

Abstract
Microglia are tissue macrophages of the central nervous system (CNS) that control tissue homeostasis. Microglia dysregulation is thought to be causal for a group of neuropsychiatric, neurodegenerative and neuroinflammatory diseases, called “microgliopathies”. However, how the intracellular stimulation machinery in microglia is controlled is poorly understood. Here, we identified the ubiquitin-specific protease (Usp) 18 in white matter microglia that essentially contributes to microglial quiescence. We further found that microglial Usp18 negatively regulates the activation of Stat1 and concomitant induction of interferon-induced genes, thereby terminating IFN signaling. The Usp18-mediated control was independent from its catalytic activity but instead required the interaction with Ifnar2. Additionally, the absence of Ifnar1 restored microglial activation, indicating a tonic IFN signal which needs to be negatively controlled by Usp18 under non-diseased conditions. These results identify Usp18 as a critical negative regulator of microglia activation and demonstrate a protective role of Usp18 for microglia function by regulating the Ifnar pathway. The findings establish Usp18 as a new molecule preventing destructive microgliopathy.
Keywords: EAE, microglia, multiple sclerosis, type I interferon, Usp18
See also: K Takata & F Ginhoux (June 2015)
Introduction
Microglia are the tissue macrophages of the brain, crucially involved in the scavenging of dying cells, pathogens and molecules through phagocytosis/endocytosis and the use of pathogen-associated molecular pattern (PAMPs) receptors (Hanisch & Kettenmann, 2007; Ransohoff & Perry, 2009). Moreover, dysregulation of microglia activation is nowadays considered the pathogenetic basis for a group of neurodegenerative and neuroinflammatory conditions, called “microgliopathies” (Prinz & Priller, 2014). These include roles for several microglia molecules such as Csf1r in hereditary diffuse leukoencephalopathy with spheroids (Rademakers et al, 2012), CD33 in Alzheimer's disease (Hollingworth et al, 2011; Naj et al, 2011), Trem2 in frontotemporal dementia (Guerreiro et al, 2013), and Tnfrsf1a and Irf8 in multiple sclerosis (De Jager et al, 2009).
In general, engagement of recognition receptors initiates a complex machinery of various signaling pathways that lead to the induction of inflammatory cytokines and type I interferons such as interferon-α (IFN-α) and IFN-β, which are critical for inhibiting early viral replication in the host (Gonzalez-Navajas et al, 2012). The induction of such inflammatory mediators is controlled in a multifaceted fashion at the transcriptional level (Gonzalez-Navajas et al, 2012). These activation mechanisms have to be tightly regulated to prevent harmful tissue damage caused by hyperinflammatory reactions. Type I interferons signal through a common heterodimeric receptor known as the IFN-α/β receptor (Ifnar), which is expressed by nearly all cell types (Gonzalez-Navajas et al, 2012). This receptor consists of two subunits—Ifnar1 and Ifnar2—that are associated with Janus kinase 1 (Jak1) (Honda et al, 2006). Upon Jak1 activation, several signal transducer and activator of transcription (Stat) family members, such as Stat1, are activated that finally induce the induction of a plethora of interferon-induced genes (ISGs) such as Isg15,2′5′Oas,Mx1 and many more (Honda et al, 2006). Recent data have also uncovered potentially harmful sides of type I IFNs, including roles in inflammatory diseases such as autoimmunity and diabetes (Gonzalez-Navajas et al, 2012; Gough et al, 2012). For example, mutations in the human 3′ repair exonuclease 1 (Trex1) gene cause Aicardi-Goutières syndrome (AGS), an IFN-associated inflammatory disorder found in the brains of infants that suffer from epileptic seizures, intracerebral calcifications and leukodystrophy (Gall et al, 2012; Prinz & Knobeloch, 2012; Crow, 2015). On the other side, constitutive type I IFN levels are important for the maintenance, maturation and mobilization of the innate immune system in the body (Gough et al, 2012). Taking into account these highly divergent effects of type I IFNs, their tight regulation is imperative for ensuring immune homeostasis.
However, it is not known yet how microglia under homeostatic conditions are kept in a quiescent state, but intracellular proteases are potential candidates for such regulatory functions. Among them, ubiquitin-specific proteases (Usps) form the largest family of deubiquitinating enzymes (Dubs) that have key functions in immune responses and many other biological processes (Hershko & Ciechanover, 1998; Liu et al, 2005). Cylindromatosis (Cyld) has been extensively studied and shown to regulate various immune functions (Sun, 2008). It is now clear that Usps like Cyld target multiple signaling molecules, such as members of the Traf (tumor necrosis factor (Tnf) receptor (Tnfr)-associated factor) family, the Ikk (inhibitor of the NF-κB (ΙκB) kinase) regulatory subunit Ikkγ (also known as Nemo) (Brummelkamp et al, 2003; Kovalenko et al, 2003; Trompouki et al, 2003), the Src protein tyrosine kinase Lck (Reiley et al, 2006), the transforming growth factor-β (Tgfβ)-activated kinase 1 (Tak1) (Reiley et al, 2007) and many more. Not surprisingly, Usps regulate diverse biological functions, including host defense against infections, immune-cell development, activation and inflammation, cell survival, cell proliferation and tumorigenesis, microtubule assembly and cell migration, mitotic cell entry, calcium-channel function, spermatogenesis, osteoclastogenesis and many more (Sun, 2008). Furthermore, individual Dubs such as A20 or Uspl1 were shown to possess additional functions besides their protease activity further extending potential functions of this class of proteins (Schulz et al, 2012; De et al, 2014).
Although the role of some Usps for the peripheral immune system is starting to emerge, the Usp family members that shape the innate immune system in the CNS, especially in microglia, have been less well studied. Here, we identified Usp18 as a novel microglia protein that is essential to prevent aberrant activation and that is required for the termination of the Ifnar2 activation signal mediated by the Ifnar1 subunit of the Ifnar heterodimer complex upon stimulation. Our data further indicate that Usp18-mediated control of the type I IFN system is critical for microglia quiescence preventing uncontrolled tissue damage. Our data additionally suggest that microglia heterogeneity in cortical and subcortical regions is determined by diverse endogenous functional programs.
Results
Usp18 silences white matter microglia under homeostatic conditions
It is not yet known how microglia are kept in a quiescent state under homeostatic conditions, but intracellular proteases are potential candidates for regulatory functions. Among them, ubiquitin-specific proteases (Usps) form the largest family of deubiquitinating enzymes (Dubs) that have key functions in immune responses and other biological processes (Hershko & Ciechanover, 1998; Liu et al, 2005).
We first performed whole-genome gene expression analysis of CD11b+CD45lo microglia from the white and gray matter and examined the expression of ubiquitin-specific proteases (Usps) as important regulators of the immune response (Sun, 2008). We found several Usps expressed in both white and gray matter microglia with only few differently expressed proteases in the gray and white matter, most prominently Usp18 (Fig1A and B). Usp18 transcripts were found to be highly expressed in unstimulated microglia with only background expression levels in other CNS cells (Fig1C). We next confirmed microglia specificity of this protease in the white matter of Usp18LacZ/LacZ (designated Usp18−/−) mice by X-gal staining (Fig1D).
Figure 1. Usp18 is a distinct feature of white matter microglia and essentially regulates microglia quiescence.

A Spatial distribution of ubiquitin-specific protease (USP) transcripts based on FACS-sorted adult microglia isolated from the white or gray matter that were subsequently examined by MouseRef-8 v2.0 Expression Bead Chip (Illumina) array analysis (Olah et al, 2012). Each USP shown exceeds a median expression value of two resulting from five mice compared to the mean expression value of the same gene in the other brain region.
B Quantitative RT–PCR of indicated genes in FACS-isolated adult microglia. Bars represent means ± s.e.m. with three mice in each group (*P < 0.05). Significant differences are determined by an unpaired t-test.
C Expression of Usp18mRNA measured by qRT–PCR in primary microglia (micro.), astrocytes (astro.), neurons (neu.) and oligodendrocytes (oli.). Bars represent means ± s.e.m with at least three samples in each group normalized to the mean expression value of Usp18 transcripts in the whole brain.
D Cell-specific expression of Usp18. Light microscopic analysis of X-gal-stained (blue) white matter brain tissue of adult Usp18LacZ/LacZ mice. Iba-1 staining (brown) reveals microglia. Inserts show microglia from the cortex. Scale bar, 20 μm.
E–G Histology of different brain areas in the cerebrum of adult Usp18+/+ and Usp18LacZ/LacZ (Usp18−/−) mice. Cortex (Co), hippocampus (Hc), thalamus (Th) and hypothalamus (Hypo) represent areas of the gray matter, whereas corpus callosum (CC) and fimbria (Fi) are defined as white matter. Scale bar, 10 μm.
H–J Histological pictures of different cerebellar regions of adult Usp18+/+ and Usp18LacZ/LacZ (Usp18−/−) mice. Molecular layer (ML) and granular layer (GL) represent areas of the gray matter, whereas arbor vitae (Arb) is part of the white matter.
