

Abstract
Craniofacial malformations are common congenital defects caused by failed midline inductive signals. These midline defects are associated with exposure of the fetus to exogenous teratogens and with inborn genetic errors such as those found in Down, Patau, Edwards' and Smith–Lemli–Opitz syndromes. Yet, there are no studies that analyze contributions of synchronous neurocranial and neural development in these disorders. Here we present the first in-depth analysis of malformations of the basicranium of a holoprosencephalic (HPE) trisomy 18 (T18; Edwards' syndrome) fetus with synophthalmic cyclopia and alobar HPE. With a combination of traditional gross dissection and state-of-the-art computed tomography, we demonstrate the deleterious effects of T18 caused by a translocation at 18p11.31. Bony features included a single developmentally unseparated frontal bone, and complete dual absence of the anterior cranial fossa and ethmoid bone. From a superior view with the calvarium plates removed, there was direct visual access to the orbital foramen and hard palate. Both the eyes and the pituitary gland, normally protected by bony structures, were exposed in the cranial cavity and in direct contact with the brain. The middle cranial fossa was shifted anteriorly, and foramina were either missing or displaced to an abnormal location due to the absence or misplacement of its respective cranial nerve (CN). When CN development was conserved in its induction and placement, the respective foramen developed in its normal location albeit with abnormal gross anatomical features, as seen in the facial nerve (CNVII) and the internal acoustic meatus. More anteriorly localized CNs and their foramina were absent or heavily disrupted compared with posterior ones. The severe malformations exhibited in the cranial fossae, orbital region, pituitary gland and sella turcica highlight the crucial involvement of transcription factors such as TGIF, which is located on chromosome 18 and contributes to neural patterning, in the proper development of neural and cranial structures. Our study of a T18 specimen emphasizes the intricate interplay between bone and brain development in midline craniofacial abnormalities in general.
Keywords: basicranium, cranial fossae development, HPE, mesoderm derivatives, neural crest derivatives, neurocranium, osteogenic–neural development, trisomy 18
Introduction
Trisomy 18 (T18), Edwards' syndrome, is one of the two most common autosomal aneuploidies in humans, second to Down syndrome (trisomy 21; Hui et al. 2012), and is often associated with malformations of midline structures. In 1960, Edwards et al. first described a newborn with multiple malformations and cognitive impairments. However, due to difficulties in differentiating the pair of autosomal chromosomes, the syndrome was given the ambiguous name of ‘17–18 trisomy’ (Edwards et al. 1960; Hui et al. 2012). Shortly thereafter, it was shown that an additional copy of chromosome 18 would later form several other chromosomal constitutions associated with Edwards’ syndrome, such as T18 mosaicism, double aneuploidy (i.e. T18 associated with other numerical changes of autosomal and sexual chromosomes), as well as translocations (Smith et al. 1960; Rosa et al. 2013).
The prevalence of T18 is relatively high: studies from populations in North America, Europe and Australia estimate its incidence to be between 1 : 3600 and 1 : 8500 of live births (Weber et al. 1967; Young et al. 1986; Embleton et al. 1996; Root & Carey, 1994; Rasmussen et al. 2003). With the increased use of prenatal diagnosis, more recent accounting for non-viable fetus and voluntary pregnancy termination cases place this overall incidence closer to 1 : 2500–1 : 2600 (Crider et al. 2008; Irving et al. 2011; Cereda & Carey, 2012). Patients born with T18 have a considerably diminished survival rate that averages approximately eight days (Rosa et al. 2013). Although three copies of chromosome 18 is the classic definition of T18, a free trisomy of chromosome 18 associated with nondisjunction during maternal gametogenesis is often presented (Rosa et al. 2013). Although most cases of T18 occur as a result of de novo meiotic nondisjunction of the maternal meiosis phase II (Rosa et al. 2013), it is important to note that T18 can also occur as a post-zygotic event caused mainly by mitotic nondisjunction, possible at any phase of embryogenesis or development (Paskulin et al. 2011). Furthermore, T18 shows a female bias, with an incidence ratio of roughly one male to two females (Weber et al. 1967; David & Glew, 1980; Lin et al. 2006).
Cyclopia and holoprosencephaly (HPE) are intriguing phenomena caused by severe disruption of genes necessary for the specification of midline and anterior-most cephalic structures. Because the brain develops in close apposition to the skull, we posit that in T18, failed interactions between bone development of skull and neural development may elicit severe cranial malformations. To evaluate this hypothesis, we investigated the interior of the cranium of a T18 human fetus at 28 weeks compared with a non-trisomy 29 week old, with particular focus on the skeletal structure of the cranial fossae, the presence of cranial nerves (CNs), as well as their respective cranial foramina. With fine gross dissections of the cranium and facial skeleton combined with three-dimensional (3D) reconstructions of computed tomography (CT) scans, we demonstrate severe primary malformations in bone development. This study is the first of its kind to analyze in detail the features of the interior of the skull in a case of HPE and synophthalmia, a form of cyclopia. Additionally, we provide comments on osteogenic–neural relationships in normal development.
Materials and methods
Specimen
Only cadaveric specimens were used in the current study and, as such, no human subject approvals from the Institutional Review Board are required. Nonetheless, these specimens were handled with the utmost care and attention to the rules governing scientific research. Although the fetus showed external characteristics consistent with a 25-week-old fetus, the gestational age was determined based on the last menstrual period of the mother, which was 28 weeks. The age-matched 29-week-old control fetus was graciously provided by Dr Rui Diogo's laboratory in the Department of Anatomy, Howard University College of Medicine.
Genotype
Fresh tissue was referred to Quest Diagnostics for analysis by standard cytogenetic analysis and by fluorescence in situ hybridization. The fetus was diagnosed as having a pseudoisodicentric chromosome 18 around band 18p11.31 with a loss of short arm material from 18p11.31 to the p terminus.
CT scans and 3D reconstructions
Computed tomography images were acquired using a GE LightSpeed VCT system (General Electric Company, CT, USA). One scan was acquired consisting of 80 contiguous 3.75 mm slices, 15 × 15 cm field of view, 512 × 512 matrix, 120 kVp, 100 mA. Images were reconstructed for 3D analysis with the NIH ImageJ software and with the VCT software. Image intensity thresholds were adjusted to allow for bone-only reconstruction.
Dissection
Dissections were carried out using gross dissection and micro-dissection tools, a three-power-lighted lens and, as needed, a binocular dissecting microscope (Nikon SMZ1500). The calvarium was carefully opened using scissors, and the brain and brainstem were removed according to the dissection instructions in the Grant's Dissector (Tank, 2013). Meninges were removed using Watchmaker forceps, and CNs were observed leaving the cranial fossae through foramina/fissures. Terminology follows Grant's Atlas of Anatomy, (Agur and Dalley, 2008). Dissections were recorded by photographs (Nikon digital camera D90). A standard centimeter was included in each photograph for comparative measurement of dimensions.
Results
In the normal fetus, the cranial fossa can be divided into anterior, middle and posterior cranial fossae (Fig.1A). A border defines each cranial fossa anteriorly and posteriorly. For the anterior cranial fossa, the frontal bone defines the anterior border and the lesser wing of the sphenoid bone defines the posterior. The middle cranial fossa is bounded by the posterior margins of the lesser wings of the sphenoid bone and the petrous portion of the temporal bone posteriorly. The posterior cranial fossa is defined anteriorly by the clivus and posteriorly by the occipital bone (Fig.1A).
Figure 1.
