

Abstract
Experimental studies on the interactions of the positive strand RNA virus hepatitis C virus (HCV) with the host have contributed to several discoveries in the field of antiviral innate immunity. These include revealing the antiviral sensing pathways that lead to the induction of type I interferon (IFN) during HCV infection and also the importance of type III IFNs in the antiviral immune response to HCV. These studies on HCV/host interactions have contributed to our overall understanding of viral sensing and viral evasion of the antiviral intracellular innate immune response. In this review, I will highlight how these studies of HCV/host interactions have led to new insights into antiviral innate immunity. Overall, I hope to emphasize that studying antiviral immunity in the context of virus infection is necessary to fully understand antiviral immunity and how it controls the outcome of viral infection.
Keywords: hepatitis C virus, antiviral immunity, NS3/4A, MAVS, interferon, innate immunity
Introduction
HCV is a positive sense, single-stranded (ss) RNA virus of the genus Hepacivirus and family Flaviviridae. HCV infects and replicates in hepatocytes within the human liver. HCV infection can result in liver disease, including fibrosis and cirrhosis, can cause hepatocellular carcinoma, and is the leading indicator for liver transplantation (1). There is no vaccine for HCV; however, recently developed, direct acting antiviral drugs (DAAs) are showing high efficacy towards HCV, although they are incredibly cost-prohibitive (2). HCV isolates have been classified into 7 different genetic groups, referred to as genotypes, based on their sequences and display sequence diversity of greater than 30% (3, 4). The previous standard of care for hepatitis C was treatment with pegylated IFN-α plus ribavirin and resulted in cure rates of only 40–50% for the most difficult to treat HCV genotypes (1 and 4) (5). However, the newest standards of care for HCV involve treatment with these newly developed DAAs, sometimes in IFN-free combinations, leading to cure rates of up to 95% in the controlled settings of clinical trials (2). It is currently unknown if these high cure rates will be maintained following widespread usage of these DAAs. Further, how antiviral resistance will be managed under widespread usage is unknown and needs to be carefully considered.
HCV infection is sensed as foreign or non-self by the host through the antiviral innate immune response. This immune response is triggered shortly after infection in a cell-intrinsic manner by host proteins called pattern recognition receptors (PRRs) that detect specific pathogen-associated molecular patterns (PAMPs) in the virus to activate downstream signaling cascades that drive immunity, including expression of antiviral genes and various cytokines, such as the type I and III interferons and IL-1β. HCV is sensed by multiple PRRs, including members of the RIG-I (retinoic acid-inducible gene I)-like receptors (RLRs), the toll-like receptors (TLRs), and the nucleotide oligomerization domain-like receptors (NLRs) (6). While the subsequent downstream antiviral response can be directly antiviral to limit virus replication and spread, it can also provide signals to the adaptive immune response for full induction of immunity to virus infection. As many viruses, including HCV, have developed effective countermeasures to inactivate this antiviral response, it is clear that the innate immune response plays an important role in determining the outcome of virus infection (7).
The HCV RNA genome is 9.6 kilobases in length and encodes for a single polyprotein that is processed by host and viral proteases into the 10 structural and non-structural proteins of the virus (Figure 1). HCV was discovered in 1989 using modern molecular biology approaches and was found to be the causative agent of non-A non-B hepatitis, first described over ten years earlier (8). Since the initial discovery of HCV, many aspects of the viral life cycle have now been revealed; some of which are now targets for DAAs (9). The initial studies to define the virology of HCV took time to develop because HCV is very difficult to grow in cell culture. For example, it took 10 years after the discovery of HCV to be able to study replicating HCV RNA in a cell culture system (10), and a fully infectious clone of HCV to be used in cell culture was only developed within the last 10 years (11, 12). Current systems for studying HCV have expanded from studying the virus in Huh7 human hepatoma cell lines to using primary human hepatocytes, mice with a chimeric human liver, or mice engineered with various human factors that promote HCV infection (13, 14). Utilization of these systems, including emerging non-chimpanzee animal models for HCV infection (13), will expand our knowledge of the full complement of HCV-host interactions that dictate the outcome of infection.
Figure 1. The HCV proteins.
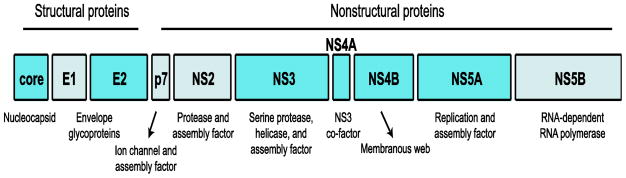
The HCV polyprotein is processed into the structural and nonstructural proteins of the virus, as shown here. The HCV proteins that have been implicated in antiviral innate immune evasion, including core, E2, NS3-NS4A, NS4B, and NS5A, are highlighted in blue.
While we now know many of the important features of both the innate and adaptive immune response to HCV (15), many of these features were unknown when HCV/host interactions were first being studied. These early studies of HCV and antiviral innate immunity were limited by the viral tools and the knowledge of innate immunity available at the time. Even today, we still do not have a full understanding of the complex interactions that govern HCV interactions with the host innate immune response in the infected liver. This review will feature several key discoveries on antiviral innate immunity in the context of HCV to further illustrate how virology research can elucidate fundamental aspects of host cell biology and antiviral immunity (16).
Early studies on antiviral immunity to HCV focus on PKR
The first hepatitis C therapies utilized the well-known antiviral cytokine IFN-α, which along with IFN-β is a member of the type I IFN family. In cell culture, type I IFN effectively limits HCV replication, however as a therapy in patients, type I IFN-based therapies have varying levels of effectiveness (5). Type I IFN signals through the IFN receptor (IFNAR1 and IFNAR2) to drive JAK/STAT signaling that activates the expression of hundreds of IFN-stimulated genes (ISGs) whose encoded proteins limit virus replication and spread. The antiviral mechanisms of action for many of these ISGs, including how they might be antiviral towards HCV, have not yet been fully described (17–19). The first work demonstrating that HCV induces an innate immune response came from HCV-infection studies in chimpanzees, which found elevated ISGs in the infected chimpanzee livers (20). Not long after that, the first studies suggesting that HCV might have a way to evade some aspects of this host innate immune system were published. These studies evaluating IFN treatment outcomes in Japanese patients infected with a genotype 1b virus found that sequence heterogeneity within the viral NS5A protein at the interferon sensitivity-determining region (ISDR) could predict IFN treatment outcomes (21, 22). While today we know that the NS5A protein plays diverse roles in the viral life cycle, including regulating HCV assembly versus replication (23), these studies on the NS5A ISDR were the first to reveal a virologic function for the NS5A protein. While it’s not entirely clear how the sequence variation at the ISDR in NS5A contributes to IFN-based therapy responses amongst the different HCV genotypes or in human populations of different ancestries (24), these studies set the stage for the subsequent work that identified the mechanisms of how HCV antagonizes the antiviral response.