K, L Quantification of Iba-1+ microglia in the different areas of Usp18LacZ/LacZ (Usp18−/−) mice. Microglia numbers are normalized to that found in Usp18+/+ littermates and are displayed as % of control. At least five mice per genotype were counted. Significant differences are determined by an unpaired t-test or Mann–Whitney U-test and marked with asterisks (*P < 0.05, **P < 0.01). Bars represent means ± s.e.m.
M Immunofluorescence of white matter and gray matter microglia (Iba-1, red) in adult Usp18−/− animals (green, scale bar, 10 μm). Three animals per genotype were examined. One characteristic picture is shown.
N Transmission electron microscopy of myelin-phagocytosing microglia in adult Usp18LacZ/LacZ (Usp18−/−) mice. Scale bars, 1 μm (overview) and 250 nm (zoom).
To further investigate the physiological role of Usp18 for the CNS, we performed a thorough histopathological analysis of different brain areas of adult Usp18LacZ/LacZ mice. While there were no obvious histopathological abnormalities in the gray matter, a significant increase of Iba-1+ microglia numbers was detectable in several white matter regions in Usp18-deficient mice (FigE–L). Furthermore, only subcortical white matter microglia but not cortical microglia exhibited strong expression of the activation marker MHC class II and of MAC-3 (LAMP2, CD107b, Fig1M). Transmission electron microscopy was used to confirm that myelin debris was visible in phagocytotically active microglia (Fig1N). Importantly, Usp18−/− brains did not show any infiltrating lymphocytes or monocytes (Supplementary Fig S1), indicating a sole microglia activation phenotype that we defined as “white matter microglia activation” (WMMA).
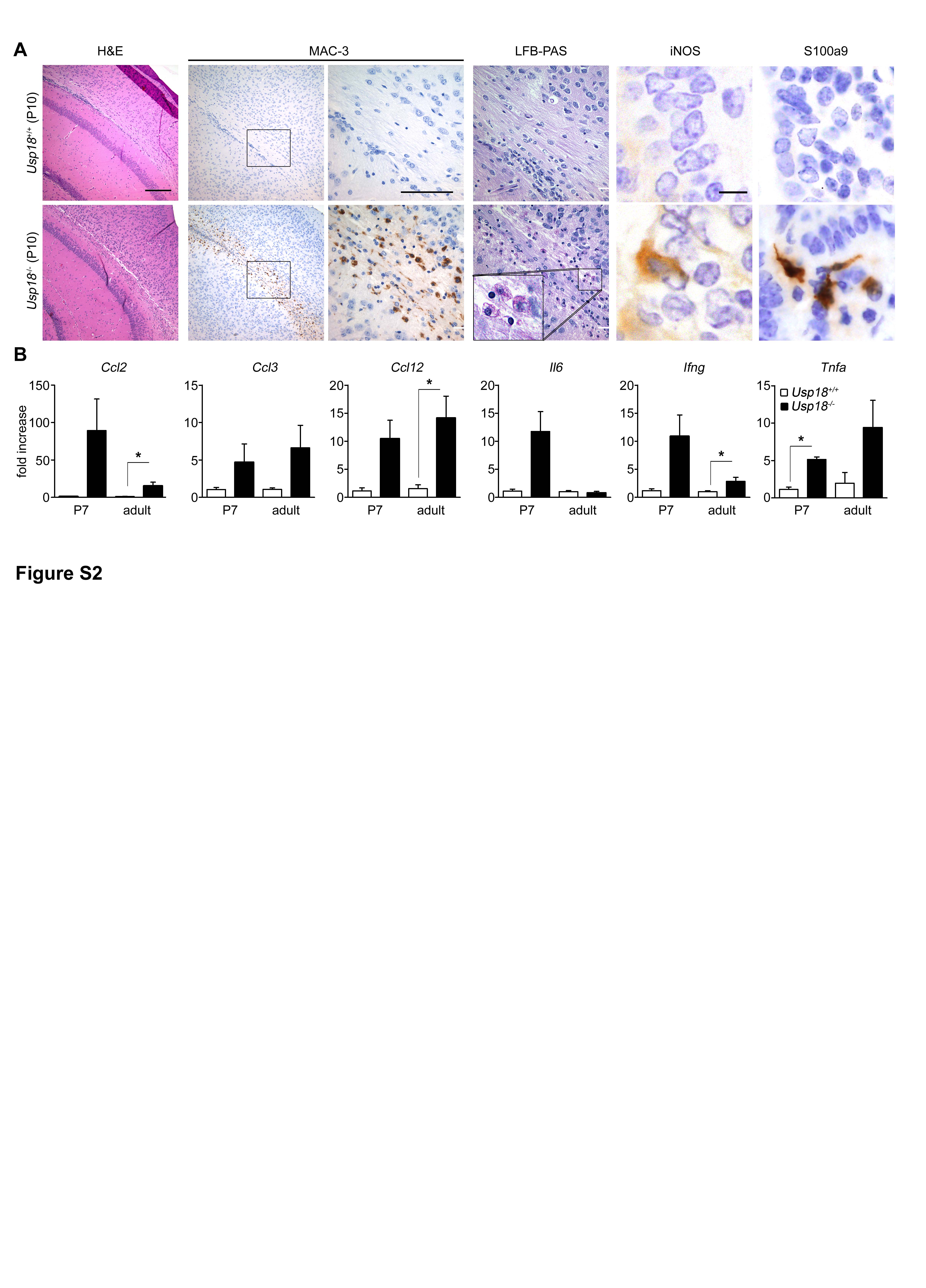
We were next interested at what developmental stage the WMMA is first detectable. Hardly any MAC-3+ microglia were visible on postnatal day P0 in both genotypes (FigA). On postnatal day P4, when microglia expansion occurs in foci within the periventricular subcortical white matter called “fountains of microglia” (Hristova et al, 2010), we observed no changes in the number of MAC-3+ microglia in mice lacking Usp18 (Fig2B and E). However, from P10 onwards until adulthood, Usp18-deficient mice exhibited accumulations of MAC-3+ microglia in the white matter that expressed the activation marker inducible nitric oxide synthase (iNOS) and S100 calcium binding protein a9 (S100a9) (Fig2C–E, Supplementary Fig S2A). Notably, in vitro oligodendrocyte differentiation as well as overall oligodendrocyte numbers was found to be independent of the presence of Usp18 (Supplementary Figs S3 and S4). In order to examine the underlying mechanisms of WMMA, we analyzed brain homogenates on P4 and P7 when Usp18−/− mice were still histologically undistinguishable from their Usp18+/+ littermates (FigF, Supplementary Fig S2B). We found a robust induction of the myeloattracting and myeloactivating chemokines Ccl2,Ccl3,Ccl5 and Cxcl10, and a slight induction of the cytokines Tnf-α and Il-6, which was also present in adulthood. In situ hybridizations of P4 brain sections revealed Ccl2 mRNA expression in white matter Iba-1+ microglia (Fig2G). Interestingly, adult Usp18−/− mice occasionally displayed intracerebral calcifications reminiscent to human brains suffering from interferonopathies (Fig2H) (Crow, 2015).
Figure 2. WMMA starts at early postnatal stages.

A–D Histology of brain sections of newborn (P0), 4-day-old (P4), 10-day-old (P10) and adult Usp18+/+ and Usp18LacZ/LacZ (Usp18−/−) mice revealing an early white matter microglia activation (WMMA). Hematoxylin and eosin (H&E), MAC-3 for activated microglia. Scale bars, 100 μm (overviews in H&E and MAC-3) and 50 μm (insert in MAC-3). Data are representative of two experiments with two mice each.
E Quantification of MAC-3-labeled microglia of P4 and adult Usp18+/+ and Usp18LacZ/LacZ (Usp18−/−) mice. At least three animals per genotype were examined. Bars represent means ± s.e.m. Significant differences are determined by an unpaired t-test and marked with an asterisk (*P < 0.05).
F Gene expression levels of Ccl5 and Cxcl10mRNA in the brains of P4, P7 and adult Usp18+/+ (white bars) and Usp18LacZ/LacZ (Usp18−/−, black bars) mice. Data are expressed as the ratio of induced factors normalized to endogenous Gapdh compared to Usp18+/+ mice and expressed as mean ± s.e.m. At least three mice were used in two independent experiments. Significant differences are determined by an unpaired t-test and marked with asterisks (*P < 0.05, **P < 0.01).
G In situ hybridization of Ccl2mRNA displays co-labeling in Iba-1+ white matter microglia of Usp18LacZ/LacZ (Usp18−/−) mice. Scale bar, 20 μm. Two mice were used in two independent experiments.
H Occurrence of cerebral calcifications in adult Usp18LacZ/LacZ (Usp18−/−) mice. Scale bars, 200 μm (overview) and 50 μm (zoom).
To test whether microglia activation found in the WMMA of Usp18-deficient mice is induced by cell autonomous or non-autonomous mechanisms, we targeted the Usp18 locus and generated Usp18 floxed animals (Usp18fl/fl, Fig3A and B). We next crossed these newly generated Usp18fl/fl mice with a transgenic line expressing the Cre recombinase under the control of the CX3CR1 promoter that drives recombination in myeloid cells in general (Yona et al, 2013) including microglia in the brain (Goldmann et al, 2013; Wieghofer et al, 2015). Resulting Cx3cr1Cre:Usp18fl/fl mice showed an Usp18 deletion in microglia but not in neuroectodermal cells of the CNS (Fig3C). Strikingly, the brains of adult Cx3cr1Cre:Usp18fl/fl mice exhibited marked microgliosis with activated amoboid Iba-1+Mac3+ microglia that precisely mirrored the WMMA pathology observed in adult Usp18−/− mice (Fig3D–J).
