

Abstract
In response to DNA damage, checkpoint signalling protects genome integrity at the cost of repressing cell cycle progression and DNA replication. Mechanisms for checkpoint down-regulation are therefore necessary for proper cellular proliferation. We recently uncovered a phosphatase-independent mechanism for dampening checkpoint signalling, where the checkpoint adaptor Rad9 is counteracted by the repair scaffolds Slx4-Rtt107. Here, we establish the molecular requirements for this new mode of checkpoint regulation. We engineered a minimal multi-BRCT-domain (MBD) module that recapitulates the action of Slx4-Rtt107 in checkpoint down-regulation. MBD mimics the damage-induced Dpb11-Slx4-Rtt107 complex by synergistically interacting with lesion-specific phospho-sites in Ddc1 and H2A. We propose that efficient recruitment of Dpb11-Slx4-Rtt107 or MBD via a cooperative ‘two-site-docking’ mechanism displaces Rad9. MBD also interacts with the Mus81 nuclease following checkpoint dampening, suggesting a spatio-temporal coordination of checkpoint signalling and DNA repair via a combinatorial mode of BRCT-domains interactions.
Keywords: BRCT domain, checkpoint, DNA damage, Dpb11, Slx4
Introduction
The ability of eukaryotic cells to properly respond to genotoxic insults heavily relies on DNA damage checkpoint (DDC) signalling. Upon detection of a DNA lesion, triggering of a checkpoint response results in inhibitory effects on cell cycle progression (Weinert & Hartwell, 1988) and DNA synthesis (Santocanale & Diffley, 1998; Lopez-Mosquedaet al, 2010; Zegerman & Diffley, 2010), accompanied by transcriptional reprogramming (Allenet al, 1994; Huanget al, 1998; Bastos de Oliveiraet al, 2012; Travesaet al, 2012) and striking elevations in dNTP levels (Zhou & Elledge, 1993; Zhaoet al, 2001; Zhao & Rothstein, 2002; Leeet al, 2008; Davidsonet al, 2012). More recently, DDC has been proposed to modulate the action of nucleases such as Exo1 and Mus81 (Kaiet al, 2005; Morinet al, 2008; Szakal & Branzei, 2013). Collectively, checkpoint functions help protect replication fork integrity and coordinate replication and DNA repair with other cell cycle events such as chromosome segregation (Branzei & Foiani, 2009). In budding yeastSaccharomyces cerevisiae, DDC is orchestrated mainly through the action of the upstream sensor kinase Mec1 (mammalian ATR), which then activates the downstream kinase Rad53 (functional analog of both mammalian CHK1/CHK2) for enforcement of a canonical checkpoint response. Transduction of signalling from Mec1 to Rad53 is mediated by protein adaptors, such as Rad9 and Mrc1, whose phosphorylation by Mec1 creates a docking site for the recruitment of Rad53 to DNA lesions (Sunet al, 1998; Durocheret al, 2000; Alcasabaset al, 2001; Schwartzet al, 2002; Pellicioli & Foiani, 2005). Similarly, in mammals, signalling from ATR and the related kinase ATM to CHK1/CHK2 also requires the action of checkpoint adaptors such as MDC1, BRCA1 and Claspin (Cimprich & Cortez, 2008). Of importance, mutations in checkpoint kinases and adaptors have been associated with tumorigenesis and neurological disorders (Shiloh, 2003).
Recruitment of checkpoint adaptors to sites of lesions is a key step in mounting a checkpoint response. Differently than the Mec1/ATR kinase, which is recruited via interaction of its cofactor Ddc2/ATRIP with ssDNA-bound RPA, recruitment of checkpoint adaptors is a more complex and regulated process that involves multiple, and partially redundant, mechanisms (Ohouo & Smolka, 2012). For example, recruitment of the yeast Rad9 adaptor has been shown to be mediated by three distinct mechanisms: (i) binding of the tudor domain of Rad9 to methylated histone H3K79 (Wysockiet al, 2005), (ii) binding of BRCT (for BRCA1 C-Terminal homology) domains of Rad9 to histone H2A phosphorylated at serine 129 (phospho-H2A) (Hammetet al, 2007) and (iii) recognition of the phosphorylation sites in Rad9 (serine 462 and threonine 474) by BRCT domains 1/2 in Dpb11 (Pfander & Diffley, 2011). Notably, these three modes of adaptor recruitment are spatiotemporally distinct. H3K79 methylation is a constitutive mark spread throughout euchromatin (Nguyen & Zhang, 2011); phospho-H2A is induced by DNA damage and, in the case of double-strand break (DSB), has been shown to spread kilobases around the lesion (Shroffet al, 2004); and the Dpb11 adaptor appears to be more specifically located at the site of lesion, by binding to a damage-induced phosphorylation in the Ddc1 subunit of the 9-1-1 complex (Pudduet al, 2008). This arrangement of anchoring points likely allows recruitment of Rad9 to be highly regulated, such that activation of DDC can be fine-tuned to match the extent and type of DNA lesion.
We have recently proposed that regulation of Rad9 displacement from the site of lesion is also used as a regulatory mechanism, in this case to modulate down-regulation of DDC (Ohouoet al, 2013). We have shown that the Slx4 and Rtt107 repair scaffolds counteract Rad9-dependent activation of Rad53 and proposed that the Slx4-Rtt107 complex balances the engagement of Rad9 at sites of lesions by interacting with phospho-H2A and Dpb11 thus displacing Rad9. By competing with Rad9 that is engaged at a lesion site, Slx4 and Rtt107 dampen DDC signalling, providing a phosphatase-independent mechanism for down-regulating Rad53 activation that we named DAMP (dampens adaptor-mediated phospho-signalling). Interestingly, several of the protein–protein interactions important for establishing the DAMP mechanism appear to involve BRCT domains in at least three different proteins: Rad9, Dpb11 and Rtt107 (Fig1A). BRCT domains often recognize phosphorylated motifs in targeted proteins and are commonly found in proteins involved in DNA damage signalling and repair (Reinhardt & Yaffe, 2013).
Figure 1.
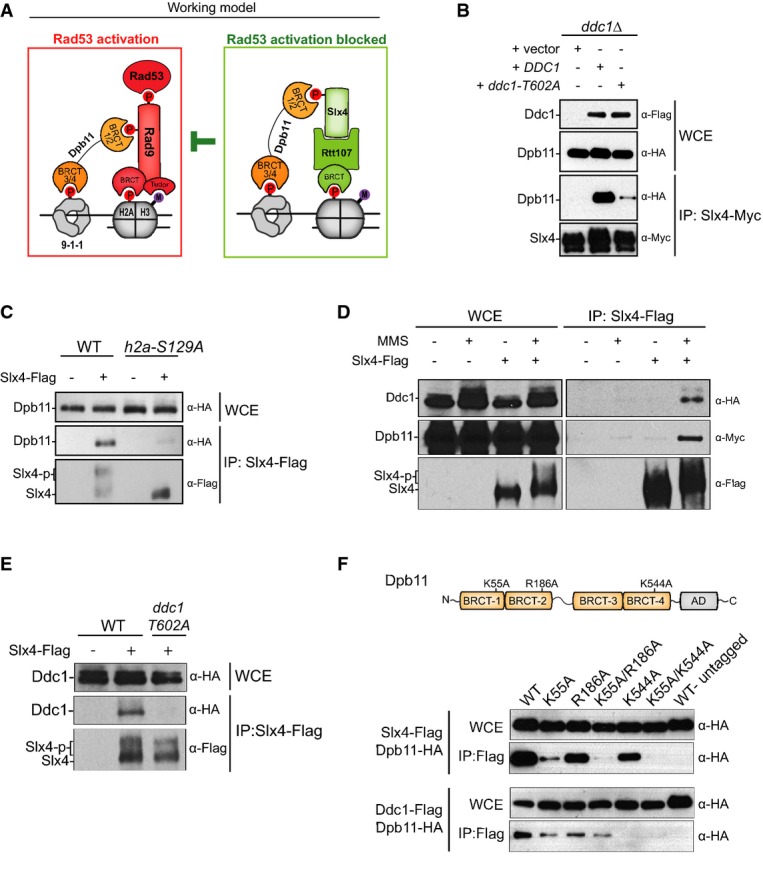
Dpb11 bridges Slx4 to the 9-1-1 complex
A Working model for how the Slx4-Rtt107 complex modulates Rad53 signalling.
B Coimmunoprecipitation (CoIP) between Slx4-Myc and Dpb11-HA in MMS-treated cells lacking Ddc1 or expressing either wild-type or the T602A mutant form of Ddc1.