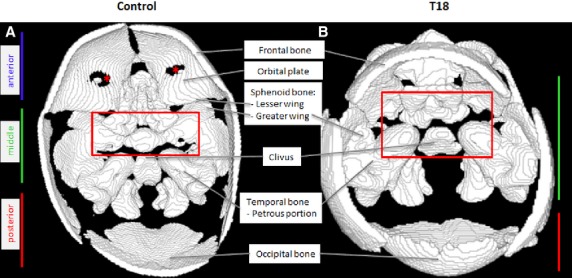
Reconstructed CT scans showing a superior view of the cranial floor in a control and a T18 fetus. The heterotopic shift of the temporal bone anteriorly is due to a shift in the greater wing of the sphenoid bone. *Holes appear due to thinness of the bone, this is an artifact of the 3D reconstruction.
Remarkably, several hallmark features of the anterior cranial fossa were absent in T18 (Fig.1B). The significant lack of features in the anterior portion of the cranium, in T18, results in the complete elimination of an anterior cranial fossa (Table1). The cribriform plate of the ethmoid bone and orbital part of the frontal bone are absent in T18 (Fig.2A). Normally, these two bony structures function as the support for the overlying prefrontal cortex. Although some middle cranial fossa features were observed, they showed a unique unilateral presentation, as evidenced in the size and shape of the superior orbital fissure. Abnormal bony features are summarized in Table1.
Table 1.
Summary of cranial fossae features and findings in the T18 fetus at 28 weeks
| Cranial fossa | T18 Cranial fossae shifted anteriorly |
|---|---|
| Anterior cranial fossa | |
| Sphenoid bone | |
| Lesser wing | Absent on right side, reduced on left side |
| Ethmoid bone | |
| Crista galli | Absent |
| Cribriform plate | Absent |
| Orbital part of the frontal bone (forms roof of orbit) | Absent |
| Middle cranial fossa | |
| Sphenoid bone | |
| Superior orbital fissure | Present on left side, absent on right |
| Hypophyseal fossa | Cartilaginous, incomplete |
| Anterior clinoid process | Highly reduced |
| Optic canal | Singular, centrally located |
| Posterior clinoid process | Highly reduced |
| Dorsum sellae | Diminutive |
| Greater wing of sphenoid bone | |
| Foramen rotundum | Size reduced |
| Foramen ovale | Size reduced |
| Temporal bone | |
| Squamous part | Underdeveloped, anteriorly located |
| Superior border of petrous part | Malformed, highly enlarged |
| Trigeminal ganglion | Highly reduced |
| Internal carotid artery | Not observed in gross view |
| Internal acoustic meatus | Enlarged, bony protuberance |
All structures were present in the control fetus at 29 weeks.
Figure 2.

Superior view of gross dissected interior of the cranium. Malformations in the cranial floor of T18 with orbital foramen exposing the hard palate (B) compared with control (A). (A and C) vs. (B and D). (C) and (D) represent regions depicted in figures. Scale bar: 1 cm.
The majority of the cranial base in T18 appeared soft and translucent, indicative of cartilage. The T18 cranial base also lacks well-defined anterior and middle cranial fossae. By contrast, the cranial base in the control is bone/partially bony and has clearly completed the endochondral ossification process. As a component of the posterior cranial fossa, the temporal bone normally exhibits a smooth appearance and defines the lateral aspects of the fossa. However, in T18, there is a heterotopic displacement of the temporal bone anteriorly (Fig.1B, red box) compared with the control (Fig.1A, below the red box).
Prominent anterior cranial fossa structures such as the orbital plates of the frontal bone, crista galli and cribriform plate of the ethmoid bone are absent in T18 (Fig.2B). Thus, a defined anterior cranial fossa fails to form in T18. Instead, a single orbital foramen is present in the interior of the cranium (Fig.2B). The right lesser wing of the sphenoid bone (Fig.2B), which normally forms the boundary of the anterior cranial fossa, is absent. The absence of the lesser wing on the right side and a malformed lesser wing on the left side creates a prominent open space in the region of the orbital fossa (Fig.2B). The middle cranial fossa is shifted anteriorly in T18 where the anterior cranial fossa should have been. This shift contributes to the shortening of the T18 cranial base along the antero-posterior axis; as evidenced by a T18 middle cranial fossa that occupies 75% of a 4 cm cranium compared with a control middle cranial fossa that takes up 46% of a 6.5 cm cranium from anterior to posterior (Fig.2B). Without the orbital plates of the frontal bone, which normally form the roof of the orbit (Fig.3A,C), the hard palate is exposed directly under the orbital foramen (Figs2B and 3D). Figure2C and D illustrates the regions analyzed in Figs7.
Figure 3.
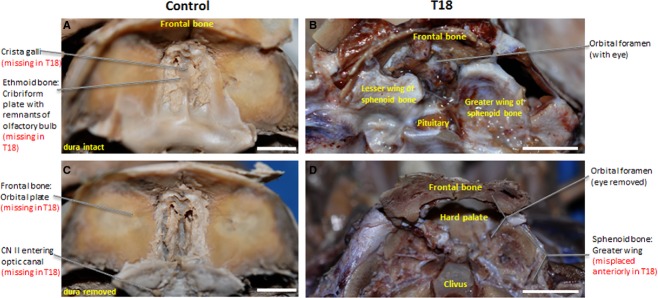
Superior view of anterior cranial fossa. The cribriform plate of the ethmoid bone (A) and the orbital plate of the frontal bone (C) are fully formed in the anterior cranial fossa, whereas a single orbital foramen (B) and clivus (D) display an anterior displacement in T18. Scale bar: 1 cm.
Figure 7.
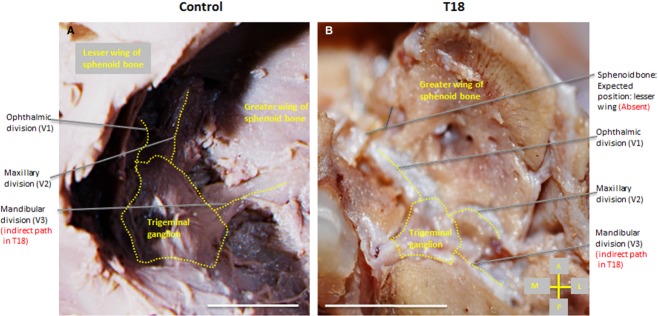
Superior view of the trigeminal ganglion (surrounded by dotted line), right side of the cranium. Branches (dotted) of the trigeminal ganglion exhibit a normal branching pattern (A), whereas the maxillary (CNV2) and mandibular division (CNV3) follow an indirect path (B) to the foramen rotundum and ovale, respectively, indicated by dashed lines in T18. Scale bar: 1 cm.
Malformations are also present in the sella turcica, located in the sphenoid bone, the cornerstone of the middle cranial fossa. Usually, the infundibulum of the pituitary gland pierces the well-formed sellar diaphragm (Fig.4A), but the sellar diaphragm is absent in T18, allowing for the pituitary gland to directly contact neural tissue (Fig.4A vs. B). Compared with the sharp angle characteristic of the greater wing of the sphenoid bone, the curvature is more shallow in T18, indicated by the dashed line in Fig.4A, B. Posteriorly, aberrations in the cranial fossa of T18 also exhibited a sharp angle in the clivus compared with the typical shallow depression in the control (Fig.4C, D). Due to asymmetrical lesser wings of the sphenoid bone, the sella turcica fails to form anteriorly, while posteriorly the dorsum sellae remains (not shown).
Figure 4.