Figure 3. WMMA due to Usp18 deletion occurs in a cell-autonomous manner.

A Targeting strategy for the conditional mutagenesis of the Usp18 gene. A targeting vector was used to modify the Usp18 gene locus. Upon homologous recombination and elimination of the frt-flanked selection marker (neo), the third exon of the gene was flanked by loxP sites allowing Cre-mediated deletion of Usp18.
B Homologous recombination in ES cells was detected by genomic Southern blot analysis. As depicted in (A), probe A detects a diagnostic 3.5-kb band upon KpnI restriction digest diagnostic for the mutated Usp18 allele.
C PCR analysis of the Usp18 deletion in primary microglia, astrocytes, oligodendrocytes or neurons of Usp18fl/fl, Cx3cr1Cre:Usp18fl/fl and wild-type mice. Recombination is only taking place in microglia but not in astrocytes, oligodendrocytes or neurons. One representative experiment out of two performed is shown.
D, E Histology of different brain areas in the cerebrum of adult Usp18fl/fl and Cx3cr1Cre:Usp18fl/fl mice. Cortex (Co), hippocampus (Hc), thalamus (Th) and hypothalamus (Hypo) represent areas of the gray matter, whereas corpus callosum (CC) and fimbria (Fi) are parts of the white matter. Scale bar = 10 μm.
F, G Histological pictures of different cerebellar regions of adult Usp18fl/fl and Cx3cr1Cre:Usp18fl/fl mice. Molecular layer (ML) and granular layer (GL) represent areas of the gray matter, whereas arbor vitae (Arb) is part of white matter.
H, I Quantification of Iba-1+ cells in Cx3cr1Cre:Usp18fl/fl mice. Microglia numbers are normalized to that found in Usp18+/+ littermates and are displayed as % of control. At least five mice per genotype were counted. Significant differences are determined by an unpaired t-test or Mann–Whitney U-test and marked with asterisks (*P < 0.05, **P < 0.01, ***P < 0.0001). Bars represent means ± s.e.m.
J Immunofluorescence of white and gray matter microglia (Iba-1, red) in adult Usp18−/− animals (green, scale bar, 10 μm). Three animals per genotype were examined. One characteristic sample is shown.
Microglia activation in the absence of Usp18 is due to prolonged STAT1 phosphorylation
We subsequently isolated microglia, and whole-genome gene expression was determined. Investigation of the gene ontology enrichment network revealed that mostly type I IFN-regulated molecules were affected in Usp18−/− microglia (Fig4A). IFN-β treatment induced Usp18 expression in a time-dependent and dose-independent fashion in primary microglia (Fig4B). Constitutive type I IFN levels have been shown for some tissues (Gough et al, 2012) but have never been reported in the healthy murine brain. We used Mx1Cre mice intercrossed with the R26-confetti reporter line to examine the constitutive expression of the classical type I IFN-induced gene Mx1 and found Iba-1+Mx1+ microglia in white matter under SPF conditions indicating the presence of constitutive IFN signaling in these cells.
Figure 4. Lack of Usp18 enhances type I IFN gene expression in microglia due to prolonged Stat1 phosphorylation.

A Gene ontology enrichment network on differentially expressed genes in microglia from unstimulated Usp18+/+ and Usp18LacZ/LacZ (Usp18−/−) microglia on the basis of an Affymetrix DNA microarray analysis. Diagram depicts results of GO clustering through GORilla. Only very highly significantly overrepresented GO terms are included with P-values ranging from P < 10−9 (yellow) to P < 10−24 (red).
B Quantitative RT–PCR for Usp18 transcripts in primary microglia stimulated for the designated time points with 100 U/ml of IFN-β (left panel) or with 10, 100 or 1,000 U/ml of IFN-β and measured after 4 h (right panel). Bars represent means ± s.e.m with three to four samples in each group. Data are representative of two independently performed experiments.
C Fluorescence microscopy of the white matter (corpus callosum) of Mx1Cre:R26-confettimice raised under specific pathogen-free conditions. Recombination of GFP, RFP, CFP or YFP (combined into one channel to XFP and displayed in red) was found in Iba-1+ microglia of the white matter. Scale bars: 20 μm (overview) and 10 μm (zoom).
D Flow cytometric quantification of Stat1 phosphorylation in BV-2 microglial cells transfected with control siRNA (siRNA co) or siRNA against Usp18. Representative dot blots of IFN-β-treated cells at indicated time points are shown that were obtained from two independent experiments. FSC: forward scatter.
E Immunoblot analysis of type I IFN signaling in microglia lacking Usp18. Upper panel: Absence of Usp18 protein leads to prolonged Stat1 activation upon IFN-β challenge (500 U/ml) of microglia from Usp18+/+ and Usp18LacZ/LacZ (Usp18−/−) mice. Gapdh is shown as a loading control. Lower panel: Altered IFN signaling in the microglia cell line BV-2 transfected with control siRNA (siRNA co) or siRNA against Usp18. Quantification of band intensities is depicted next to the blots. Representative Western blots of three to four independently performed experiments are shown.
F Brain histology of the white matter reveals increased pStat1 and interferon-induced gene (ISG) 15 levels in white matter microglia in adult Cx3cr1Cre:Usp18fl/fl and Usp18LacZ/LacZ (Usp18−/−) mice but not in Usp18+/+ individuals. Quantification of Isg15+ cells in the gray (GM) and white matter (WM) is presented next to the respective histological images. Scale bars: 200 μm (overview) and 10 μm (insert). Each symbol indicates the mean of one mouse. Error bars represent s.e.m. Significant differences are determined by an unpaired t-test and marked with an asterisk (*P < 0.05).
G Heat map (standardized and scaled to log2 expression) of non-stimulated conditions (0 h) or after IFN-β (500 U/ml for 6 h and 24 h) treatment in primary microglia from Usp18LacZ/LacZ (Usp18−/−) and Usp18+/+ mice or the microglia cell line BV-2 (transfected with control siRNA [siRNA co] or siRNA against Usp18). Expression profile of top 50 induced genes in Usp18+/+ or siRNA co upon 6 and 24 h IFN-β is shown.
To clarify the cause of enhanced type I IFN target gene expression in Usp18−/− microglia, we examined the kinetics of Stat1 phosphorylation. Deletion of Usp18 led to a robust prolongation of Stat1 phosphorylation (FigD and E). Concordantly, examination of the CNS revealed activated subcortical amoboid Iba-1+pSTAT1+microglia resulting in cell-specific Isg15 expression, a key feature of WMMA (Fig4F). We next challenged cells with IFN-β and investigated the transcriptional profile (Fig4G). Most importantly, Usp18-deficient microglia failed to downregulate interferon-induced genes, suggesting that the termination of type I IFN signaling is severely impaired (Fig4G, Supplementary Figs S5 and S6).
The Ifnar2 interaction domain rather than the protease function of Usp18 controls microglia activation
Usp18 is a molecule with dual functions (Malakhova et al, 2003). First, it closely interacts with the Ifnar subunit 2 by competing with JAK1 for receptor binding, as shown in 293 T cells, and secondly, it is the major protease for Isg15 to deconjugate Isg15 (deISGylate) from substrate proteins (Malakhova et al, 2006) (Fig5A). In the murine Usp18, the cysteine at position 61 (Cys61) is essential for protease activity (Malakhov et al, 2002). To test whether the catalytic domain of Usp18 regulates microglia activation in vivo, we employed a novel mouse strain recently generated in our lab, in which the endogenous Usp18 gene locus was mutated to express an Usp18 protein selectively lacking its isopeptidase activity due to a single amino acid exchange from cysteine to alanine at position 61 (Usp18-C61A) (Ketscher et al, 2015). Usp18-C61A microglia neither changed the kinetics of phosphorylated Stat1 protein (Fig5B) nor altered the gene expression profile upon stimulation with IFN-β (Fig5C). Gene profiles of Usp18-C61A microglia were indistinguishable from those in Usp18+/+ microglia and showed no alteration in termination of IFN signaling. Importantly, WMMA was absent in brain sections of adult Usp18-C61A mice, clearly showing that USP18-mediated microglia activation is mediated in an isopeptidase-independent manner (Fig5D).
Figure 5. Usp18-mediated microglia activation is independent of its catalytic activity.

A Scheme of Usp18 interactions with either cytoplasmatic substrate/interacting proteins or trans-membrane anchored type I interferon receptor (Ifnar) and its subunits 1 and 2. The red cross indicates the genetically inactivated motif in Usp18-C61A mutant mice.
B Immunoblot of primary microglia from Usp18+/+ and catalytically inactive Usp18-C61A mutant mice reveals normal pStat1 kinetics at different time points after IFN-β (500 U/ml) challenge. Gapdh is shown as a loading control. Quantification of band intensities is depicted next to the blots. One representative data set out of three independent experiments is illustrated.
