

Summary
Bacterial infections associated with methicillin-resistant Staphylococcus aureus (MRSA) are a major economic burden to hospitals and confer high rates of morbidity and mortality amongst those infected. Exploitation of novel therapeutic targets is thus necessary to combat this dangerous pathogen. Here we report on the identification and characterization, including crystal structures, of two nitric oxide synthase (NOS) inhibitors that function as antimicrobials against MRSA. These data provide the first evidence that bacterial NOS (bNOS) inhibitors can work synergistically with oxidative stress to enhance MRSA killing. Crystal structures show that each inhibitor contacts an active site Ile residue in bNOS that is Val in the mammalian NOS isoforms. Mutagenesis studies show that the additional nonpolar contacts provided by the Ile in bNOS contributes to tighter binding of the bacterial enzyme.
Graphical abstract

Introduction
As bacterial pathogens continually acquire resistance to commonly used antibiotics, it has become clear that novel therapeutic strategies are required to combat serious infections (Talbot et al., 2006). In particular, there is an urgent need for the development of new pharmaceuticals that target the preeminent Gram-positive human bacterial pathogen methicillin-resistant Staphylococcus aureus (MRSA). MRSA, a Gram-positive pathogen resistant to common β-lactam antibiotics (Loomba et al., 2010), was first reported in 1961(Jevons et al., 1961) and remains one of the most costly bacterial infections worldwide (Diekema et al., 2001). MRSA is a major threat to public health because of the high prevalence among nosocomial infections and the emergence of highly virulent community-associated strains and their varying epidemiology (Stefani et al., 2012). In recent years, the threat of MRSA has been heightened by reports of strains resistant to vancomycin, as this agent is often considered the drug of last resort (Gardete and Tomasz, 2014). Characterization and exploitation of alternative bacterial drug targets will be essential for future management of MRSA infections.
Recent gene deletion experiments in S. aureus, Bacillus anthracis, and B. subtilis have implicated bacterial nitric oxide synthase (bNOS) as a potential drug target, since this enzyme provides the bacterial cell a protective defense mechanism against oxidative stress and select antibiotics (Gusarov et al., 2009; Shatalin et al., 2008; van Sorge et al., 2013). In Gram-positive pathogens, it has been proposed that bacterial NO functions to remove damaging peroxide species by activating catalase and to limit damaging Fenton chemistry by nitrosylating thioredoxins involved in recycling the Fenton reaction (Gusarov and Nudler, 2005; Shatalin et al., 2008). We recently provided an initial proof of principle regarding pharmacological targeting of bNOS, as growth of the nonpathogenic model organism B. subtilis was severely perturbed in response to combination therapy with an active site NOS inhibitor and an established antimicrobial (Holden et al., 2013).
Design and development of a potent bNOS inhibitor against bone fide pathogens such as MRSA is complicated by the active site structural homology shared with the three mammalian NOS (mNOS) isoforms (Pant et al., 2002): neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS). It is especially important not to inhibit eNOS given the critical role eNOS plays in maintaining vascular tone and blood-pressure (Yamamoto et al., 2001). Selectivity over nNOS may represent less of an immediate problem, since many of the polar NOS inhibitors characterized thus far are not very effective at crossing the blood-brain barrier (Silverman, 2009). Recent structure-based studies utilizing B. subtilis NOS (bsNOS) as a model system for bNOS suggest that specificity can be achieved through targeting the pterin-binding site (Holden et al., 2013; Holden et al., 2014), as the bNOS and mNOS pterin binding sites are quite different.
To quickly identify potent bNOS inhibitors we screened a diverse set of NOS inhibitors (Figure 1) using a novel chimeric enzyme recently reported for bNOS activity analysis (Holden et al., 2014). From this high-throughput analysis we were able to identify two potent and chemically distinct bNOS inhibitors. Crystal structures and binding analyses of these inhibitors revealed both to bind a hydrophobic patch within the bNOS active site. Moreover, both compounds possess antimicrobial activity against S. aureus, suggesting that these NOS inhibitors could represent viable new drug leads against this foremost human pathogen so frequently resistant to current antimicrobials.
Figure 1.

NOS inhibitor library used in this study. The inhibitor KS values, determined from an imidazole displacement assay are reported in µM for each inhibitor of bsNOS. Isolation and characterization of NOS inhibitors marked by α were previously reported in (Delker et al., 2010), β in (Huang et al., 2013), γ in (Huang et al., 2014), δ in (Holden et al., 2013), ξ in (Jing et al., 2014), π in (Holden et al., 2013), σ in (Huang et al., 2012), φ in (Huang et al., 2014), ψ (Holden et al., 2014), ϑ in (Cinelli et al., 2014), ϕ in (Kang et al., 2015, unpublished data), and θ reported here.