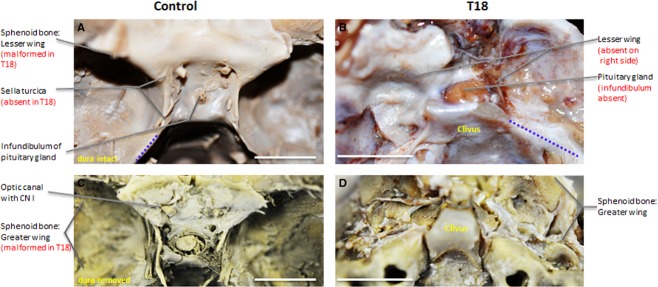
Superior view of middle cranial fossa. Infundibulum of the pituitary gland piercing sellar diaphragm (A) of well-formed sella turcica (C), while sellar diaphragm and sella turcica are absent in T18 (B) and (D), respectively. Scale bar: 1 cm.
The T18 viscerocranium lacks a glabella, a hallmark feature of the underlying frontal bone normally found above the nasal bone, which is absent in T18 (Fig.5A). Moreover, the palatine process of the maxilla, which contributes to the hard palate, sits immediately inferior to the orbit (Fig.5B), rather than inferior to the nasal cavity as would be expected in normal fetuses. Remarkably, there are no anterior nasal apertures in T18, and the anterior nasal spine shows a minimal distance to the alveolar process of the maxilla (Fig.6B).
Figure 5.
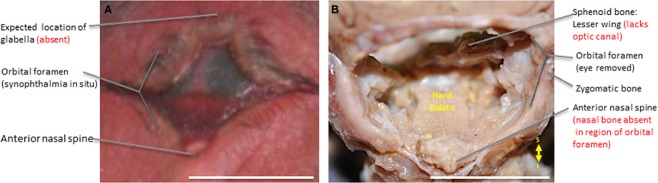
Frontal view of synophthalmia in situ (A) and hard palate revealed with prominent anterior nasal spine in dissected T18 (B). Scale bar: 1 cm.
Figure 6.
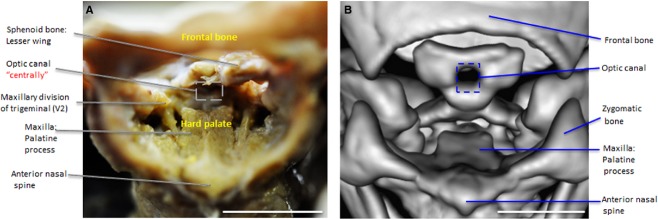
Frontal view of orbital foramen. Maxillary division of trigeminal (V2) on either side of the hard palate (A) courses inferiorly to a single, centrally located optic canal (B) in the dorsum sella of T18. Scale bar: 1 cm.
The morphological development of CNs in T18 displayed variability even as some foramina were conserved (Table2). CNs I, II, V, VI, VII and VIII were either grossly abnormal or absent. Only a single, centrally located optic canal developed (Fig.6A,B), and the optic nerve (CN II) appeared as a fused, single nerve. The maxillary division of the trigeminal nerve (CNV2) was detected coursing via a circuitous path to the foramen rotundum (Fig.7). We further observed its exit from this foramen. After its exit, CNV2 does not course through the pterygopalatine fossa as expected nor does it branch. Rather, CNV2 descends directly on either side of the hard palate (Fig.6A). The mandibular division of the trigeminal nerve (CNV3) in T18 follows an indirect path to foramen ovale (indicated by dashed lines, Fig.7B) as it branches from the trigeminal ganglion (Fig.7B). Yet, the inferior alveolar nerve was noted as retaining a normal course inferiorly toward the mandible during dissection (not shown).
Table 2.
Presence or absence of CNs and their respective foramina in T18 at 28 weeks
| CN* | Presence | Foramen | Comments |
|---|---|---|---|
| Olfactory nerve (I) | N | N | CN I not observed due to the absent cribriform plate of the ethmoid bone |
| Optic nerve (II) | X | X | CN II fused, forming a single nerve and is in direct contact with neural tissue due to absent orbital part of the frontal bone Optic canal is a single foramen |
| Oculomotor nerve (III) | X | X | Superior orbital fissure on the right side not bordered superiorly by lesser wing of sphenoid bone |
| Trochlear nerve (IV) | N/O | X | N/O |
| Trigeminal nerve (V) | X | X | Branches are thin compared with the control |
| Ophthalmic division (V1) | |||
| Maxillary division (V2) | V3 has indirect course to foramen ovale | ||
| Mandibular division (V3) | |||
| Abducent nerve (VI) | N | X | N/O |
| Facial nerve (VII) | X | X | Internal acoustic meatus – large and pronounced within the temporal bone presenting bilaterally |
| Vestibulocochlear nerve (VIII) | X | X | CN VIII in close association with CN VIII upon entry into the foramen |
| Glossopharyngeal nerve (IX) | X | X | Jugular foramen size is significantly reduced, partially occluded by dense petrous bone |
| Vagus nerve (X) | X | X | CN X rootlets are in close association with CN IX |
| Accessory nerve (XI) | N | X | CN XI, specifically the cervical root, obstructed due to the poorly defined foramen magnum in the posterior cranial fossa Detection of cervical root in jugular foramen obstructed, tightly packed |
| Hypoglossal nerve (XII) | N/O | X | N/O |
N = structure absent.
X = structure present. CN, cranial nerve.
All CNs and foramina are as expected in control. Abnormalities of T18 are only shown above.
In the temporal bone, T18 displays an elevated and grossly enlarged internal acoustic meatus that measures four times larger at 118 mm compared with 28 mm in the control (Fig.2A, B). The petrous part of the temporal bone appears as a protuberance and lacks smooth, rounded margins like the control (Fig.8A,B). Although the internal acoustic meatus does display a nerve entering the foramen, the facial nerve (CNVII) and vestibulocochelar nerve (CNVIII) are indistinguishable and underdeveloped compared with the clear division of the CNs in the control (Fig.8A,B).
Figure 8.
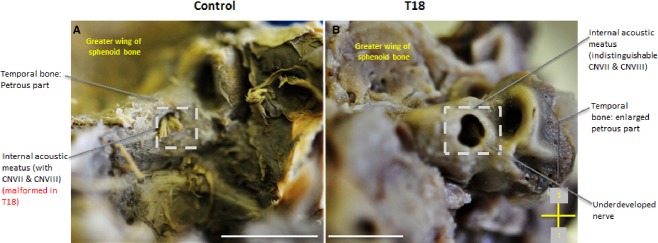
Posterior view of the internal acoustic meatus. T18 displays an abnormally pronounced internal acoustic meatus (B) within the petrous part of the temporal bone. Scale bar: 1 cm.
Discussion
This study provides a novel assessment of the anatomical findings of the cranial floor of a T18 fetus with synophthalmic cyclopia and alobar HPE. With CTs and gross dissections, we demonstrated adverse effects in the cranial and neural patterning of the T18 genetic anomaly, including, but not limited to, the following lesions: absent anterior cranial fossa with missing orbital plates of the frontal bone; exposed underlying hard palate; anteriorly shifted middle cranial fossa; fused eyes; absent sella turcica; and a pituitary gland in direct contact with overlying neural tissue. The gross dissection revealed underdeveloped CNs and uncharacteristic foramina. Abnormalities involving CNs of the anterior and posterior cranium varied in severity.