C Gene expression scatter plot of Affymetrix oligo-array data depicting different gene expression patterns in microglia from Usp18 mutants. The relative mRNA levels from wild-type microglia (Usp18+/+, x-axis) are normalized to Usp18LacZ/LacZ (Usp18−/−) microglia (y-axis, red dots) and catalytically inactive Usp18-C61A (y-axis, black dots) microglia under non-stimulated conditions and after IFN-β (500 U/ml) exposure. Pooled data from two independent experiments are shown.
D CNS histology of adult Usp18-C61A mice discloses unchanged microglial cells with normal morphological appearance (Iba-1) but no MAC-3+ amoboid microglia. Quantification of Isg15+ cells in the GM and WM is shown next to the respective histological images. Three mice per genotype were examined. Scale bars: 200 μm (overview), 10 μm (zoom).
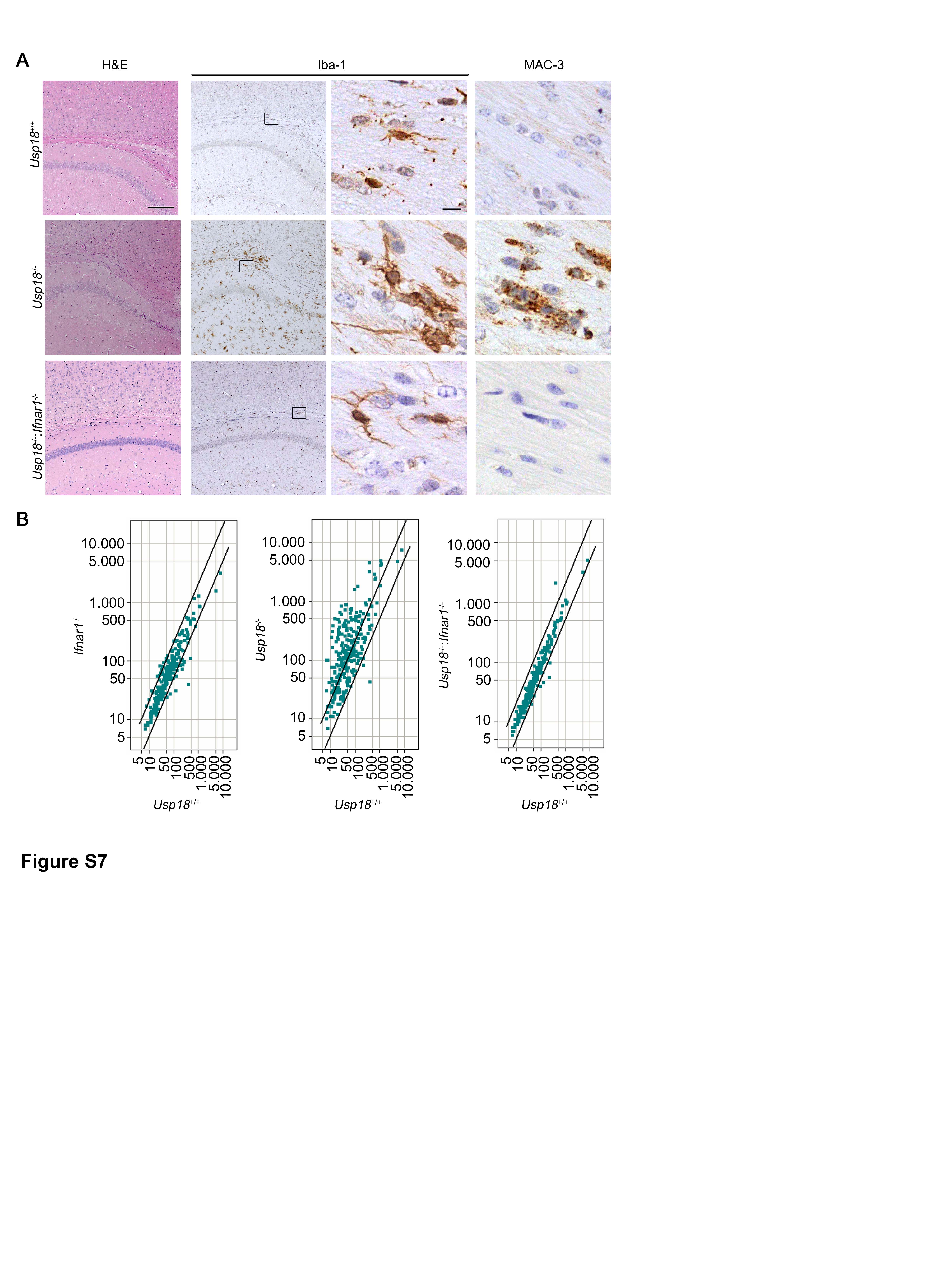
As deISGylation does not play a role for Usp18-induced Stat1 activation in microglia, it is conceivable that expression of Usp18 may affect signaling via the Ifnar. We therefore tested whether Usp18 interacts with the subunit 2 of Ifnar by immunoprecipitating Ifnar2 and found physical interaction of both partners (FigA). Close proximity of Usp18 with Ifnar2 could further be confirmed by proximity ligation (Duolink) (Fig6B). To prove the in vivo relevance of the Usp18 interaction with Ifnar2, we next took advantage of a mouse mutant called Usp18-L361F, which was recently identified in an ENU screen (Richer et al, 2010) (Fig6C and D). Using in vitro interaction studies, we were able to confirm the functionality of the interaction domain. The replacement of Leu361 by Phe in Usp18 abolished the binding affinity to Ifnar2 (Fig6E). In fact, Usp18-L361F mutant microglia revealed stronger and prolonged phosphorylation of Stat1 (Fig6F), mirroring the IFN signaling alteration detected in Usp18-deficient microglia. Accordingly, Usp18-L361F mutant adult brains histologically showed a plethora of activated amoboid microglia in subcortical white matter regions reminiscent of WMMA (Fig6G). To examine the impact of Ifnar1 on Usp18-regulated microglia activation, we crossed Usp18−/− mice with the Ifnar1−/− line and analyzed the brains of their progeny (Supplementary Fig S7A). Intriguingly, lack of Ifnar1 completely rescued this microglia activation phenotype, as seen histologically, and normalized the gene expression profile (Supplementary Fig S7B).
Figure 6. WMMA is regulated by interaction of Usp18 with the Ifnar2 domain.

A Co-immunoprecipitation (Co-IP) of flag-tagged Usp18 with the GST-Ifnar2 subunit upon overexpression in HEK293T cells. FLAG-Usp18 was precipitated using anti-FLAG beads, and Ifnar2 was detected by anti-GST immunoblotting revealing direct interaction of Ifnar2 with USsp18. Input is shown as transfection control.
B Proximity ligation assay for the co-localization of Usp18 with Inar2 in the microglial cell line BV-2. S-tagged Usp18 (red) associates with V5-tagged Ifnar2 (green). Close proximity of both proteins is shown by fluorescence dots (red) using a Duolink® probe. Scale bar, 10 μm.
C Scheme of Usp18 interactions with either cytoplasmatic substrate/interacting proteins or trans-membrane anchored type I interferon receptor (Ifnar) and its subunits 1 and 2. The red cross designates the genetically inactivated motif in Usp18-L361F mutant animals.
D Molecular model of Usp18 wild-type and Usp18-L361F variant. The replacement of Leu at position 361 by Phe results in a sterical clash with Phe271 and might disturb the conformation of the surface loop comprising residues 268–275.
E Binding analysis of USP18 and USP18-L361F with the intracellular domain of Ifnar2 by microscale thermophoresis. USP18 binds with high affinity (Kd = 54 ± 5 nM) to the intracellular domain of Ifnar2, whereas no binding is observed for USP18-L361F in the same concentration range. Data represent mean ± s.e.m. of two independent experiments.
F Immunoblot of microglia from Usp18+/+ and Usp18-L361F mutant mice demonstrates prolonged Stat1 phosphorylation at defined time points after IFN-β (500 U/ml) exposition. Gapdh is presented as a loading control. Quantification of band intensities is shown next to the blots. One representative data set out of three independent experiments is illustrated.
G Robust microgliosis (Iba-1), presence of amoboid MAC-3+ microglia and accumulation of Isg15 in adult Usp18-L361F mutant but not in Usp18+/+ mice. Quantification of Isg15+ cells in the GM and WM is depicted next to the respective histological images. Five mice per genotype were examined. Scale bars: 200 μm (overview), 10 μm (insert) Each symbol indicates the mean of one mouse. Error bars represent s.e.m. Significant differences are determined by an unpaired t-test and marked with an asterisk (*P < 0.05).
Myeloid-specific Usp18 shapes autoimmune inflammation in the CNS
To investigate whether Usp18 and the Stat1 pathways are induced upon autoimmune inflammation, we analyzed CNS samples from animals with autoimmune encephalomyelitis (EAE), a widely accepted mouse model for multiple sclerosis (MS). We observed a profound induction of Usp18 mRNA (Fig7A), elevated Stat1 protein levels and enhanced phosphorylation of the Stat1 protein, respectively (Fig7B). Of note, active Stat1 signaling in CD68+ macrophages/microglia was detectable on CNS sections in EAE as well as in human MS tissue samples, indicating that this pathway is also activated in MS patients (Fig7C). We then investigated the involvement of Usp18 in myeloid cells for CNS autoimmune diseases. The mice developed EAE with a similar incidence and mean disease onset (Fig7D). However, in Cx3cr1Cre:Usp18fl/fl mice, the effector phase of disease was changed with a significant augmented mean maximal clinical score compared to Usp18fl/fl mice.
