288(19):13563–74. doi: 10.1074/jbc.M113.460436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schallus T, Fehér K, Sternberg U, Rybin V, Muhle-Goll C. Analysis of the specific interactions between the lectin domain of malectin and diglucosides. Glycobiology. 2010;20(8):1010–20. doi: 10.1093/glycob/cwq059. [DOI] [PubMed] [Google Scholar]
- 24.Galli C, Bernasconi R, Solda T, Calanca V, Molinari M. Malectin participates in a back up glycoprotein quality control pathway in the Mammalian ER. PLoS One. 2011;6(1):e16304. doi: 10.1371/journal.pone.0016304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Hu D, Yabe R, Tateno H, Qin S-Y, Matsumoto N, et al. Role of malectin in Glc2Man9GlcNAc2-dependent quality control of a1-antitrypsin. Molecular biology of the cell. 2011;22(19):3559–70. doi: 10.1091/mbc.E11-03-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin SY, Hu D, Matsumoto K, Takeda K, Matsumoto N, Yamaguchi Y, et al. Malectin forms a complex with ribophorin I for enhanced association with misfolded glycoproteins. The Journal of biological chemistry. 2012;287(45):38080–9. doi: 10.1074/jbc.M112.394288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trombetta ES, Simons JF, Helenius A. Endoplasmic Reticulum Glucosidase II Is Composed of a Catalytic Subunit, Conserved from Yeast to Mammals, and a Tightly Bound Noncatalytic HDEL-containing Subunit. Journal of Biological Chemistry. 1996;271(44):27509–16. doi: 10.1074/jbc.271.44.27509. [DOI] [PubMed] [Google Scholar]
- 28.Treml K, Meimaroglou D, Hentges A, Bause E. The α- and β-subunits are required for expression of catalytic activity in the hetero-dimeric glucosidase II complex from human liver. 2000:493–502. doi: 10.1093/glycob/10.5.493. [DOI] [PubMed] [Google Scholar]
- 29.Stigliano ID, Caramelo JJ, Labriola CA, Parodi AJ, D’Alessio C. Glucosidase II β Subunit Modulates N-Glycan Trimming in Fission Yeasts and Mammals. Molecular biology of the cell. 2009;20(17):3974–84. doi: 10.1091/mbc.E09-04-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammond C, Braakman I, Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proceedings of the National Academy of Sciences. 1994;91(3):913–7. doi: 10.1073/pnas.91.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ora A, Helenius A. Calnexin Fails to Associate with Substrate Proteins in Glucosidase-deficient Cell Lines. Journal of Biological Chemistry. 1995;270(44):26060–2. doi: 10.1074/jbc.270.44.26060. [DOI] [PubMed] [Google Scholar]
- 32.Hebert DN, Foellmer B, Helenius A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell. 1995;81(3):425–33. doi: 10.1016/0092-8674(95)90395-x. [DOI] [PubMed] [Google Scholar]
- 33.Cannon KS, Helenius A. Trimming and Readdition of Glucose to N-Linked Oligosaccharides Determines Calnexin Association of a Substrate Glycoprotein in Living Cells. Journal of Biological Chemistry. 1999;274(11):7537–44. doi: 10.1074/jbc.274.11.7537. [DOI] [PubMed] [Google Scholar]
- 34.Kaushal GP, Pastuszak I, Hatanaka K, Elbein AD. Purification to homogeneity and properties of glucosidase II from mung bean seedlings and suspension-cultured soybean cells. Journal of Biological Chemistry. 1990;265(27):16271–9. [PubMed] [Google Scholar]
- 35.Totani K, Ihara Y, Matsuo I, Ito Y. Substrate Specificity Analysis of Endoplasmic Reticulum Glucosidase II Using Synthetic High Mannose-type Glycans. Journal of Biological Chemistry. 2006;281(42):31502–8. doi: 10.1074/jbc.M605457200. [DOI] [PubMed] [Google Scholar]
- 36.Grinna LS, Robbins PW. Substrate specificities of rat liver microsomal glucosidases which process glycoproteins. Journal of Biological Chemistry. 1980;255(6):2255–8. [PubMed] [Google Scholar]
- 37.Stigliano ID, Alculumbre SG, Labriola CA, Parodi AJ, D’Alessio C. Glucosidase II and N-glycan mannose content regulate the half-lives of monoglucosylated species in vivo. Molecular biology of the cell. 2011;22(11):1810–23. doi: 10.1091/mbc.E11-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Helenius A. How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Molecular Biology of the Cell. 1994;5(3):253–65. doi: 10.1091/mbc.5.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wada I, Rindress D, Cameron PH, Ou WJ, Doherty JJ, Louvard D, et al. SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. Journal of Biological Chemistry. 1991;266(29):19599–610. [PubMed] [Google Scholar]