Given that midline malformations of the skull and central nervous system are among the most frequent phenotypic characteristics of the T18 syndrome in particular, and HPE in general, it was of particular interest to examine the cartilaginous portion of the neurocranium. Neural crest cells give rise to the cranium, which can be divided into neurocranium, i.e. calvaria, the case around the brain, and viscerocranium, i.e. the skeleton of the face and mandible (Santagati & Rijli, 2003). The base of the skull begins to develop as separate cartilages. As those cartilages fuse and ossify by endochondral ossification, the cranial fossae become defined. Cranial fossae receive contributions from mesodermal sclerotomes and neural crest cells, and are defined by their anterior or posterior proximity to the level of the pituitary gland in the center of the sella turcica (Sadler, 2006). The cartilage/skeletal structures of the cranial base anterior to the pituitary gland are derived from neural crest cells and form the prechordal chondrocranium, whereas those posterior to the pituitary are derived from occipital sclerotomes that arise from paraxial mesoderm and form the chordal chondrocranium (Sadler, 2006; Fig.9A,B). The components formed anterior (prechordal) and posterior (chordal) to the pituitary gland, fuse at the sella turcica around the midsphenoidal synchondroses (Lieberman et al. 2000a,b; Jeffery & Spoor, 2004). Thus, the fetal cartilaginous neurocranium, which includes the basicranium (cranial base), develops into the scaffolding for the rapidly growing brain.
Figure 9.
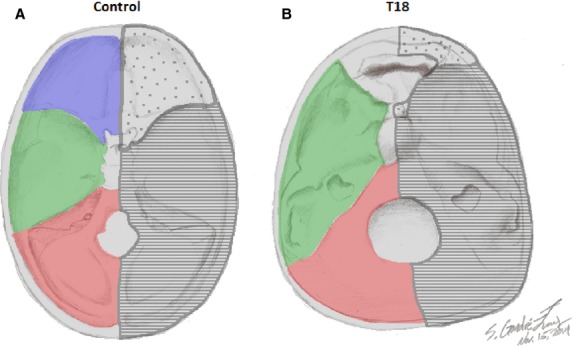
Schematic reconstruction of the boundaries of cranial fossae and developmental cellular origins. Schema of anterior (blue), middle (green) and posterior (red) cranial fossae and their cell origins (neural crest cells, dotted; occipital sclerotomes, striped) defined in proximity to the center of the sella turcica in the control (A) vs. T18 (B).
The reciprocal interplay between bone development of the skull and the inductive signaling that occurs in the development of the human brain offers some insight into the malformations we have observed in the T18 specimen. This cross-talk between skull and brain development has been noted in several other syndromes, e.g. trisomy 13 (Patau syndrome), trisomy 21 (Down syndrome), Apert syndrome, Treacher–Collins syndrome, and craniosynostosis (Cohen, 2000). This has raised a deeper question concerning the influence of skeletal structure, particularly the cranial base, in relationship with the developmental tempo of neural tissue. In general, the cranial base is influenced by complex and multifactorial processes, including brain morphogenesis, and muscle development. As a consequence, the three endocranial fossae are not strictly integrated according to specific morphological schemes, but rather are influenced by distinct and local factors (Bruner & Ripani, 2008).
It has been suggested that the brain and connective tensors of the dura layers drive neurocranial morphogenesis. They induce the neurocranial bones to undergo structural arrangements at the outer and inner surface by deposition and absorption of cartilage (Enlow, 1990), possibly via soft-tissue-mediated signal transduction at the suture boundaries (Ogle et al. 2004). Delimited by these suture boundaries, local factors and their properties independently impact the overall structure of the endocranial base by forming different functional modules (Stringer, 2002). For instance, cranial midline structures are relatively independent of the lateral structures due to these boundaries (Bastir & Rosas, 2005).
The findings presented in our work have expanded the discussion of the pathology involved in T18 to include reciprocal osteogenic–neural interplay. Importantly, the cranial base plays a major architectural role in primate ontogeny and evolution (Lieberman et al. 2000a,b), and cranial morphogenesis is particularly sensitive to bone deposition and resorption associated with growth fields of osteoblasts and osteoclasts (Martínez-Maza & Rosas, 2006). The case presented in this paper will add to our understanding of human evolutionary development.
Development of the cranium
The cranial base has a multifactorial establishment involving developmental, phylogenetic and functional interactions. The anatomy of the cranial base is a major determinant of the cranial architecture in primates, and a major constraint of the overall cranial form (Lieberman et al. 2000a,b). Variations in cranial base shape have been documented in several syndromes, including: Cri-du-chat syndrome, Apert syndrome and Pfeiffer syndrome, in which shortness of the anterior cranial fossa is observed (Cohen & Shiota, 2002). The cranial base formation results from an intricate relationship between proliferation of migrating neural crest cells and the regression of ectodermal and mesodermal derived tissue. Neural crest cells begin to migrate before neural folds fuse anteriorly to form the neural tube (Jiang et al. 2002; McBratney-Owen et al. 2008). Neural crest cells deriving from the anterior-most regions of the neural tube (anterior to the notochord) give rise to the entire viscerocranium (face) and the rostral part of the basicranium (Fig.9A) from the frontal bone to the midsphenoidal synchondroses within the sella turcica (Nie, 2005; McBratney-Owen et al. 2008). By contrast, the posterior cranial base originates from the mesoderm, and is demarcated anteriorly by the dorsum sellae of the sphenoid bone and apex of the petrous temporal bone (Couly et al. 1993; Evans & Noden, 2006; McBratney-Owen, 2008; Balczerski et al. 2012; Fig.9A,B).
It is well documented that ossification of the cranial base is initiated during week 5 of embryological development (Vermeij-Keers, 1990), forming from at least 41 ossification centers (Bruner & Ripani, 2008). Bilateral symmetry of ossification is regulated by the centrally localized chordal cartilage, which produces chordin, a regulator of bone morphogenetic protein 7 (BMP-7; Santaolalla-Montoya et al. 2012). In rare cases, fibrous dysplasia alters ossification of the skull, resulting in replacement of the bone structure with fibrous tissue (Davies & Macpherson, 1991; Bowers et al. 2014). This fibrous tissue may have been highly produced within the cranial fossae in the T18 specimen, and could account for the relative soft, non-ossified texture of the bone.
The development of the cranial base is regulated by a variety of genes, for example, matrix metallopeptidase 9 (MMP 9), Indian hedgehog (IHH) and Sonic hedgehog (SHH; Riccomagno et al. 2002; Nie et al. 2005a,b; Young et al. 2006; Balczerski et al. 2012). During normal embryonic development, it was suggested that cranial base chondrogenesis is delayed compared with that of the axial skeleton due to its unresponsiveness to SHH signaling (Balczerski et al. 2012). However, the timing of SHH signaling required in formation of the cranial base and its specific role is still unknown. Yet, it has been shown that in the absence of SHH, the cranial base does not undergo chondrogenic commitment as determined by the loss of Sox 9 expression (Balczerski et al. 2012). There is such a fine biomechanical balance associated with morphogenesis that small changes in the biochemistry or structure of connective, osseous or neural elements of the brain can exert direct influence on the spatial relationship of endocranial components (Bruner et al. 2014). However, none of the genes mentioned here is located on chromosome 18. Yet, genes on this chromosome (i.e. TGIF) are likely involved in the complex regulatory network necessary for normal cranial and neural development via dysregulation of SHH signaling.
In our specimen, there was an anterior shift of the lesser wings and highly reduced expansion of the greater wings of the sphenoid bone laterally. Indeed, this specimen has severe reduction in cortical development indicative of partial anencephaly. Nonetheless, the specimen holds an alobar HPE classification, in which midline cleavage of the embryonic forebrain (prosencephalon) fails to occur, yielding undifferentiated cerebral hemispheres, telencephalon, diencephalon, olfactory tracts and bulbs. It should be noted that HPE is causally heterogeneous and pathogenically variable (Cohen & Shiota, 2002). There is a strong consensus that mutations in several genes that control inductive signaling during development directly result in HPE. These genes include SHH (Roessler et al. 1996, 1997; Nanni et al. 1999) and its receptor, patched (PTCH; Ming et al. 1998), that is master regulators of neural, skeletal and muscle precursors at the midline. Additional transcription factors implicated in HPE are TGIF and CRIPTO, necessary for neural patterning (Gripp et al. 2000; Muenke & Beachy, 2001), ZIC2 and SIX3 for specific induction of the telencephalon (Wallis & Muenke, 1999; Brown et al. 1998; Brown et al. 2001), and DHCR7 for regulating cholesterol homeostasis (Kelley et al. 1996; Gondre-Lewis et al. 2006). Mutations in SHH account for about 17% of familial cases and about 3.7% of all cases of HPE (Cohen & Shiota, 2002). This case of T18 is due to a pseudoisodicentric chromosomal translocation at 18p11.31; the location of TGIF (Genecards, 2014). It was shown that mutations in the human TGIF1 gene were associated with HPE (Gripp et al. 2000). Thus, the HPE with cyclopia is caused by both the partial loss of 18p where the HPE4 locus resides (Nanni et al. 2000), and a triplication of 18q genes.