To identify how HCV antagonized the antiviral innate immune response, studies focused on the antiviral effector proteins that had been characterized to date, including the Mx proteins, 2′-5′ oligoadenylate synthestase, RNAseL, and the double-stranded (ds) RNA-activated protein kinase R (PKR). At the time, PKR was the most extensively studied of these antiviral effector proteins. The antiviral activity of PKR is activated by dsRNA, which stimulates its dimerization, autophosphorylation, and phosphorylation of eIF2α, resulting in a global block to cellular translation (25). We now know that PKR-sensing of dsRNA also activates a kinase-independent function of PKR that induces the antiviral IFN response (26). Even in the late 1990s, viral antagonizers of PKR had been described (27). Therefore, because genetic variation within the HCV NS5A protein predicted the IFN-sensitivity of HCV, it seemed likely that HCV also encoded a viral antagonizer of PKR. The most probable candidate was the HCV NS5A protein. Indeed, the NS5A protein did interact with PKR to disrupt its dimerization and ability to catalyze eIF2α phosphorylation (28, 29). Subsequently, it was also shown that the HCV E2 protein also inhibited PKR activation by acting as a pseudosubstrate through its encoded PKR-eIF2α phosphorylation homology domain (30).
The fact that HCV encodes at least two strategies to restrict PKR function would seem to suggest that preventing the inhibition of translation by PKR would be required for effective HCV replication. However, PKR activation does not actually directly regulate HCV translation because translation of the HCV polyprotein is unaffected by eIF2α phosphorylation (31–34). This is because its HCV translation is directed by an internal ribosome entry site (IRES) within its 5′ untranslated region (UTR) that can use eIF2A for translation initiation instead of eIF2α (35). While HCV RNA translation is unaffected by PKR activation and eIF2α phosphorylation, we know that during HCV infection the translation of ISGs and/or IFN is suppressed by PKR activation and the subsequent eIF2α phosphorylation (31, 32). Therefore, in the context of an activated IFN system, PKR activation by HCV may allow HCV to evade the antiviral function of ISGs and therefore be a positive regulator of HCV replication. Based on these findings, there appears to be an unexplained role for NS5A and E2 inhibition of PKR function during HCV infection. As HCV encodes these two PKR-antagonizers, PKR suppression must have some beneficial role in the virus life cycle. It is possible that at early times after infection, before a potent IFN signaling response has been activated, PKR inhibition by HCV proteins could relieve the translational suppression of critical host factors required to promote viral replication. However, we know that at later times after infection when the IFN signaling response is activated, PKR is no longer repressed by the E2 and NS5A proteins perhaps because they are involved in other aspects of the viral life cycle, such as viral assembly. Therefore, at these later times after infection, PKR would be activated and could contribute to the translational suppression of ISGs. More recent work has suggested that PKR, through a kinase-independent mechanism, is also involved in the induction of the signaling cascade that induces type I IFN (26). Therefore, it remains possible that the mechanism for HCV NS5A and/or E2 evasion of PKR has more to do with bypassing this innate immune signaling rather than circumventing the translational suppression function of PKR. Even though PKR was the first antiviral protein studied in the context of HCV, it seems that we still have much more to learn about the role of PKR during HCV infection.
During the initial studies on HCV and PKR, the virologic tools to study if the dsRNA generated during HCV replication was specifically activating PKR and the antiviral immune response had not yet been developed, and so it was unknown if HCV dsRNA was an actual PAMP that activated antiviral immunity. The ability to study HCV replication and how it impacted antiviral immunity finally became possible following the groundbreaking work of the Bartenschlager and Rice labs in developing the HCV subgenomic RNA replicon systems ((10, 36) and reviewed in (14)). These studies utilizing these HCV RNA replicons did not immediately find the dsRNA sensor/PRR for HCV, but rather found that replicating HCV RNA actually inhibited IRF-1 and IRF-3-dependent signaling induced by transfected dsRNA (37, 38). IRF-1 and IRF-3 are transcription factors that contribute to the induction of IFN-β, although IRF-1 is not essential for this induction (39). The HCV block to IRF-I activation was directed by NS5A inhibition of the PKR/IRF-1 signaling axis (37). On the other hand, the HCV-mediated block to IRF-3 activation, which is a seminal finding in the study of HCV/host interactions that regulated innate immunity, was directed by the actions of the HCV NS3/4A serine protease (40). The multifunctional HCV NS3/4A protease is a protein complex between NS3, which contains a serine protease domain and an NTPase/RNA helicase domain, and its 54-amino acid cofactor NS4A, the membrane targeting subunit of the protease complex (41). NS3/4A is essential for HCV replication, viral polyprotein processing, and even viral assembly (42). NS3/4A blocked IRF-3 activation by preventing both the phosphorylation and its downstream signaling to IFN-β (40). Importantly, it was found that the protease activity of NS3 was responsible for the block in IRF-3 signaling because protease inactivation by mutation or treatment with an NS3-specific protease inhibitor relieved the block to virus-induced antiviral signaling through IRF-3 (40). This data suggested very clearly that NS3/4A was cleaving a host factor to prevent signal transduction to IRF-3 signaling, but the identity of this host factor remained unknown for several more years. Indeed, at this time the PRRs that initiated this signaling had yet to be described, and the virus-activated kinases that phosphorylated IRF-3 (now known as TBK1 and IKK-ε) were just being revealed (43, 44). Nonetheless, these early studies on HCV-innate immune interactions regulated by NS3/4A paved the way for the discovery of the PRRs RIG-I and MDA5, as well as their signaling adaptor protein MAVS, which was eventually found to be the proteolytic target of NS3/4A.
Antiviral innate immune sensing of HCV by RIG-I and MDA5
While PKR was the first described PRR for HCV (and for viruses in general), studies of PKR-deficient mice revealed that PKR-independent antiviral sensing mechanisms existed (45, 46). Soon after these observations, the PRRs that sensed extracellular dsRNA (TLR3)(47) and viral ssRNA (TLR7/8)(48–50) were discovered. However, there still existed an unidentified antiviral PRR, as type I IFN was still induced in response to virus infection in mice and cell lines lacking these TLRs or PKR (51–55). In 2004, Dr. Takashi Fujita and colleagues published their findings on the identification of RIG-I, which is one of the PRRs for intracellular dsRNA (56). RIG-I contains a caspase activation and recruitment domain (CARD) and a DexD/H box RNA helicase that binds to cytoplasmic dsRNA to drive signaling to NF-κB and IRF-3 (reviewed in (57)). A database search of GenBank for other proteins containing CARD motifs led to the discovery of MDA5, another IFN-inducible CARD-containing helicase protein that senses viral dsRNA. The presence of the CARD in RIG-I and MDA5 suggested that these PRRs may propagate their signals through interaction with another CARD-containing protein, as CARD-CARD interactions between proteins were known to regulate apoptotic and innate immune signaling (58). Indeed, the discovery of MAVS, a CARD-containing protein found to interact with both RIG-I and MDA5 to drive antiviral signaling through IRF-3 to IFN-β, proved this hypothesis to be correct (59–62). Importantly for HCV research, MAVS was the sought-after proteolytic target of NS3/4A, and it was found to be cleaved both in human hepatoma Huh7 cell lines and in livers of HCV-infected patients (60, 63–66).