C CoIP between Slx4-Flag and Dpb11-HA in MMS-treated cells expressing either wild-type or the S129A mutant form of histone H2A.
D, E CoIP between Slx4-Flag and Ddc1-HA in MMS-treated cells. In (D), a plasmid was used for ectopic expression of Dpb11-Myc via anADH1 promoter. In (E), Dpb11 was expressed from the endogenousDPB11 locus and driven by its own promoter.
F Dpb11 containing the mutations indicated in the schematic illustration (see also Supplementary Fig S2) was expressed from a plasmid in yeast cells containing Flag-tagged Slx4 or Ddc1 for CoIP experiments. AD: activation domain.
Data information: For (B, C), cells were treated with MMS 0.02% for 2 h. For (D, E, F), cells were incubated with MMS 0.033% for 2 h where indicated. Source data are available online for this figure.
Failure to down-regulate the DDC strongly represses cell proliferation (Clericiet al, 2001). In yeast for example, Rad53 hyperactivation caused by lack of the Rad53 phosphatase Pph3 or by lack of Slx4 has been shown to increase sensitivity to genotoxins (O'Neillet al, 2007; Ohouoet al, 2010). Given the importance of proper checkpoint down-regulation, it is not surprising that multiple mechanisms for down-regulating checkpoint exist. While the role of phosphatases such as Pph3, Ptc2 and Ptc3 in down-regulating Rad53 signalling has been well documented [for review, see Heidekeret al (2007)], much less is known about the DAMP mechanism. Here, we uncovered the molecular determinants of the DAMP mechanism and show that it is established via cooperative interactions mediated by BRCT domains in Rtt107 and Dpb11. We have engineered a minimal multi-BRCT-domain (MBD) module that fully recapitulates the action of the Dpb11-Slx4-Rtt107 complex in dampening checkpoint signalling. Efficient recruitment of Dpb11-Slx4-Rtt107 or MBD via a cooperative ‘two-site-docking’ mechanism explains how these BRCT-domain modules can displace the checkpoint adaptor Rad9 and down-regulate Rad53 signalling. We propose that this two-site-docking mechanism may be a general strategy for efficient and timely recruitment of multi-BRCT-domain protein complexes to DNA lesion sites.
Results
A working model for Slx4-Rtt107-dependent dampening of Rad53 signalling
We have previously shown that the Slx4-Rtt107 complex down-regulates Rad53 signalling by counteracting the Rad9 adaptor (Ohouoet al, 2013). We proposed that Slx4 and Rtt107 counteract Rad9 by binding to the Dpb11 scaffold and to phosphorylated histone H2A, both of which also interact with Rad9 to promote Rad53 activation at DNA lesions (Fig1A). In our proposed model, BRCT domains of Dpb11 play a central role in mediating the competitive recruitment of Rad9 or Slx4-Rtt107 to the site of lesion where the 9-1-1 complex is loaded. While the pair of BRCT domains 3/4 of Dpb11 binds to a phosphorylated residue in the 9-1-1 complex (threonine 602 in the Ddc1 subunit) (Pudduet al, 2008), the pair of BRCT domains 1/2 of Dpb11 was found to bind to phosphorylation sites in either Rad9 (Pfander & Diffley, 2011) or Slx4 (Ohouoet al, 2013). Here, we thoroughly tested the model depicted in Fig1A and provide multiple lines of evidence supporting it. First, we predicted that efficient formation of the damage-induced Dpb11-Slx4-Rtt107 complex at lesion sites would be mediated, cooperatively, by Mec1 phosphorylation sites on H2A and Ddc1, which are expected to form anchoring points at the site of lesion. Consistent with this model, we found that mutation of either of these phosphorylation sites significantly reduced the Slx4-Dpb11 interaction (Fig1B and C). Next, to test whether Dpb11 may indeed bridge Slx4 to Ddc1, as predicted in our model, we pulled down Slx4 and monitored the recovery of Ddc1. As shown in Fig1D, we were able to specifically recover Ddc1 from Slx4 immunoprecipitates, consistent with our bridging model shown in Fig1A. Noteworthy, the formation of the ternary complex (Ddc1-Dpb11-Slx4) is dependent on DNA damage (Fig1D) and on phosphorylation of threonine 602 in Ddc1 (Fig1E) previously shown to be recognized by BRCT 3/4 of Dpb11 (Wang & Elledge, 2002; Pudduet al, 2008). While these results support the model that Dpb11 bridges Slx4 to Ddc1 by recognizing these proteins via BRCT 1/2 and BRCT 3/4, respectively, a recent report showed that Dpb11 interaction with Slx4 can also be mediated by BRCT 3/4 (Gritenaiteet al, 2014). Therefore, we performed experiments to check which BRCT domain plays a major role in mediating the Slx4-Dpb11 interaction in MMS-treated cells. As shown in Supplementary Fig S1, while we could indeed detect Slx4 in a pull-down using BRCT 3/4, we were able to detect significantly more Slx4 by pulling down BRCT 1/2. In addition, we constructed a series of Dpb11 mutants predicted to disrupt different BRCT domains [K55A in BRCT-1; R186A in BRCT-2 and K544A in BRCT-4; see Supplementary Fig S2 and Quet al (2013) for details] and tested their binding to Slx4 when expressed in yeast cells following MMS treatment. Combined mutations in BRCT domains 1 and 2 (K55A/R186A) almost completely abolished the interaction with Slx4 (Fig1F), further supporting that BRCT 1/2 plays a major role in the Slx4 interaction. Mutation in BRCT-4 moderately reduced interaction of Dpb11 with Slx4, which is also consistent with our model, as we showed in Fig1B that recruitment of Dpb11 to Ddc1 helps promote robust Dpb11 interaction with Slx4. On the other hand, interaction of Dpb11 with Ddc1 was strongly affected by mutation in BRCT-4, as expected, and only mildly reduced by mutations in BRCT 1/2, which could also play a role in promoting efficient recruitment of Dpb11 to Ddc1 based on our model. Collectively, these results support the idea that Dpb11 functions to bridge Slx4 to the 9-1-1 complex, providing the basis for a competition-based model as illustrated in Fig1A. We do not exclude the possibility that Dpb11 may also engage into distinct modes of BRCT-mediated interactions with Rad9 or Slx4, as recently proposed for the Crb2-Rad4 (orthologs of Rad9 and Dpb11, respectively) interaction inSchizosaccharomyces pombe (Quet al, 2013).
Functional rescue ofslx4Δ cells via an Slx4-Dpb11 chimera
To functionally test our model and determine the minimal requirements for the ability of Slx4-Rtt107 to counteract Rad9 at lesion sites, we used a Dpb11-Slx4 chimera by fusing full-length Dpb11 with the C-terminal portion of Slx4 containing the S486A mutation as well as mutation of 7 SQ/TQ sites (7MUT) (Fig2A). We have previously shown that the S486A and the 7MUT mutations in Slx4 impair binding to Dpb11 and cause Rad53 hyperactivation and MMS sensitivity (Ohouoet al, 2010, 2013). If the major role of the Dpb11-Slx4 interaction is indeed to bridge Slx4-Rtt107 to 9-1-1, the expectation is that the Dpb11-Slx4S486A/7MUT chimera will rescue MMS sensitivity ofslx4Δ cells back to wild-type levels. As predicted, this chimera was able to rescue the MMS sensitivity of anslx4Δ strain, bypassing the inability of Slx4S486A/7MUT to bind to Dpb11 (Fig2B) (see also Gritenaiteet al, 2014). Interestingly, MMS sensitivity was rescued by a chimera carrying a mutation in BRCT-1 of Dpb11 (K55A) but not by a chimera with mutated BRCT-4 of Dpb11 (K544A). These results are consistent with the model that the Dpb11-Slx4S486A/7MUT chimera rescues the MMS sensitivity ofslx4Δ cells by bridging Rtt107 to 9-1-1 and displacing the Dpb11-Rad9 complex. Further supporting this model, we observed that both the Dpb11-Slx4S486A/7MUT and Dpb11K55A-Slx4S486A/7MUT chimeras underwent extensive MMS-induced phosphorylation, as measured by reduced electrophoretic mobility, while the Dpb11K544A-Slx4S486A/7MUT chimera did not undergo reduced mobility shift (Fig2C). We interpret this MMS-induced phosphorylation as an indication of recruitment of the chimera to sites of DNA lesions, as we previously showed that Mec1 and Rad53 extensively phosphorylate Dpb11 and Slx4 (Ohouoet al, 2010). Of importance, increased phosphorylation of the Dpb11-Slx4S486A/7MUT chimera inversely correlated with phosphorylation of endogenous Dpb11 (Fig2C), in agreement with the notion that recruitment of the chimera and the Dpb11-Rad9 complex is mutually exclusive. Finally, mutation of the phosphorylation site in H2A (S129) or in Ddc1 (T602), which are directly recognized by BRCT domains 5/6 of Rtt107 or BRCT domains 3/4 of Dpb11, respectively, significantly reduced the MMS-induced mobility shift of the chimera (Fig2D and E). Taken together, these results are consistent with our proposed model for how Slx4-Rtt107 engage at sites of lesions.