Cranial fossae
During human evolution, cranial base morphology is deeply influenced by both facial and brain variations (Strait, 1999; Bastir et al. 2010). It has been hypothesized that dysmorphologies and craniosynostoses are actually the result of morphogenetic imbalance between the cranial base and connective tensors of the dura layers (Moss, 1959). These connective tensors serve as a principal source of structural integration between the endocranial elements and, if redistribution of the tensile forces and growth vectors should occur, will spur osteogenic responses, leading to an altered balance in growth and development of the fossae (Bruner & Ripani, 2008).
The three endocranial fossae display weak reciprocal integration and relative independence from one another in terms of morphology (Bruner & Ripani, 2008). Hence, the morphology of a specific area of the cranial base is not necessarily informative on the possible morphology of other areas, because its morphogenesis is not channeled through global processes, but rather molded by many local influences (Di Ieva et al. 2014). For instance, the anterior cranial fossa is strongly constrained by the orbital and upper face structures (Enlow, 1990; Bruner, 2007; Masters, 2012). Proper formation of the anterior cranial fossa is achieved through the rotation of the face inferiorly during development (Lieberman et al. 2000a,b; Bruner, 2007). As the rotation of the face occurs, the frontal lobes enlarge. Abnormalities in the anterior cranial fossa will be associated with both the prefrontal cerebral cortex and with spatial interactions of the underlying facial system (Bruner & Ripani, 2008). Indeed, in the case presented here, the most severe malformations occurred anteriorly with an unseparated frontal bone and absent orbital plates of the frontal bone (Fig.2B).
By contrast, the morphology of the middle cranial fossa is associated with the anatomy and biomechanics of the underlying mandibular ramus (Bastir & Rosas, 2005). The posterior cranial fossa is sensitive to the parieto-occipital integration schemes in the upper cranial vault and cerebro-cerebellar dynamics (Jeffery, 2002; Gunz & Harvati, 2007). In normal development, the three cranial fossae and their components develop in a tight hand-and-glove configuration, i.e. the anterior cranial fossa with prefrontal cortex, the middle with the temporal lobes, the clivus with brainstem structures, and the posterior fossa with cerebellar lobes; thus these brain regions exert significant local influence on the bony features of each respective fossa of the neurocranium (Richtsmeier and Flaherty, 2013).
Much of the development of the cranial base occurs in an antero-posterior wave of maturation. This is an important consideration in malformations, because structures that develop anteriorly may constrain the development of those that develop downstream. In terms of shape, the bones at the midline in the middle cranial fossa mature first. From the midline, the progression of maturation normally occurs in a medio-lateral fashion (Bruner & Ripani, 2008). In this specimen, the lesser wings of the sphenoid bone were oriented antero-posteriorly (Fig.3D), while the greater wings of the sphenoid bone were reduced from left to right on the horizontal plane (Fig.3D). The part of the temporal bone housing the internal acoustic meatus was positioned far more anteriorly in the T18 specimen compared with control (Fig.4D). Thus, we postulate that the absence of the anterior cranial fossa, i.e. the loss of physical constraint, caused the middle and posterior fossae to develop unhindered anteriorly. The clivus was vertically oriented at a sharp angle and accompanied by a laterally widened posterior cranial fossa. (Figs1B and 2B).
Eye/orbit development
Many ocular complications have been reported in patients with T18, but major ocular defects are present in < 10% of cases (Geiser et al. 1986). The T18 fetus in the present study has synophthalmic cyclopia, where the eye fields fail to fully separate. Synophthalmia is due to a loss of midline tissue, likely initiated between days 19 and 21 of gestation when facial development begins (Sadler, 2006). Although true cyclopia is even more rare than synophthalmic cyclopia, many of the findings reported here are consistent with true cyclopic features involving symmetric deformities of the nose, skull, orbits and brain (Deftereou et al. 2013), and suppression of the organogenic development of the two separate eyes (Garzozi & Barkay, 1985).
Sonic hedgehog is expressed by the prechordal plate and notochord in the ventral midline. It has a fundamental role in the growth, differentiation and patterning of the developing central nervous system, viscera and craniofacial structures, and is crucial for the separation of the single eye field into two optic primordia (Ingham & McMahon, 2001; Chong et al. 2012). Based on the observation that separate eye fields were present but fused, we posit that SHH signaling was perturbed, resulting in synophthalmia. This may be related to TGIF, as TGIF regulates SHH and is located at 18p11.31 where the T18 has its translocation.
The failure in proper eye development likely influences the overlying anterior cranial fossa, which forms the superior border of the bony orbit. In T18, the orbit sits in the presumptive space of the anterior cranial fossa with no overlying frontal orbital plates separating the eye from the forebrain. The only contributions to the orbit in this specimen are the left lesser wing of the sphenoid, and the zygomatic and frontal bones (Fig.5B). This fetus exhibits features consistent with a failure of the face to rotate properly as the anterior cranial fossa forms.
Pituitary gland and sella turcica
We focus on this region of the middle cranial fossa because it defines the borders between the anterior and middle cranial fossae and the degree of severity of malformations of the interior of the cranium in T18. The development of the pituitary gland and sella turcica also appear to be highly influenced by an osteogenic–neural relationship, in which the morphology of the sella turcica is highly dependent upon the proper formation of the pituitary gland. Normally, the sella turcica develops from the most rostral part of the area where the notochord ends cranially (Müller & O'Rahilly, 2003). Whereas this bone's development begins at week 9 (O'Rahilly & Müller, 1999), development of the pituitary gland precedes that of the cartilaginous sella turcica and occurs at 4–5 weeks gestation (Sheng & Westphal, 1999). The pituitary gland develops from two cell populations: the more posterior neuropituitary is of neural origin; whereas the anterior adenopituitary gland is from an evagination of oral ectodermal cells, known as Rathke's pouch.
Similarly, the walls of the sella turcica are derived from two distinct developmental origins: the anterior wall from neural crest cells; and the posterior wall (dorsum sella) from para-notochordal (mesodermal) tissue (Fig.9A). The posterior wall is under direct influence of the notochord, which secretes SHH to program ventral neural and skeletal structures (Kjaer et al. 2002). SHH secreted from the rostral end of the notochord induces formation of the midaxial part of the cranium extending from the anterior wall of the sella turcica just anterior to the hypophyseal fossa (Schoenwolf et al. 2009). Because the pituitary gland must be fully formed prior to sella turcica development, deviations in the tempo of pituitary gland development, as is likely in T18 and HPEs in general, will greatly influence the morphology of the sella turcica (Kjaer I, 2012). In this case, the dorsum sella is the only structure present, and the superior, lateral and anterior boundaries of it and the cavernous sinus are absent (Fig.4B). Thus, the undifferentiated pituitary gland was exposed to the overlying brain with no apparent infundibulum, and was held on dura mater, but not in bony material.