- 40.Schrag JD, Bergeron JJ, Li Y, Borisova S, Hahn M, Thomas DY, et al. The structure of calenxin, an ER chaperone involved in quality control of protein folding. Molecular Cell. 2001;8(3):633–44. doi: 10.1016/s1097-2765(01)00318-5. [DOI] [PubMed] [Google Scholar]
- 41.Ellgaard L, Bettendorff P, Braun D, Herrmann T, Fiorito F, Jelesarov I, et al. NMR structures of 36 and 73-residue fragments of the calreticulin P-domain. J Mol Biol. 2002;322:773–84. doi: 10.1016/s0022-2836(02)00812-4. [DOI] [PubMed] [Google Scholar]
- 42.Frickel EM, Riek R, Jelesarov I, Helenius A, Wuthrich K, Ellgaard L. TROSY-NMR reveals interaction between ERp57 and the tip of the calreticulin P-domain. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:1954–9. doi: 10.1073/pnas.042699099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vassilakos A, Michalak M, Lehrman MA, Williams DB. Oligosaccharide binding characteristics of the molecular chaperones calnexin and calreticulin. Biochemistry. 1998;37(10):3480–90. doi: 10.1021/bi972465g. [DOI] [PubMed] [Google Scholar]
- 44.Thomson SP, Williams DB. Delineation of the lectin site of the molecular chaperone calreticulin. Cell stress & chaperones. 2005;10(3):242–51. doi: 10.1379/CSC-126.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kozlov G, Pocanschi CL, Rosenauer A, Bastos-Aristizabal S, Gorelik A, Williams DB, et al. Structural basis of carbohydrate recognition by calreticulin. The Journal of biological chemistry. 2010;285(49):38612–20. doi: 10.1074/jbc.M110.168294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wijeyesakere SJ, Rizvi SM, Raghavan M. Glycan-dependent and -independent Interactions Contribute to Cellular Substrate Recruitment by Calreticulin. Journal of Biological Chemistry. 2013;288(49):35104–16. doi: 10.1074/jbc.M113.507921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daniels R, Kurowski B, Johnson AE, Hebert DN. N-Linked Glycans Direct the Cotranslational Folding Pathway of Influenza Hemagglutinin. Molecular Cell. 2003;11(1):79–90. doi: 10.1016/s1097-2765(02)00821-3. [DOI] [PubMed] [Google Scholar]
- 48.Wang N, Glidden EJ, Murphy SR, Pearse BR, Hebert DN. The Cotranslational Maturation Program for the Type II Membrane Glycoprotein Influenza Neuraminidase. Journal of Biological Chemistry. 2008;283(49):33826–37. doi: 10.1074/jbc.M806897200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pearse BR, Hebert DN. Lectin chaperones help direct the maturation of glycoproteins in the endoplasmic reticulum. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2010;1803(6):684–93. doi: 10.1016/j.bbamcr.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roderick HL, Lechleiter JD, Camacho P. Cytosolic Phosphorylation of Calnexin Controls Intracellular Ca2+ Oscillations via an Interaction with Serca2b. The Journal of Cell Biology. 2000;149(6):1235–48. doi: 10.1083/jcb.149.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multi- process calcium-buffering chaperone of the endoplasmic reticulum. The Biochemical journal. 2009;417(3):651–66. doi: 10.1042/BJ20081847. [DOI] [PubMed] [Google Scholar]
- 52.Helenius A, Trombetta ES, Hebert DN, Simons JF. Calnexin, calreticulin and the folding of glycoproteins. Trends in Cell Biology. 7(5):193–200. doi: 10.1016/S0962-8924(97)01032-5. [DOI] [PubMed] [Google Scholar]
- 53.Peterson JR, Ora A, Van PN, Helenius A. Transient, lectin-like association of calreticulin with folding intermediates of cellular and viral glycoproteins. Molecular biology of the cell. 1995;6(9):1173–84. doi: 10.1091/mbc.6.9.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zapun A, Petrescu SM, Rudd PM, Dwek RA, Thomas DY, Bergeron JJM. Conformation-Independent Binding of Monoglucosylated Ribonuclease B to Calnexin. Cell. 1997;88(1):29–38. doi: 10.1016/s0092-8674(00)81855-3. [DOI] [PubMed] [Google Scholar]
