

Abstract
A-Kinase Anchoring Proteins (AKAPs) coordinate complex signaling events by serving as spatiotemporal modulators of cAMP-dependent protein kinase activity in cells. Although AKAPs organize a plethora of diverse pathways, their cellular roles are often elusive due to the dynamic nature of these signaling complexes. AKAPs can interact with the type I or type II PKA holoenzymes by virtue of high-affinity interactions with the R-subunits. As a means to delineate AKAP-mediated PKA signaling in cells, we sought to develop isoform-selective disruptors of AKAP signaling. Here, we report the development of conformationally constrained peptides named RI-STapled Anchoring Disruptors (RI-STADs) that target the docking/dimerization domain of the type 1 regulatory subunit of PKA. These high-affinity peptides are isoform-selective for the RI isoforms, can outcompete binding by the classical AKAP disruptor Ht31, and can selectively displace RIα, but not RIIα, from binding the dual-specific AKAP149 complex. Importantly, these peptides are cell-permeable and disrupt Type I PKA-mediated phosphorylation events in the context of live cells. Hence, RI-STAD peptides are versatile cellular tools to selectively probe anchored type I PKA signaling events.
Generation of the second messenger molecule 3′-5′-cyclic adenosine monophosphate (cAMP) triggers a variety of downstream cellular and physiological events.1 After extracellular stimulation, intracellular cAMP levels rise and activate several targets. Protein kinase A (PKA) is the principal effector that is activated in response to cAMP. This broad specificity protein kinase regulates a multitude of diverse processes such as cell proliferation, cell difierentiation, cell death, metabolism and immune responses.2–4 When intracellular levels of cAMP are low, PKA is maintained in an inactive tetrameric holoenzyme complex that is composed of a regulatory subunit (R) dimer and two catalytic subunits (C). Upon stimulated cAMP production, the R-subunits bind two molecules each of cAMP and undergo conformational changes to release and activate the C-subunits. Since PKA phosphorylates numerous substrates, signaling specificity is partially conferred by different R-subunits (RIα, RIβ, RIIα, and RIIβ). Each regulatory subunit isoform varies in their holoenzyme structure, cAMP responsiveness, tissue distribution, and subcellular localization.5 While both RII isoforms are precisely localized in subcellular compartments,6 the RI isoforms are more diffusely dispersed throughout the cytoplasm.7–9
Despite similar domain organization, the R-subunit isoforms of PKA have distinct biological profiles. For example, RI isoform expression is enhanced when the C-subunit is overexpressed, whereas RII seems to be less inducible. 10–13 While RIα is constitutively expressed by all cell types, RIβ is primarily expressed in neurons and brain tissue.14 Moreover, misregulation of RI isoforms has been implicated in a variety of diseases. Altered expression of RIα is believed to play a role in malignant transformation,15 and constitutive overexpression of RIα occurs in several cancers.16 This leads to tumor growth, changes in cell morphology, and poor patient prognosis.16 Thus, while RI is critical regulator in cells, many questions remain about the mechanistic role of RI in the pathological conditions outlined above. At a subcellular level, RIα is found in the cytoplasm, whereas RIβ has been detected more prominently at the mitochondria.9 Moreover, structural studies suggest that RIα and RIβ may impose different modes of allosteric regulation on PKA.9,17–19 Thus, it would appear that both RI isoforms perform unique roles in the cellular environment.
Regulation can be achieved in part through their interaction with a diverse family of proteins called A-Kinase Anchoring Proteins (AKAPs).20 AKAPs facilitate spatial and temporal regulation of PKA through R-subunit interactions.20 AKAPs are also scaffolding proteins that constrain macromolecular complexes within the cell. Although AKAPs are divergent in both structure and function, they share the common feature of directly binding to the R-subunit of PKA. AKAPs anchor PKA in proximity to other proteins and enzymes including other kinases, phosphatases, adenylyl cyclases, phosphodiesterases, and substrate targets.21–24 By forming these dynamic complexes, AKAPs coordinate signaling events by localizing cAMP-responsive signaling complexes to specific sites within the cell.25 Under these conditions, the scaffolded fraction of PKA has restricted access to potential substrates and therefore provides a mechanism for signaling specificity.26,27
While the vast majority of AKAPs selectively bind the RII isoform,22 RI-selective AKAPs including SKIP and smAKAP have been identified.18,19,28 A third class of AKAPs, termed dual-specific AKAPs, can bind both the RI and RII isoforms, yet their preference for binding the RII isoform strongly predominates.29–31 On the basis of the identification of these classes, sequence preferences for docking were defined for each R-subunit isoform.32–34 NMR and crystal structures of A Kinase Binding (AKB) sequences bound to the docking/dimerization (D/D) domain of either RI or RII has provided further structural insight into docking interactions and isoform specificity.33,35–37 While both isoforms form a hydrophobic binding surface for AKAP binding, RI has a deeper, more rigid binding cleft and is therefore more discriminating for binding. Indeed, introduction of single point mutations at most positions in the binding interface of an AKB sequence results in abolishment of binding to RIα.38 Furthermore, an aromatic group in the first helical turn of the AKB binding interface appears to favor binding of RI over RII (Figure 1a) since this side chain complements a cavity on the docking surface of RI which is absent in RII.35
Figure 1.