The PRRs RIG-I and MDA5 are now known to differentially recognize distinct PAMPs within RNA viruses (67, 68). In particular, we now know that RIG-I is activated by short dsRNA containing either a 5′ triphosphate motif or a 5′ diphosphate motif (69, 70), while MDA5 senses long dsRNA or even higher order RNA structures that could be viral replication intermediates (71–73). In fact, both RIG-I and MDA5 have now been shown to be important for the innate immune response to HCV (74, 75). The studies that identified RIG-I and MDA5 as sensors of HCV have led to the discovery of several key properties of antiviral innate immune sensing by these PRRs, described below.
HCV replicates poorly in cell culture, and efficient replication requires cell culture adaptive mutations in the viral RNA (36, 76). Therefore, to study HCV replication it was necessary to identify cell lines highly permissive for HCV replication. To identify these HCV-permissive cell lines, cell lines stably replicating HCV RNA were cured of the replicating HCV RNA using IFN, after which the ability of these IFN-cured cell lines to support HCV replication was tested (77). One particular Huh7 cell line, called Huh7.5, supported very high levels of HCV replication (77). This Huh7.5 cell line was unable to make certain ISGs in response to Sendai virus, which is a paramyxovirus that is normally a potent inducer of type I IFN (74). To identify the missing innate immune factor, a genetic complementation assay to identify cDNA products that could restore antiviral activation of IRF-3 was performed. This assay identified RIG-I as being one of the factors deficient in Huh7.5 cells, and found that Huh7.5 cells contained a single amino acid change in the CARD of RIG-I (T55I) that acts as a dominant negative of RIG-I signaling (74). This study identified RIG-I as a protein that controls innate immunity and permissiveness to HCV infection. The PAMPs that activate RIG-I have now been well-described (reviewed in (78–80)). Studies in HCV contributed to this understanding of the RIG-I PAMPs, as the poly U/UC region at the 3′UTR region of the HCV genome was found to be the HCV PAMP sensed by RIG-I, revealing that RIG-I can bind to RNA in a sequence-specific manner (81, 82). Taken together, HCV studies of innate immunity contributed to the discovery of RIG-I as an antiviral sensor and identified that RIG-I activation by PAMP RNA occurs in an RNA sequence-specific manner.
While RIG-I is essential for the antiviral innate immune response to HCV, a role for MDA5 had been largely over-looked. This was in part because it was known that transfection of in vitro transcribed HCV RNA induced signaling to IFN-β in an MDA5-independent manner (81). However, there were several hints suggesting a role for MDA5 in the antiviral response to HCV. The first hint came from the finding that HCV replication was enhanced in the presence of the paramyxovirus V protein, which inhibits MDA5, but not RIG-I function (83–85). In addition, MDA5 gene polymorphisms were found to be associated with spontaneous resolution of HCV infection (86). Now, studies using individual knockdowns of RIG-I and MDA5 have convincingly revealed a role for MDA5 (and confirmed the role of RIG-I) in sensing HCV to activate as antiviral innate immune response (75). Therefore HCV, similar to West Nile virus, is sensed by both MDA5 and RIG-I (87). In the future, it will be important to define the HCV PAMPs sensed by these PRRs during infection and how sensing by both of these PRRs contributes to the resolution of HCV infection. In addition to RIG-I and MDA5, other PRRs have now been identified that sense HCV infection, either directly in the infected cells or in other cells within the liver (88–90). Future studies will undoubtedly provide insights into how these PRRs activate innate immunity to HCV and contribute to the inflammatory responses found in the infected liver.
Studies on HCV evasion of antiviral immunity reveal key features of the anti-HCV innate immune response
While HCV infection is sensed by multiple PRRs that drive induction of the antiviral response and inflammation, HCV has evolved several mechanisms to block these antiviral responses. It is quite likely that this innate immune regulation by HCV contributes to its remarkable success as a viral pathogen by either promoting a cellular state conducive to viral replication or by dysregulating priming of a successful adaptive immune response to HCV (15). HCV has several mechanisms to block antiviral immunity, and this section will focus on the mechanisms by which the HCV NS3/4A protease blocks innate immunity and how this has impacted our understanding of antiviral immunity to HCV (Figure 2). A full review of HCV evasion of innate immunity, including how it inhibits IFN signaling, can be found elsewhere (91–94).
Figure 2. The HCV NS3/4A protein regulates the antiviral innate immune response to HCV.

Following HCV infection of hepatocytes, viral pathogen-associated molecular patterns (PAMPs) present in the HCV RNA released into the cytosol are sensed by pattern-recognition receptors, including RIG-I, MDA5, and TLR3 (shown in blue) that signal through the adaptor proteins MAVS and TRIF, respectively, to induce the transcriptional antiviral response (shown in green). Full activation of RIG-I is regulated in part by Riplet, which mediates K63-linked ubiquitination of the repressor domain of RIG-I. The HCV NS3/4A protein blocks this antiviral signaling through cleavage of the host proteins MAVS, Riplet, and TRIF (shown in red).
The discovery that the HCV NS3/4A protease cleaves the host protein MAVS to block innate immunity strongly supported the finding of several groups that MAVS is a key antiviral signaling adaptor molecule, especially during HCV infection (59–66). Further studies on NS3/4A have revealed that it cleaves several additional host proteins, highlighting the prominent role of NS3/4A in HCV pathogenesis (42, 95, 96). Importantly, three of these proteins (Riplet, MAVS, and TRIF) targeted by NS3/4A are known innate immune signaling proteins, further supporting the idea that HCV evasion of antiviral immunity is necessary to establish successful infection.