Figure 2.
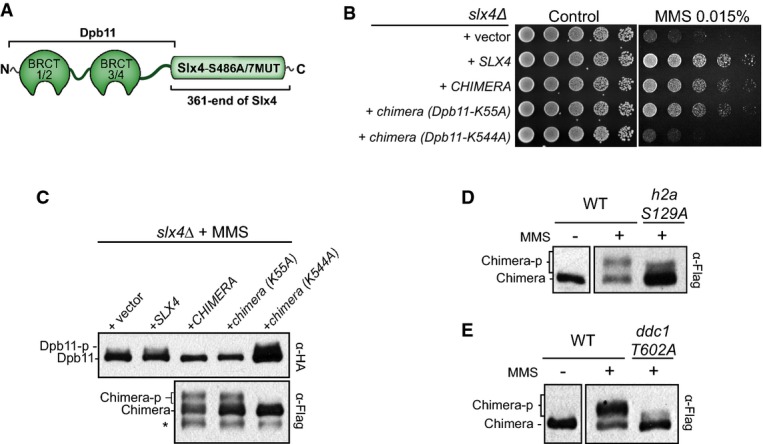
A Dpb11-Slx4S486A/7MUT chimera confers resistance to MMS treatment
A Schematic illustration of the Dpb11-Slx4S486A/7MUT chimera.
B Effect of the Dpb11-Slx4S486A/7MUT chimera and the indicated mutants in the MMS sensitivity of anslx4Δ strain. Fourfold serial dilutions were spotted on SC-URA plates and grown for 2–3 days at 30°C.
C Immunoblots showing the phosphorylation status of Dpb11-HA (endogenously expressed copy) and Dpb11-Slx4S486A/7MUT chimera-Flag after MMS treatment (0.02% for 2 h).
D, E Immunoblots showing the phosphorylation status of the Dpb11-Slx4S486A/7MUT chimera-Flag in the indicated strains after MMS treatment (MMS 0.01% for 2 h).
Source data are available online for this figure.
A minimal multi-BRCT-domain module recapitulates the function of Slx4-Rtt107 in dampening Rad53 signalling
As shown in our working model (Fig1A), the Dpb11-Rad9 and the Dpb11-Slx4-Rtt107 complexes are both anchored at a DNA lesion via two Mec1-targeted phospho-sites, Ddc1 T602 and H2A S129. To further test the notion that a key role of the Dpb11-Slx4-Rtt107 complex is to bridge 9-1-1 to H2A and compete out Rad9 from sites of lesions, we fused BRCT 5/6 of Rtt107 to BRCT 3/4 of Dpb11 (Fig3A) with the prediction that this minimal multi-BRCT-domain module (herein referred as MBD) should compete out Rad9 and, consequently, rescue to some extent the MMS sensitivity of a strain lacking Slx4. Remarkably, expression of MBD fully rescued the MMS sensitivity ofslx4Δ cells (Fig3B), which correlated with decreased phosphorylation of Rad53, Dpb11 and Rad9, as well as reduced Dpb11-Rad9 interaction (Fig3C and D). Of note, the mild sensitivity ofslx4Δ cells to the UV-mimetic 4-NQO was also fully rescued by MBD (Supplementary Fig S3). Importantly, in these experiments, MBD expression was driven by the relatively weakDPB11 promoter [Dpb11 protein is expressed at around 400 copies per cell (Mantieroet al, 2011)]. Furthermore, a point mutation in either pairs of BRCT domains in MBD was sufficient to prevent the ability of MBD to rescue the MMS sensitivity ofslx4Δ cells (Fig3E) and an alternative chimera containing the BRCT domain 1/2 of Dpb11 (instead of the BRCT domain 3/4) fused to BRCT 5/6 of Rtt107, herein referred as alt-MBD, could not rescue the MMS sensitivity ofslx4Δ cells (Supplementary Fig S4). As predicted by our model, MBD was capable of robustly interacting with Ddc1, but not Slx4, in a CoIP experiment (Fig3F). Finally, as Slx4 also plays established roles in DNA repair in conjunction with the structure-specific nuclease Slx1 (Fricke & Brill, 2003), we addressed whether MBD specifically rescues the checkpoint regulatory function of Slx4 but not the Slx4 function in Slx1-mediated DNA repair. For that, we examined whether MBD is able to rescue the lethality caused by deletion of bothSLX4 andSGS1, a helicase also known to display synthetic lethality with Slx1 (Mullenet al, 2001). Our prediction was that MBD would not rescue this synthetic lethality, for the following reasons: (i) while deletion ofSLX1 also results in synthetic lethality withsgs1Δ, Slx1 has no role in the DAMP mechanism (Ohouoet al, 2013); (ii) lack of Rtt107, a protein that is required for DAMP (Fig1A), does not result in lethality insgs1Δ cells (Zappullaet al, 2005). Indeed, we found that expression of MBD does not rescue the lethality of cells lackingSLX4 andSGS1 (Fig3G). Taken together, these results support our model that Slx4 has an important role in checkpoint down-regulation that is independent from its role in Slx1-mediated DNA repair and show that the MBD-mediated rescue is dependent on the cooperative action of both BRCT 3/4 of Dpb11 and BRCT 5/6 of Rtt107, mimicking what occurs by the phospho-mediated assembly of the Dpb11-Slx4-Rtt107 complex. We therefore propose that a ‘two-site-docking’ mechanism established by cooperative interactions of two pairs of BRCT domains with two lesion-specific anchoring points (Ddc1-T602 and H2A-S129) promotes efficient recruitment of the Dpb11-Slx4-Rtt107 complex to sites of lesions to displace Rad9. The results are consistent with the idea that the cooperative nature of multi-BRCT domain interactions, as shown for MBD, is important for these modules to have a robust and highly specific effect at sites of lesions.
Figure 3.
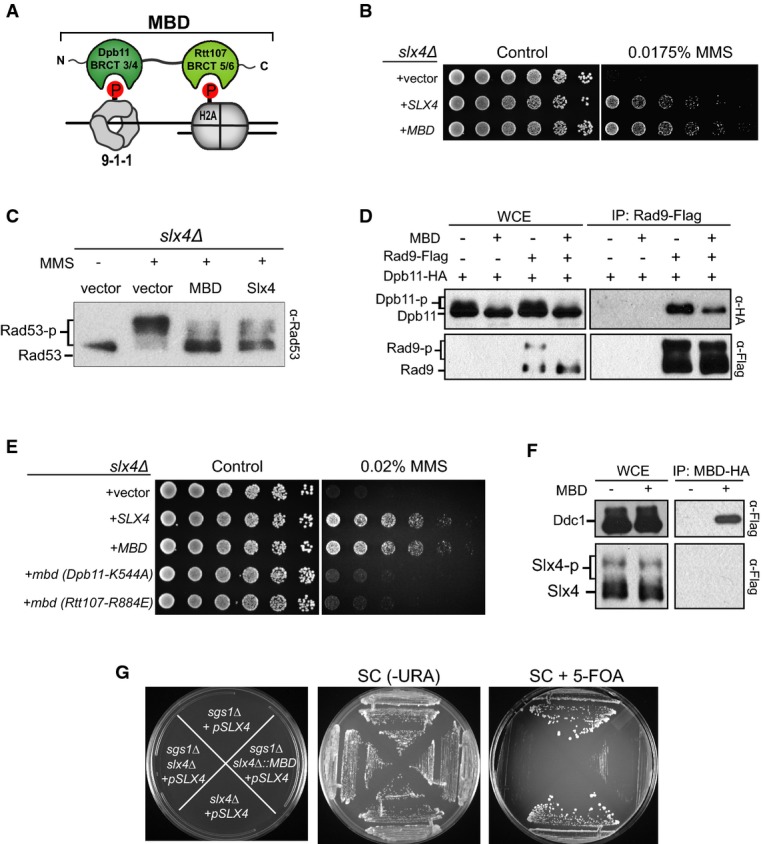
- Schematic illustration of the MBD (Minimal multi-BRCT-domain module) chimera.