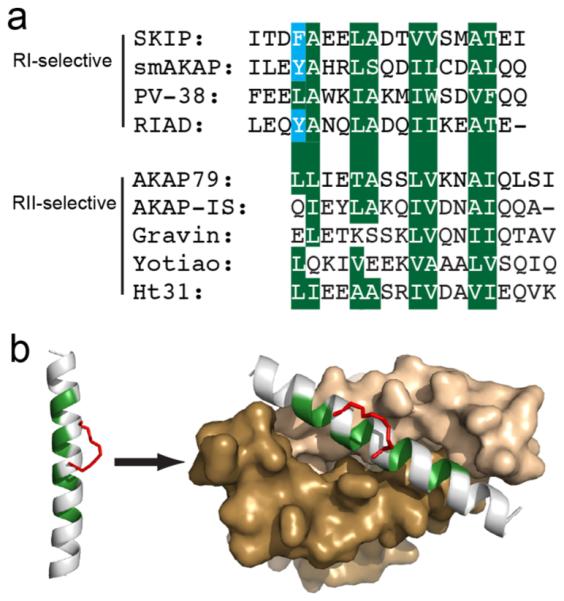
RIAD serves as a template for developing stapled RI-anchoring disruptors. (a) Alignment of various RI- and RII-specific AKAP or AKAP-mimicking sequences. The four hydrophobic registers shared by the docking sequences are highlighted in green. The aromatic residues shared by RI-specific sequences are highlighted in blue. (b) The hydrophobic surface of the α-helical docking peptide is highlighted in green. The hydrocarbon staple is introduced on the solvent-exposed surface and is shown in red. The right panel models the stapled peptide bound to the surface of the docking/dimerization (D/D) domain of PKA-RI (brown and tan) with its hydrophobic surface highlighted in green. The structure was rendered in PyMol using PDB ID 3IM4.
Despite having an improved understanding of the physical interactions involved in the engagement of PKA, the functional significance of AKAPs on signaling in normal and disease-state cells remains elusive. One means to study the role of PKA anchoring on cellular processes has been the development of peptide-based inhibitors. Peptides were designed from AKB sequences that were empirically derived from various AKAPs.38–43 The AKB domain is an amphipathic helix that is formed by 16–20 residues which target a binding groove formed on the surface of the D/D domain (Figure 1).44 The original RII-selective AKAP disruptor, Ht31, was derived from AKAP-Lbc (AKAP13).39 Since then, multiple high-affinity, RII-selective disruptors were identified including AKAP-IS,40 SuperAKAP-IS,41 and STAD-2.42 Although several RII disruptors were developed, it took over a decade to develop the first RI-selective disruptors. The first RI-specific disruptor, PV-38, was derived from peptide array screening using the AKB domain of AKAP10 (D-AKAP2) as a starting template.38 Shortly after this, a generic RI-selective disruptor, RIAD, emerged.43 RIAD was subsequently modified to incorporate non-natural amino acids that improved proteolytic stability.45 However, major drawbacks that remain with the currently available RI-selective compounds are that they are hydrophobic and lack appreciable cell permeability. Accordingly, the utility of these reagents in cellulo remains limited. Intrinsic disadvantages of nonmodified peptides include compromised secondary structure in solution, insuficient cellular permeability in intact cell membranes, and susceptibility to proteolytic degradation.46
Although current AKAP disruptor peptides are proficient tools for studying local PKA signaling, there remains a significant need for cell-permeable and RI-selective AKAP disruptors. Consequently, the biology of local type I PKA-mediated signaling remains less well understood. To address this gap in our knowledge, we sought to develop RI-selective reagents that can selectively disrupt RI interactions with AKAPs without requiring further manipulation to cells. Here, we report a series of RI-STapled Anchoring Disruptors (RI-STADs) that are cell-permeable and selectively bind the RI subunit and disrupt RI-AKAP anchoring in live cells. These high-affinity compounds preferentially bind RIα by 1 to 2 orders of magnitude over the RII isoforms. Moreover, these peptides can effectively outcompete Ht31 binding, demonstrating that the RI-STADs target the canonical D/D binding site on the R-subunit. Furthermore, RI-STAD-2 was found to have nanomolar affinities for both type I R-subunits (RIα and RIβ) and could interrupt protein–protein interactions between the dual-specific anchoring protein AKAP149 (D-AKAP1) and RIα but not RIIα.