Studies on the molecular mechanisms of how NS3/4A regulates innate immunity have revealed critical aspects of the antiviral response to HCV. NS3/4A cleavage of MAVS during infection inhibits antiviral signaling because this cleavage occurs proximal to the transmembrane domain of MAVS, releasing it from intracellular membranes. The resulting cytoplasmic MAVS is unable to transduce RIG-I/MDA5 signals (reviewed in (42, 94)). Confocal imaging studies have revealed that the multifunctional NS3/4A has several different subcellular localizations, including at the mitochondria, peroxisomes, ER, and mitochondrial-associated ER membranes (MAM) (42, 97, 98). Indeed, we now know that MAVS is also localized to some of these same organelles as NS3/4A. While early studies showed that MAVS was localized to mitochondria (61), in more recent years, we and others have shown that MAVS is not exclusively mitochondrial, but that it is also localized on the MAM and on peroxisomes (98, 99). Quite unexpectedly, in an analysis of the localization of MAVS in isolated subcellular fractions during HCV replication, we found that MAM-localized MAVS was cleaved by NS3/4A, while the mitochondrial-MAVS remained uncleaved during HCV replication (98). While we were unable to determine specifically if NS3/4A targets MAVS localized to peroxisomes, the fact that our confocal imaging analyses revealed that a portion of NS3/4A could be localized to peroxisomes (98), strongly suggests that NS3/4A could also cleave peroxisomal-localized MAVS. This possible cleavage of MAVS at peroxisomes could abrogate peroxisomal-MAVS signaling, recently suggested to induce type III IFNs (100). Further studies to understand why NS3/4A does not target MAVS on the mitochondria are required, and it could be that HCV uses other strategies independent of NS3/4A to down regulate MAVS signaling of innate immunity from the mitochondria, such as the induction of mitochondrial fission and mitophagy (101).
Why does NS3/4A specifically target the MAM-localized MAVS? Our previous work revealed that MAM/mitochondrial interactions regulate signaling to IFN-β (98). Therefore, it could be that NS3/4A-cleavage of the MAM-localized MAVS blocks antiviral signaling by also regulating membrane interactions between these organelles. MAVS oligomerization, which occurs through the CARD motifs, is also blocked by NS3/4A (102–104). Therefore, we hypothesize that NS3/4A cleavage of MAVS prevents interactions between the CARDs of the MAM-associated MAVS and the mitochondrial-associated MAVS to inhibit antiviral signaling during HCV infection. By studying how the HCV NS3/4A protease targets MAVS, we have learned about new subcellular localizations and signaling functions for MAVS and also about membrane/organelle interactions that regulate the antiviral response to RNA viruses.
Following the discovery that NS3/4A blocked IRF-3 activation of innate immunity, it was also shown that NS3/4A cleaves TRIF (105), the TLR3 adaptor protein that directs dsRNA-induced IRF-3-signaling to IFN-β (106). This cleavage of TRIF prevents polyI:C signaling of the TLR3 pathway, both in vitro and during infection (105, 107). The fact that HCV has a mechanism to cleave TRIF and prevents its signaling, strongly suggests that regulation of TRIF must be important for successful HCV infection. This cleavage of TRIF by NS3/4A could prevent TLR3 activation and the resulting inflammation observed during HCV infection (88) or it could alleviate TLR3-independent TRIF signaling of innate immunity (108).
Riplet, another protein in the RIG-I/MAVS signaling pathway is also cleaved by NS3/4A during HCV infection (96). Riplet is an E3 ubiquitin ligase that mediates K63-linked polyubiquitination of the repressor domain of RIG-I to activate RIG-I (109). Riplet contains a canonical NS3/4A serine protease cleavage site, and mutational inactivation of this site prevents its cleavage by NS3/4A (96). This cleavage of Riplet by NS3/4A functionally blocks the Riplet-mediated ubiquitination of RIG-I, inhibiting full RIG-I activation and downstream signaling. As HCV replication is increased following Riplet depletion, Riplet plays an essential function in the antiviral response to HCV (96). Riplet is also inactivated during influenza virus infection, suggesting that Riplet may be a potent antiviral signaling factor to a number of RNA viruses (110). It is not clear why HCV needs to encode a strategy to target Riplet when it already targets MAVS (downstream of Riplet in the antiviral signaling cascade). Understanding the mechanisms that regulate this differential targeting of Riplet and MAVS by NS3/4A will be an important area of future research.
Genome-wide association studies (GWAS) reveal the importance of type III IFNs during HCV infection
It has long been known that hepatitis C patients have varying ability to naturally resolve infection and to respond to IFN-based therapies for HCV (1). Some of these differences can be attributed to the HCV genotype present in the infection. Patients infected with HCV genotypes 2 and 3 have the highest response rates to IFN-based therapies, while those infected with genotypes 1 and 4 have the lowest response rates (111). However, a large part of the variation in natural and treatment-induced clearance has been attributed to human genetic variation. Ground-breaking GWAS have identified single nucleotide polymorphisms (SNPs) in and around the gene encoding IFNL3 that predict both natural and treatment-induced clearance of HCV (112–116). IFNL3 is a member of the type III IFNs, also known as the IFN-λs, which consist of 4 antiviral cytokines (IFNL 1–4). The type III IFNs signal through their receptor (IL10R2 and IFNLR1) (117) to the JAK/STAT pathway to induce transcriptional activation of ISGs (reviewed in (118)). The type III IFNs appear to drive a more prolonged antiviral response than type I IFN (118, 119). Importantly, the type III IFNs have potent antiviral activity towards HCV (120). Prior to the GWAS that identified SNPs near IFNL3, not much was known about the role of the type III IFNs in antiviral immunity. However, the results of the GWAS strongly implicate type III IFNs as having very important antiviral roles to HCV. These findings propelled the study of the role of type III IFNs in the antiviral response, especially during HCV infection.
While it was initially debated if a SNP in the IFNL3 locus affected IFNL3 protein expression, multiple studies have now found that the unfavorable haplotype at the IFNL3 locus results in decreased IFNL3 expression (115, 116, 121–125). Further demonstrating that the protective allele affects the antiviral response, studies in HCV-infected primary human hepatocytes isolated from different donors have found that those that have the protective IFNL3 allele have an increased ISG response that limits HCV infection (126). In addition to having an obvious role in the innate antiviral response, the type III IFNs have also been recently implicated in contributing to the adaptive immune response (125). A full description of the mechanisms underlying how genetic variation at the IFNL3 locus impacts HCV clearance is beyond the scope of this article, but I will point out that several candidate functional SNPs have been identified that may regulate IFNL function, either of IFNL3 or the newly described IFNL4 (127–131).
The pioneering HCV GWAS highlight a role for the type III IFNs in the antiviral response during HCV infection. While we know a great deal about how type I IFNs are activated and regulated during HCV infection, we know comparatively little about the type III IFNs. We do know that type III IFN is the predominant IFN made during acute HCV infection in hepatocytes (132–134). Additionally, these studies and others have revealed that the Huh7 liver hepatoma cell lines that support HCV replication in cell culture do not actually make much of a type III IFN response following HCV infection (133). A recently described HepG2 cell line does secrete type III IFNs in response to HCV infection, and these type III IFNs limit HCV replication in cell culture (75). Therefore, these studies have revealed that the Huh7-based cell lines that we have used for the last 15 years to study HCV/innate immune interactions actually lack some part of the innate immune response that drives type III IFN induction in response to HCV! It is therefore not that surprising that the HCV field is only now beginning to understand how type III IFNs are induced during HCV infection.