Deviations in the morphology of the sella turcica appear to vary according to the syndrome. As opposed to the aberrations in the posterior wall of the sella turcica seen in this example of a severe case of T18, the anterior wall of sella turcica can be affected to different degrees in other conditions, especially trisomy 21, ranging from a slight depression in the lower aspect of the anterior wall to more severe cases where the anterior wall is completely separated from the posterior wall (Kjaer et al. 1998a,b). This supports a closely coordinated relationship between the pituitary gland and sella turcica where divergent morphologies of the pituitary gland influence malformations in the sella turcica.
Cranial foramina
There were obvious abnormalities in the presence, location and size of foramina through which CNs enter and exit the basicranium. Developing CNs follow paths set by neural crest cells through the dense mesenchyme (Freter et al. 2013), and cartilage development through mesenchymal condensation takes place after CNs and blood vessels have developed. This results in the demarcation of a specific location for foramina before bones are fully formed (McBratney-Owen et al. 2008). Overlying neural tissue, such as CNs, provide inductive signals to the surrounding cartilage to form osteogenic centers that will then ossify, forming a foramen. Without proper CN development, guidance in formation of the foramina is impaired. In addition, differentiating afferent and efferent neural tissue also provide guidance for bone development from cartilaginous precursors. As previously stated (see above: eye development), we found a single optic foramen, and spaces that could represent an absent superior orbital fissure on one side and an enlarged superior orbital fissure on the other, resembling an open space, i.e. not demarcated by the lesser wing of the sphenoid bone, where the optic canal is usually found. Furthermore, both foramen rotundum and foramen ovale are displaced in the greater wing of the sphenoid bone, which may be due to the absence of the constraints that would have been provided by the sphenoid bone, producing meandering paths of the maxillary (CNV2) and mandibular (CNV3) divisions of the trigeminal nerve, respectively. The petrous part of the temporal bone was abnormally large and dense in the CT scans compared with controls, with enlarged internal acoustic meatus detected in gross dissections. Compared with the controls, with a tight fit of CNVII and CNVIII in the foramenal aperture, the T18 had small, seemingly not fully differentiated nerves located in the internal acoustic meatus opening (Fig.8B). The more posterior hypoglossal canal, although medially located, seemed no different than the control (not shown), consistent with only mild defects in the posterior cranial skeleton. The position of the jugular foramen was also conserved in T18 compared with the control. Much like the pituitary–sella turcica interaction, it seems neural formation dictates osteogenic activity for formation of foramina. Notably, the posterior cranial fossa formed and was bordered by a widened foramen magnum that measured two–three times the width of the age-matched control fetus, along with sparse occipital shelves.
Conclusion
The complex osteogenic–neural dynamics in craniofacial development are integral in the understanding of the pathophysiology of genetic malformations. The cranial base is crucial for craniofacial patterning due to its remarkable interactions with the bordering and resident brain, pituitary gland, and eye. Its intricacy may be attributed to the confluence of signaling pathways and the vigorous interactions that drive their functional and morphological properties. In our presentation of a specimen with synophthalmia and alobar HPE in a T18 background, we have given further insight into the involvement of these multifaceted interactions on the interplay between bone and brain. This specimen with a translocation at 18p11.31 provides further evidence that transcription factors involved in neural patterning, such as TGIF, are crucial for proper neural and cranial development, and for the complexities of HPE.
Acknowledgments
This research was supported in part by NIH/NIMHD grant #G12 MD007597. The authors thank Drs Greene and Fidelia-Lambert and the Department of Pathology for providing the opportunity to analyze the T18 specimen, Dr Rui Diogo for use of the control fetus, the Radiology department for the CT scans, and Mr Temitayo Gboluage for assistance with CT scan reconstructions. The authors also thank Mr Steven Gondré-Lewis for his artwork in the Fig.9 schematic.
Author contributions
SNR performed the dissections, photographed the dissections and wrote the manuscript. JMZ helped guide the dissections, contributed to data interpretation and writing of the manuscript. MGL designed the study, contributed to data analysis and interpretation, and wrote the manuscript.
References
- Agur AMR, Dalley AF. Grant's Atlas of Anatomy. 12th edn. Baltimore: Lippincott Williams & Wilkins; 2008. [Google Scholar]
- Balczerski B, Zakaria S, Tucker AS. Distinct spatiotemporal roles of hedgehog signaling during chick and mouse cranial base and axial skeleton development. Dev Biol. 2012;371:203–214. doi: 10.1016/j.ydbio.2012.08.011. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastir M, Rosas A. Hierarchical nature of morphological integration and modularity in the human posterior face. Am J Phys Anthropol. 2005;128:26–34. doi: 10.1002/ajpa.20191. [DOI] [PubMed] [Google Scholar]
- Bastir M, Rosas A, Stringer C. Effects of brain and facial size on basicranial form in human and primate evolution. J Hum Evol. 2010;58:424–431. doi: 10.1016/j.jhevol.2010.03.001. , et al. ( [DOI] [PubMed] [Google Scholar]
- Bowers CA, Taussky P, Couldwell WT. Surgical treatment of craniofacial fibrous dysplasia in adults. Neurosurg Rev. 2014;37:47–53. doi: 10.1007/s10143-013-0500-z. [DOI] [PubMed] [Google Scholar]
- Brown SA, Warburton D, Brown LY. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–183. doi: 10.1038/2484. , et al. ( [DOI] [PubMed] [Google Scholar]
- Brown LY, Odent S, David V. Holoprosencephaly due to mutations in ZIC2: alanine tract expansion mutations may be caused by parental somatic recombination. Hum Mol Genet. 2001;10:791–796. doi: 10.1093/hmg/10.8.791. , et al. ( [DOI] [PubMed] [Google Scholar]
- Bruner E. Cranial shape and size variation in human evolution: structural and functional perspectives. Childs Nerv Syst. 2007;23:1357–1365. doi: 10.1007/s00381-007-0434-2. [DOI] [PubMed] [Google Scholar]
- Bruner E, de la Cuétara JM, Masters M. Functional craniology and brain evolution: from paleontology to biomedicine. Front Neuroanat. 2014;8:19. doi: 10.3389/fnana.2014.00019. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner E, Ripani M. A quantitative and descriptive approach to morphological variation of the endocranial base in modern humans. Am J Phys Anthropol. 2008;137:30–40. doi: 10.1002/ajpa.20837. [DOI] [PubMed] [Google Scholar]