- 55.Otteken A, Moss B. Calreticulin Interacts with Newly Synthesized Human Immunodeficiency Virus Type 1 Envelope Glycoprotein, Suggesting a Chaperone Function Similar to That of Calnexin. Journal of Biological Chemistry. 1996;271(1):97–103. doi: 10.1074/jbc.271.1.97. [DOI] [PubMed] [Google Scholar]
- 56.Danilczyk UG, Williams DB. The Lectin Chaperone Calnexin Utilizes Polypeptide-based Interactions to Associate with Many of Its Substrates in Vivo. Journal of Biological Chemistry. 2001;276(27):25532–40. doi: 10.1074/jbc.M100270200. [DOI] [PubMed] [Google Scholar]
- 57.Sandhu N, Duus K, Jørgensen CS, Hansen PR, Bruun SW, Pedersen LØ, et al. Peptide binding specificity of the chaperone calreticulin. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2007;1774(6):701–13. doi: 10.1016/j.bbapap.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 58.Brockmeier A, Brockmeier U, Williams DB. Distinct Contributions of the Lectin and Arm Domains of Calnexin to Its Molecular Chaperone Function. Journal of Biological Chemistry. 2009;284(6):3433–44. doi: 10.1074/jbc.M804866200. [DOI] [PubMed] [Google Scholar]
- 59.Hebert DN, Zhang J-X, Chen W, Foellmer B, Helenius A. The Number and Location of Glycans on Influenza Hemagglutinin Determine Folding and Association with Calnexin and Calreticulin. The Journal of cell biology. 1997;139(3):613–23. doi: 10.1083/jcb.139.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harris MR, Yu YYL, Kindle CS, Hansen TH, Solheim JC. Calreticulin and Calnexin Interact with Different Protein and Glycan Determinants During the Assembly of MHC Class I. The Journal of Immunology. 1998;160(11):5404–9. [PubMed] [Google Scholar]
- 61.Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, et al. Contrasting Functions of Calreticulin and Calnexin in Glycoprotein Folding and ER Quality Control. Molecular cell. 2004;13(1):125–35. doi: 10.1016/s1097-2765(03)00494-5. [DOI] [PubMed] [Google Scholar]
- 62.Wada I, Imai S-i, Kai M, Sakane F, Kanoh H. Chaperone Function of Calreticulin When Expressed in the Endoplasmic Reticulum as the Membrane-anchored and Soluble Forms. Journal of Biological Chemistry. 1995;270(35):20298–304. doi: 10.1074/jbc.270.35.20298. [DOI] [PubMed] [Google Scholar]
- 63.Pieren M, Galli C, Denzel A, Molinari M. The Use of Calnexin and Calreticulin by Cellular and Viral Glycoproteins. Journal of Biological Chemistry. 2005;280(31):28265–71. doi: 10.1074/jbc.M501020200. [DOI] [PubMed] [Google Scholar]
- 64.Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause K-H, et al. Calreticulin Is Essential for Cardiac Development. The Journal of cell biology. 1999;144(5):857–68. doi: 10.1083/jcb.144.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Denzel A, Molinari M, Trigueros C, Martin JE, Velmurgan S, Brown S, et al. Early Postnatal Death and Motor Disorders in Mice Congenitally Deficient in Calnexin Expression. Molecular and cellular biology. 2002;22(21):7398–404. doi: 10.1128/MCB.22.21.7398-7404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jessop CE, Tavender TJ, Watkins RH, Chambers JE, Bulleid NJ. Substrate specificity of the oxidoreductase ERp57 is determined primarily by its interaction with calnexin and calreticulin. J Biol Chem. 2009;284:2194–202. doi: 10.1074/jbc.M808054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kozlov G, Bastos-Aristizabal S, Maattanen P, Rosenauer A, Zheng F, Killikelly A, et al. Structural basis of cyclophilin B binding by the calnexin/calreticulin P-domain. J Biol Chem. 2010;285:35551–7. doi: 10.1074/jbc.M110.160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meunier L, Usherwood YK, Chung KT, Hendershot LM. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol Biol Cell. 2002;13:4456–69. doi: 10.1091/mbc.E02-05-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang J, Herscovitz H. Nascent lipidated apolipoprotein B is transported to the Golgi as an incompletely folded intermediate as probed by its association with network of endoplasmic reticulum molecular chaperones, GRP94, ERp72, BiP, calreticulin, and cyclophilin B. J Biol Chem. 2003;278:7459–68. doi: 10.1074/jbc.M207976200. [DOI] [PubMed] [Google Scholar]