The most relevant cell types that drive expression of the type III IFNs during infection are unknown, and how innate immune sensing of HCV drives type III IFN induction is only beginning to be elucidated. While overexpression of the HCV PAMP (the poly U/UC region in the 3′ UTR of HCV) can induce expression of the type III IFNs in plasmacytoid dendritic cells (135), it is unknown if this PAMP also induces the type III IFNs in primary human hepatocytes during infection. It appears that the signal transduction cascade that activates type III IFNs in response to HCV in hepatocytes is dependent on both of the PRRs RIG-I and MDA5 (75); and also the signaling adaptor protein MAVS (136). Further, there is evidence that HCV may suppress the long-term induction of the type III IFNs, although this doesn’t appear to be as efficient as the ability of the HCV to suppress type I IFN induction in the infected cell (75). Because it has been proposed that MAVS signaling from peroxisomes induces the type III IFNs during virus infection (100), and as we know that some NS3/4A protein is localized to peroxisomes (98), it is possible that NS3/4A could cleave MAVS from peroxisomes to block MAVS signaling to the type III IFNs. Alternatively, HCV induction of host miRNAs (miR-208b and miR-499a-5p) that target the 3′ UTR of IFNL3 mRNA could prevent full expression of IFNL3 protein during infection (127). Thus, through this strategy HCV could encode a second mechanism to prevent full induction of the type III IFNs. However, this mechanism of IFN-regulation by HCV may be variable depending on the host genetic background, as a SNP (rs4803217) exists within the miRNA targeting site in the 3′ UTR of IFNL3 that would prevent targeting by the HCV-induced miRNAs (127). Regardless, the fact that HCV seems to have more than one strategy to block type III IFN induction and/or expression supports the idea that viral evasion of the type III IFNs is critical for HCV infection (126, 132).
Summary
Studies of HCV/host interactions during the antiviral innate immune response have led to important discoveries and insights into the field of innate immunity. They have revealed complex roles for PKR in the antiviral response, including a kinase-independent signaling function for PKR. They have contributed to the discovery of RIG-I and provided a more detailed understanding into nature of the RIG-I-activating PAMP RNA. They have solidified the roles of MAVS, TRIF, and Riplet in the innate immune response to HCV, as all three of these proteins are targeted for inactivation by the HCV NS3/4A protease. Studies into the molecular mechanisms of how NS3/4A regulates antiviral signaling have identified new subcellular localizations and signaling functions for MAVS that are leading to a greater understanding of the cell biological organization of the antiviral innate immune response. Finally, HCV/host interaction studies at both the cellular and genetic levels have implicated the type III IFNs as playing a major role in the antiviral response to HCV. I would argue that some of these discoveries would not have been possible, would have been delayed, or their importance would not have been fully realized without HCV research. There are more hidden facets of innate immune signaling that have yet to be discovered, and experimental studies using HCV (and other viruses!) will inform us of the most important aspects of the antiviral innate immune response. If a virus targets it, it must be important! Therefore, it is clear that not only is the field of virology alive and well (16), the field of hepatitis C virology must not be abandoned. Continued research into this fascinating, rapidly evolving RNA virus will undoubtedly lead to more discoveries in innate immunity and also in basic cell biology that will have broad scientific implications.
Acknowledgments
Research in the Horner lab is supported by an NIH/NIAID Research Scholar Development Award (K22 AI100935), the Duke University Center for AIDS Research (CFAR), an NIH funded program (5P30 AI064518), and a Duke School of Medicine Whitehead Scholarship. I would like to thank the members of the lab for critical reading of this article and helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Seeff LB. The history of the “natural history” of hepatitis C (1968–2009) Liver Int. 2009;29(Suppl 1):89–99. doi: 10.1111/j.1478-3231.2008.01927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawlotsky JM. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology. 2014;146:1176–1192. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Kuiken C, Simmonds P. Nomenclature and numbering of the hepatitis C virus. Methods Mol Biol. 2009;510:33–53. doi: 10.1007/978-1-59745-394-3_4. [DOI] [PubMed] [Google Scholar]
- 4.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. Expanded classification of hepatitis C Virus into 7 genotypes and 67 Subtypes: updated criteria and assignment web resource. Hepatology. 2013 doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soriano V, Peters MG, Zeuzem S. New therapies for hepatitis C virus infection. Clin Infect Dis. 2009;48:313–320. doi: 10.1086/595848. [DOI] [PubMed] [Google Scholar]
- 6.Wilkins C, Gale M., Jr Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 9.Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 11.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 12.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Billerbeck E, de Jong Y, Dorner M, de la Fuente C, Ploss A. Animal models for hepatitis C. Curr Top Microbiol Immunol. 2013;369:49–86. doi: 10.1007/978-3-642-27340-7_3. [DOI] [PubMed] [Google Scholar]
- 14.Steinmann E, Pietschmann T. Cell culture systems for hepatitis C virus. Curr Top Microbiol Immunol. 2013;369:17–48. doi: 10.1007/978-3-642-27340-7_2. [DOI] [PubMed] [Google Scholar]
- 15.Park SH, Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity. 2014;40:13–24. doi: 10.1016/j.immuni.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimaio D. Is virology dead? mBio. 2014;5:e01003–01014. doi: 10.1128/mBio.01003-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metz P, Dazert E, Ruggieri A, Mazur J, Kaderali L, Kaul A, Zeuge U, Trippler M, Lohmann V, Binder M, Frese M, Bartenschlager R. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology. 2012 doi: 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- 18.Metz P, Reuter A, Bender S, Bartenschlager R. Interferon-stimulated genes and their role in controlling hepatitis C virus. J Hepatol. 2013 doi: 10.1016/j.jhep.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 19.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato T, Esumi M, Yamashita S, Abe K, Shikata T. Interferon-inducible gene expression in chimpanzee liver infected with hepatitis C virus. Virology. 1992;190:856–860. doi: 10.1016/0042-6822(92)90925-f. [DOI] [PubMed] [Google Scholar]
- 21.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Izumi N, Marumo F, Sato C. Comparison of full-length sequences of interferon-sensitive and resistant hepatitis C virus 1b. Sensitivity to interferon is conferred by amino acid substitutions in the NS5A region. J Clin Invest. 1995;96:224–230. doi: 10.1172/JCI118025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Ogura Y, Izumi N, Marumo F, Sato C. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N Engl J Med. 1996;334:77–81. doi: 10.1056/NEJM199601113340203. [DOI] [PubMed] [Google Scholar]