Results and Discussion
Identification of Potent bNOS Inhibitors
Rapid identification of molecular fragments that function as potent bNOS inhibitors is a key initial step toward the design and characterization of future bNOS inhibitors. To carry this out, we adapted a bNOS activity assay (Holden et al., 2014) to screen through a series of NOS inhibitors using a single time point approach (Fig 2). Concurrently, we measured the KS for each inhibitor using the imidazole displacement assay. In both of these studies bsNOS was utilized as a model system, since bsNOS assays are well developed and bsNOS shares high active site sequence homology with S. aureus and B. anthracis NOS enzymes. While all inhibitors bound to bsNOS in the µM range, the most potent bsNOS inhibitors identified from the activity analysis were calculated to have KS values in the low µM to nM range. Using the single time point approach in combination with the imidazole displacement assay, we identified compounds that were both potent inhibitors and tight binders to the active site. Since L-NNA is an excellent inhibitor analog of the NOS substrate L-Arg, the potency of L-NNA at 40.9 ± 5.3% nitrite (Fig 2) was established as an arbitrary threshold for identifying designer molecules with increased potency. Using L-NNA as a benchmark led us to classify several NOS inhibitors as potent bNOS inhibitors. This group includes three aminoquinoline inhibitors, two 6-benzyl aminopyridine inhibitors, and two aminopyridine inhibitors. Of the two aminopyridine inhibitors, 7 was previously described as a NOS inhibitor with antimicrobial properties (Holden et al., 2013). Since we previously characterized the binding of aminopyridine inhibitors to bsNOS, we selected the most potent aminoquinoline and 6-benzylaminopyridine based inhibitors, 19 and 32, respectively, for further analysis. Compounds 19 and 32 were also the two most potent inhibitors of the 37 NOS inhibitors evaluated using the bsNOS single time point analysis at 6.1% nitrite and 13.2% nitrite, respectively. In addition, inhibitor potency of 19 and 32 was a direct result of competing with substrate at the active site, as neither compound influenced electron transfer rates or the Griess reaction chemistry used to measure bNOS activity (Fig S1 and Table S1).
Figure 2.
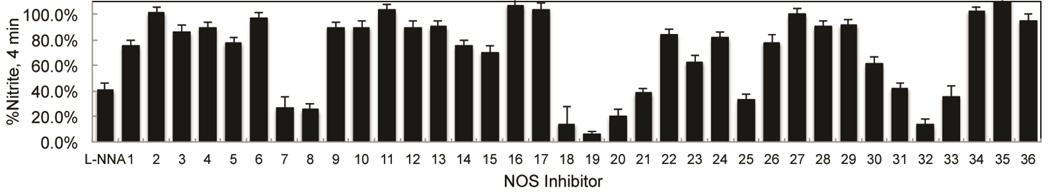
On the basis of a single time point analysis using the bBiDomain for bacterial NOS inhibition, NOS inhibitors have varying potency toward bacterial NOS. Nitrite concentrations were measured after a 4 min incubation. Error bars represent the average ± the SEM for three separate experiments.
Isoform Selectivity of NOS Inhibitors
Compounds 19 and 32 were next assayed separately against purified NOS isoforms at varying concentrations (Holden et al., 2014). Even though the IC50s for both mNOS and bsNOS were measured by complimentary methods, both methods allowed for an excellent comparison of inhibitor potency, as IC50 was used to calculate Ki using the Cheng-Prusoff equation (Cheng and Prusoff, 1973). From our Ki analysis (Table 1), it is clear that both 19 (269 nM) and 32 (1940 nM) function as potent bNOS inhibitors and demonstrate excellent selectivity over both iNOS and eNOS (Table 1). Though selectivity over nNOS remains an issue it is unclear whether cross-reactivity with nNOS expressed in neuronal tissues would represent an important limiting factor for these drugs during short course antibacterial therapy unless blood-brain penetration was high; indeed, nNOS inhibition itself has been examined as a treatment for Parkinson’s disease in a rat model (Yuste et al., 2012).
Table 1.
Inhibition of NOS isoforms by inhibitors 32 and 19. The bBiDomain construct was used to evaluate inhibitor Ki against bsNOS.
| Inhibitor | Ki bBiDomain (nM) | Ki nNOS (nM) | Ki iNOS (nM) | Ki eNOS (nM) |
|---|---|---|---|---|
| 19 | 269 | 164 | 31900 | 7250 |
| 32 | 1940 | 525 | 6440 | 2870 |
To better understand the structural basis for inhibitor potency and selectivity we solved inhibitor bound crystal structures of 19 and 32 (Fig 3, Table 2). Both 19 and 32 were co-crystallized in the presence of the pterin molecule H4B. However, the physiological pterin group for bNOS remains unclear as many bNOS containing bacteria do not contain the biosynthetic machinery required for H4B synthesis (Pant et al., 2002). Previous work showed the ubiquitous pterin, tetrahydrofolate, supports NO production by bNOS (Adak et al., 2002; Reece et al., 2009). In NOS crystal structures, H4B binding is stabilized by a H-bond to heme propionate D, a H-bond with a conserved Arg residue, and a π-π stacking interaction with a conserved Trp residue (Fig 3). The function of pterins in bNOS is unclear, although spectroscopic studies indicate pterins are not required for stability, as in mNOS; pterins are required for electron transfer in all NOS isoforms (Chartier and Couture, 2004).
Figure 3.