Figure 7. Myeloid-specific Usp18 deficiency shapes clinical course and pathology of autoimmune CNS inflammation.

A Quantitative RT–PCR for Usp18mRNA in the spinal cord of healthy control and EAE-diseased mice. Data are expressed as ratio of Usp18 expression versus endogenous Gapdh relative to control and shown as mean ± s.e.m. Each symbol represents one mouse. (**P < 0.01). Significant differences are determined by a Mann-Whitney U-test and marked with a asterisk (**P < 0.01).
B Immunoblot for pStat1 and Stat1 in spinal cord lysates of diseased or control mice. Gapdh is shown as a loading control. Data are representative of three independent experiments performed.
C Left panel: immunohistochemistry for phosphorylated Stat1 (pStat1) in the border region of demyelinating lesions in spinal cord EAE samples (above) and brain samples from a patient with multiple sclerosis (MS). Scale bars: 100 μm (overview) and 25 μm (zoom). Four EAE-diseased animals and three biopsies of MS patients were examined, and one representative picture is shown. Right panel: immunofluorescence of pSTAT1 (red) and CD68 (green) in demyelinating lesions of MS patients. Quantification of pSTAT1+CD68+ and pSTAT1+CD68− cells are shown next to the respective histological images. Each symbol represents on patient sample. Error bars represent s.e.m. Significant differences are determined by an unpaired t-test and marked with an asterisk (*P < 0.05).
D EAE was induced by active immunization of Cx3cr1Cre:Usp18fl/fl (n = 9, filled squares) and Usp18fl/fl (n = 15, open squares) mice, and disease was scored. Each data point represents the mean ± s.e.m. Significant differences are determined by a Mann–Whitney U-test and marked with asterisks (*P < 0.05, **P < 0.01). The data shown are the mean from two independent experiments.
E Normal recall assay in Cx3cr1Cre:Usp18fl/fl mice. Lymph node T cells were collected and cultured for 48 h at the indicated MOG35–55 concentrations. Proliferation was measured by BrdU incorporation for 16 h (left). IL-17 (middle) and IFN-γ (right) release were measured by ELISA. Data represent mean ± s.e.m. of at least three animals per group. Results are representative of two independent experiments.
F, G Histology of cerebral, cerebellar and spinal cord sections from diseased mice using CD3 for T lymphocytes (F) and amyloid precursor protein (APP, arrowhead) for axonal damage (G). Scale bars, 100 μm. Quantification of T-cell infiltrates and axonal damages are depicted below the respective histological images. Each symbol indicates the mean of one mouse. Significant differences are determined by an unpaired t-test and marked with asterisks (*P < 0.05, **P < 0.01). Error bars represent s.e.m.
H–J Gene expression levels of TH1- (H), TH17- (I) and TH2-linked factors (J) in isolated mononuclear cells from the cerebrum (cere.) or the spinal cord (sc) of Cx3cr1Cre:Usp18fl/fl (black bars; n = 7) or Usp18fl/fl (white bars; n = 7) mice. Data are normalized to endogenous Gapdh, expressed as fold increase of diseased Usp18fl/fl mice and displayed as mean ± s.e.m. Significant differences are determined by an unpaired t-test and marked with an asterisk (*P < 0.05).
Efficient priming of T lymphocytes is a critical requirement for the induction of CNS inflammation and concomitant pathology. We therefore isolated lymphocytes from immunized mice and tested the recall response upon secondary exposure to MOG35–55 peptide in vitro (FigE). T cells derived from Cx3cr1Cre:Usp18fl/fl animals did not show an increased recall response compared to those of control littermates, suggesting that the increased disease burden is not caused by enhanced peripheral T-cell responses. We next examined different CNS regions by immunohistochemistry (Fig7F and G, Supplementary Fig S8). In control mice, we detected many CD3+ T cells and B220+ B cells in the spinal cord parenchyma and its adjacent meninges. In comparison, Cx3cr1Cre:Usp18fl/fl and Usp18fl/fl mice had similar inflammatory cells and the same degree of axonal (APP) and myelin damage (LFB-PAS) in the spinal cord. However, Cx3cr1Cre:Usp18fl/fl mice revealed significantly more lymphocytes, and increased axonal damage in the upper white matter regions of the CNS, namely cerebellum and cerebrum. We next quantified the expression of T helper cell (TH)17, TH1- and TH2-linked factors in several CNS regions (Fig7H–J). All investigated factors showed similar expression levels in the spinal cord, whereas in the cerebrum of Cx3cr1Cre:Usp18fl/fl animals, the TH1-related gene Ifng was significantly increased. Therefore, the increased EAE severity in the absence of Usp18 in myeloid cells is potentially linked to locally pre-activated microglia in the WMMA.
Discussion
Here, we identified Usp18 as a critical gatekeeper of white matter microglia activation under homeostatic conditions. Usp18-mediated maintenance of microglia quiescence strictly relied on the interaction with Ifnar2, whereas the catalytic function of this isopeptidase was found to be dispensable to control microglia activation. In addition, the presence of the Ifnar1 subunit was mandatory to uphold constitutive type I IFN signaling in microglia, which needs to be tightly regulated by Usp18 to prevent fatal activation in white matter.
Our data are of great importance because, for the first time, provide evidence for a constitutively active type I IFN system in microglia cells in the healthy brain. The significance of constitutive IFN-β in maintaining immune homeostasis has been revealed by studies examining the aberrant phenotype of mice lacking type I IFN receptors. Ifnar-deficient mice have decreased splenic NK cells and increased Gr-1+CD11c+ myeloid cells (Hwang et al, 1995; Swann et al, 2007). Constitutive IFN-β signaling augments myeloid cell function and macrophage homeostasis, as shown by analysis of macrophages from C3H-HeJ mice, which are incapable of inducing IFN in response to LPS because of a defect in the Tlr4 gene (Poltorak et al, 1998). Culturing C3H-HeJ macrophages with supernatants from wild-type C3H-HeN macrophages that express constitutive IFN-β enhanced their phagocytic potential. A similar effect was obtained by adding low “priming” concentrations of IFN to C3H-HeJ macrophages (Vogel & Fertsch, 1984). Conversely, phagocytic capacity was attenuated when C3H-HeN macrophages were incubated with IFN-α- and IFN-β-neutralizing antibodies (Vogel & Fertsch, 1984), documenting the requirement for constitutive IFN in preserving macrophage function. The physiologic consequence of the importance of constitutive IFN for macrophage function may be reflected by the influence of the gut microbiota on hematopoietic homeostasis through basal TLR signaling (Musso et al, 2011). Whether intestinal colonization is also able to tune constitutive IFN responses in myeloid cells inside the brain remains to be determined. Although the constitutive activation of type I IFNs in microglia has not been addressed so far under germ-free conditions to assess a potential role for commensal microorganisms, we clearly detected Mx1-positive microglia in brains from healthy mice kept in a specific pathogen-free environment. However, it also becomes clear from our experiments that a strict control of constitutive IFN signaling in white matter microglia is necessary to keep these myeloid cells under resting conditions to avoid cellular hyperactivation with detrimental consequences for the tissue environment. On the other hand, constitutive IFN signaling might be important for priming microglia to maintain a rapid and robust innate and adaptive immune response to subsequent challenge. Our data provide evidence that this tight balance is secured by Usp18.
Our data on the specific expression of Usp18 in white matter rather than in cortical microglia highlight the regional heterogeneity of these glial cells in the CNS. It is unknown so far whether microglia heterogeneity is a result of irreversible subtype-specific differentiation or a consequence of continuous but reversible induction of diverse functional programs (Hanisch, 2013). Local tissue-derived signals are thought to control functional polarization of microglia, but their identity remains largely unknown. Our data on the region-specific upregulation of Usp18 in microglia, however, suggest that cell autonomous programs exist that may correlate with their regional-specific function. Microglia seem to assume also particular function in the normal turnover of myelin (Fitzner et al, 2011). Oligodendrocytes create well-organized and also tremendously elaborate myelin structures that require constant upkeep. Apparently, they thereby rely on the assistance of microglia. Oligodendrocytes can wrap myelin material from their turnover process in exosomes and deliver them to the microglia in their surrounding (Fitzner et al, 2011), indicating that subpopulations of microglia for myelin clearance do exist.