- 23.Lindenbach BD, Rice CM. The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol. 2013;11:688–700. doi: 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El-Shamy A, Hotta H. Impact of hepatitis C virus heterogeneity on interferon sensitivity: an overview. World J Gastroenterol. 2014;20:7555–7569. doi: 10.3748/wjg.v20.i24.7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 26.Arnaud N, Dabo S, Akazawa D, Fukasawa M, Shinkai-Ouchi F, Hugon J, Wakita T, Meurs EF. Hepatitis C virus reveals a novel early control in acute immune response. PLoS Pathog. 2011;7:e1002289. doi: 10.1371/journal.ppat.1002289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gale M, Jr, Katze MG. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol Ther. 1998;78:29–46. doi: 10.1016/s0163-7258(97)00165-4. [DOI] [PubMed] [Google Scholar]
- 28.Gale M, Jr, Blakely CM, Kwieciszewski B, Tan SL, Dossett M, Tang NM, Korth MJ, Polyak SJ, Gretch DR, Katze MG. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol Cell Biol. 1998;18:5208–5218. doi: 10.1128/mcb.18.9.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gale MJ, Jr, Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE, Polyak SJ, Gretch DR, Katze MG. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 30.Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 31.Arnaud N, Dabo S, Maillard P, Budkowska A, Kalliampakou KI, Mavromara P, Garcin D, Hugon J, Gatignol A, Akazawa D, Wakita T, Meurs EF. Hepatitis C virus controls interferon production through PKR activation. PLoS ONE. 2010;5:e10575. doi: 10.1371/journal.pone.0010575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe. 2009;6:513–522. doi: 10.1016/j.chom.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimoike T, McKenna SA, Lindhout DA, Puglisi JD. Translational insensitivity to potent activation of PKR by HCV IRES RNA. Antiviral Res. 2009;83:228–237. doi: 10.1016/j.antiviral.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Koev G, Duncan RF, Lai MM. Hepatitis C virus IRES-dependent translation is insensitive to an eIF2alpha-independent mechanism of inhibition by interferon in hepatocyte cell lines. Virology. 2002;297:195–202. doi: 10.1006/viro.2002.1455. [DOI] [PubMed] [Google Scholar]
- 35.Terenin IM, Dmitriev SE, Andreev DE, Shatsky IN. Eukaryotic translation initiation machinery can operate in a bacterial-like mode without eIF2. Nat Struct Mol Biol. 2008;15:836–841. doi: 10.1038/nsmb.1445. [DOI] [PubMed] [Google Scholar]
- 36.Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 37.Pflugheber J, Fredericksen B, Sumpter R, Jr, Wang C, Ware F, Sodora DL, Gale M., Jr Regulation of PKR and IRF-1 during hepatitis C virus RNA replication. Proc Natl Acad Sci U S A. 2002;99:4650–4655. doi: 10.1073/pnas.062055699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fredericksen B, Akkaraju GR, Foy E, Wang C, Pflugheber J, Chen ZJ, Gale M., Jr Activation of the interferon-beta promoter during hepatitis C virus RNA replication. Viral Immunol. 2002;15:29–40. doi: 10.1089/088282402317340215. [DOI] [PubMed] [Google Scholar]
- 39.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 40.Foy E, Li K, Wang C, Sumpter R, Jr, Ikeda M, Lemon SM, Gale M., Jr Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 41.Brass V, Berke JM, Montserret R, Blum HE, Penin F, Moradpour D. Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc Natl Acad Sci U S A. 2008;105:14545–14550. doi: 10.1073/pnas.0807298105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morikawa K, Lange CM, Gouttenoire J, Meylan E, Brass V, Penin F, Moradpour D. Nonstructural protein 3-4A: the Swiss army knife of hepatitis C virus. J Viral Hepat. 2011;18:305–315. doi: 10.1111/j.1365-2893.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- 43.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 44.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 45.Zhou A, Paranjape JM, Der SD, Williams BR, Silverman RH. Interferon action in triply deficient mice reveals the existence of alternative antiviral pathways. Virology. 1999;258:435–440. doi: 10.1006/viro.1999.9738. [DOI] [PubMed] [Google Scholar]
- 46.Abraham N, Stojdl DF, Duncan PI, Methot N, Ishii T, Dube M, Vanderhyden BC, Atkins HL, Gray DA, McBurney MW, Koromilas AE, Brown EG, Sonenberg N, Bell JC. Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J Biol Chem. 1999;274:5953–5962. doi: 10.1074/jbc.274.9.5953. [DOI] [PubMed] [Google Scholar]
- 47.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 48.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 49.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 50.Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reis e Sousa C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 52.Smith EJ, Marie I, Prakash A, Garcia-Sastre A, Levy DE. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or Ikappa B kinase but is blocked by Vaccinia virus E3L protein. J Biol Chem. 2001;276:8951–8957. doi: 10.1074/jbc.M008717200. [DOI] [PubMed] [Google Scholar]
- 53.Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB. Does Toll-like receptor 3 play a biological role in virus infections? Virology. 2004;322:231–238. doi: 10.1016/j.virol.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 54.Malmgaard L, Melchjorsen J, Bowie AG, Mogensen SC, Paludan SR. Viral activation of macrophages through TLR-dependent and -independent pathways. J Immunol. 2004;173:6890–6898. doi: 10.4049/jimmunol.173.11.6890. [DOI] [PubMed] [Google Scholar]
- 55.Lopez CB, Moltedo B, Alexopoulou L, Bonifaz L, Flavell RA, Moran TM. TLR-independent induction of dendritic cell maturation and adaptive immunity by negative-strand RNA viruses. J Immunol. 2004;173:6882–6889. doi: 10.4049/jimmunol.173.11.6882. [DOI] [PubMed] [Google Scholar]
- 56.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 57.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bouchier-Hayes L, Martin SJ. CARD games in apoptosis and immunity. EMBO Rep. 2002;3:616–621. doi: 10.1093/embo-reports/kvf139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 60.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 61.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 62.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 63.Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, Carney DS, Wang T, Ishida H, Yoneyama M, Fujita T, Saito T, Lee WM, Hagedorn CH, Lau DT, Weinman SA, Lemon SM, Gale M., Jr Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin R, Lacoste J, Nakhaei P, Sun Q, Yang L, Paz S, Wilkinson P, Julkunen I, Vitour D, Meurs E, Hiscott J. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J Virol. 2006;80:6072–6083. doi: 10.1128/JVI.02495-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bellecave P, Sarasin-Filipowicz M, Donze O, Kennel A, Gouttenoire J, Meylan E, Terracciano L, Tschopp J, Sarrazin C, Berg T, Moradpour D, Heim MH. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology. 2010;51:1127–1136. doi: 10.1002/hep.23426. [DOI] [PubMed] [Google Scholar]