Inhibitor bound NOS crystal structures with select side chains colored white, heme group colored salmon, and both the active site inhibitor and H4B molecule colored yellow. For bsNOS inhibitor bound structures there is a chlorine ion bound at the carboxylate-binding site of L-Arg, which is shown as a green sphere. Both 19 and 32 bind to nNOS and bsNOS. In the nNOS structures (A and B) the fluorinated-benzyl group binds to a hydrophobic patch that is not present in bsNOS, adjacent to the heme propionate and composed of Y706, L337 and M336. At the NOS active sites both 19 and 32 bind in similar orientations to form a network of H bonds indicated by dashed lines. For the bsNOS structures, both 19 and 32 are within a hydrophobic contact of bsNOS I218. A) 19 bound to nNOS (PDB 4CAO). B) 32 bound to nNOS with the FO-FC map contoured at 4.0σ. C) Chemical representations of 19 and 32. D) 19 bound to bsNOS with the FO-FC map contoured at 3.0σ. E) 32 bound to bsNOS with the FO-FC map contoured at 3.0σ. F) 32 bound to I218V bsNOS with the FO-FC map contoured at 3.0σ.
Table 2.
Data collection, processing and refinement statistics of the NOS inhibitor bound structures
| PDB Code | bsNOS-19 4D7H |
bsNOS-32 4D7J |
bsNOS-32 4D7I |
|---|---|---|---|
| Data Collection | |||
| Wavelength (Å) | 0.976484 | 0.918370 | 0.999746 |
| Space group | P21212 | P21212 | P21212 |
| No. unique reflections | 32128 (2261) | 70341 (3408) | 48394 (2575) |
| Cell dimensions | |||
| a, b, c (Å) | 80.9 94.7 62.8 | 80.5 94.8 62.8 | 80.6 95.0 61.6 |
| α, β, γ (°) | 90 90 90 | 90 90 90 | 90 90 90 |
| Resolution (Å) | 49.62 - 2.02 (2.07 - 2.02) | 37.06 - 1.55 (1.58 - 1.55) | 48.94 - 1.96 (2.01 - 1.96) |
| Rmerge | 0.128 (0.570) | 0.052 (2.522) | 0.135 (1.518) |
| RPIM | 0.078 (0.530) | 0.033 (1.599) | 0.096 (1.074) |
| CC1/2 | 0.997 (0.834) | 1.000 (0.528) | 0.992 (0.558) |
| I / σI | 10.1 (1.6) | 18.0 (0.6) | 7.3 (1.0) |
| Completeness (%) | 99.5 (97.5) | 99.8 (99.8) | 99.6 (100.0) |
| Redundancy | 5.2 (3.0) | 6.5 (6.7) | 4.3 (4.4) |
| Refinement | |||
| Resolution (Å) | 49.62 - 2.02 (2.092 - 2.02) | 37.061 - 1.550 (1.605 - 1.55) | 48.94 - 1.96 (2.03 - 1.96) |
| No. reflections used | 31936 | 70050 | 34419 |
| Completeness (%) | 98.8 | 99.45 | 99.23 |
| Rwork | 0.1849 (0.2734) | 0.173 (0.3612) | 0.1893 (0.3501) |
| Rfree | 0.2377 (0.3350) | 0.2035 (0.3715) | 0.2352 (0.3622) |
| No. of atoms | 3257 | 3468 | 3253 |
| macromolecules | 2952 | 2950 | 2940 |
| ligands | 101 | 121 | 92 |
| solvent | 204 | 397 | 221 |
| Average B-factor | 41.4 | 28.7 | 41.1 |
| macromolecules | 41.4 | 27.7 | 41.2 |
| ligands | 42.8 | 29.5 | 34.5 |
| solvent | 41.9 | 36.4 | 42.9 |
| R.m.s deviations | |||
| Bond lengths (Å) | 0.007 | 0.007 | 0.008 |
| Bond angles (°) | 1.177 | 1.195 | 1.19 |
Values in parentheses are for highest-resolution shell.
Although 19 and 32 are chemically quite different, they both bind to the active site Glu-243 through a series of H-bonds, and do not interact with H4B. For the nNOS inhibitor-bound crystal structures, the fluorinated-benzyl group of both 19 and 32 bound to a hydrophobic pocket adjacent to the heme propionate group. This hydrophobic pocket is composed of residues Leu-337 and Met-336 from the N-terminal Zn2+ binding motif and Tyr-706 (Figure 3A and 3B). Unlike nNOS, bNOS does not contain an N-terminal Zn2+ binding motif, and therefore does not contain an analogous hydrophobic pocket adjacent to the heme propionate. Despite slight differences in binding of the fluorinated-benzyl group, in both NOS isoforms binding of 19 and 32 was further stabilized by H-bonds between the secondary amine of each inhibitor and the heme propionate groups (Figures 3D and 3E). Direct comparison of the bsNOS-19 and the previously reported nNOS-19 (Cinelli et al., 2014) structures revealed the binding mode of 19 to be unchanged between the two NOS isoforms. However, the binding mode in bsNOS was further stabilized by the hydrophobic contact between Ile-218 and the aminoquinoline group of 19. Since Ile-218 is within van der Waals contact of 19 and the analogous residue in nNOS is Val-567, our data suggest that the slight differences in hydrophobicity between Ile and Val allow for improved binding of 19 to bsNOS.