- Effect of MBD expression on the MMS sensitivity ofslx4Δ cells.
- Immunoblot showing Rad53 phosphorylation status inslx4Δ cells expressing either MBD or Slx4 from apRS416 plasmid.
- CoIP between Dpb11 and Rad9 in wild-type cells after MMS treatment, in the presence or absence of a plasmid expressing MBD.
- Effect of expressing MBD or the indicated mutants on the MMS sensitivity ofslx4Δ cells. Mutations in MBD indicated in parenthesis correspond to the equivalent residue in the full-length Dpb11 or Rtt107 proteins.
- CoIP of MBD-HA with Ddc1-Flag and Slx4-Flag upon MMS treatment.
- Counter-selection of indicated strains bearing an Slx4-expressing plasmid (pMBS213) containing theURA3 gene in 5-FOA plates.
Modulation of Rad53 signalling via MBD is dosage dependent, localized and specific for the Rad9 branch of DDC signalling
To better understand additional features of MBD-mediated modulation of DDC signalling, we asked whether MBD action is dosage dependent. We expressed MBD with theCPY andADH1 promoters (Fig4A), which are stronger than theDPB11 promoter, and are referred here as C-MBD and A-MBD, respectively. As shown in Fig4B, while C-MBD could still efficiently rescue MMS sensitivity ofslx4Δ cells, A-MBD could only provide partial rescue. The high level of MBD expression through theADH1 promoter was correlated with stronger reduction in Rad53 and Rad9 phosphorylation (Fig4C), and expression of A-MBD conferred enhanced MMS sensitivity in wild-type cells (Fig4D). Also, we observed reduced levels of checkpoint-dependent Slx4 phosphorylation in wild-type cells expressing A-MBD (Supplementary Fig S5). Taken together, these results show that modulation of DDC signalling via MBD is dosage dependent and that increasing levels of MBD prevent a proper level of Rad53 activation to be reached.
Figure 4.

- Immunoblot showing MBD-HA expression under the control of different promoters.
- Effect of increased MBD expression on the MMS sensitivity ofslx4Δ cells.
- Immunoblots showing the effect of increased MBD expression on Rad53 and Rad9 phosphorylation status after MMS treatment.
- Effect of increased MBD expression on the MMS sensitivity of wild-type cells.
- Effect of MBD expression levels on the number of Dpb11, Rad9 or Slx4 foci. Foci were scored manually, and for the nuclear foci formation experiments, the means and standard deviations from three independent experiments are shown.
- Effect of C-MBD expression on the sensitivity ofmrc1Δ cells to replication stress.
- Same number of WT ormrc1Δ cells were transformed with equimolar concentrations of vector control or vector for the expression of A-MBD and directly grown on SC-URA plates.
Next, to address how specific the action of MBD is at DNA lesions, we monitored the effect of MBD expression on localization of Slx4, Dpb11 and Rad9 to sub-nuclear foci. Interestingly, MBD expression resulted in a decrease in the number of Dpb11 foci but did not affect the number of Rad9 or Slx4 foci (Fig4E). This result is consistent with the notion that MBD is specifically impairing the recruitment of Dpb11, which is dependent on the 9-1-1 complex that is loaded at the 5′ end of a double-/single-stranded DNA junction. On the other hand, MBD expression does not impact Slx4-Rtt107 and Rad9 as they can also be recruited via phospho-H2A (Hammetet al, 2007; Liet al, 2012), which likely forms a long platform spreading kilobases around the site of lesion (Downset al, 1999; Shroffet al, 2004). In addition, Rad9 can also be recruited via methylated H3K79 that is believed to be constitutively present throughout chromatin (Grenonet al, 2007). These findings reveal the specificity of MBD action and support the model that the Dpb11-mediated engagement of Rad9 or Slx4 to the 9-1-1 complex is the key point of regulation during checkpoint dampening. Consistent with this model, we could detect a robust interaction between MBD and Ddc1 in a CoIP experiment (Fig3F). Finally, to confirm the prediction that MBD is specifically and efficiently affecting the Rad9 branch of the pathway for Rad53 activation, we expressed MBD inmrc1Δ cells and noticed that it rendered cells extremely sensitive to replication stress (Fig4F). It is well established that inmrc1Δ cells, the activation of the checkpoint is dependent on Rad9 (Alcasabaset al, 2001). Remarkably, overexpression of MBD with theADH1 promoter resulted in a strong growth defect inmrc1Δ cells even in the absence of genotoxins (Fig4G), suggesting that the essential function of Rad53 is being affected. Collectively, these data further support the initial model depicted in Fig1A and reveal key features of checkpoint dampening via a multi-BRCT-module that mimics the action of the Dpb11-Slx4-Rtt107 complex.
MBD modulates Rad53 signalling independently of Mus81-Mms4
Activation of Rad53 has been proposed to antagonize the function of the Mus81 nuclease (Szakal & Branzei, 2013). In this manner, Rad53 hyper-activation inslx4Δ cells would lead to persistent repression of Mus81 function. Consistent with this notion, we found that expression of MBD inslx4Δ cells rescues chromosomal defects seen by electrophoretic analysis of chromosome plugs after transient exposure to MMS (Fig5A). This defect is characteristic ofmus81Δ cells (Saugaret al, 2013), and in cells lackingSLX4, such defect has been attributed to inefficient resolution of joint molecules (JMs) formed between sister chromatids, with the idea that Slx4 helps facilitate Mus81 function (Gritenaiteet al, 2014). Consistent with this view, we found that expression of MBD could not provide any rescue of MMS sensitivity in cells lacking both Slx4 and Mus81 (Fig5B). Our findings therefore support that MBD is promoting Mus81 function by dampening Rad53 activation. It has been proposed that inhibition of Mus81 by Rad53 signalling is established indirectly, via regulation of the CDK and Cdc5 kinases (Szakal & Branzei, 2013). However, a recent report showed that Dpb11 can physically interact with the Mms4 subunit of the Mus81-Mms4 complex specifically during G2/M (Gritenaiteet al, 2014). The authors proposed that formation of an Slx4-Rtt107-Dpb11-Mms4-Mus81 complex has a role in directly regulating the action of Mus81-Mms4 for the resolution of DNA repair intermediates formed in response to MMS treatment. We found that MBD can interact with Mms4 in G2/M cells (Fig5C). Of note, a point mutation in MBD corresponding to K544A in Dpb11 (BRCT-4 of Dpb11) significantly reduced interaction of MBD with Mms4, indicating that the pair of BRCT 3/4 of Dpb11 possibly recognizes phosphorylated Mms4. Strengthening this notion, we could not detect Mms4 when pulling down alt-MBD (which has Dbp11 BRCT 1/2 instead of 3/4) (Supplementary Fig S6). These results, in combination with our data showing that Dpb11 BRCT 1/2 plays a prominent role in mediating the interaction with Slx4, help explain how an Slx4-Rtt107-Dpb11-Mms4-Mus81 complex is formed in G2/M. Furthermore, these results reveal the highly coordinated nature of the cell cycle-specific interactions that are mediated by the BRCT domains of Dpb11. Importantly, we decided to rule out the possibility that regulation of Rad53 activation via MBD expression could somehow be mediated by the interaction of MBD with Mms4. We monitored Rad53 activation in cells lacking Mus81 and/or Slx4 and found that MBD could still dampen checkpoint signalling (Fig5D). These results show that lack of Mus81 does not lead to Rad53 hyperactivation and that dampening of checkpoint signalling via MBD is independent of Mus81. Altogether, these findings support our working model of how Dpb11-Slx4-Rtt107 complex and MBD promote the DAMP mechanism. It remains to be determined whether the ability of MBD to interact with Mms4 has any role in the resolution of MMS-induced JMs independently of Rad53 regulation.
Figure 5.