- 70.Jansen G, Maattanen P, Denisov AY, Scarffe L, Schade B, Balghi H, et al. An interaction map of endoplasmic reticulum chaperones and foldases. Molecular & cellular proteomics : MCP. 2012;11:710–23. doi: 10.1074/mcp.M111.016550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ellgard L, Ruddock L. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO reports. 2005;6(1):28–32. doi: 10.1038/sj.embor.7400311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kozlov G, Maattanen P, Thomas DY, Gehring K. A structural overview of the PDI family of proteins. The FEBS journal. 2010;277:3924–36. doi: 10.1111/j.1742-4658.2010.07793.x. [DOI] [PubMed] [Google Scholar]
- 73.Galligan JJ, Petersen DR. The human protein disulfide isomerase gene family. Human genomics. 2012;6:6. doi: 10.1186/1479-7364-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Appenzeller-Herzog C, Ellgaard L. In vivo reduction-oxidation state of protein disulfide isomerase: the two active sites independently occur in the reduced and oxidized forms. Antioxidants & redox signaling. 2008;10:55–64. doi: 10.1089/ars.2007.1837. [DOI] [PubMed] [Google Scholar]
- 75.Denisov AY, Maattanen P, Dabrowski C, Kozlov G, Thomas DY, Gehring K. Solution structure of the bb’ domains of human protein disulfide isomerase. The FEBS journal. 2009;276:1440–9. doi: 10.1111/j.1742-4658.2009.06884.x. [DOI] [PubMed] [Google Scholar]
- 76.Pirneskoski A, Klappa P, Lobell M, Williamson RA, Byrne L, Alanen HI, et al. Molecular characterization of the principal substrate binding site of the ubiquitous folding catalyst protein disulfide isomerase. J Biol Chem. 2004;279:10374–81. doi: 10.1074/jbc.M312193200. [DOI] [PubMed] [Google Scholar]
- 77.Byrne LJ, Sidhu A, Wallis AK, Ruddock LW, Freedman RB, Howard MJ, et al. Mapping of the ligand-binding site on the b’ domain of human PDI: interaction with peptide ligands and the x-linker region. Biochem J. 2009;423:209–17. doi: 10.1042/BJ20090565. [DOI] [PubMed] [Google Scholar]
- 78.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxidants & redox signaling. 2009;11:2807–50. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 79.Roos G, Garcia-Pino A, Van Belle K, Brosens E, Wahni K, Vandenbussche G, et al. The conserved active site proline determines the reducing power of Staphylococcus aureus thioredoxin. J Mol Biol. 2007;368:800–11. doi: 10.1016/j.jmb.2007.02.045. [DOI] [PubMed] [Google Scholar]
- 80.Mohorko E, Owen RL, Malojcic G, Brozzo MS, Aebi M, Glockshuber R. Structural basis of substrate specificity of human oligosaccharyl transferase subunit N33/Tusc3 and its role in regulating protein N-glycosylation. Structure. 2014;22:590–601. doi: 10.1016/j.str.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 81.Oliver JD, van der Wal FJ, Bulleid NJ, High S. Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science. 1997;275:86–8. doi: 10.1126/science.275.5296.86. [DOI] [PubMed] [Google Scholar]
- 82.Ellgaard L, Frickel EM. Calnexin, calreticulin, and ERp57: teammates in glycoprotein folding. Cell biochemistry and biophysics. 2003;39:223–47. doi: 10.1385/CBB:39:3:223. [DOI] [PubMed] [Google Scholar]
- 83.Russell SJ, Ruddock LW, Salo KE, Oliver JD, Roebuck QP, Llewellyn DH, et al. The primary substrate binding site in the b’ domain of ERp57 is adapted for endoplasmic reticulum lectin association. J Biol Chem. 2004;279:18861–9. doi: 10.1074/jbc.M400575200. [DOI] [PubMed] [Google Scholar]
- 84.Kozlov G, Maattanen P, Schrag JD, Pollock S, Cygler M, Nagar B, et al. Crystal structure of the bb’ domains of the protein disulfide isomerase ERp57. Structure. 2006;14:1331–9. doi: 10.1016/j.str.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 85.Freedman RB, Klappa P, Ruddock LW. Protein disulfide isomerases exploit synergy between catalytic and specific binding domains. EMBO Rep. 2002;3:136–40. doi: 10.1093/embo-reports/kvf035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nigam SK, Goldberg AL, Ho S, Rohde MF, Bush KT, Sherman M. A set of endoplasmic reticulum proteins possessing properties of molecular chaperones includes Ca(2+)-binding proteins and members of the thioredoxin superfamily. J Biol Chem. 1994;269:1744–9. [PubMed] [Google Scholar]