Similar to 19, crystal structure analysis of 32 demonstrates the inhibitor-binding mode to be further stabilized by the hydrophobic contact between the inhibitor and Ile-218 (Fig 3C and Fig S1). In both the nNOS-32 and I218V-bsNOS-32 crystal structures (Figs 3E and 3F, respectively), the inhibitor-binding mode of 32 is unchanged by the Ile/Val difference, as compared to WT bsNOS. To evaluate the contribution of Ile-218 to the inhibitor-binding mode we measured inhibitor binding using the imidazole displacement assay. From this analysis we found the inhibitor binding of both 19 and 32 to be ~5–6 fold tighter to Ile-218 over I218V (Table 3). Between the crystal structures and binding assay results, our data suggest that the increased hydrophobicity of Ile-218 over the analogous mNOS Val residue improves inhibitor binding to bNOS. This is partly observed in the crystal structures, as binding of 19 or 32 induces an alternative rotameric position in Ile-218 to form a hydrophobic contact with both 19 and 32 (Fig S1). Considering that Ile-218 is conserved across all bNOS enzymes (Wang et al., 2004), future inhibitors designed to target bNOS should continue to exploit Ile-218 by using the scaffolds of 19 and 32.
Table 3.
Calculated KS values by imidazole displacement for NOS ligands to bsNOS.
| Ligand | WT -KS (µM) | I218V - KS (µM) |
|---|---|---|
| L-Arg | 4.8 ± 0.1 (Wang et al., 2004) | 2.0 ± 0.2 (Wang et al., 2004) |
| 19 | 3.6 ± 0.8 | 18 ± 2 |
| 32 | 8.9 ± 2.0 | 58 ± 4 |
Anti-MRSA Activity of NOS inhibitors
To evaluate the antibacterial potential of NOS inhibitors 19 and 32 on bacterial growth, we utilized the highly virulent CA-MRSA strain UAMS118 (wt) representative of the USA300 clonal lineage and a previously engineered isogenic NOS deletion mutant (van Sorge et al., 2013). Since previous experiments have shown bacterial Δnos strains are more susceptible to H2O2-mediated killing (Holden et al., 2013; Shatalin et al., 2008; van Sorge et al., 2013), we measured the effect of NOS inhibitors and H2O2 on S. aureus (Fig 4). Our results both confirm that the Δnos strain is more susceptible to H2O2-mediated killing compared to the wt strain and further demonstrate that co-treatment of S. aureus with H2O2 and a NOS inhibitor significantly increases the H2O2-mediated killing of the bacteria. Interestingly, both 19 and 32 exhibit some direct bacteria toxicity at 200 µM as demonstrated by the modest decrease in bacterial survival for both wt and Δnos when treated with inhibitor alone (Fig 4). For example, 19 alone decreases growth by about 3-fold but with peroxide 19 decreases growth 30-fold. While this indicates a modest effect on non-NOS targets, the primary effect of 19 is to impart far greater sensitivity to oxidative stress and is consistent with 19 operating primarily by inhibiting bNOS. We also evaluated the toxicity of 19 and 32 using mouse embryonic fibroblast cells and found the IC50 values for 19 and 32 to be 5.84 µM and 11.86 µM (Table S2), respectively. These data indicate that toxicity of NOS inhibitors towards mammalian cells needs to be lowered for further consideration as a therapeutic agent.
Figure 4.

NOS inhibitors and peroxide work synergistically to eliminate S. aureus over time. Colonies of S. aureus observed after A) 30 min and B) 60 min exposure to 200 µM 19 and/or 5 mM H2O2. Similarly, S. aureus viability was also measured at C) 30 min and D) 60 min following exposure to 200 µM 32 and/or 5 mM H2O2. Error bars represent the mean ± SD of 3 replicates. Students t test gives ***P < 0.001, **P < 0.01 and *P < 0.05.
The major effect of 19 and 32 is to work synergistically with H2O2 to significantly limit bacterial growth, most likely by limiting NO production. These results are consistent with previous results indicating that blocking of NO signaling increases bacterial susceptibility to oxidative stress(Gusarov and Nudler, 2005; Holden et al., 2013) and indicate 19 and 32 could perhaps function as antimicrobials to increase susceptibility to innate immune clearance via an oxidative burst. Furthermore, considering that many existing pharmaceutical antibiotics function through an oxidative mechanism (Kohanski et al., 2007), bNOS inhibitors like 19 and 32 could theoretically synergize to increase the killing efficiency of such agents.
Significance
Nitric oxide (NO) generated by bacterial nitric oxide synthase (bNOS) helps to protect certain gram positive bacteria from oxidative stress including antibiotic-induced oxidative stress (Gusarov and Nudler, 2005; Gusarov et al., 2009; van Sorge et al., 2013). In earlier work, we found that a small number of inhibitors developed for selective nNOS inhibition also improved the efficacy of antimicrobials, suggesting that bNOS might be a viable drug target (Holden et al., 2013). In the present study we sought to achieve two goals. The first was to identify bNOS-selective inhibitors with antimicrobial activity against the important human pathogen, MRSA. Of the many compounds screened two were found to bind well to bNOS and exhibit antimicrobial activity with selectivity over eNOS and iNOS. Selectivity over eNOS is more important since interfering with eNOS will adversely effect the critical role that eNOS derived NO plays in maintaining vascular tone and blood pressure (Yamamoto et al., 2001). The second goal was to use crystallography to identify subtle differences between the bNOS and mNOS active sites to exploit for future inhibitor design. Ile-218 (Val in mNOS) contacts the inhibitors and that the I218V mutant exhibits about a 6-fold lower affinity than wild type. Although this is a rather modest difference, we have also found that several NOS inhibitors more readily bind to the pterin site in bNOS (Holden et al., 2015). Given the lower affinity of pterins for bNOS compared to mNOS, this is another important binding site difference between bNOS and mNOS. The Ile vs. Val active site difference together with the larger structural differences in the pterin site are critical molecular features that could be exploited in future inhibitor design efforts.