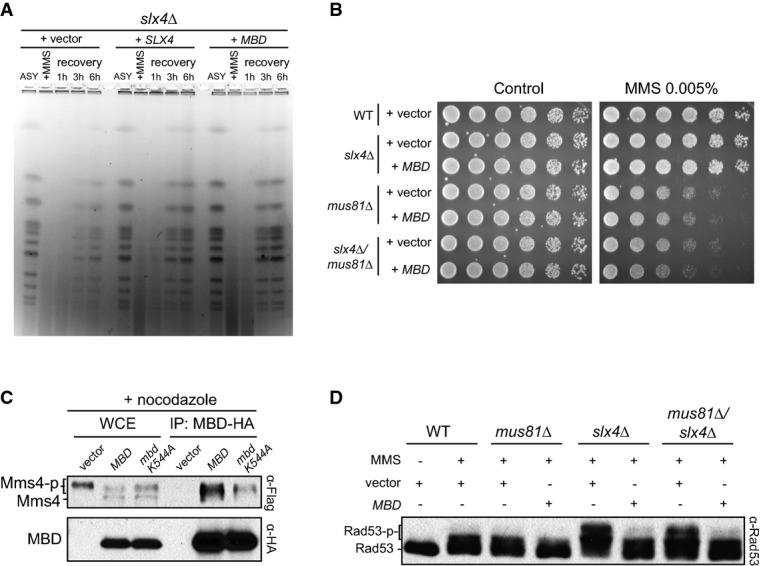
- Asynchronous (ASY) cells were treated with 0.033% MMS for 2 h and then released in MMS-free media. Recovery of fully replicated chromosomes was analysed by pulse-field gel electrophoresis (PFGE).
- Effect of MBD expression on MMS sensitivity of indicated cells.
- CoIP experiment showing the binding of Mms4 to MBD or MBD containing a mutation (corresponding to K544A in Dpb11) shown to destabilize BRCT-4. Cells were grown until log phase and then arrested in metaphase with nocodazole.
- Immunoblot showing Rad53 phosphorylation status after MMS treatment (0.01% for 2 h) in the indicated strains expressing or not MBD.
Temporal dynamics of DAMP
While both MBD and the ‘physiological’ Dpb11-Slx4-Rtt107 complex are able to promote the DAMP mechanism, a key difference between them is that formation of the later module is regulated. The Slx4-Dpb11 interaction is dependent on CDK and checkpoint-mediated phosphorylation events (Ohouoet al, 2010, 2013; Gritenaiteet al, 2014), which are expected to impact establishment of the cooperative BRCT-mediated interactions and recruitment to sites of lesions. For example, stronger binding of Slx4 to Dpb11 should result in more cooperative binding to phosphorylated Ddc1-T602 and H2A-S129 at lesions and more efficient recruitment. On the other hand, MBD does not require phosphorylation to assemble a module capable of docking at both phosphorylated Ddc1-T602 and H2A-S129. As Slx4 requires CDK phosphorylation that builds up during S-phase to interact with Dpb11, we predicted that ‘physiological’ DAMP (via phospho-dependent Slx4-Dpb11 interaction) occurs later than a ‘synthetic’ DAMP (via MBD). Temporal analysis of the dynamics of Rad53 activation during S-phase progression in the presence of MMS showed that this is indeed the case, as Rad53 activation was delayed in MBD expressing cells (Fig6A). Notably, overexpression of MBD led to a stronger impairment of Rad53 activation.
Figure 6.
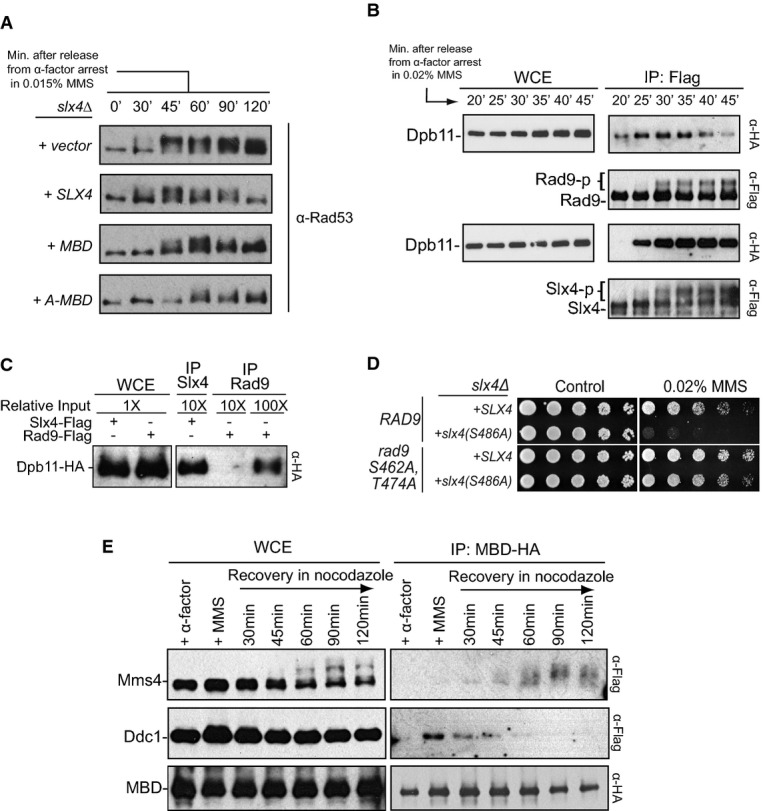
- Dynamics of Rad53 activation during S-phase in the presence of MBD.slx4Δ cells carrying plasmids expressing Slx4, MBD or A-MBD were arrested with alpha-factor and then released in media containing 0.015% MMS. Cells were collected at different time points following release.
- Temporal dynamics of the Dpb11-Rad9 and Dpb11-Slx4 interactions during early S-phase. Cells were arrested with alpha-factor and then released in media containing 0.02% MMS. Cells were collected starting 20 min following release, which coincides with the start of DNA synthesis.
- CoIP between Dpb11 and Rad9 or Slx4 from cells treated with 0.04% MMS for 1 h.
- Effect of the S462A and T474A mutations in Rad9 (known to disrupt Rad9 binding to Dpb11) on the MMS sensitivity of cells expressing theslx4-S486A mutant.
- Alpha-factor-arrested cells were released in MMS 0.02% for 45 min and then transferred to MMS-free media containing nocodazole for the indicated times. MBD was expressed from a plasmid (pMBS798) and monitored for its interaction with Ddc1 and Mms4 using CoIP.
To gain a better understanding of the temporal coordination of the events during ‘physiological’ DAMP, we monitored the Dpb11-Rad9 and Dpb11-Slx4 interactions in synchronized cells during replication stress response. We were able to detect the Rad9-Dpb11 interaction very early in S-phase, as early as 20 min following release of cells from an alpha-factor arrest (Fig6B). On the other hand, we could only detect the Dpb11-Slx4 interaction 25 min after release. Importantly, at later time points, we could observe a decrease in the Dpb11-Rad9 interaction, whereas the Dpb11-Slx4 interaction was sustained. This result suggests that there is a temporal coordination of Dpb11 interactions as replication forks encounter MMS-induced lesions. A conceivable model is that Dpb11 first binds to Rad9 to activate Rad53 at fork-proximal regions, and as forks bypass and progress far from the lesion site, Slx4 and Rtt107 are then recruited to displace Rad9 that is engaged onto the 9-1-1 complex (see working model in Supplementary Fig S7). Consistent with this model and the idea that Slx4 displaces Rad9, we could detect that the amount of Dpb11 co-immunoprecipitated with Slx4 is at least 10 times higher than the amount of Dpb11 co-immunoprecipitated with Rad9 after MMS treatment for 60 min (Fig6C). The overall abundance of Rad9 in the cell is higher than that of Slx4 (see input in Supplementary Fig S1A), strengthening the notion that 60 min in the replication stress response, Slx4 more avidly interacts with Dpb11 than Rad9 does. The importance of Slx4 in counteracting the action of the Dpb11-Rad9 complex is further supported by the fact that mutation of the two phospho-sites in Rad9 (S462 and T474) that mediate interaction with Dpb11 leads to the rescue of the MMS sensitivity of cells carrying the S486 mutation in Slx4 (Fig6D). Following checkpoint dampening, additional coordination of Dpb11 interactions is apparently established, as the Dpb11-Slx4-Rtt107 complex was reported to interact with Mus81-Mms4 in G2/M (Gritenaiteet al, 2014). As we have shown that both Ddc1 and Mms4 are recognized by BRCT 3/4 of Dpb11 (for Ddc1 see Supplementary Fig S1B and Fig1F, for Mms4 see Fig5C), we predicted that as the Dpb11-Mms4 interaction increases in G2/M, the Dpb11-Ddc1 interaction should decrease. To test this, we used MBD (which only has BRCT 3/4, but lacks BRCT 1/2 of Dpb11) as bait in experiments with cells arrested in MMS and released from MMS treatment in media containing nocodazole. While we could detect an increase in the MBD-Mms4 interaction following release of cells from MMS treatment into media containing nocodazole, the MBD-Ddc1 interaction was reduced (Fig6E). This is consistent with the model that these Dpb11 interactions are mutually exclusive. Overall, the findings reported here support the working model depicted in Fig1A for how the DAMP mechanism is established and help elucidate how Dpb11 may coordinate DDC signalling and DNA repair (Fig7).