RESULTS AND DISCUSSION
Design of Stapled RI-Selective AKAP Disruptors
Since there are notable differences in AKAP docking interactions between the RI and RII isoforms, we reasoned that these variations could be exploited to design improved isoformselective disruptors for PKA anchoring. The peptide sequence of the RI-selective inhibitor, RIAD (LEQYANQLADQ-IIKEATE), is approximately 50-fold more selective for RIα over RIIα.43 RIAD is an amphipathic helix with hydrophobic residues aligned on one face so as to form a hydrophobic patch that complements the hydrophobic binding surface of RI while the polar and charged residues are on the opposing, solvent-exposed face (Figures 1 and 2a). Using RIAD as a template, we sought to engineer improved biophysical and biochemical properties into this sequence by adopting a helix-stabilizing strategy coined hydrocarbon “stapling”.47,48 This synthetic modification not only stabilizes the α-helical structure but also improves cell permeability and proteolytic stability of the peptide.46,48 In order to chemically constrain the α-helical conformation, a pair of non-natural olefinic amino acids ((S)-2-(4′-pentenyl)alanine), abbreviated as S5, were introduced into the sequence. The S5 residues were positioned on the solvent-exposed face so that the staple would not interfere with the protein–peptide interface (Figure 1b). A small library was generated where the staple position was varied so as to surround each individual hydrophobic register along the sequence, thereby stabilizing a single helical turn (Figure 2b). Ring-closing metathesis was subsequently performed to covalently cross-link the olefinic side chains to form the hydrophobic staple (Figure 1b and Figures S1-S3). Since RIAD is hydrophobic and the hydrocarbon staple provides additional hydrophobicity, all peptides were conjugated with an N-terminal (PEG)3 moiety to promote suitability for aqueous-based experiments.
Figure 2.
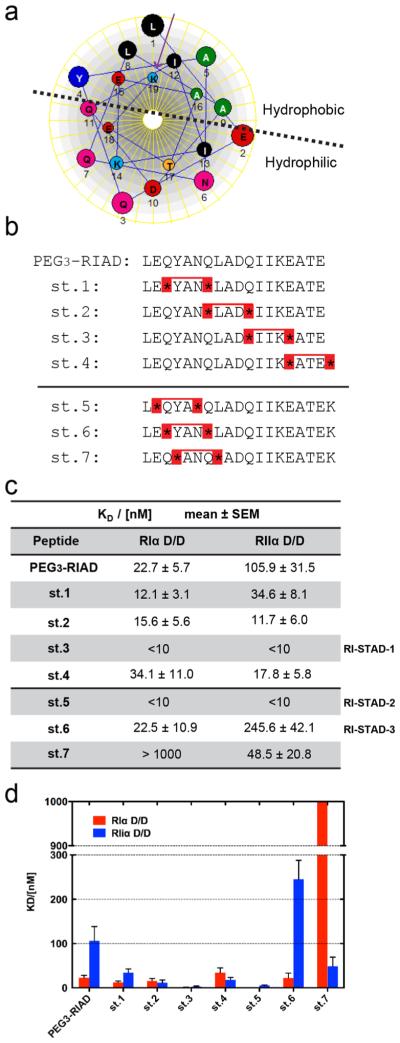
Development of RI-STAD peptides. (a) The amphipathic RIAD sequence is represented as a helical wheel. The addition of a terminal Lys (K19) is included in the wheel (as indicated by a purple arrow). The helical wheel was created using DNASTAR. (b) Sequences for two generations of peptide candidates are listed. The second-generation peptides, st.5–st.7, contain the addition of a terminal Lys residue. S5 is represented by asterisk symbol, and a red bridge represents the staple. (c) Fluorescence polarization (FP) was used to determine the dissociation constant of the fluorescein-labeled peptides using D/D domain constructs from either PKA-RIα or PKA-RIIα. Dissociation constants were calculated using nonlinear regression and are presented as the mean ± SEM from triplicate experiments. (d) Graphic presentation of the dissociation constants from the FP binding assay.