Experimental Procedures
Molecular Biology
Active site mutation I218V was introduced to Bacillus subtilis NOS (bsNOS) by site directed mutagenesis using PfuTrubo (Agilent). Both WT and I218V bsNOS were expressed and purified from E. coli as previously described for bsNOS (Pant et al., 2002). YumC and bBiDomain were also purified from E. coli and used for activity analysis (Holden et al., 2014). Recombinant rat nNOS and murine iNOS were expressed in E. coli and isolated as reported(Hevel et al., 1991; Roman et al., 1995).
Bacterial NOS Activity Inhibition
Reactions containing both bBiDomain (a chimera of bsNOS and redox partner YkuN) and YumC were initiated with NADPH and run for 4 min at 35 °C as previously described(Holden et al., 2014). Substrate N-omega-hydroxy-L-arginine (NOHA) and NOS inhibitor were included in each reaction at 200 µM and 30 µM, respectively. The Griess reaction was used to measure nitrite levels as a function of NOS activity. %Nitrite was calculated for each reaction as the concentration of nitrite detected in the presence of inhibitor divided by the concentration of nitrite detected without inhibitor present. Each reaction was measured in duplicate for three separate trials.
Ki Determination
The Ki was calculated from the half maximal inhibitor concentration (IC50) and KD of L-NOHA using the Cheng-Prusoff equation (Cheng and Prusoff, 1973). For bBidomain the previously reported KD of L-NOHA at 23.5 µM (Hannibal et al., 2011) was used to calculate Ki. IC50 was measured for bsNOS using bBiDomain and YumC as previously described (Holden et al., 2014). Cross reactivity of inhibitors 19 and 32 was checked over a concentration range of 0.01 to 50 µM inhibitor with the Griess reagents, and neither compound interfered or contributed towards the Griess reaction. IC50 for mammalian NOS was determined using the oxyhemoglobin assay as previously described (Huang et al., 2014).
Cytochrome-c oxidase Activity
Horse heart cytochrome C oxidase reduction was evaluated as previously described (Holden et al., 2014) using Δε550= 21 mM−1cm−1 (Martasek et al., 1999) and NADPH at 100 nM to initiate the reaction. For individual reactions containing a NOS inhibitor, inhibitor concentrations were set at 1 µM, 10 µM, and 50 µM inhibitor. Each reaction contained bBiDomain and YumC at 100 nM and 1 µM, respectively.
Crystallization and Structure Determination
Although the target of this study is S. aureus, we utilized bsNOS owing to the better diffraction power of bsNOS crystals. In fact, bsNOS and S. aureus NOS(Bird et al., 2002) are very similar and the crystal structures superimpose with a 0.55 Å root-meansquare- deviation of alpha carbon atoms. In addition, 32 of 33 residues within 10 Å of the heme iron and 14 of 17 residues within 10 Å of the pterin cofactor are identical. As a result structural insights gained from bsNOS are directly applicable to saNOS. Crystals of bsNOS and the I218V mutant were prepared using the hanging drop method by mixing protein at 18 mg/mL and well solution in a 1:1 ratio. Prior to crystallization the protein was stored in a buffer composed of 25 mM Bis-Tris methane at pH 7.4, 75 mM NaCl, 2% (vol/vol) glycerol, 0.5% (w/vol) PEG 3350, and 1 mM DTT. The well solution used for crystallization was composed of 60 mM Bis-Tris methane, 40 mM citric acid, 15% (w/vol) PEG3350, and 1.9% (vol/vol) 1-propanol at pH 7.6. Crystals grew overnight after seeding with old crystals. Crystals were cryoprotected in the well solution supplemented with 30% (vol/vol) glycerol, 2 mM H4B, and 5–10 mM inhibitor prior to being flash frozen at 100 K. Crystals of rat nNOS oxygenase domain were prepared and flash frozen as previously described (Li et al., 2014). Data were collected under cryogenic conditions on individual crystals at both the Advanced Light Source (Berkeley, CA) and Stanford Synchrotron Radiation Lightsource (Menlo Park, CA). The raw data frames were indexed and integrated using either iMOSFLM (Battye et al., 2011) or XDS (Kabsch, 2010). The program Aimless was then used to scale the data sets (Evans, 2006). Inhibitor bound structures were refined using either PHENIX (Adams et al., 2009) and Refmac (Vagin et al., 2004) with inhibitor restraints built using PRODRG (Schuttelkopf and van Aalten, 2004).