- 87.Baksh S, Burns K, Andrin C, Michalak M. Interaction of calreticulin with protein disulfide isomerase. J Biol Chem. 1995;270:31338–44. doi: 10.1074/jbc.270.52.31338. [DOI] [PubMed] [Google Scholar]
- 88.Pollock S, Kozlov G, Pelletier MF, Trempe JF, Jansen G, Sitnikov D, et al. Specific interaction of ERp57 and calnexin determined by NMR spectroscopy and an ER two- hybrid system. EMBO J. 2004;23:1020–9. doi: 10.1038/sj.emboj.7600119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sakono M, Seko A, Takeda Y, Ito Y. PDI family protein ERp29 forms 1:1 complex with lectin chaperone calreticulin. Biochem Biophys Res Commun. 2014;452:27–31. doi: 10.1016/j.bbrc.2014.08.041. [DOI] [PubMed] [Google Scholar]
- 90.Van der Wal FJ, Oliver JD, High S. The transient association of ERp57 with N- glycosylated proteins is regulated by glucose trimming. European journal of biochemistry / FEBS. 1998;256:51–9. doi: 10.1046/j.1432-1327.1998.2560051.x. [DOI] [PubMed] [Google Scholar]
- 91.Zapun A, Darby NJ, Tessier DC, Michalak M, Bergeron JJ, Thomas DY. Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J Biol Chem. 1998;273:6009–12. doi: 10.1074/jbc.273.11.6009. [DOI] [PubMed] [Google Scholar]
- 92.Antoniou AN, Ford S, Alphey M, Osborne A, Elliott T, Powis SJ. The oxidoreductase ERp57 efficiently reduces partially folded in preference to fully folded MHC class I molecules. EMBO J. 2002;21:2655–63. doi: 10.1093/emboj/21.11.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Molinari M, Helenius A. Glycoproteins form mixed disulphides with oxidoreductases during folding in living cells. Nature. 1999;402(6757):90–3. doi: 10.1038/47062. [DOI] [PubMed] [Google Scholar]
- 94.Oliver JD, Roderick HL, Llewellyn DH, High S. ERp57 Functions as a Subunit of Specific Complexes Formed with the ER Lectins Calreticulin and Calnexin. Molecular biology of the cell. 1999;10(8):2573–82. doi: 10.1091/mbc.10.8.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jessop CE, Chakravarthi S, Garbi N, Hämmerling GJ, Lovell S, Bulleid NJ. ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. The EMBO Journal. 2006;26(1):28–40. doi: 10.1038/sj.emboj.7601505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007;87:1377–408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- 97.Soldà T, Garbi N, Hämmerling GJ, Molinari M. Consequences of ERp57 Deletion on Oxidative Folding of Obligate and Facultative Clients of the Calnexin Cycle. Journal of Biological Chemistry. 2006;281(10):6219–26. doi: 10.1074/jbc.M513595200. [DOI] [PubMed] [Google Scholar]
- 98.Garbi N, Tanaka S, Momburg F, Hammerling GJ. Impaired assembly of the major histocompatibility complex class I peptide-loading complex in mice deficient in the oxidoreductase ERp57. Nature immunology. 2006;7(1):93–102. doi: 10.1038/ni1288. [DOI] [PubMed] [Google Scholar]
- 99.Di Martino GP, Masetti M, Cavalli A, Recanatini M. Mechanistic insights into Pin1 peptidyl-prolyl cis-trans isomerization from umbrella sampling simulations. Proteins. 2014 doi: 10.1002/prot.24650. [DOI] [PubMed] [Google Scholar]
- 100.Kiefhaber T, Quaas R, Hahn U, Schmid FX. Folding of ribonuclease T1. 2. Kinetic models for the folding and unfolding reactions. Biochemistry. 1990;29:3061–70. doi: 10.1021/bi00464a024. [DOI] [PubMed] [Google Scholar]
- 101.Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature. 1989;337:476–8. doi: 10.1038/337476a0. [DOI] [PubMed] [Google Scholar]
- 102.Stocki P, Chapman DC, Beach LA, Williams DB. Depletion of cyclophilins B and C leads to dysregulation of endoplasmic reticulum redox homeostasis. J Biol Chem. 2014;289:23086–96. doi: 10.1074/jbc.M114.570911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Price ER, Zydowsky LD, Jin MJ, Baker CH, McKeon FD, Walsh CT. Human cyclophilin B: a second cyclophilin gene encodes a peptidyl-prolyl isomerase with a signal sequence. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:1903–7. doi: 10.1073/pnas.88.5.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fischer G, Aumuller T. Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. Reviews of physiology, biochemistry and pharmacology. 2003;148:105–50. doi: 10.1007/s10254-003-0011-3. [DOI] [PubMed] [Google Scholar]