- 67.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 68.Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M., Jr Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baum A, Sachidanandam R, Garcia-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A. 2010;107:16303–16308. doi: 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, Dermody TS, Hartmann G, Reis ESC. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature. 2014 doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol. 2009;83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Triantafilou K, Vakakis E, Kar S, Richer E, Evans GL, Triantafilou M. Visualisation of direct interaction of MDA5 and the dsRNA replicative intermediate form of positive strand RNA viruses. J Cell Sci. 2012;125:4761–4769. doi: 10.1242/jcs.103887. [DOI] [PubMed] [Google Scholar]
- 74.Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Israelow B, Narbus CM, Sourisseau M, Evans MJ. HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection. Hepatology. 2014 doi: 10.1002/hep.27227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lohmann V, Korner F, Dobierzewska A, Bartenschlager R. Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J Virol. 2001;75:1437–1449. doi: 10.1128/JVI.75.3.1437-1449.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rehwinkel J, Reis e Sousa C. RIGorous detection: exposing virus through RNA sensing. Science. 2010;327:284–286. doi: 10.1126/science.1185068. [DOI] [PubMed] [Google Scholar]
- 79.Rehwinkel J, Reis ESC. Targeting the viral Achilles’ heel: recognition of 5′-triphosphate RNA in innate anti-viral defence. Curr Opin Microbiol. 2013 doi: 10.1016/j.mib.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schlee M, Hartmann G. The chase for the RIG-I ligand--recent advances. Mol Ther. 2010;18:1254–1262. doi: 10.1038/mt.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M., Jr Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Uzri D, Gehrke L. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J Virol. 2009;83:4174–4184. doi: 10.1128/JVI.02449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Parisien JP, Bamming D, Komuro A, Ramachandran A, Rodriguez JJ, Barber G, Wojahn RD, Horvath CM. A shared interface mediates paramyxovirus interference with antiviral RNA helicases MDA5 and LGP2. J Virol. 2009;83:7252–7260. doi: 10.1128/JVI.00153-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Andrus L, Marukian S, Jones CT, Catanese MT, Sheahan TP, Schoggins JW, Barry WT, Dustin LB, Trehan K, Ploss A, Bhatia SN, Rice CM. Expression of paramyxovirus V proteins promotes replication and spread of hepatitis C virus in cultures of primary human fetal liver cells. Hepatology. 2011;54:1901–1912. doi: 10.1002/hep.24557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hoffmann F, Schmidt A, Dittmann Chevillotte M, Wisskirchen C, Hellmuth JC, Willms S, Gilmore RH, Glas J, Folwaczny M, Müller T, Berg T, Spengler U, Fitzmaurice K, Kelleher D, Reisch N, Rice CM, Endres S, Rothenfusser S. Polymorphisms in MDA-5 link protein function to clearance of hepatitis C virus. Hepatology. 2014:n/a–n/a. doi: 10.1002/hep.27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M., Jr The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J Virol. 2013;87:11416–11425. doi: 10.1128/JVI.01488-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li K, Li NL, Wei D, Pfeffer SR, Fan M, Pfeffer LM. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology. 2012;55:666–675. doi: 10.1002/hep.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, Delker DA, Jo J, Bertoletti A, Hagedorn CH, Gale M., Jr IL-1beta Production through the NLRP3 Inflammasome by Hepatic Macrophages Links Hepatitis C Virus Infection with Liver Inflammation and Disease. PLoS Pathog. 2013;9:e1003330. doi: 10.1371/journal.ppat.1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M, Chisari FV. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci U S A. 2010;107:7431–7436. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gokhale NS, Vazquez C, Horner SM. Hepatitis C virus: Strategies to evade antiviral responses. Future Virology. 2014;9:1061–1075. doi: 10.2217/fvl.14.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Horner SM, Gale M., Jr Regulation of hepatic innate immunity by hepatitis C virus. Nat Med. 2013;19:879–888. doi: 10.1038/nm.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Heim MH. Innate immunity and HCV. J Hepatol. 2013;58:564–574. doi: 10.1016/j.jhep.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 94.Schoggins JW, Rice CM. Innate immune responses to hepatitis C virus. Curr Top Microbiol Immunol. 2013;369:219–242. doi: 10.1007/978-3-642-27340-7_9. [DOI] [PubMed] [Google Scholar]
- 95.Kang X, Chen X, He Y, Guo D, Guo L, Zhong J, Shu HB. DDB1 is a cellular substrate of NS3/4A protease and required for hepatitis C virus replication. Virology. 2013;435:385–394. doi: 10.1016/j.virol.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 96.Oshiumi H, Miyashita M, Matsumoto M, Seya T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013;9:e1003533. doi: 10.1371/journal.ppat.1003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wolk B, Sansonno D, Krausslich HG, Dammacco F, Rice CM, Blum HE, Moradpour D. Subcellular localization, stability, and trans-cleavage competence of the hepatitis C virus NS3-NS4A complex expressed in tetracycline-regulated cell lines. J Virol. 2000;74:2293–2304. doi: 10.1128/jvi.74.5.2293-2304.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Horner SM, Liu HM, Park HS, Briley J, Gale M., Jr Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Odendall C, Dixit E, Stavru F, Bierne H, Franz KM, Durbin AF, Boulant S, Gehrke L, Cossart P, Kagan JC. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol. 2014 doi: 10.1038/ni.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim SJ, Syed GH, Khan M, Chiu WW, Sohail MA, Gish RG, Siddiqui A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci U S A. 2014;111:6413–6418. doi: 10.1073/pnas.1321114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Baril M, Racine ME, Penin F, Lamarre D. MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J Virol. 2009;83:1299–1311. doi: 10.1128/JVI.01659-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tang ED, Wang CY. MAVS self-association mediates antiviral innate immune signaling. J Virol. 2009;83:3420–3428. doi: 10.1128/JVI.02623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang N, Liang Y, Devaraj S, Wang J, Lemon SM, Li K. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J Virol. 2009;83:9824–9834. doi: 10.1128/JVI.01125-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 107.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Jr, Lemon SM. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, Ghaffari AA, Qin J, Cheng G, Liu YJ. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity. 2011;34:866–878. doi: 10.1016/j.immuni.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J Biol Chem. 2009;284:807–817. doi: 10.1074/jbc.M804259200. [DOI] [PubMed] [Google Scholar]