In Vitro Characterization of RI-Stapled Anchoring Disruptors (RI-STADs)
As an initial step to characterize isoform specificity, fluorescence polarization (FP) assays were performed to determine the binding affinity of each peptide toward the D/D domains of RIα and RIIα (Figures 2c,d and S4). The D/D domains were plated over a concentration range of 100 pM to 100 μM using 10 nM of each fluorescently labeled peptide tested. The stapled (PEG)3-RIAD peptides (st.1–3) showed increased binding affinities for both RIα and RIIα D/D as compared to that of the unstapled (PEG)3-RIAD sequence. In contrast, st.4 had a slightly weaker affinity for RIα. Generally, the dissociation constants (KD) of the peptides binding to RIIα had a larger decrease as compared to RIα. The improved affinity for RIIα is perhaps not too surprising given that the D/D of RIIα is more tolerant to changes in the AKB domain of AKAPs38 and peptide stapling can improve binding affinities.46 Importantly, st.1 and st.3 maintained high-affinity binding for RIα in the low nanomolar range. Furthermore, st.1 maintained selectivity toward RIα over RIIα.
Although st.1 demonstrated the highest isoform selectivity for RIα (~3-fold preference), it also exhibited notably limited cell permeability (Figure S5b). Therefore, we further modified this sequence by shifting the position of the staple to the N-or C-terminus by a single residue to determine the optimal staple position over this hydrophobic patch (st.5–7, Figure 2b). In addition, an extra lysine was added to the C-terminus of each st.1-derived sequence to increase the overall net charge in an effort to remedy the poor cellular uptake demonstrated by st.1.49 Binding affinities were measured by fluorescence polarization (FP), and st.5 was found to have improved KD values for both RIα and RIIα. On the other hand, st.7 had no appreciable binding to RIα. This may be due to the fact that st.7 lacks an aromatic residue in the first hydrophobic register, and this is a hallmark feature often found in RI-specific AKB sequences.50 Furthermore, as compared to st.1, st.6 showed an increase in KD values for the D/D domain of both isoforms, particularly RIIα. However, st.6 still maintained approximately 10-fold isoform selectivity toward RI. On the basis of their overall properties, three peptides were selected as candidates for RI-selective disruption and were designated RI-STapled Anchoring Disruptors (st.3, RI-STAD-1; st.5, RI-STAD-2; and st.6, RI-STAD-3).
Next, binding affinities were measured using full-length versions of all four human PKA isoforms (Figure 3a,b). The R-subunits were tested over a concentration range of 60 pM to 10 μM using 4 nM of each fluorescently labeled peptide. All three peptides demonstrated nanomolar affinities for RIα (KD = 5.8, 6.2, and 7.3 nM for RI-STAD-1, -2, and -3, respectively) and similar KD values for RIβ ranging from 12 to 35 nM. Consistent with our previous findings using the D/D domain construct, RI-STAD-1 was less isoform-selective and also bound tightly to RIIα, with a KD value of 19.4 nM. However, RI-STAD-1 had a lower affinity for RIIβ, with a KD value in the midnanomolar range (166 nM), indicating that RI-STAD-1 selectively binds all isoforms with high affinity except for RIIβ. RI-STAD-2 and RI-STAD-3, on the other hand, were both notably selective for the RI isoforms. Relative to the RIα isoform, RI-STAD-2 was 26-fold less selective for RIIα and 79-fold less selective for RIIβ. Of the three RI-STAD peptides, RI-STAD-3 demonstrated the highest selectivity for the RI isoforms, with 58-fold selectivity for RIα over RIIα and 89-fold selectivity over RIIβ. Thus, our data shows that RI-STAD-2 and RI-STAD-3 have notable isoform selectivity for RI over RII by 1 to 2 orders of magnitude. We observed a considerable increase in isoform selectivity for the RI disruptor peptides when using the full-length R-subunits. Although full-length crystal structures are not available for each of the R-subunit isoforms, it has been proposed that each RI isoform may exhibit slightly different modes of interaction for dimerization.9 Hence, long-range allosteric effects may play a key role in regulating engagement with AKAP proteins, and this mode of regulation may provide enhanced isoform selectivity.