- 105.Colley NJ, Baker EK, Stamnes MA, Zuker CS. The cyclophilin homolog ninaA is required in the secretory pathway. Cell. 1991;67:255–63. doi: 10.1016/0092-8674(91)90177-z. [DOI] [PubMed] [Google Scholar]
- 106.Feige MJ, Groscurth S, Marcinowski M, Shimizu Y, Kessler H, Hendershot LM, et al. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol Cell. 2009;34:569–79. doi: 10.1016/j.molcel.2009.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chevet E, Wong HN, Gerber D, Cochet C, Fazel A, Cameron PH, et al. Phosphorylation by CK2 and MAPK enhances calnexin association with ribosomes. The EMBO journal. 1999;18(13):3655–66. doi: 10.1093/emboj/18.13.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lynes EM, Raturi A, Shenkman M, Sandoval CO, Yap MC, Wu J, et al. Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. Journal of cell science. 2013;126(17):3893–903. doi: 10.1242/jcs.125856. [DOI] [PubMed] [Google Scholar]
- 109.Cameron PH, Chevet E, Pluquet O, Thomas DY, Bergeron JJ. Calnexin phosphorylation attenuates the release of partially misfolded alpha1-antitrypsin to the secretory pathway. The Journal of biological chemistry. 2009;284(50):34570–9. doi: 10.1074/jbc.M109.053165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lakkaraju AK, Abrami L, Lemmin T, Blaskovic S, Kunz B, Kihara A, et al. Palmitoylated calnexin is a key component of the ribosome-translocon complex. The EMBO journal. 2012;31(7):1823–35. doi: 10.1038/emboj.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sousa M, Parodi AJ. The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO journal. 1995;14(17):4196–203. doi: 10.1002/j.1460-2075.1995.tb00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Arnold SM, Kaufman RJ. The Noncatalytic Portion of Human UDP- glucose:Glycoprotein Glucosyltransferase I Confers UDP-glucose Binding and Transferase Function to the Catalytic Domain. Journal of Biological Chemistry. 2003;278(44):43320–8. doi: 10.1074/jbc.M305800200. [DOI] [PubMed] [Google Scholar]
- 113.Wada I, Kai M, Imai S, Sakane F, Kanoh H. Promotion of transferrin folding by cyclic interactions with calenxin and carleticulin. The EMBO Journal. 1997;16(17):5420–32. doi: 10.1093/emboj/16.17.5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tannous A, Patel N, Tamura T, Hebert DN. Reglucosylation by UDP-glucose: glycoprotein glucosyltransferase 1 delays glycoprotein secretion but not degradation. Molecular biology of the cell. 2014 doi: 10.1091/mbc.E14-08-1254. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Keith N, Parodi AJ, Caramelo JJ. Glycoprotein Tertiary and Quaternary Structures Are Monitored by the Same Quality Control Mechanism. Journal of Biological Chemistry. 2005;280(18):18138–41. doi: 10.1074/jbc.M501710200. [DOI] [PubMed] [Google Scholar]
- 116.Soldà T, Galli C, Kaufman RJ, Molinari M. Substrate-specific requirements for UGT1-dependent release from calnexin. Molecular Cell. 2007;27(2):238–49. doi: 10.1016/j.molcel.2007.05.032. [DOI] [PubMed] [Google Scholar]
- 117.Molinari M, Galli C, Vanoni O, Arnold SM, Kaufman RJ. Persistent Glycoprotein Misfolding Activates the Glucosidase II/UGT1-Driven Calnexin Cycle to Delay Aggregation and Loss of Folding Competence. Molecular Cell. 2005;20(4):503–12. doi: 10.1016/j.molcel.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 118.Pearse BR, Tamura T, Sunryd JC, Grabowski GA, Kaufman RJ, Hebert DN. The role of UDP-Glc:glycoprotein glucosyltransferase 1 in the maturation of an obligate substrate prosaposin. The Journal of cell biology. 2010;189(5):829–41. doi: 10.1083/jcb.200912105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ferris SP, Jaber NS, Molinari M, Arvan P, Kaufman RJ. UDP- glucose:glycoprotein glucosyltransferase (UGGT1) promotes substrate solubility in the endoplasmic reticulum. Molecular biology of the cell. 2013;24(17):2597–608. doi: 10.1091/mbc.E13-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Taylor SC, Thibault P, Tessier DC, Bergeron JJ, Thomas DY. Glycopeptide specificity of the secretory protein folding sensor UDP-glucose glycoprotein:glucosyltransferase. EMBO reports. 2003;4(4):405–11. doi: 10.1038/sj.embor.embor797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ritter C, Helenius A. Recognition of local glycoprotein misfolding by the ER folding sensor UDP:glucose glycoprotein glucosyltransferase. Nature Structural Biology. 2000;7(4):270–80. doi: 10.1038/74035. [DOI] [PubMed] [Google Scholar]