Imidazole Displacement
Purified bsNOS was diluted to 2 µM into a buffered solution containing 50 mM Tris (pH 7.6), 10 mM NaCl, 100 µM dithiothreitol, and 1 mM imidazole to generate a low spin heme. NOS inhibitors were titrated into the bsNOS-buffered solution, and the conversion of the heme group from low spin to high spin was monitored using a Cary 3E UV-visible spectrophotometer. The KS was calculated as previously described from the KS,app (Holden et al., 2013; Roman et al., 1995) using the bsNOS KD imidazole at 384 µM and the bsNOS-I218V KD imidazole at 506 µM (Wang et al., 2004).
Effect of Antimicrobial Induced Stress and NOS Inhibitors on S. aureus
Creation of the S. aureus UAMS1182 nos isogenic knockout is described in a previous report (van Sorge et al., 2013). Parent (wild type, wt) and knockout (Δnos) were cultured in cation-adjusted Mueller Hinton broth (CAMHB). Prior to H2O2 assays, strains were cultured overnight at 37 °C then subcultured at a 1/20 dilution in fresh CAMHB. Strains were grown to mid-log phase (OD600 ~0.4), pelleted by centrifugation, washed twice in CAMHB, and diluted in CAMHB to a predetermined concentration approximating 2×107 colony forming units per mL (CFU/mL). Volumes of 25 µL (5×105 CFU) were dispensed to 96 well plates (Corning Life Sciences) in 200 µL aliquots of CAMHB and CAMHB with amendments including 5 mM H2O2 (Sigma), 200 µM 19, 200 µM 32, and equivalent control volumes of 19/32 solvent. Plates were incubated at 37 °C with shaking. Cultures were sampled at 30 min intervals by removing 25 µL for serial dilution in CAMHB and spot plating on Todd Hewitt agar (Becton Dickinson). Plates were incubated overnight and culture CFU/mL was calculated by enumerating counted colonies and multiplying back through the dilution factor. All conditions were sampled in triplicate; values presented are mean ± standard deviation. Statistical analysis was performed in Excel (Microsoft) using the Student’s t-test.
NOS inhibitor cytotoxicity in mammalian cell culture
Cell toxicity assays were performed on mouse embryonic fibroblasts (MEF), which were maintained in DMEM (Corning Cell Gro, USA) media supplemented with 10% fetal calf serum (Sigma-Aldrich, USA) and 1% penicillin-streptomycin (Mediatech, Corning, USA) at ~70% confluency. Cell Titer Glo assays (Cell Titer Glo Luminescent Cell Viability Assay kit, Promega Corporation) were performed in a clear bottom 96-well black cell culture plates (Greiner Bio- One, NC, USA). After plating at 250 cells/well in a volume of 100 µl, cells were left undisturbed for at least 24 h before NOS inhibitor addition. NOS inhibitors 19, 32, and L-NAME (Enzo Life Sciences) were added to the MEF’s at 40 µM, 20 µM, 10 µM, 5 µM, 2.5 µM, 1.25 µM, 0.625 µM and 0.3125 µM. Cells were prepared for analysis 72 h after NOS inhibitors were added by addition of 10 µl of 0.1% Triton-X100 in PBS with shaking for 1 min at room temperature (RT). Cell Titer-Glo lysis reagent (20 µl) was then added followed by 1 min of shaking and a 10 min incubation in the dark at RT. Luminescence was detected using an IVIS imaging system (IVIS Lumina II, Perkin Elmer, USA). IC50 values were determined using the GraphPad Prism software (GraphPad Software, Inc, La Jolla, CA).
Chemical library preparation
Since bacterial NOS-selective inhibitors had not yet been identified, we collected a diverse set of NOS inhibitors (1–25) from our previous NOS studies (Holden et al., 2013; Huang et al., 2012; Huang et al., 2014; Jing et al., 2014; Kang et al., 2014; Kohanski et al., 2007) as well as several newly synthesized molecules (26–36). The collected small molecule library (1–36) was composed of a chemically diverse set of aminopyridine derivatives (aminopyridinyl-2-ethyl, aminopyridinyl-2-benzyl, aminopyridinyl-2-phenyl), 7-azaindoles, thiopheneamidines, and 2-aminoquinolines. In general, inhibitors 26–36 generally have arylalkyl side chains or a N1,N2-dimethylethane-1,2-diamine tail. Chemical syntheses and spectral validation of the NOS inhibitors are included in the Supplemental Information.
Chemical Synthesis
Details of the synthesis are provided in the Supplemental Material.
Supplementary Material
Highlights.
Inhibitors selective toward bacterial nitric oxide synthase have been identified.
These inhibitors are antimicrobial against MRSA.
Crystallography reveals the structural basis for selectivity.
NOS inhibitor library to identify molecules with antimicrobial function.
Acknowledgment
This work was supported by National Institutes of Health grants GM57353 (TLP), GM49725 (RBS), HD071600 (VN), and AI057153 (VN). We also thank the beamline staff at SSRL and ALS for their assistance during the remote X-ray diffraction data collections.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession codes
Coordinate and structure factor files were deposited in the Protein Data Bank with the following accession codes: 4D7H, 4D7I, 4D7J, and 4D7O
Author contributions JKH designed and carried out the crystallographic and enzyme assay experiments; SK and MAC did the chemical synthesis in the lab of RBS; HL assisted with X-ray data collection; DD assisted JKH with protein preparation; FCB carried out the MRSA experiments in the lab of VN; SGR evaluated NOS inhibitor cytotoxicity in the lab of ALE; JKH and TLP wrote the paper and VN and RBS edited the paper.