- Cereda A, Carey JC. The trisomy 18 syndrome. Orphanet J Rare Dis. 2012;7:81. doi: 10.1186/1750-1172-7-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong HJ, Young NM, Hu D. Signaling by SHH rescues facial defects following blockade in the brain. Dev Dyn. 2012;241:247–256. doi: 10.1002/dvdy.23726. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MM., Jr . Epidemiology of craniosynostosis. In: Cohen MM Jr, MacLean RE, editors. Craniosynostosis: Diagnosis, Evaluation, and Management. 2nd edn. New York: Oxford Press; 2000. pp. 112–118. [Google Scholar]
- Cohen MM, Jr, Shiota K. Teratogenesis of holoprosencephaly. Am J Med Genet. 2002;109:1–15. doi: 10.1002/ajmg.10258. [DOI] [PubMed] [Google Scholar]
- Couly GF, Coltey PM, Le Douarin NM. The triple origin of skull in higher vertebrates: a study in quail-chick chimeras. Development. 1993;117:409–429. doi: 10.1242/dev.117.2.409. [DOI] [PubMed] [Google Scholar]
- Crider KS, Olney RS, Cragan JD. Trisomies 13 and 18: population prevalences, characteristics, and prenatal diagnosis, metropolitan Atlanta, 1994–2003. Am J Med Genet. 2008;146A:820–826. doi: 10.1002/ajmg.a.32200. [DOI] [PubMed] [Google Scholar]
- David TJ, Glew S. Morbidity of trisomy 18 includes delivery by caesarean section. Lancet. 1980;2:1295. doi: 10.1016/s0140-6736(80)92353-3. [DOI] [PubMed] [Google Scholar]
- Davies ML, Macpherson P. Fibrous dysplasia of the skull: disease activity in relation to age. Br J Radiol. 1991;64:576–579. doi: 10.1259/0007-1285-64-763-576. [DOI] [PubMed] [Google Scholar]
- Deftereou TE, Tsoulopoulos V, Alexiadis G. Congenital disorder of true cyclopia with polydactylia: case report and review of the literature. Clin Exp Obstet Gynecol. 2013;3:420–422. , et al. ( [PubMed] [Google Scholar]
- Di Ieva A, Bruner E, Haider T. Skull base embryology: a multidisciplinary review. Childs Nerv Syst. 2014;6:991–1000. doi: 10.1007/s00381-014-2411-x. , et al. ( [DOI] [PubMed] [Google Scholar]
- Edwards JH, Harnden DG, Cameron AH. A new trisomic syndrome. Lancet. 1960;1:787–790. doi: 10.1016/s0140-6736(60)90675-9. , et al. ( [DOI] [PubMed] [Google Scholar]
- Embleton ND, Wyllie JP, Wright MJ. Natural history of Trisomy 18. Arch Dis Child Fetal Neonatal Ed. 1996;75:F38–F41. doi: 10.1136/fn.75.1.f38. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enlow DH. Facial Growth. Philadelphia: WB Saunders; 1990. [Google Scholar]
- Evans DJ, Noden DM. Spatial relations between avian craniofacial neural crest and paraxial mesoderm cells. Dev Dyn. 2006;235:1310–1325. doi: 10.1002/dvdy.20663. [DOI] [PubMed] [Google Scholar]
- Freter S, Fleenor SJ, Freter R. Cranial neural crest cells form corridors prefiguring sensory neuroblast migration. Development. 2013;140:3595–3600. doi: 10.1242/dev.091033. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzozi HJ, Barkay S. Case of true cyclopia. Br J Ophthalmol. 1985;69:307–311. doi: 10.1136/bjo.69.4.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser SC, Carey JC, Apple DJ. Human chromosomal disorders and the eye. In: Renie WA, editor. Goldberg's Genetic and Metabolic Eye Disease. Boston: Little, Brown; 1986. pp. 185–240. [Google Scholar]
- GeneCards. 2014. ) Available from: < http://www.genecards.org/cgibin/carddisp.pl?gene=TGIF1&search=19fdafb9744386ff595c1f1c89991232 (28 December 2014)
- Gondre-Lewis MC, Petrache HI, Wassif CA. Abnormal sterols in cholesterol-deficiency diseases cause secretory granule malformation and decreased membrane curvature. J Cell Sci. 2006;119:1876–1885. doi: 10.1242/jcs.02906. , et al. ( [DOI] [PubMed] [Google Scholar]
- Gripp KW, Wotton D, Edwards MC. Mutations in TGIF cause holoprosencephaly and link NODAL signaling to human neural axis determination. Nat Genet. 2000;25:205–208. doi: 10.1038/76074. , et al. ( [DOI] [PubMed] [Google Scholar]
- Gunz P, Harvati K. The Neanderthal ‘chignon’: variation, integration, and homology. J Hum Evol. 2007;52:262–274. doi: 10.1016/j.jhevol.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Hui L, Slonim DK, Wick HC. Novel neurodevelopmental information revealed in amniotic fluid supernatant transcripts from fetuses with trisomies 18 and 21. Hum Genet. 2012;11:1751–1759. doi: 10.1007/s00439-012-1195-x. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Jeffery N. Differential regional brain growth and rotation of the prenatal human tentorium cerebelli. J Anat. 2002;200:135–144. doi: 10.1046/j.0021-8782.2001.00017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery N, Spoor F. Ossification and midline shape changes of the human fetal cranial base. Am J Phys Anthropol. 2004;123:78–90. doi: 10.1002/ajpa.10292. [DOI] [PubMed] [Google Scholar]
- Jiang X, Iseki S, Maxson RE. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241:106–116. doi: 10.1006/dbio.2001.0487. , et al. ( [DOI] [PubMed] [Google Scholar]
- Irving C, Richmond S, Wren C. Changes in fetal prevalence and outcome for trisomies 13 and 18: a population-based study over 23 years. J Matern Fetal Neonatal Med. 2011;24:137–141. doi: 10.3109/14767051003758879. , et al. ( [DOI] [PubMed] [Google Scholar]
- Kelley RI, Roessler E, Hennekam RCM. Holoprosencephaly in RSH/Smith-Lemli-Opitz syndrome: does abnormal cholesterol metabolism affect the function of Sonic Hedgehog? Am J Med Genet. 1996;66:478–484. doi: 10.1002/(SICI)1096-8628(19961230)66:4<478::AID-AJMG22>3.0.CO;2-Q. , et al. ( [DOI] [PubMed] [Google Scholar]
- Kjær I. Sella turcica morphology and the pituitary gland – a new contribution to craniofacial diagnostics based on histology and neuroradiology. Eur J Orthod. 2012;37:28–36. doi: 10.1093/ejo/cjs091. [DOI] [PubMed] [Google Scholar]
- Kjaer I, Keeling JW, Fischer Hansen B. Midline skeletodental morphology in holoprosencephaly. Cleft Palate Craniofac J. 2002;39:357–363. doi: 10.1597/1545-1569_2002_039_0357_msmih_2.0.co_2. , et al. ( [DOI] [PubMed] [Google Scholar]
- Kjaer I, Keeling JW, Reintoft I. Pituitary gland and sella turcica in human trisomy 18 fetuses. Am J Med Genet. 1998a;76:87–92. , et al. ( [PubMed] [Google Scholar]
- Kjaer I, Keeling JW, Reintoft I. Pituitary gland and sella turcica in human trisomy 21 fetuses related to axial skeletal development. Am J Med Genet. 1998b;80:494–500. doi: 10.1002/(sici)1096-8628(19981228)80:5<494::aid-ajmg11>3.0.co;2-7. , et al. ( [DOI] [PubMed] [Google Scholar]
- Lieberman DE, Pearson OM, Mowbray KM. Basicranial influence on overall cranial shape. J Hum Evol. 2000a;38:291–315. doi: 10.1006/jhev.1999.0335. [DOI] [PubMed] [Google Scholar]
- Lieberman DE, Ross CF, Ravosa MJ. The primate cranial base: ontogeny, function, and integration. Am J Phys Anthropol Suppl. 2000b;31:117–169. doi: 10.1002/1096-8644(2000)43:31+<117::aid-ajpa5>3.3.co;2-9. [DOI] [PubMed] [Google Scholar]