- 122.Taylor SC, Ferguson AD, Bergeron JJ, Thomas DY. The ER protein folding sensor UDP-glucose glycoprotein-glucosyltransferase modifies substrates distant to local changes in glycoprotein conformation. Nature Structural & Molecular Biology. 2004;11(2):128–34. doi: 10.1038/nsmb715. [DOI] [PubMed] [Google Scholar]
- 123.Trombetta SE, Bosch M, Parodi AJ. Glucosylation of Glycoproteins by Mammalian, Plant, Fungal, and Trypanosomatid Protozoa Microsomal Membranes. Biochemistry. 1989;28(20):8108–16. doi: 10.1021/bi00446a022. [DOI] [PubMed] [Google Scholar]
- 124.Sousa MC, Ferrero-Garcia MA, Parodi AJ. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. Biochemistry. 1992;31(1):97–105. doi: 10.1021/bi00116a015. [DOI] [PubMed] [Google Scholar]
- 125.Fernández FS, Trombetta SE, Hellman U, Parodi AJ. Purification to homogeneity of UDP-glucose:glycoprotein glucosyltransferase from Schizosaccharomyces pombe and apparent absence of the enzyme fro Saccharomyces cerevisiae. Journal of Biological Chemistry. 1994;269(48):30701–6. [PubMed] [Google Scholar]
- 126.Parker CG, Fessler LI, Nelson RE, Fessler JH. Drosophila UDP- Glucose:glycoprotein glucosyltransferase: sequence and characterization of an enzyme that distinguishes between denatured and native proteins. The EMBO Journal. 1995;14(7):1294–303. doi: 10.1002/j.1460-2075.1995.tb07115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Labriola C, Cazzulo JJ, Parodi AJ. Trypanosoma cruzi Calreticulin Is a Lectin That Binds Monoglucosylated Oligosaccharides but Not Protein Moieties of Glycoproteins. Molecular Biology of the Cell. 1999;10(5):1381–94. doi: 10.1091/mbc.10.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Trombetta ES, Helenius A. Conformational Requirements for Glycoprotein Reglucosylation in the Endoplasmic Reticulum. The Journal of cell biology. 2000;148(6):1123–30. doi: 10.1083/jcb.148.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Caramelo JJ, Castro OA, Alonso LG, de Prat-Gay G, Parodi AJ. UDP- Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proceedings of the National Academy of Sciences. 2003;100(1):86–91. doi: 10.1073/pnas.262661199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Caramelo JJ, Castro OA, de Prat-Gay G, Parodi AJ. The Endoplasmic Reticulum Glucosyltransferase Recognizes Nearly Native Glycoprotein Folding Intermediates. Journal of Biological Chemistry. 2004;279(44):46280–5. doi: 10.1074/jbc.M408404200. [DOI] [PubMed] [Google Scholar]
- 131.Arnold SM, Fessler LI, Fessler JH, Kaufman RJ. Two homologues encoding human UDP-glucose: glycoprotein glucosyltransferase differ in mRNA expression and enzymatic activity. Biochemistry. 2000;39(9):2149–63. doi: 10.1021/bi9916473. [DOI] [PubMed] [Google Scholar]
- 132.Takeda Y, Seko A, Hachisu M, Daikoku S, Izumi M, Koizumi A, et al. Both isoforms of human UDP-glucose:glycoprotein glucosyltransferase are enzymatically active. Glycobiology. 2014;24(4):344–50. doi: 10.1093/glycob/cwt163. [DOI] [PubMed] [Google Scholar]
- 133.Ermonval M, Kitzmüller C, Mir AM, Cacan R, Ivessa NE. N-glycan structure of a short-lived variant of ribophorin I expressed in the MadIA214 glycosylation-defective cell line reveals the role of a mannosidase that is not ER mannosidase I in the process of glycoprotein degradation. Glycobiology. 2001;11(7):565–76. doi: 10.1093/glycob/11.7.565. [DOI] [PubMed] [Google Scholar]
- 134.Olivari S, Cali T, Salo KEH, Paganetti P, Ruddock LW, Molinari M. EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding- defective polypeptides and by inhibiting their covalent aggregation. Biochemical and biophysical research communications. 2006;349(4):1278–84. doi: 10.1016/j.bbrc.2006.08.186. [DOI] [PubMed] [Google Scholar]