Figure 7.
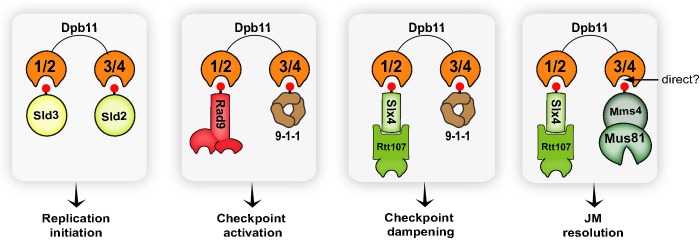
Dpb11 as a central multi-BRCT-domain module in the coordination of replication initiation, checkpoint signalling and DNA repair
See Discussion for details.
Discussion
Despite the importance of DDC for cell survival and genome maintenance, the inability to properly down-regulate a checkpoint response can severely compromise cell growth. Given the importance of DDC down-regulation, multiple mechanisms exist to modulate and fine-tune the activation state of checkpoint kinases. Our recent finding of the DAMP mechanism provides an example of a phosphatase-independent mechanism for counteracting checkpoint signalling (Ohouoet al, 2013). Here, we have examined the molecular requirements for this new mechanism of checkpoint regulation. We demonstrate that we can engineer a minimal BRCT-domain module that recapitulates the action of the Dpb11-Slx4-Rtt107 complex in down-regulating Rad53 signalling. The availability of this minimal module allowed us to rigorously test our working model for how DAMP works. Our findings support the existence of a competition-based mechanism through which Slx4 and Rtt107 counteract the checkpoint adaptor Rad9 to dampen checkpoint signalling.
DAMP is established via the cooperative action of BRCT domains
The fact that the minimal MBD module, which lacks anySLX4 sequence, can fully rescue the MMS sensitivity ofslx4Δ cells (Fig3B and E) strongly supports that BRCT domain-mediated interactions are the minimal requirement for establishing a DAMP mechanism. Our results are consistent with the notion that the well-established interactions of Rtt107's BRCT 5/6 with phospho-H2A (Williamset al, 2010; Liet al, 2012) and Dpb11's BRCT 3/4 with phospho-Ddc1 (Wang & Elledge, 2002; Pudduet al, 2008; Pfander & Diffley, 2011) are directly enforcing DAMP. Importantly, point mutations that disrupt only one of these BRCT interactions severely compromise the ability of the Dpb11-Slx4S486A/7MUT chimera to be robustly phosphorylated and to prevent phosphorylation of endogenous Dpb11 in response to DNA damage (Fig2C–E), likely reflecting the inability of the mutated chimera to be efficiently recruited to the site of lesion. Furthermore, disruption of one of the BRCT domains of the MBD is enough to abolish its ability to down-regulate Rad53 signalling (Fig3E). We therefore concluded that the combined action of these BRCT domain interactions confer the ability of MBD to efficiently compete with Rad9 and down-regulate DDC. In line with these findings, physiological DAMP would be established by the formation of a Dpb11-Slx4-Rtt107 complex capable of interacting with the phospho-H2A and phospho-Ddc1 anchoring points in a cooperative manner, similar to MBD. It is tempting to speculate that the coordination of these interactions provides advantages for precise spatio-temporal control of Slx4-Rtt107 recruitment. Because phospho-H2A may spread through kilobases around the site of lesion, while 9-1-1 is specifically loaded at the 5′ end of primer-template junction at the lesion site, we speculate that a long tract of phospho-H2A at chromatin serves as a platform for initial recruitment and to guide Dpb11-Slx4-Rtt107 (or MBD) to the lesion site. Interestingly, Rad9 is recruited through a similar mechanism, but because the Dpb11-Rad9 interaction is presumably more transient than the Dpb11-Slx4 interaction, the latter will be able to be more efficiently recruited.
A spatio-temporal model for DAMP
It is important to mention that the work presented here is focused on the response to MMS treatment, which generates replication blocks that can be bypassed by a moving replication fork, leaving the lesion and an ssDNA gap behind the fork (Branzei & Foiani, 2010). As exemplified in our detailed working model (Supplementary Fig S7), we propose that early in the response, as forks encounter a lesion, the Rad9 adaptor is rapidly recruited to the site of lesion to mediate Rad53 activation. At this initial stage, key events include the recruitment of Mec1 to the lesion and subsequent phosphorylation of histone H2A and Ddc1 by Mec1 to create the anchoring sites for the recruitment of Dpb11 and Rad9. We speculate that the ability of Rad9 to be constitutively bound to chromatin via the H3K79 methylation mark enables Rad9 to be rapidly recruited to the phospho-H2A platform and, subsequently, to the site of lesion. In contrast, Slx4 and Rtt107 do not recognize methylated H3K79, which may explain why these proteins are recruited later on (Fig6B). Following checkpoint activation, we propose that Slx4 and Rtt107 become more efficiently recruited and able to compete out Rad9 due to a presumably stronger interaction with Dpb11 (Fig6C). The rapid formation of the Rad53-activating complex (Ddc1-Dpb11-Rad9) at the fork is likely important for rapidly inhibiting nucleases, such as Exo1, from processing fork structures (Morinet al, 2008), in addition to arresting the cell cycle and inhibiting late origin firing. Once the replication fork bypasses the lesion and recombination-dependent intermediates accumulate (such as intermediates derived from template-switching events), checkpoint down-regulation would be important to allow cell cycle progression and proper execution of mitotic events such as the resolution of JMs via the Mus81 nuclease. Therefore, we propose that DAMP occurs post-replicatively to disengage Rad9 from the site of lesion and disrupt the Ddc1-Dpb11-Rad9 complex. Consistent with this idea, recent ChIP-seq data show that Slx4 and Rtt107 accumulate on chromatin behind replication forks as they progress through the genome of MMS-treated cells (A Balint, T Kim, D Gallo, JR Cussiol, FB Oliveira, A Yimit, J Ou, R Nakato, A Gurevich, K Shirahige, MB Smolka, Z Zhang, G Brown, manuscript submitted).
The Slx4-Dpb11 interaction facilitates Mus81-Mms4 action
Cells that lack DAMP (such asslx4Δ cells) are hypersensitive to MMS and accumulate JMs (Mullenet al, 2001; Gritenaiteet al, 2014). This is consistent with the idea that a major deleterious effect of Rad53 hyperactivation upon MMS treatment is the mis-regulation of the Mus81 nuclease, which has a prominent role in the resolution of MMS-induced JMs (Saugaret al, 2013; Szakal & Branzei, 2013). Indeed, DDC has been shown to inhibit Mus81 action, with the prevailing model that it negatively affects the ability of the Cdc5 polo-like kinase to phosphorylate Mms4, a cofactor of Mus81, and activate the nuclease activity of Mus81. In this scenario, the DAMP mechanism would facilitate Mus81 action by alleviating the inhibitory action of DDC on Cdc5. Recently, it has been shown that Dpb11 can interact with Mms4 specifically in G2/M phase (Gritenaiteet al, 2014), suggesting an additional regulatory mechanism by which the Dpb11-Slx4 interaction may help promote the resolution of JMs following DNA replication. Here, we showed that the DAMP mechanism is independent of Mus81 as MBD can down-regulate Rad53 activation even inmus81Δ cells (Fig5D). However, we also detected an interaction between MBD and Mms4 (Fig5C), supporting that Mus81-Mms4 may also be regulated by an interaction with Dpb11. Nonetheless, the extent by which the Dpb11-Mms4 interaction contributes to promoting Mus81 action is currently unclear, as a separation of function mutant of Mms4 that cannot interact with Dpb11 has not been generated. Further more, it remains to be defined whether BRCT 3/4 of Dpb11 is directly recognizing phosphorylated motifs in Mms4 or is actually binding Mms4 via another protein.