References
- Adak S, Aulak KS, Stuehr DJ. Direct evidence for nitric oxide production by a nitric-oxide synthase-like protein from Bacillus subtilis. J Biol Chem. 2002;277:16167–16171. doi: 10.1074/jbc.M201136200. [DOI] [PubMed] [Google Scholar]
- Adams PD, Mustyakimov M, Afonine PV, Langan P. Generalized X-ray and neutron crystallographic analysis: more accurate and complete structures for biological macromolecules. Acta Crystallogr D Biol Crystallogr. 2009;65:567–573. doi: 10.1107/S0907444909011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird LE, Ren J, Zhang J, Foxwell N, Hawkins AR, Charles IG, Stammers DK. Crystal structure of SANOS, a bacterial nitric oxide synthase oxygenase protein from Staphylococcus aureus. Structure. 2002;10:1687–1696. doi: 10.1016/s0969-2126(02)00911-5. [DOI] [PubMed] [Google Scholar]
- Chartier FJ, Couture M. Stability of the heme environment of the nitric oxide synthase from Staphylococcus aureus in the absence of pterin cofactor. Biophys J. 2004;87:1939–1950. doi: 10.1529/biophysj.104.042119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cinelli MA, Li H, Chreifi G, Martasek P, Roman LJ, Poulos TL, Silverman RB. Simplified 2-aminoquinoline-based scaffold for potent and selective neuronal nitric oxide synthase inhibition. J Med Chem. 2014;57:1513–1530. doi: 10.1021/jm401838x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delker SL, Xue F, Li H, Jamal J, Silverman RB, Poulos TL. Role of zinc in isoformselective inhibitor binding to neuronal nitric oxide synthase. Biochemistry. 2010;49:10803–10810. doi: 10.1021/bi1013479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekema DJ, Pfaller MA, Schmitz FJ, Smayevsky J, Bell J, Jones RN, Beach M, Group SP. Survey of infections due to Staphylococcus species: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997–1999. Clin Infect Dis. 2001;32(Suppl 2):S114–S132. doi: 10.1086/320184. [DOI] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Gardete S, Tomasz A. Mechanisms of vancomycin resistance in Staphylococcus aureus. J Clin Invest. 2014;124:2836–2840. doi: 10.1172/JCI68834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarov I, Nudler E. NO-mediated cytoprotection: Instant adaptation to oxidative stress in bacteria. Proc Natl Acad Sci USA. 2005;102:13855–13860. doi: 10.1073/pnas.0504307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarov I, Shatalin K, Starodubtseva M, Nudler E. Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science. 2009;325:1380–1384. doi: 10.1126/science.1175439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannibal L, Somasundaram R, Tejero J, Wilson A, Stuehr DJ. Influence of hemethiolate in shaping the catalytic properties of a bacterial nitric-oxide synthase. J Biol Chem. 2011;286:39224–39235. doi: 10.1074/jbc.M111.286351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevel JM, White KA, Marletta MA. Purification of the inducible murine macrophage nitric oxide synthase. Identification as a flavoprotein. J Biol Chem. 1991;266:22789–22791. [PubMed] [Google Scholar]
- Holden JK, Kang S, Hollingsworth SA, Li H, Lim N, Chen S, Huang H, Xue F, Tang W, Silverman RB, et al. Structure-based design of bacterial nitric oxide synthase inhibitors. J Med Chem. 2015;58:994–1004. doi: 10.1021/jm501723p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden JK, Li H, Jing Q, Kang S, Richo J, Silverman RB, Poulos TL. Structural and biological studies on bacterial nitric oxide synthase inhibitors. Proc Natl Acad Sci USA. 2013;110:18127–18131. doi: 10.1073/pnas.1314080110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden JK, Lim N, Poulos TL. Identification of Redox Partners and Development of a Novel Chimeric Bacterial Nitric Oxide Synthase for Structure Activity Analyses. J Biol Chem. 2014;289:29437–29445. doi: 10.1074/jbc.M114.595165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Ji H, Li H, Jing Q, Labby KJ, Martasek P, Roman LJ, Poulos TL, Silverman RB. Selective monocationic inhibitors of neuronal nitric oxide synthase. Binding mode insights from molecular dynamics simulations. J Am Chem Soc. 2012;134:11559–11572. doi: 10.1021/ja302269r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Li H, Martasek P, Roman LJ, Poulos TL, Silverman RB. Structure-guided design of selective inhibitors of neuronal nitric oxide synthase. J Med Chem. 2013;56:3024–3032. doi: 10.1021/jm4000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Li H, Yang S, Chreifi G, Martasek P, Roman LJ, Meyskens FL, Poulos TL, Silverman RB. Potent and selective double-headed thiophene-2-carboximidamide inhibitors of neuronal nitric oxide synthase for the treatment of melanoma. J Med Chem. 2014;57:686–700. doi: 10.1021/jm401252e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevons MP, Rolinson GN, Knox R. Celbenin-Resistant Staphylococci. Brit Med J. 1961;1:124-&. [Google Scholar]