Figure 3.

In vitro characterization using full-length human R-subunits. (a) Normalized fluorescence polarization (FP) is shown for each of the full-length PKA R-subunit isoforms with the indicated fluorescently labeled peptides RI-STAD-1, -2, and -3. PKA-RI is represented in red (closed circles, α; open circles, β), and PKA-RII is shown in blue (closed triangles, α; open triangles, β). Peptides were plated at a final concentration of 4 nM, and the PKA R-subunits were tested over a concentration range of 60 pM to 10 μM. (b) Comparison of KD values of FP binding assays. RI-STAD-2 and RI-STAD-3 have higher selectivity for PKA-RI over PKA-RII. (c) FP competition spectra are shown for PKA-RIα with the three indicated RI-STAD peptides. The assay was performed using a final concentration of 4 nM RI-STADs and 5 nM RIα. The competitor peptide, Ht31, was tested over a concentration range of 15 nM to 30 μM. (d) Apparent EC50 values of FP competition assays with RIα. All FP data were collected in triplicates for each concentration measurement.
The AKAP family is structurally and functionally diverse.51 This extends to the R-subunit docking interface between AKAPs. In fact, not all AKAPs exclusively bind to the D/D domain of PKA using an AKB docking helix. Some of these noncanonical AKAPs include pericentrin, which binds the RII subunit using a nonhelical, leucine-rich region that spans over 100 amino acids.52 Another example is α4-integrin, which is an RI-specific AKAP that is insensitive to traditional amphipathic peptide disruptors and therefore may dock through a different site on the R-subunit.53 Furthermore, several conventional AKAPs also utilize an additional binding surface that has been denoted as the RI specifier region (RISR).54 This region is proximal or distal to the traditional amphipathic AKB domain and appears to facilitate the stabilization of AKAP-RI complex. Therefore, competition assays were performed using a prototypic AKAP disrupter, Ht31, to ensure that the RI-STAD peptides are indeed targeting RI through its conventional binding site on the D/D domain. The competition experiment was performed by mixing 4 nM of fluorescently labeled RI-STAD peptides with a concentration range of unlabeled Ht31, followed by a 5 min incubation period with RIα before measuring fluorescence polarization. The calculated EC50 values of Ht31 for RI-STAD-1, -2, and -3 are 1.9, 2.1, and 1.5 μM, respectively (Figure 3c,d). These values correlate with the reported KD value of 2.1 μM for the measured binding affinity between Ht31 and RIα.55 As a control, a competition assay was performed using RI-STAD-2 and a concentration range of Ht31P (Figure S6). Ht31P is an inactive analog of Ht31 where two isoleucines are substituted with prolines, thereby disrupting the essential helical conformation that is required for binding the D/D domain.39,56 Even at concentrations up to 30 μM, Ht31P did not displace RI-STAD-2 from RIα. Together, this data suggests that the RI-STAD peptides do indeed bind the D/D domain of RIα by mimicking the amphipathic helical AKB of canonical AKAPs. Furthermore, it appears that these peptides have the potential to act as disruptors to displace AKAPs that interface with type I PKA holoenzymes.
RI-STAD-2 Is a High-affinity Binder to the RI Isoforms
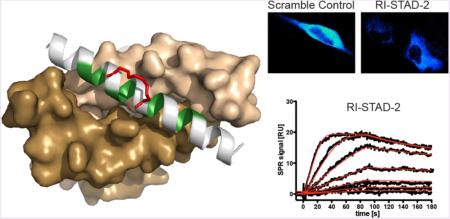
To quantify the interaction of RI-STAD-2 with recombinant human RI subunits, Biacore technology was employed. RIα and RIβ were captured on 8-AHA-cAMP sensor surfaces at surface concentrations of 800 resonance units (see Supporting Information, Methods, for details). RI-STAD-2 was injected as an analyte at different concentrations varying from 4.8 to 312.5 nM RI-STAD-2 (Figure 4a,b). Association and dissociation rate constants were determined. Using these rate constants, RI-STAD-2 was calculated to have an equilibrium binding constant of 5.7 nM on an RIα surface and 19 nM on an RIβ surface (Table 1). Notably, RI-STAD-2 associates with fast on-rate kinetics; however, it displays very slow off rates. Thus, RI-STAD-2 binds both of the RI isoforms with high affinity, and once the complex is formed, it demonstrates slow dissociation from the RI complex.