Coordination of DDC signalling and DNA repair via combinatorial Dpb11 interactions
Collectively, the findings reported here provide insights on how Dpb11 functions in the coordination of DDC and DNA repair. By defining which pairs of BRCT domains of Dpb11 recognize the Slx4-Rtt107 and the Mus81-Mms4 complexes, our work reveals the architecture of distinct Dpb11-mediated complexes (Fig7). Interestingly, our findings imply that several Dpb11 interactions are mutually exclusive and we speculate that this feature is important for Dpb11 to ensure that distinct cellular events, such as checkpoint signalling and Mus81-mediated JM resolution, are executed in a mutually exclusive manner. As shown here, Dpb11 binds Rad9 and Slx4 through the same pair of BRCT domains, preventing Slx4 from participating in Rad53 activation. In this scenario, the Dpb11-Slx4 interaction establishes checkpoint dampening in preparation for JM resolution. It is tempting to further speculate that the mutually exclusive Ddc1-Dpb11 and Mms4-Dpb11 interactions (established via BRCT 3/4) also provide an additional safeguard mechanism to spatio-temporally separate checkpoint signalling from JM resolution. Such separation would be important to prevent Mus81 from prematurely acting on replication forks structures or DNA repair intermediates during S-phase, as recently proposed (Szakal & Branzei, 2013; Gritenaiteet al, 2014). Furthermore, Dpb11 has been previously shown to interact with Sld3 and Sld2 via BRCT domains 1/2 and 3/4, respectively, to initiate replication (Taket al, 2006; Tanakaet al, 2007; Zegerman & Diffley, 2007). Therefore, it is also conceivable that mutually exclusive Dpb11-Sld3 and Dpb11-Rad9 interactions, established via BRCT 1/2, play a role in ensuring that origin firing and checkpoint activation occur in a mutually exclusive manner. Interestingly, the Dpb11 ortholog in mammals, TOPBP1, is also involved in replication initiation and checkpoint signalling (Wardlawet al, 2014), and the TOPBP1-SLX4 interaction was recently found to be conserved in humans (Gritenaiteet al, 2014). It will be interesting to extend the findings from our work to understand how TOPBP1 operates in the coordination of replication initiation, DDC and DNA repair in mammals. Finally, the implications of this work extend beyond TOPBP1, as the results could help understand the mechanism for spatio-temporal control of other multi-BRCT complexes.
Materials and Methods
Yeast strains and plasmids
Strains generated in this study were derived either from MBS164 or MBS191 (both congenic to S288C), BY4741 or W303 (where indicated). All tags were inserted at the C-terminus of the corresponding genes by homologous recombination at the genomic locus and were verified by Western blotting. Tagged strains were assayed for sensitivity to MMS to ensure they behaved similarly to the wild-type strain. Standard cloning methods were used to generate the plasmids for this study.DPB11 orMBD constructs containing aCPY or anADH1 promoter were generated by fusing the respective promoters (800 base pairs upstream of the start codon) to the corresponding open reading frame. The resulting PCR products were subsequently cloned into thepRS416 vector (pMBS148). All point mutations were generated by site-directed mutagenesis using either the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene) or the Primestar® Max DNA Polymerase (Takara). All yeast strains and plasmids used in this study are described in Supplementary Tables S1 and S2, respectively.
Western blot analysis
Rad53 and epitope-tagged proteins were probed using specific monoclonal antibodies: anti-Rad53 (EL7, gift from Dr. Achille Pellicioli), anti-FLAG (M2; Sigma), anti-HA (12CA5; Roche), anti-Myc (9E10; Santa Cruz Biotechnology), ECL HRP-Linked Secondary Antibodies (NA931-GE).
Cell culture and pull-down procedure for co-immuno-precipitation experiments
Cells with the indicated epitope tags were grown until log phase in YPD and treated with MMS (concentration and incubation time used for each experiment are described in the figure legends). In case of strains carrying thepRS416 plasmids, cells were grown in synthetic complete medium lacking uracil (SC –URA). After centrifugation, pellets were washed with TE + PMSF and kept at −80°C prior to cell lysis. Approximately 0.1 g of cell pellet of each strain was lysed by bead beating at 4°C in 1 ml of lysis buffer [50 mM Tris-HCl pH 7.5, 0.2% Tergitol, 150 mM NaCl, 5 mM EDTA, Complete EDTA-free protease inhibitor cocktail (Roche), 5 mM sodium fluoride, 10 mM B-glycerol-phosphate]. Samples were normalized by protein concentration, and lysates were incubated with either anti-HA, anti-Flag or anti-Myc agarose resin (Sigma) for 2 h at 4°C. After three washes with lysis buffer, bound proteins were eluted with three resin volumes of elution buffer (for anti-HA and anti-Myc: 100 mM Tris-HCl pH 8.0, 1% SDS; for anti-FLAG: 0.5 μg/ml of FLAG peptide in 100 mM Tris, 0.2% Tergitol).
5-FOA counter-selection
Cells carrying a plasmid containing theURA3 auxotrophic marker were grown in SC–URA media until saturation. Cells were then normalized by OD, streaked onto SC + 5-FOA plates (1 mg/ml, Zymo Research F9001-1) and allowed to grow for 48 h at 30°C.
Cell cycle synchronization
Cells were grown in YPD or SC–URA media at 30°C until log phase. For arrest of cells in G1, α-factor (Zymo Research) was added to a final concentration of 30 ng/ml (forbar1Δ background strains) and incubated for 2 h. To release cells from G1 arrest, cells were centrifuged and resuspended in fresh media in the presence of pronase (50 μg/ml). For arrest of cells in G2/M, nocodazole (Calbiochem #487928) was added to a final concentration of 7.5 μg/ml and cells were incubated for 2 h. To attest whether the arrest was successful, flow cytometry (FACS) analysis of yeast cell DNA content were performed using a C6 Flow Cytometer® (BD Biosciences).
Pulse-field gel electrophoresis
Cells were transformed with either an empty vector [pRS416 (pMBS148)],pSLX4 (pMBS213) orpMBD (pMBS798) plasmids and allowed to reach log phase in SC–URA media. An untreated, asynchronous sample (ASY) was taken for control. Cells were then treated with 0.033% MMS for 2 h and then recovered in MMS-free media for up to 6 h. At each indicated time point, 50 mg of cells were collected. Pulse-field gel electrophoresis (PFGE) analysis was adapted from Arguesoet al (2008).
For more details of PFGE, confocal fluorescence microscope analysis and pull-down of recombinant BRCT domains, please see Supplementary Materials and Methods.
Acknowledgments
We thank Beatriz S. Almeida for technical support, Dr. Lucas Argueso (Colorado State University) for advice with PFGE and Dr. Achille Pellicioli (University of Milan) for the EL7 antibody. We also thank Dr. Daniel Durocher for reading the manuscript. This work is supported by an M.B.S. grant from the National Institute of Health (R01-GM097272), G.W.B. grant from the Canadian Cancer Society Research Institute (702310) and the Cancer Research Society.