- Lin HY, Lin SP, Chen YJ. Clinical characteristics and survival of trisomy 18 in a medical center in Taipei, 1988–2004. Am J Med Genet A. 2006;140:945–951. doi: 10.1002/ajmg.a.31173. , et al. ( [DOI] [PubMed] [Google Scholar]
- Martínez-Maza C, Rosas G-VS. Bone paleohistology and human evolution. J Anthropol Sci. 2006;84:33–52. [Google Scholar]
- Masters MP. Relative size of the eye and orbit: an evolutionary and craniofacial constraint model for examining the etiology and disparate incidence of juvenile-onset myopia in humans. Med Hypotheses. 2012;78:649–656. doi: 10.1016/j.mehy.2012.02.002. [DOI] [PubMed] [Google Scholar]
- McBratney-Owen B, Iseki S, Bamforth SD. Development and tissue origins of the mammalian cranial base. Dev Biol. 2008;322:121–132. doi: 10.1016/j.ydbio.2008.07.016. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming JE, Kaupas ME, Roessler E. Mutations of PATCHED in holoprosencephaly. Am J Hum Genet. 1998;63:A27. doi: 10.1007/s00439-002-0695-5. , et al. ( [DOI] [PubMed] [Google Scholar]
- Moss ML. The pathogenesis of premature cranial synostosis in man. Acta Anat. 1959;37:351–370. doi: 10.1159/000141479. [DOI] [PubMed] [Google Scholar]
- Muenke M, Beachy PA. Holoprosencephaly. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Basis of Inherited Disease. 8th edn. Vol. 4. New York: McGraw-Hill; 2001. pp. 6203–6230. ). In:, Vol. [Google Scholar]
- Müller F, O'Rahilly R. The prechordal plate, the rostral end of the notochord and nearby median features in staged human embryos. Cells Tissues Organs. 2003;173:1–20. doi: 10.1159/000068214. [DOI] [PubMed] [Google Scholar]
- Nanni L, Ming JE, Bocian M. The mutational spectrum of the Sonic Hedgehog gene in holoprosencephaly SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum Mol Genet. 1999;8:2479–2488. doi: 10.1093/hmg/8.13.2479. , et al. ( [DOI] [PubMed] [Google Scholar]
- Nanni L, Schelper RL, Muenke M. Molecular genetics of holoprosencephaly. Front Biosci. 2000;5:334–342. doi: 10.2741/nanni. [DOI] [PubMed] [Google Scholar]
- Nie X. Cranial base in craniofacial development: developmental features, influence on facial growth, anomaly, and molecular basis. Acta Odontol Scand. 2005;63:127–135. doi: 10.1080/00016350510019847. [DOI] [PubMed] [Google Scholar]
- Nie X, Luukko K, Fjeld K. Developmental expression of Dkk1-3 and Mmp9 and apoptosis in cranial base of mice. J Mol Histol. 2005a;36:419–426. doi: 10.1007/s10735-005-9014-5. , et al. ( [DOI] [PubMed] [Google Scholar]
- Nie X, Luukko K, Kvinnsland IH. Developmentally regulated expression of Shh and Ihh in the developing mouse cranial base: comparison with Sox9 expression. Anat Rec A Discov Mol Cell Evol Biol. 2005b;286:891–898. doi: 10.1002/ar.a.20231. , et al. ( [DOI] [PubMed] [Google Scholar]
- Ogle RC, Tholpady SS, McGlynn KA. Regulation of cranial suture morphogenesis. Cells Tissues Organs. 2004;176:54–66. doi: 10.1159/000075027. , et al. ( [DOI] [PubMed] [Google Scholar]
- O'Rahilly R, Müller F. Minireview: summary of the initial development of the human nervous system. Teratology. 1999;60:39–41. doi: 10.1002/(SICI)1096-9926(199907)60:1<39::AID-TERA11>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Paskulin GA, Lorenzen MB, Rosa RF. Importance of the fibroblast chromosomal analysis in suspected cases of mosaicism: experience of a clinical genetics service. Rev Paul Pediatr. 2011;29:73–79. , et al. ( [Google Scholar]
- Rasmussen SA, Wong L, Yang Q. Population-based analyses of mortality in trisomy 13 and trisomy 18. Pediatrics. 2003;111:777–784. doi: 10.1542/peds.111.4.777. , et al. ( [DOI] [PubMed] [Google Scholar]
- Riccomagno MM, Martinu L, Mulheisen M. Specification of the mammalian cochlea is dependent on Sonic hedgehog. Genes Dev. 2002;16:2365–2378. doi: 10.1101/gad.1013302. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richtsmeier JT, Flaherty K. Hand in glove: brain and skull in development and dysmorphogenesis. Acta Neuropathol. 2013;125:469–489. doi: 10.1007/s00401-013-1104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. , et al. ( [DOI] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K. Mutations in the C-terminal domain of Sonic Hedgehog cause holoprosencephaly. Hum Mol Genet. 1997;6:1847–1853. doi: 10.1093/hmg/6.11.1847. , et al. ( [DOI] [PubMed] [Google Scholar]
- Root S, Carey JC. Survival in trisomy 18. Am J Med Genet. 1994;49:170–174. doi: 10.1002/ajmg.1320490203. [DOI] [PubMed] [Google Scholar]
- Rosa RF, Rosa RC, Zen PR. Trisomy 18: review of the clinical, etiologic, prognostic, and ethical aspects. Rev Paul Pediatr. 2013;1:111–120. doi: 10.1590/s0103-05822013000100018. , et al. ( [DOI] [PubMed] [Google Scholar]
- Sadler TW. Langman's Medical Embryology. 10th edn. Baltimore: Lippincott Williams and Wilkins; 2006. pp. 125–128. , 333. [Google Scholar]
- Santaolalla-Montoya F, Martinez-Ibarguen A, Sanchez-Fernandez JM. Principles of cranial base ossification in humans and rats. Acta Otolaryngol. 2012;132:349–354. doi: 10.3109/00016489.2011.642814. , et al. ( [DOI] [PubMed] [Google Scholar]
- Santagati F, Rijli FM. Cranial neural crest and the building of the vertebrate head. Nature Rev Neurosci. 2003;4:806–818. doi: 10.1038/nrn1221. [DOI] [PubMed] [Google Scholar]
- Schoenwolf GC, Bleyl SB, Brauer PR. Larsen's Human Embryology. Philadelphia: Churchill Livingstone; 2009. , et al. ( [Google Scholar]
- Sheng HZ, Westphal H. Early steps in pituitary organogenesis. Trends Genet. 1999;15:236–240. doi: 10.1016/s0168-9525(99)01742-4. [DOI] [PubMed] [Google Scholar]
- Smith DW, Patau K, Therman E. A new autosomal trisomy syndrome: multiple congenital anomalies caused by an extra chromosome. J Pediatr. 1960;57:338–345. doi: 10.1016/s0022-3476(60)80241-7. , et al. ( [DOI] [PubMed] [Google Scholar]
- Strait DS. The scaling of basicranial flexion and length. J Hum Evol. 1999;37:701–719. doi: 10.1006/jhev.1999.0314. [DOI] [PubMed] [Google Scholar]
- Stringer C. Modern human origins: progress and prospects. Phil Trans R Soc London B. 2002;357:563–579. doi: 10.1098/rstb.2001.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank PW. Grant's Dissector. 15th edn. Walters Kluwer: Lippincott Williams & Wilkins; 2013. [Google Scholar]
- Vermeij-Keers C. Craniofacial Embryology and Morphogenesis: Normal and Abnormal. Edinburgh: Churchill Livingstone; 1990. [Google Scholar]
- Wallis DE, Muenke M. Molecular mechanisms of holoprosencephaly. Mol Genet Metabol. 1999;68:126–138. doi: 10.1006/mgme.1999.2895. [DOI] [PubMed] [Google Scholar]
- Weber WW. Survival and the sex ratio in trisomy 17–18. Am J Hum Genet. 1967;19:369–377. [PMC free article] [PubMed] [Google Scholar]
- Young B, Minugh-Purvis N, Shimo T. Indian and sonic hedgehogs regulate synchondrosis growth plate and cranial base development and function. Dev Biol. 2006;299:272–282. doi: 10.1016/j.ydbio.2006.07.028. , et al. ( [DOI] [PubMed] [Google Scholar]
- Young ID, Cook JP, Mehta L. Changing demography of trisomy 18. Arch Dis Child. 1986;61:1035–1036. doi: 10.1136/adc.61.10.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]