Figure 4.

Binding analysis to human PKA type I R-subunits. Interaction of RI-STAD-2 with RIα and RIβ (for rate and equilibrium binding constants, see Table 1). (a, b) RIα (800 RU) or RIβ (800 RU) subunits were captured on 8-AHA-cAMP sensor chips, and RI-STAD-2 was injected at the concentrations indicated. Black curves represent the measured responses, whereas red lines reflect the applied global fit using the 1:1 Langmuir interaction model. Association and dissociation phases were monitored for 180 s. RU, response units.
Table 1.
Apparent Rate and Equilibrium Binding Constants of RI-STAD-2 Binding to the RI Isoforms
| ka (M−1 s−1) | kd (s−1) | KD (nM) | ||
|---|---|---|---|---|
| RI-STAD-2 | RIα | 8.8 × 105 | 5.0 × 10−3 | 5.7 |
| RIβ | 2.9 × 105 | 5.5 × 10−3 | 19.0 |
RI-STAD-2 Outcompetes RI-AKAP Interactions
As a strategy to measure whether RI-STAD-2 could effectively outcompete interactions between an AKAP and the R-subunits, binding studies were performed using the dual-specific anchoring protein AKAP149 (Figure 5). Competition experiments were performed with either RI-STAD-2 or the RIAD control peptide. Binding interactions were measured by immobilizing either RIα or RIβ to the Biacore chip surface at concentrations of approximately 2500 RU. A fragment of AKAP149 (285–387) was then injected in the presence or absence of 5 μM RIAD or RI-STAD-2 (see Supporting Information, Methods, for details). Both RIAD and RI-STAD-2 could efficiently outcompete interactions between RIα and AKAP149. On the other hand, binding of the AKAP149 construct to RIIα was not affected by the presence of these AKAP disruptor peptides. No AKAP149 (285–387) binding was detected when RIβ was captured as described previously.34 Thus, RI-STAD-2 appears to have high specificity for the RI complex and does not appear to disrupt AKAP binding interactions at the RII complex.
Figure 5.
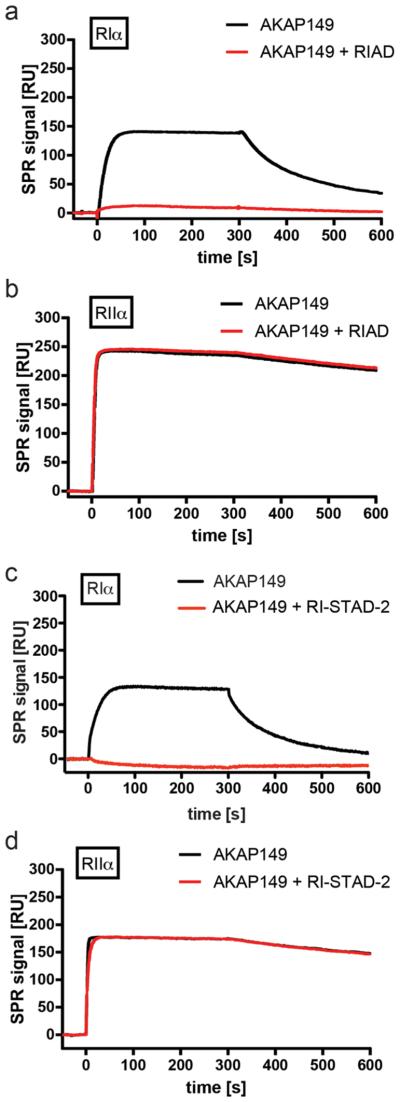
Qualitative surface competition experiments. RIα (a, c) and RIIα (b, d) were immobilized to an 8-AHA-cAMP surface at resonance levels of 2300 or 2500 RUs, respectively. AKAP149 (285–387, 500 nM) was injected in the absence or presence of 5 μM RIAD (a, b) or RI-STAD-2 (c, d), and the residual binding to the R-subunits was determined. Both RIAD and RI-STAD-2 specifically inhibit binding of AKAP149 (285–387) to RIα, whereas no competitive effect was found for the interaction between RIIα and AKAP149 (285–387). Since AKAP disruptor peptides bind to the immobilized R-subunits, a small increase in mass was expected. Therefore, control experiments were performed using 5 μM of RIAD or RI-STAD-2 without AKAP149 (285–387) present. RU, response units.