Author contributions
MBS and JRC conceived and designed experiments. JRC, CMJ and AY performed the experiments. MBS, JRC and GWB analysed the data. MBS and JRC wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, Furuya K, Diffley JF, Carr AM, Elledge SJ. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol. 2001;3:958–965. doi: 10.1038/ncb1101-958. [DOI] [PubMed] [Google Scholar]
- Allen JB, Zhou Z, Siede W, Friedberg EC, Elledge SJ. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev. 1994;8:2401–2415. doi: 10.1101/gad.8.20.2401. [DOI] [PubMed] [Google Scholar]
- Argueso JL, Westmoreland J, Mieczkowski PA, Gawel M, Petes TD, Resnick MA. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc Natl Acad Sci USA. 2008;105:11845–11850. doi: 10.1073/pnas.0804529105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Harris MR, Brazauskas P, de Bruin RA, Smolka MB. Linking DNA replication checkpoint to MBF cell-cycle transcription reveals a distinct class of G1/S genes. EMBO J. 2012;31:1798–1810. doi: 10.1038/emboj.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Foiani M. The checkpoint response to replication stress. DNA Repair (Amst) 2009;8:1038–1046. doi: 10.1016/j.dnarep.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Paciotti V, Baldo V, Romano M, Lucchini G, Longhese MP. Hyperactivation of the yeast DNA damage checkpoint by TEL1 and DDC2 overexpression. EMBO J. 2001;20:6485–6498. doi: 10.1093/emboj/20.22.6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson MB, Katou Y, Keszthelyi A, Sing TL, Xia T, Ou J, Vaisica JA, Thevakumaran N, Marjavaara L, Myers CL, Chabes A, Shirahige K, Brown GW. Endogenous DNA replication stress results in expansion of dNTP pools and a mutator phenotype. EMBO J. 2012;31:895–907. doi: 10.1038/emboj.2011.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JA, Lowndes NF, Jackson SP. A role forSaccharomyces cerevisiae histone H2A in DNA repair. Nature. 1999;408:1001–1004. doi: 10.1038/35050000. [DOI] [PubMed] [Google Scholar]
- Durocher D, Taylor IA, Sarbassova D, Haire LF, Westcott SL, Jackson SP, Smerdon SJ, Yaffe MB. The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol Cell. 2000;6:1169–1182. doi: 10.1016/s1097-2765(00)00114-3. [DOI] [PubMed] [Google Scholar]
- Fricke WM, Brill SJ. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 2003;17:1768–1778. doi: 10.1101/gad.1105203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenon M, Costelloe T, Jimeno S, O'Shaughnessy A, Fitzgerald J, Zgheib O, Degerth L, Lowndes NF. Docking onto chromatin via theSaccharomyces cerevisiae Rad9 Tudor domain. Yeast. 2007;24:105–119. doi: 10.1002/yea.1441. [DOI] [PubMed] [Google Scholar]
- Gritenaite D, Princz LN, Szakal B, Bantele SC, Wendeler L, Schilbach S, Habermann BH, Matos J, Lisby M, Branzei D, Pfander B. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014;28:1604–1619. doi: 10.1101/gad.240515.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammet A, Magill C, Heierhorst J, Jackson SP. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 2007;8:851–857. doi: 10.1038/sj.embor.7401036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heideker J, Lis ET, Romesberg FE. Phosphatases, DNA damage checkpoints and checkpoint deactivation. Cell Cycle. 2007;6:3058–3064. doi: 10.4161/cc.6.24.5100. [DOI] [PubMed] [Google Scholar]
- Huang M, Zhou Z, Elledge SJ. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell. 1998;94:595–605. doi: 10.1016/s0092-8674(00)81601-3. [DOI] [PubMed] [Google Scholar]
- Kai M, Boddy MN, Russell P, Wang TS. Replication checkpoint kinase Cds1 regulates Mus81 to preserve genome integrity during replication stress. Genes Dev. 2005;19:919–932. doi: 10.1101/gad.1304305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YD, Wang J, Stubbe J, Elledge SJ. Dif1 is a DNA-damage-regulated facilitator of nuclear import for ribonucleotide reductase. Mol Cell. 2008;32:70–80. doi: 10.1016/j.molcel.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Liu K, Li F, Wang J, Huang H, Wu J, Shi Y. Structure of C-terminal tandem BRCT repeats of Rtt107 protein reveals critical role in interaction with phosphorylated histone H2A during DNA damage repair. J Biol Chem. 2012;287:9137–9146. doi: 10.1074/jbc.M111.311860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Mosqueda J, Maas NL, Jonsson ZO, Defazio-Eli LG, Wohlschlegel J, Toczyski DP. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature. 2010;467:479–483. doi: 10.1038/nature09377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Mackenzie A, Donaldson A, Zegerman P. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J. 2011;30:4805–4814. doi: 10.1038/emboj.2011.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin I, Ngo HP, Greenall A, Zubko MK, Morrice N, Lydall D. Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J. 2008;27:2400–2410. doi: 10.1038/emboj.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase inSaccharomyces cerevisiae. Genetics. 2001;157:103–118. doi: 10.1093/genetics/157.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25:1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Almeida BS, Smolka MB. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell. 2010;39:300–306. doi: 10.1016/j.molcel.2010.06.019. [DOI] [PubMed] [Google Scholar]
- Ohouo PY, Smolka MB. The many roads to checkpoint activation. Cell Cycle. 2012;11:4495. doi: 10.4161/cc.22933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Liu Y, Ma CJ, Smolka MB. DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature. 2013;493:120–124. doi: 10.1038/nature11658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill BM, Szyjka SJ, Lis ET, Bailey AO, Yates JR, 3rd, Aparicio OM, Romesberg FE. Pph3-Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. Proc Natl Acad Sci USA. 2007;104:9290–9295. doi: 10.1073/pnas.0703252104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Foiani M. Signal transduction: how rad53 kinase is activated. Curr Biol. 2005;15:R769–771. doi: 10.1016/j.cub.2005.08.057. [DOI] [PubMed] [Google Scholar]
- Pfander B, Diffley JF. Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J. 2011;30:4897–4907. doi: 10.1038/emboj.2011.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puddu F, Granata M, Di Nola L, Balestrini A, Piergiovanni G, Lazzaro F, Giannattasio M, Plevani P, Muzi-Falconi M. Phosphorylation of the budding yeast 9-1-1 complex is required for Dpb11 function in the full activation of the UV-induced DNA damage checkpoint. Mol Cell Biol. 2008;28:4782–4793. doi: 10.1128/MCB.00330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu M, Rappas M, Wardlaw CP, Garcia V, Ren JY, Day M, Carr AM, Oliver AW, Du LL, Pearl LH. Phosphorylation-dependent assembly and coordination of the DNA damage checkpoint apparatus by Rad4(TopBP(1)) Mol Cell. 2013;51:723–736. doi: 10.1016/j.molcel.2013.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt HC, Yaffe MB. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat Rev Mol Cell Biol. 2013;14:563–580. doi: 10.1038/nrm3640. [DOI] [PubMed] [Google Scholar]
- Santocanale C, Diffley JF. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. 1998;395:615–618. doi: 10.1038/27001. [DOI] [PubMed] [Google Scholar]
- Saugar I, Vazquez MV, Gallo-Fernandez M, Ortiz-Bazan MA, Segurado M, Calzada A, Tercero JA. Temporal regulation of the Mus81-Mms4 endonuclease ensures cell survival under conditions of DNA damage. Nucleic Acids Res. 2013;41:8943–8958. doi: 10.1093/nar/gkt645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF. Rad9 phosphorylation sites couple Rad53 to theSaccharomyces cerevisiae DNA damage checkpoint. Mol Cell. 2002;9:1055–1065. doi: 10.1016/s1097-2765(02)00532-4. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol. 2004;14:1703–1711. doi: 10.1016/j.cub.2004.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Hsiao J, Fay DS, Stern DF. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science. 1998;281:272–274. doi: 10.1126/science.281.5374.272. [DOI] [PubMed] [Google Scholar]
- Szakal B, Branzei D. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 2013;32:1155–1167. doi: 10.1038/emboj.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak Y-SS, Tanaka Y, Endo S, Kamimura Y, Araki H. A CDK-catalysed regulatory phosphorylation for formation of the DNA replication complex Sld2-Dpb11. EMBO J. 2006;25:1987–1996. doi: 10.1038/sj.emboj.7601075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Umemori T, Hirai K, Muramatsu S, Kamimura Y, Araki H. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature. 2007;445:328–332. doi: 10.1038/nature05465. [DOI] [PubMed] [Google Scholar]
- Travesa A, Kuo D, de Bruin RA, Kalashnikova TI, Guaderrama M, Thai K, Aslanian A, Smolka MB, Yates JR, 3rd, Ideker T, Wittenberg C. DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1. EMBO J. 2012;31:1811–1822. doi: 10.1038/emboj.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Elledge SJ. Genetic and physical interactions between DPB11 and DDC1 in the yeast DNA damage response pathway. Genetics. 2002;160:1295–1304. doi: 10.1093/genetics/160.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw CP, Carr AM, Oliver AW. TopBP1: a BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair (Amst) 2014;22:165–174. doi: 10.1016/j.dnarep.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Weinert TA, Hartwell LH. The RAD9 gene controls the cell cycle response to DNA damage inSaccharomyces cerevisiae. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- Williams JS, Williams RS, Dovey CL, Guenther G, Tainer JA, Russell P. gammaH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J. 2010;29:1136–1148. doi: 10.1038/emboj.2009.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki R, Javaheri A, Allard S, Sha F, Cote J, Kron SJ. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol. 2005;25:8430–8443. doi: 10.1128/MCB.25.19.8430-8443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappulla DC, Maharaj AS, Connelly JJ, Jockusch RA, Sternglanz R. Rtt107/Esc4 binds silent chromatin and DNA repair proteins using different BRCT motifs. BMC Mol Biol. 2005;7:40. doi: 10.1186/1471-2199-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegerman P, Diffley JF. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature. 2007;445:281–285. doi: 10.1038/nature05432. [DOI] [PubMed] [Google Scholar]
- Zegerman P, Diffley JF. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature. 2010;467:474–478. doi: 10.1038/nature09373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Chabes A, Domkin V, Thelander L, Rothstein R. The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J. 2001;20:3544–3553. doi: 10.1093/emboj/20.13.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Rothstein R. The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc Natl Acad Sci USA. 2002;99:3746–3751. doi: 10.1073/pnas.062502299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Elledge SJ. DUN1 encodes a protein kinase that controls the DNA damage response in yeast. Cell. 1993;75:1119–1127. doi: 10.1016/0092-8674(93)90321-g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.