One may speculate that a disturbance of physiological myelin processing by microglia could be detrimental, and there might be functional links to the triggering of myelin-afflicting autoimmune diseases such as MS. Just recently, vulnerability of microglia has been connected to several neurological and psychiatric disorders that are characterized by primary or secondary “microgliopathies” (Prinz & Priller, 2014). In fact, recent genomewide association studies with thousands of MS patients revealed the existence of some microglia-related factors for disease pathogenesis such as interferon regulatory factor (Irf-) 8 and Tnfrsf1a (De Jager et al, 2009). In a recent study, two Usp18-related polymorphisms, one intronic and another located in the promoter region of the Usp18 gene, were found to be associated with MS susceptibility (Malhotra et al, 2013). Importantly, haplotypic analysis revealed one haplotype, which correlated with lower Usp18 gene expression in peripheral blood mononuclear cells and higher clinical disease activity (Malhotra et al, 2011). These clinical observations fit to our experimental findings with an augmented EAE course in myeloid-specific Usp18 knockout animals. However, this clinical study included only a limited number of MS patients. Therefore, additional functional studies will be needed to delineate the role of microglia-specific Usp18 in MS. Our current study, however, suggests a potential role of microglia dysregulation in the pathogenesis of white matter diseases.
Further studies indicated broader effects of Usp18 that may be targeted to treat other autoimmune diseases as well. It was previously reported that Usp18 regulates T-cell activation and Th17 cell differentiation. Usp18-deficient T cells were defective in Th17 differentiation, and Usp18−/− mice consecutively showed alterations in CNS inflammation (Liu et al, 2013). Usp18 has further been identified as a candidate gene for type I diabetes (Santin & Eizirik, 2013; Altorok et al, 2014), and active hypomethylated CpG sites of Usp18 were found to be expressed in patients with primary Sjögren's syndrome (Altorok et al, 2014). Furthermore, Usp18 may also be involved in viral infections that are often associated with a break of immunological tolerance and subsequent induction of autoimmune diseases. In fact, it was recently shown that in a mouse model of autoimmune diabetes, Usp18-enforced viral replication evoked a break of immunological tolerance (Honke et al, 2013). Interestingly, the presence of intracranial calcifications as well as an increased type IFN-signature due to reduced protein stability of Usp18 was described recently in Isg15-deficient patients (Zhang et al, 2015).
In sum, we report here a crucial function of Usp18 in microglia activation during health and disease and provide evidence for Usp18-related regulation of constitutive IFN activity in the CNS. These results may potentially assist our understanding and treatment of microglia-associated brain diseases in general and inflammatory diseases in particular.
Materials and Methods
Human samples
Active demyelinating MS samples that were histologically classified as described before (Lucchinetti et al, 2000). Samples were collected according to the approval by the Ethics Committee of the Albert-Ludwigs University Freiburg, Germany.
Mice
All animal experiments were approved by the Federal Ministry for Nature, Environment and Consumers' Protection of the state of Baden-Württemberg (35-9185.81/G12/71) and were performed in accordance with the respective national, federal and institutional regulations. We bred Usp18+/− heterozygous mice on a mixed background (C57/Bl6 × 129/S) to obtain Usp18−/− and Usp18+/+ mice (Ritchie et al, 2002). To generate mice allowing conditional Usp18 depletion, a target vector was constructed as depicted in detail in Fig2A. After transfection of ES cells and upon homologous recombination, Exon 3 was flanked by loxP sites. ES cells carrying the desired mutant gene locus were detected by Southern analysis of genomic DNA digested with KpnI due to the appearance of a diagnostic 3.5- and 7.5-kb band recognized by 5′probe A and 3′probe B, respectively. Germline chimeras were generated from these ES cells by morula injection. Resulting mutant offspring was crossed to a flp-deleter strain to excise the frt-flanked pgk-neo selection marker from the genome yielding mice with the loxP-flanked Usp18 gene (Usp18fl/+). Usp18fl/+ mice were backcrossed for > 9 generations onto the C57Bl/7 background and finally crossed to Cx3cr1Cre (Yona et al, 2013). Usp18fl/fl mice were genotyped by PCR using the following primers 5′-cacctccatttggtttcagg-3′ and 5′-aactccttcctctggcttcc-3′ that amplify a 250-bp fragment from the transgenic region. Usp18-C61A mice were generated by us and bred on a C57/Bl6 background (Ketscher et al, 2015). Usp18-L361F mice were kept under 129/S1 background (Richer et al, 2010). Usp18+/− were crossed to Ifnar1−/− (Prinz et al, 2008) in order to generate Usp18−/−:Ifnar1−/− double mutants. Mx1Cre were crossed to R26-confetti mice (Snippert et al, 2010). In all experiments, littermates carrying the respective loxP-flanked alleles but lacking expression of Cre recombinase were used as controls.
Cell culture
Primary cultures from microglia, astrocytes and oligodendrocytes were prepared from newborn mice; neurons were prepared from E16 embryos and cultured as described previously (Raasch et al, 2011). HEK293T and BV-2 cells were grown in DMEM GlutaMAX (Invitrogen) supplemented with 10% fetal calf serum (PAA) and 1% penicillin/streptomycin (PAA) at 37°C and 5% CO2. Transfections of plasmid DNA and siRNA in HEK 293T and BV-2 cells were performed using X-tremeGENE HP (Roche) FuGENE (Promega) and Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol.
Induction of EAE
Female mice from each group, aged 6–8 weeks, were immunized subcutaneously with 200 μg of MOG35–55 peptide emulsified in CFA containing 1 mg of Mycobacterium tuberculosis (H37RA; Difco Laboratories, Detroit, Michigan, USA). The mice received intraperitoneal injections with 250 ng pertussis toxin (Sigma-Aldrich, Deisenhofen, Germany) at the time of immunization and 48 h later.
Clinical evaluation
Mice were scored daily as follows: 0, no detectable signs of EAE; 0.5 distal limb tail; 1.0, complete limp tail; 1.5, limp tail and hind limb weakness; 2, unilateral partial hind limb paralysis; 2.5, bilateral partial hind limb paralysis; 3, complete bilateral hind limb paralysis; 3.5, complete hind limb paralysis and unilateral forelimb paralysis; and 4, total paralysis of fore and hind limbs.
Recall assay
On day 7 after immunization, the draining axillary and inguinal lymph nodes (LN) were removed from MOG35–55-immunized mice and single-cell suspensions were prepared. 6 × 105 LN cells were placed as triplicates in a 96-well plate and pulsed with the indicated dosages of MOG peptide. BrdU uptake was measured for 16 h to determine proliferation (Cell Proliferation ELISA colorimetric, Roche Applied Science) according to the manufacturer's protocol. RPMI 1,640 (Invitrogen) and Dulbecco's modified Eagle's medium (Invitrogen) each supplemented with 10% (v/v) fetal calf serum (FCS), 3 mM l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (all from Sigma-Aldrich) were used. For cytokine analysis, sister cultures supernatants were analyzed by ELISA for IFN-γ and IL-17 (R&D Systems, Wiesbaden, Germany).
Isolation of mononuclear cells
For gene expression, samples of the cerebrum and spinal cord were prepared by density gradient separation as described previously (Mildner et al, 2009).
Histology
Histology was performed as described recently (Goldmann et al, 2013). Cerebrum, cerebellum and spinal cord were removed on day 26 after immunization and fixed in 4% buffered formalin. Then, tissue samples were embedded in paraffin before staining with H&E, luxol fast blue (LFB-PAS) to assess the degree of demyelination, Mac-3 for activated macrophages/microglia, CD3 for T cells, B220 for B cells, APP for indication of axonal damage, Iba-1 for microglia, Nogo for oligodendrocytes, iNOS and S100a9 for microglia activation, pStat1 and Isg15 as representative IFN-induced genes. For the β-galactosidase staining, tissue was directly frozen unfixed in tissue tec on dry ice. Cryosections were postfixed using 0.2% glutaraldehyde and stained by X-gal staining solution. Tissue sections were evaluated on Olympus BX-61 microscope using the cell-P software (Olympus).
Fluorescence microscopy
After transcardial perfusion with phosphate-buffered saline (PBS), brains were fixed in 4% PFA and embedded. 14-μm cryosections were obtained as described previously (Goldmann et al, 2013). Sections were then blocked with PBS containing 5% bovine serum albumin and permeabilized with 0.1% Triton-X 100 in blocking solution. Primary antibodies were added overnight at a dilution of 1:500 for Iba-1(019-19741, WACO, Japan), 1:50 for pStat1 (# 9167, Cell Signaling, Danvers, USA), 1:250 for MAC-3 (ab13524, Abcam, Cambridge, UK) and 1:100 for Mhc class II (ab23990, Abcam), at 4°C. Secondary antibodies were added as follows: Alexa Flour® 488 1:500, Alexa Flour® 555 1:500 and Alexa Fluor® 568 1:500 for 2 h at RT. Nuclei were counterstained with DAPI. The examined area was determined microscopically using a conventional fluorescence microscope (Olympus BX-61), and the confocal pictures were taken with Fluoview FV 1000 (Olympus).
Electron microscopy
Samples for electron microscopy were prepared as previously described (Kierdorf et al, 2013). Briefly, tissue was fixed in 3% glutaraldehyde in Sörensen phosphate buffer and embedded in araldite, and subsequently, ultra-thin sections were cut. Ultrastructural analysis was performed using CM100 transmission electron microscope (Philips).