- 135.Geva Y, Schuldiner M. The Back and Forth of Cargo Exit from the Endoplasmic Reticulum. Current Biology. 2014;24(3):R130–R6. doi: 10.1016/j.cub.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 136.Balch WE, Gist Farquhar M. Beyond bulk flow. Trends in Cell Biology. 1995;5(1):16–9. doi: 10.1016/s0962-8924(00)88928-x. [DOI] [PubMed] [Google Scholar]
- 137.Kamiya Y, Kamiya D, Yamamoto K, Nyfeler B, Hauri H-P, Kato K. Molecular Basis of Sugar Recognition by the Human L-type Lectins ERGIC-53, VIPL, and VIP36. Journal of Biological Chemistry. 2008;283(4):1857–61. doi: 10.1074/jbc.M709384200. [DOI] [PubMed] [Google Scholar]
- 138.Reiterer V, Nyfeler B, Hauri HP. Role of the lectin VIP36 in post-ER quality control of human alpha1-antitrypsin. Traffic (Copenhagen, Denmark) 2010;11(8):1044–55. doi: 10.1111/j.1600-0854.2010.01078.x. [DOI] [PubMed] [Google Scholar]
- 139.Satoh T, Suzuki K, Yamaguchi T, Kato K. Structural basis for disparate sugar- binding specificities in the homologous cargo receptors ERGIC-53 and VIP36. PloS one. 2014;9(2):e87963. doi: 10.1371/journal.pone.0087963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nyfeler B, Reiterer V, Wendeler MW, Stefan E, Zhang B, Michnick SW, et al. Identification of ERGIC-53 as an intracellular transport receptor of alpha1-antitrypsin. J Cell Biol. 2008;180(4):705–12. doi: 10.1083/jcb.200709100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vollenweider F, Kappeler F, Itin C, Hauri H-P. Mistargeting of the Lectin ERGIC- 53 to the Endoplasmic Reticulum of HeLa Cells Impairs the Secretion of a Lysosomal Enzyme. The Journal of Cell Biology. 1998;142(2):377–89. doi: 10.1083/jcb.142.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nichols WC, Seligsohn U, Zivelin A, Terry VH, Hertel CE, Wheatley MA, et al. Mutations in the ER Golgi Intermediate Compartment Protein ERGIC-53 Cause Combined Deficiency of Coagulation Factors V and VIII. Cell. 1998;93(1):61–70. doi: 10.1016/s0092-8674(00)81146-0. [DOI] [PubMed] [Google Scholar]
- 143.Appenzeller C, Andersson H, Kappeler F, Hauri H-P. The lectin ERGIC-53 is a cargo transport receptor for glycoproteins. Nat Cell Biol. 1999;1(6):330–4. doi: 10.1038/14020. [DOI] [PubMed] [Google Scholar]
- 144.Hara-Kuge S, Ohkura T, Ideo H, Shimada O, Atsumi S, Yamashita K. Involvement of VIP36 in Intracellular Transport and Secretion of Glycoproteins in Polarized Madin-Darby Canine Kidney (MDCK) Cells. Journal of Biological Chemistry. 2002;277(18):16332–9. doi: 10.1074/jbc.M112188200. [DOI] [PubMed] [Google Scholar]
- 145.Hara-Kuge S, Seko A, Shimada O, Tosaka-Shimada H, Yamashita K. The binding of VIP36 and α-amylase in the secretory vesicles via high-mannose type glycans. Glycobiology. 2004;14(8):739–44. doi: 10.1093/glycob/cwh082. [DOI] [PubMed] [Google Scholar]
- 146.Arshad N, Ballal S, Visweswariah SS. Site-specific N-Linked Glycosylation of Receptor Guanylyl Cyclase C Regulates Ligand Binding, Ligand-mediated Activation and Interaction with Vesicular Integral Membrane Protein 36, VIP36. Journal of Biological Chemistry. 2013;288(6):3907–17. doi: 10.1074/jbc.M112.413906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nishimura N, Balch WE. A Di-Acidic Signal Required for Selective Export from the Endoplasmic Reticulum. Science. 1997;277(5325):556–8. doi: 10.1126/science.277.5325.556. [DOI] [PubMed] [Google Scholar]
- 148.Appenzeller-Herzog C, Hauri H-P. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. Journal of cell science. 2006;119(11):2173–83. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- 149.Chang J, Block TM, Guo J-T. Antiviral therapies targeting host ER alpha- glucosidases: Current status and future directions. Antiviral Research. 2013;99(3):251–60. doi: 10.1016/j.antiviral.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hebert DN, Foellmer B, Helenius A. Calnexin and calreticulin promote folding, delay oligormerization and suppress degradation of influenza hemagglutinin in microsomes. The EMBO Journal. 1996;15(12):2961–8. [PMC free article] [PubMed] [Google Scholar]
- 151.Riemer J, Hansen HG, Appenzeller-Herzog C, Johansson L, Ellgaard L. Identification of the PDI-family member ERp90 as an interaction partner of ERFAD. PLoS One. 2011;6(2):e17037. doi: 10.1371/journal.pone.0017037. [DOI] [PMC free article] [PubMed] [Google Scholar]