- Jing Q, Li H, Roman LJ, Martasek P, Poulos TL, Silverman RB. Combination of chiral linkers with thiophenecarboximidamide heads to improve the selectivity of inhibitors of neuronal nitric oxide synthase. Bioorg Med Chem Lett. 2014;24:4504–4510. doi: 10.1016/j.bmcl.2014.07.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Tang W, Li H, Chreifi G, Martasek P, Roman LJ, Poulos TL, Silverman RB. Nitric oxide synthase inhibitors that interact with both heme propionate and tetrahydrobiopterin show high isoform selectivity. J Med Chem. 2014;57:4382–4396. doi: 10.1021/jm5004182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- Li H, Jamal J, Delker S, Plaza C, Ji H, Jing Q, Huang H, Kang S, Silverman RB, Poulos TL. The mobility of a conserved tyrosine residue controls isoform-dependent enzyme-inhibitor interactions in nitric oxide synthases. Biochemistry. 2014;53:5272–5279. doi: 10.1021/bi500561h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba PS, Taneja J, Mishra B. Methicillin and Vancomycin Resistant S. aureus in Hospitalized Patients. J Glob Infect Dis. 2010;2:275–283. doi: 10.4103/0974-777X.68535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martasek P, Miller RT, Roman LJ, Shea T, Masters BS. Assay of isoforms of Escherichia coli-expressed nitric oxide synthase. Methods Enzymol. 1999;301:70–78. doi: 10.1016/s0076-6879(99)01070-8. [DOI] [PubMed] [Google Scholar]
- Pant K, Bilwes AM, Adak S, Stuehr DJ, Crane BR. Structure of a nitric oxide synthase heme protein from Bacillus subtilis . Biochemistry. 2002;41:11071–11079. doi: 10.1021/bi0263715. [DOI] [PubMed] [Google Scholar]
- Reece SY, Woodward JJ, Marletta MA. Synthesis of nitric oxide by the NOS-like protein from deinococcus radiodurans: a direct role for tetrahydrofolate. Biochemistry. 2009;48:5483–5491. doi: 10.1021/bi900385g. [DOI] [PubMed] [Google Scholar]
- Roman LJ, Sheta EA, Martasek P, Gross SS, Liu Q, Masters BS. High-level expression of functional rat neuronal nitric oxide synthase in Escherichia coli. Proc Natl Acad Sci USA. 1995;92:8428–8432. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Shatalin K, Gusarov I, Avetissova E, Shatalina Y, McQuade LE, Lippard SJ, Nudler E. Bacillus anthracis-derived nitric oxide is essential for pathogen virulence and survival in macrophages. Proc Natl Acad Sci USA. 2008;105:1009–1013. doi: 10.1073/pnas.0710950105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman RB. Design of selective neuronal nitric oxide synthase inhibitors for the prevention and treatment of neurodegenerative diseases. Acc Chem Res. 2009;42:439–451. doi: 10.1021/ar800201v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani S, Chung DR, Lindsay JA, Friedrich AW, Kearns AM, Westh H, Mackenzie FM. Meticillin-resistant Staphylococcus aureus (MRSA): global epidemiology and harmonisation of typing methods. Int J Antimicrob Agents. 2012;39:273–282. doi: 10.1016/j.ijantimicag.2011.09.030. [DOI] [PubMed] [Google Scholar]
- Talbot GH, Bradley J, Edwards JE, Jr, Gilbert D, Scheld M, Bartlett JG Antimicrobial Availability Task Force of the Infectious Diseases Society of, A. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin Infect Dis. 2006;42:657–668. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- van Sorge NM, Beasley FC, Gusarov I, Gonzalez DJ, von Kockritz-Blickwede M, Anik S, Borkowski AW, Dorrestein PC, Nudler E, Nizet V. Methicillin-resistant Staphylococcus aureus bacterial nitric-oxide synthase affects antibiotic sensitivity and skin abscess development. J Biol Chem. 2013;288:6417–6426. doi: 10.1074/jbc.M112.448738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Wei CC, Sharma M, Pant K, Crane BR, Stuehr DJ. A conserved Val to Ile switch near the heme pocket of animal and bacterial nitric-oxide synthases helps determine their distinct catalytic profiles. J Biol Chem. 2004;279:19018–19025. doi: 10.1074/jbc.M311663200. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Shimamura K, Sekiguchi F, Sunano S. Effects of NG-nitro-L-arginine on the blood pressure of spontaneously hypertensive rats with different degrees of hypertension. Clin Exp Hypertens. 2001;23:533–544. doi: 10.1081/ceh-100106824. [DOI] [PubMed] [Google Scholar]
- Yuste JE, Echeverry MB, Ros-Bernal F, Gomez A, Ros CM, Campuzano CM, Fernandez- Villalba E, Herrero MT. 7-Nitroindazole down-regulates dopamine/DARPP-32 signaling in neostriatal neurons in a rat model of Parkinson's disease. Neuropharmacology. 2012;63:1258–1267. doi: 10.1016/j.neuropharm.2012.07.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.