Flow cytometry
Cells were stained with primary antibodies directed against CD11b and CD45 (eBioscience, San Diego, USA) at 4°C for 15 min. Cells were washed and sorted for microarray analyses. pStat1 (612596, BD Bioscience) staining was performed according to the manufacturer's protocol and analyzed using a FACSCanto II (Becton Dickinson). Viable cells were gated by forward and side scatter pattern. Data were acquired with FACSdiva software (Becton Dickinson). Postacquisition analysis was performed using FlowJo software (Tree Star, Inc.).
Microarray & qRT–PCR
Tissues were dissected and flushed with ice-cold HBSS. Cells were washed with PBS. RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer′s instructions. Before microarray analyses were performed, and RNA quality was assessed using Agilent 2100 bioanalyzer. Affymetrix GeneChips were used for genomewide expression analysis. Total RNA was processed using the GeneChip Expression 3′ Amplification One-Cycle Target Labeling Kit according to the manufacturer's instruction. Biotinylated cRNA was hybridized on MOE430A 2.0 GeneChips that were stained, washed and scanned following standard procedures. Cel files were normalized using robust multiarray analysis (RMA) implemented in the R affy package (http://www.bioconductor.org). The normalized expression data were then analyzed for differential gene expression using the software package LIMMA for R. For qRT–PCR, RNA-samples were treated with DNaseI (Roche, Mannheim, Germany) and 1.5 μg of RNA was transcribed into cDNA using either oligo-dT primers and the SuperScript II RT kit (Invitrogen, Carlsbad, CA) or High-Capacity RNA-to-cDNA™ kit (Life Technologies). A total of 1 μl cDNA was transferred into a 96-well Multiply® PCR plate (Sarstedt, Germany) and 11.5 μl ABsolute™ QPCR® SYBR Green master mix (Thermo Fisher). RT–PCRs were performed as described recently (Goldmann et al, 2013).
Cloning, expression and purification of recombinant proteins
Murine Usp18 was expressed in Sf21 insect cells (Basters et al, 2014). The cDNA encoding amino acid residues 264–374 of mouse Ifnar2 were amplified from a mouse cDNA clone (Source BioScience) using primers EcoRI-Ifnar2-for (AAAAAGAATTCAAACGGATTGGTTATATATGCC) and XhoI-Ifnar2-rev (TTTTTCTCGAGTCAAGCTTCATCAGATTCCTCAGC). EcoRI and XhoI restriction sites were introduced during amplification. The PCR product was digested with restriction enzymes EcoRI and XhoI and ligated into pGEX6p1 vector digested with the same enzymes. The resulting vector pGEX6p1-Ifnar2 (264–374) encoded Ifnar2 amino acid residues 264–374 fused in frame to a N-terminal GST-tag. E. coli TB1 cells were transformed with pGEX6p1-Ifnar2 (264–374) vector. E. coli cells were grown in DYT medium supplemented with 0.2% (w/v) glucose and 100 μg/ml ampicillin. At an OD600 nm = 0.6, protein expression was induced by addition of 0.5 mM IPTG to a final concentration of 0.5 mM and cells were grown at 19°C for 20 h. Cells were harvested by centrifugation, suspended in PBS supplemented with complete protease inhibitor (Roche) and disrupted by three passages through a French pressure cell at 137 Mpa. The crude extract was cleared by centrifugation at 40,000 g for 1 h, and the supernatant was applied to a 30-ml glutathione column. The bound protein was eluted with 50 mM Tris–HCl, 10 mM reduced glutathione, pH 8.0.
Microscale thermophoresis
Recombinant USP18 was labeled with fluorescence dye using the L001 Monolith NT.115 Protein Labeling Kit RED-NHS according to the instructions of the manufacturer (NanoTemper). For microscale thermophoresis analysis, the labeled protein was used at a concentration of 200 nM. GST-Ifnar2 (264–374) was added in a concentration range from 3 nM to 10 μM and equilibrated for 30 min prior analysis. The thermophoresis measurement was performed in 20 mM sodium phosphate, pH 7.4, 300 mM NaCl, 5 mM DTT and 0.05% (v/v) Tween-20 at 25°C in standard treated glass capillaries using a Monolith NT.115 Red/Blue instrument (NanoTemper).
Protein interaction
Co-immunoprecipitation was performed using anti-FLAG M2 Magnetic Bead (M8823, Sigma-Aldrich) according to the manufacturer's protocol. Briefly, transfected cells were harvested and extracts were incubated with the beads overnight. After washing in PBS, samples were separated by SDS–PAGE. Duolink in situ kit was utilized in transfected BV-2 cells using antibodies against S-tag (1:100, Novagen) and V5 (1:50, sc-81594, Santa Cruz) in compliance with the manufacturer's instructions.
Western blot analysis
Tissues or cells were harvested and extracted in RIPA buffer (25 mM Tris–HCl, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, pH 7.5). Samples were separated by SDS–PAGE and immunoblotted using antibodies to pStat1 (1:1,000, #9171, Cell signaling), Stat1 (1:1,000, #9176, Cell signaling), pStat2 (1:1,000, #4441, Cell signaling), Stat2 (1:1,000, #4597, Cell signaling), Usp18 [1:1,000 (Ketscher et al, 2015)], Gapdh (1:2,500, Mab374, Millipore, Billerica, USA), anti-GST (1:1,000, GE healthcare) and anti-FLAG (1:2,000, F1804, Sigma-Aldrich). Band intensities were quantified with ImageJ software.
Statistical analysis
No statistical methods were used to predetermine sample sizes, and exact group numbers were determined by animal availability. However, we did ensure that our sample sizes were similar to those generally employed in the field. For the sample size in EAE experiments, power analysis was performed. A sample size of at least n = 15 per group was determined by 80% power to reach statistical significance of 0.05 to detect an effect size of at least 1.06. To obtain unbiased data, experimental mice of all relevant genotypes were all processed together by technicians and cell quantifications were performed blinded to the genotype by two scientists independently and separately. Only after finalization of all quantitative measurements were the samples allocated to their genotypes.
If not otherwise stated, data were tested for normality applying the Kolmogorov–Smirnov test. If normality was given, an unpaired t-test was applied. If the data did not meet the criteria of normality, the Mann–Whitney U-test was applied. Differences were considered significant when P-value < 0.05.
Data availability
The datasets of all microarray analyses are deposited at Gene Expression Omnibus (GEO) (GSE61501).
Acknowledgments
This work is dedicated to our former friend and teacher Ivan Horak, past director of the Department of Molecular Genetics at the Leibnitz-Institute for Molecular Pharmacology, Berlin, who devoted his whole life to the exploration of the genetic basis of the immune system. We thank Maria Oberle, Margarethe Ditter, Dr. Alexandra Müller, Tina El-Gaz, Anika Lückoff, Jana Dautzenberg and Christopher Fix for technical assistance. MP was supported by the BMBF-funded competence network of multiple sclerosis (KKNMS), the DFG (SFB 992, FOR1336, ZE 894/1-1, PR 577/8-1), the Fritz-Thyssen Foundation, the Sobek Foundation, the Gemeinnützige Hertie Foundation (GHST) and ERA-Net NEURON initiative “NEURO-IFN”. This work was further supported by DFG Grants KN590/3-2 (SPP1365) and KN590/1-3 to KPK. DM was supported by the Canadian Institutes of Health Research (CTP-87520). Doron Merkler is supported by the Swiss National Science Foundation (PP00P3_152928), the Klaus-Tschira Foundation and the Gebert-Rüf Foundation.