- 110.Rajsbaum R, Albrecht RA, Wang MK, Maharaj NP, Versteeg GA, Nistal-Villan E, Garcia-Sastre A, Gack MU. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012;8:e1003059. doi: 10.1371/journal.ppat.1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pang PS, Planet PJ, Glenn JS. The evolution of the major hepatitis C genotypes correlates with clinical response to interferon therapy. PLoS ONE. 2009;4:e6579. doi: 10.1371/journal.pone.0006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, Kidd J, Kidd K, Khakoo SI, Alexander G, Goedert JJ, Kirk GD, Donfield SM, Rosen HR, Tobler LH, Busch MP, McHutchison JG, Goldstein DB, Carrington M. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, Sulkowski M, McHutchison JG, Goldstein DB. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 114.Rauch A, Kutalik Z, Descombes P, Cai T, Di Iulio J, Mueller T, Bochud M, Battegay M, Bernasconi E, Borovicka J, Colombo S, Cerny A, Dufour JF, Furrer H, Gunthard HF, Heim M, Hirschel B, Malinverni R, Moradpour D, Mullhaupt B, Witteck A, Beckmann JS, Berg T, Bergmann S, Negro F, Telenti A, Bochud PY. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138:1338–1345. 1345 e1331–1337. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 115.Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML, Bassendine M, Spengler U, Dore GJ, Powell E, Riordan S, Sheridan D, Smedile A, Fragomeli V, Muller T, Bahlo M, Stewart GJ, Booth DR, George J. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 116.Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, Nakagawa M, Korenaga M, Hino K, Hige S, Ito Y, Mita E, Tanaka E, Mochida S, Murawaki Y, Honda M, Sakai A, Hiasa Y, Nishiguchi S, Koike A, Sakaida I, Imamura M, Ito K, Yano K, Masaki N, Sugauchi F, Izumi N, Tokunaga K, Mizokami M. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 117.Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kelly C, Klenerman P, Barnes E. Interferon lambdas: the next cytokine storm. Gut. 2011;60:1284–1293. doi: 10.1136/gut.2010.222976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, MacDonald MR, Rice CM. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131:1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 120.Pagliaccetti NE, Robek MD. Interferon-lambda in the immune response to hepatitis B virus and hepatitis C virus. J Interferon Cytokine Res. 2010;30:585–590. doi: 10.1089/jir.2010.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dill MT, Duong FH, Vogt JE, Bibert S, Bochud PY, Terracciano L, Papassotiropoulos A, Roth V, Heim MH. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology. 2011;140:1021–1031. doi: 10.1053/j.gastro.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 122.Langhans B, Kupfer B, Braunschweiger I, Arndt S, Schulte W, Nischalke HD, Nattermann J, Oldenburg J, Sauerbruch T, Spengler U. Interferon-lambda serum levels in hepatitis C. J Hepatol. 2011;54:859–865. doi: 10.1016/j.jhep.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 123.Yoshio S, Kanto T, Kuroda S, Matsubara T, Higashitani K, Kakita N, Ishida H, Hiramatsu N, Nagano H, Sugiyama M, Murata K, Fukuhara T, Matsuura Y, Hayashi N, Mizokami M, Takehara T. Human blood dendritic cell antigen 3 (BDCA3)(+) dendritic cells are a potent producer of interferon-lambda in response to hepatitis C virus. Hepatology. 2013;57:1705–1715. doi: 10.1002/hep.26182. [DOI] [PubMed] [Google Scholar]
- 124.Fukuhara T, Taketomi A, Motomura T, Okano S, Ninomiya A, Abe T, Uchiyama H, Soejima Y, Shirabe K, Matsuura Y, Maehara Y. Variants in IL28B in liver recipients and donors correlate with response to peg-interferon and ribavirin therapy for recurrent hepatitis C. Gastroenterology. 2010;139:1577–1585. 1585 e1571–1573. doi: 10.1053/j.gastro.2010.07.058. [DOI] [PubMed] [Google Scholar]
- 125.Egli A, Santer DM, O’Shea D, Barakat K, Syedbasha M, Vollmer M, Baluch A, Bhat R, Groenendyk J, Joyce MA, Lisboa LF, Thomas BS, Battegay M, Khanna N, Mueller T, Tyrrell DL, Houghton M, Humar A, Kumar D. IL-28B is a Key Regulator of B- and T-Cell Vaccine Responses against Influenza. PLoS Pathog. 2014;10:e1004556. doi: 10.1371/journal.ppat.1004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sheahan T, Imanaka N, Marukian S, Dorner M, Liu P, Ploss A, Rice CM. Interferon lambda alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe. 2014;15:190–202. doi: 10.1016/j.chom.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.McFarland AP, Horner SM, Jarret A, Joslyn RC, Bindewald E, Shapiro BA, Delker DA, Hagedorn CH, Carrington M, Gale M, Jr, Savan R. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat Immunol. 2014;15:72–79. doi: 10.1038/ni.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, Hergott D, Porter-Gill P, Mumy A, Kohaar I, Chen S, Brand N, Tarway M, Liu L, Sheikh F, Astemborski J, Bonkovsky HL, Edlin BR, Howell CD, Morgan TR, Thomas DL, Rehermann B, Donnelly RP, O’Brien TR. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45:164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.de Castellarnau M, Aparicio E, Parera M, Franco S, Tural C, Clotet B, Martinez MA. Deciphering the interleukin 28B variants that better predict response to pegylated interferon-alpha and ribavirin therapy in HCV/HIV-1 coinfected patients. PLoS ONE. 2012;7:e31016. doi: 10.1371/journal.pone.0031016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bibert S, Roger T, Calandra T, Bochud M, Cerny A, Semmo N, Duong FH, Gerlach T, Malinverni R, Moradpour D, Negro F, Mullhaupt B, Bochud PY. IL28B expression depends on a novel TT/-G polymorphism which improves HCV clearance prediction. J Exp Med. 2013;210:1109–1116. doi: 10.1084/jem.20130012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lu YF, Goldstein DB, Urban TJ, Bradrick SS. Interferon-lambda4 is a cell-autonomous type III interferon associated with pre-treatment hepatitis C virus burden. Virology. 2015;476C:334–340. doi: 10.1016/j.virol.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Park H, Serti E, Eke O, Muchmore B, Prokunina-Olsson L, Capone S, Folgori A, Rehermann B. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology. 2012;56:2060–2070. doi: 10.1002/hep.25897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thomas E, Gonzalez VD, Li Q, Modi AA, Chen W, Noureddin M, Rotman Y, Liang TJ. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology. 2012;142:978–988. doi: 10.1053/j.gastro.2011.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Marukian S, Andrus L, Sheahan TP, Jones CT, Charles ED, Ploss A, Rice CM, Dustin LB. Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology. 2011;54:1913–1923. doi: 10.1002/hep.24580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stone AE, Giugliano S, Schnell G, Cheng L, Leahy KF, Golden-Mason L, Gale M, Jr, Rosen HR. Hepatitis C Virus Pathogen Associated Molecular Pattern (PAMP) Triggers Production of Lambda-Interferons by Human Plasmacytoid Dendritic Cells. PLoS Pathog. 2013;9:e1003316. doi: 10.1371/journal.ppat.1003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Okamoto M, Oshiumi H, Azuma M, Kato N, Matsumoto M, Seya T. IPS-1 is essential for type III IFN production by hepatocytes and dendritic cells in response to hepatitis C virus infection. J Immunol. 2014;192:2770–2777. doi: 10.4049/jimmunol.1301459. [DOI] [PubMed] [Google Scholar]