Author contributions
TG, NZ, JR, KK, KF, LK, AB, OS, SMB, AS, TLT, TB, GF, KB, DMa, MH and KPK conducted experiments. RL, CK, JT and GMSM analyzed gene expression data. DMe provided mice. NZ and KPK generated mice. MP and KPK supervised the project and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Altorok N, Coit P, Hughes T, Koelsch KA, Stone DU, Rasmussen A, Radfar L, Scofield RH, Sivils KL, Farris AD, Sawalha AH. Genome-wide DNA methylation patterns in naive CD4+ T cells from patients with primary Sjogren's syndrome. Arthritis Rheumatol. 2014;66:731–739. doi: 10.1002/art.38264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basters A, Geurink PP, El OF, Ketscher L, Casutt MS, Krause E, Ovaa H, Knobeloch KP, Fritz G. Molecular characterization of ubiquitin-specific protease 18 reveals substrate specificity for interferon-stimulated gene 15. FEBS J. 2014;281:1918–1928. doi: 10.1111/febs.12754. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- Crow YJ. Type I interferonopathies: Mendelian type I interferon up-regulation. Curr Opin Immunol. 2015;32C:7–12. doi: 10.1016/j.coi.2014.10.005. [DOI] [PubMed] [Google Scholar]
- De A, Dainichi T, Rathinam CV, Ghosh S. The deubiquitinase activity of A20 is dispensable for NF-kappaB signaling. EMBO Rep. 2014;15:775–783. doi: 10.15252/embr.201338305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, Piccio L, Raychaudhuri S, Tran D, Aubin C, Briskin R, Romano S, Baranzini SE, McCauley JL, Pericak-Vance MA, Haines JL, Gibson RA, Naeglin Y, Uitdehaag B, Matthews PM, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzner D, Schnaars M, van RD, Krishnamoorthy G, Dibaj P, Bakhti M, Regen T, Hanisch UK, Simons M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J Cell Sci. 2011;124:447–458. doi: 10.1242/jcs.074088. [DOI] [PubMed] [Google Scholar]
- Gall A, Treuting P, Elkon KB, Loo YM, Gale M, Jr, Barber GN, Stetson DB. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36:120–131. doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Muller PF, Wolf Y, Varol D, Yona S, Brendecke SM, Kierdorf K, Staszewski O, Datta M, Luedde T, Heikenwalder M, Jung S, Prinz M. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci. 2013;16:1618–1626. doi: 10.1038/nn.3531. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough DJ, Messina NL, Clarke CJ, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity. 2012;36:166–174. doi: 10.1016/j.immuni.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N, Dursun B, Bilgic B, Hanagasi H, Gurvit H, Emre M, Singleton A, Hardy J. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013;70:78–84. doi: 10.1001/jamaneurol.2013.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Functional diversity of microglia – how heterogeneous are they to begin with? Front Cell Neurosci. 2013;7:65. doi: 10.3389/fncel.2013.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Honke N, Shaabani N, Zhang DE, Iliakis G, Xu HC, Haussinger D, Recher M, Lohning M, Lang PA, Lang KS. Usp18 driven enforced viral replication in dendritic cells contributes to break of immunological tolerance in autoimmune diabetes. PLoS Pathog. 2013;9:e1003650. doi: 10.1371/journal.ppat.1003650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristova M, Cuthill D, Zbarsky V, Acosta-Saltos A, Wallace A, Blight K, Buckley SM, Peebles D, Heuer H, Waddington SN, Raivich G. Activation and deactivation of periventricular white matter phagocytes during postnatal mouse development. Glia. 2010;58:11–28. doi: 10.1002/glia.20896. [DOI] [PubMed] [Google Scholar]
- Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, Whitty G, Bertoncello I, Kola I. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci USA. 1995;92:11284–11288. doi: 10.1073/pnas.92.24.11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketscher L, Hannss R, Morales DJ, Basters A, Guerra S, Goldmann T, Hausmann A, Prinz M, Naumann R, Pekosz A, Utermohlen O, Lenschow DJ, Knobeloch KP. Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance. Proc Natl Acad Sci USA. 2015;112:1577–1582. doi: 10.1073/pnas.1412881112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C, Muller DN, Luckow B, Brocker T, Debowski K, Fritz G, Opdenakker G, Diefenbach A, Biber K, Heikenwalder M, Geissmann F, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16:273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424:801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- Liu YC, Penninger J, Karin M. Immunity by ubiquitylation: a reversible process of modification. Nat Rev Immunol. 2005;5:941–952. doi: 10.1038/nri1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li H, Zhong B, Blonska M, Gorjestani S, Yan M, Tian Q, Zhang DE, Lin X, Dong C. USP18 inhibits NF-kappaB and NFAT activation during Th17 differentiation by deubiquitinating the TAK1-TAB 1 complex. J Exp Med. 2013;210:1575–1590. doi: 10.1084/jem.20122327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem. 2002;277:9976–9981. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- Malakhova OA, Yan M, Malakhov MP, Yuan Y, Ritchie KJ, Kim KI, Peterson LF, Shuai K, Zhang DE. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev. 2003;17:455–460. doi: 10.1101/gad.1056303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, Shuai K, Zhang DE. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006;25:2358–2367. doi: 10.1038/sj.emboj.7601149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra S, Bustamante MF, Perez-Miralles F, Rio J, Ruiz de Villa MC, Vegas E, Nonell L, Deisenhammer F, Fissolo N, Nurtdinov RN, Montalban X, Comabella M. Search for specific biomarkers of IFNbeta bioactivity in patients with multiple sclerosis. PLoS ONE. 2011;6:e23634. doi: 10.1371/journal.pone.0023634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra S, Morcillo-Suarez C, Nurtdinov R, Rio J, Sarro E, Moreno M, Castillo J, Navarro A, Montalban X, Comabella M. Roles of the ubiquitin peptidase USP18 in multiple sclerosis and the response to interferon-beta treatment. Eur J Neurol. 2013;20:1390–1397. doi: 10.1111/ene.12193. [DOI] [PubMed] [Google Scholar]
- Mildner A, Mack M, Schmidt H, Bruck W, Djukic M, Zabel MD, Hille A, Priller J, Prinz M. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. 2009;132:2487–2500. doi: 10.1093/brain/awp144. [DOI] [PubMed] [Google Scholar]
- Musso G, Gambino R, Cassader M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu Rev Med. 2011;62:361–380. doi: 10.1146/annurev-med-012510-175505. [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K, Boddeke HW. Identification of a microglia phenotype supportive of remyelination. Glia. 2012;60:306–321. doi: 10.1002/glia.21266. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van HC, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, Merkler D, Detje C, Gutcher I, Mages J, Lang R, Martin R, Gold R, Becher B, Bruck W, Kalinke U. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity. 2008;28:675–686. doi: 10.1016/j.immuni.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Prinz M, Knobeloch KP. Type I interferons as ambiguous modulators of chronic inflammation in the central nervous system. Front Immunol. 2012;3:67. doi: 10.3389/fimmu.2012.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- Raasch J, Zeller N, van Loo G, Merkler D, Mildner A, Erny D, Knobeloch KP, Bethea JR, Waisman A, Knust M, Del Turco D, Deller T, Blank T, Priller J, Bruck W, Pasparakis M, Prinz M. I{kappa}B kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-{kappa}B in the central nervous system. Brain. 2011;134:1184–1198. doi: 10.1093/brain/awq359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, Lash J, Wider C, Wojtas A, DeJesus-Hernandez M, Adamson J, Kouri N, Sundal C, Shuster EA, Aasly J, MacKenzie J, Roeber S, Kretzschmar HA, Boeve BF, Knopman DS. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2012;44:200–205. doi: 10.1038/ng.1027. et al ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Reiley WW, Zhang M, Jin W, Losiewicz M, Donohue KB, Norbury CC, Sun SC. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat Immunol. 2006;7:411–417. doi: 10.1038/ni1315. [DOI] [PubMed] [Google Scholar]
- Reiley WW, Jin W, Lee AJ, Wright A, Wu X, Tewalt EF, Leonard TO, Norbury CC, Fitzpatrick L, Zhang M, Sun SC. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med. 2007;204:1475–1485. doi: 10.1084/jem.20062694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richer E, Prendergast C, Zhang DE, Qureshi ST, Vidal SM, Malo D. N-ethyl-N-nitrosourea-induced mutation in ubiquitin-specific peptidase 18 causes hyperactivation of IFN-alpha signaling and suppresses STAT4-induced IFN-gamma production, resulting in increased susceptibility to Salmonella typhimurium. J Immunol. 2010;185:3593–3601. doi: 10.4049/jimmunol.1000890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie KJ, Malakhov MP, Hetherington CJ, Zhou L, Little MT, Malakhova OA, Sipe JC, Orkin SH, Zhang DE. Dysregulation of protein modification by ISG15 results in brain cell injury. Genes Dev. 2002;16:2207–2212. doi: 10.1101/gad.1010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santin I, Eizirik DL. Candidate genes for type 1 diabetes modulate pancreatic islet inflammation and beta-cell apoptosis. Diabetes Obes Metab. 2013;15(Suppl. 3):71–81. doi: 10.1111/dom.12162. [DOI] [PubMed] [Google Scholar]
- Schulz S, Chachami G, Kozaczkiewicz L, Winter U, Stankovic-Valentin N, Haas P, Hofmann K, Urlaub H, Ovaa H, Wittbrodt J, Meulmeester E, Melchior F. Ubiquitin-specific protease-like 1 (USPL1) is a SUMO isopeptidase with essential, non-catalytic functions. EMBO Rep. 2012;13:930–938. doi: 10.1038/embor.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, Haegebarth A, Kasper M, Jaks V, van Es JH, Barker N, van de Wetering M, van den Born M, Begthel H, Vries RG, Stange DE, Toftgard R, Clevers H. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science. 2010;327:1385–1389. doi: 10.1126/science.1184733. [DOI] [PubMed] [Google Scholar]
- Sun SC. Deubiquitylation and regulation of the immune response. Nat Rev Immunol. 2008;8:501–511. doi: 10.1038/nri2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann JB, Hayakawa Y, Zerafa N, Sheehan KC, Scott B, Schreiber RD, Hertzog P, Smyth MJ. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol. 2007;178:7540–7549. doi: 10.4049/jimmunol.178.12.7540. [DOI] [PubMed] [Google Scholar]
- Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- Vogel SN, Fertsch D. Endogenous interferon production by endotoxin-responsive macrophages provides an autostimulatory differentiation signal. Infect Immun. 1984;45:417–423. doi: 10.1128/iai.45.2.417-423.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieghofer P, Knobeloch KP, Prinz M. Genetic targeting of microglia. Glia. 2015;63:1–22. doi: 10.1002/glia.22727. [DOI] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, Tezcan I, Rice GI, Chen C, Mansouri N, Mahdaviani SA, Itan Y, Boisson B, Okada S, Zeng L, Wang X, et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature. 2015;517:89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets of all microarray analyses are deposited at Gene Expression Omnibus (GEO) (GSE61501).