RI-STADs Are Cell-Permeable and Target the RI Isoform in Cells
To determine whether the RI-STAD peptides would be suitable for cell-based experiments, intracellular access was measured. To measure the extent of cellular uptake of the RI-STAD peptides, localization was measured using live cell imaging. Cells were treated with 5 μM of fluorescein-labeled peptides for 6 h prior to imaging. As our previous work has shown, hydrocarbon stapling drastically increased the ability of AKAP inhibitor peptides to permeate through intact cell membranes.42 Indeed, whole-field quantification of multiple fields (n = 28–32 fields representing at least 1400 cells) indicates that the RI-STAD peptides penetrated cells after this treatment period to a greater extent as compared to that of the unstapled RI-STAD-2 control (Figure 6a). While the extent of permeability varied among the peptides, the measured fluorescence per cell was statistically significant and at least roughly twice that of the control. Furthermore, there is an increase in the cytoplasmic levels of fluorescein staining for the stapled STAD peptides, demonstrating that these peptides can access this intracellular compartment.
Figure 6.
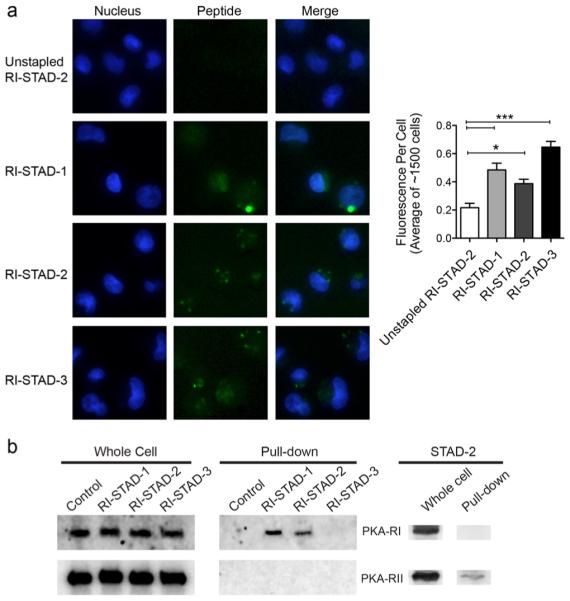
RI-STAD peptides are cell-permeable and selectively bind the RI isoform in cells. (a) Fluorescent images and quantification of live cells after treatment with FITC-labeled peptides (5 μM) for 6 h shows that RI-STAD-1, -2, and -3 demonstrate enhanced intracellular localization in cells. Quantification was performed using 28–32 fields (n = 1410–1670 cells). * p < 0.05, *** p < 0.001 relative to the unstapled control. (b) RI-STAD-1 and RI-STAD-2 bind the RI isoform, but not RII, in cells. MDA-MB-231 cell were treated with 5 μM biotin-labeled peptides for 12 h before lysis. Pulldowns were performed using avidin-coated resin, and the RI and RII isoforms were detected by immunoblotting. As a control, the peptide STAD-2 binds only the RII isoform, not RI.
Next, we wanted to verify that the RI-STAD peptides could bind their target protein, RI, within the complexity of a cellular environment. To test this, biotin pulldowns were performed. Biotin-labeled RI-STAD-1, -2, or -3 was added to the media of intact MDA-MB-231 cells for 12 h. Cells were then lysed, and pulldowns were performed using avidin-coated resin. To determine whether the peptides maintained isoform selectivity in cells, immunoblotting was performed to probe for interactions with the RI and RII isoforms. Whole-cell lysates were used as an input control, and cells that were treated only with DMSO were used as a pulldown control. As compared to the DMSO-treated control, RI-STAD-1 and RI-STAD-2 appeared to gain cytosolic access and successfully bound to PKA-RI within the intracellular environment, whereas RI-STAD-3 showed nominal interactions with the RI subunit (Figure 6b). None of the RI-STAD peptides appeared to bind to the RII subunit to a detectable level. As a control, the RII-specific AKAP disruptor STAD-242 pulled down only RII but not RI. On the basis of this study, RI-STAD-1 and -2 appear to act as promising, competitive RI-selective disruptors both in vitro and in the context of a cellular environment.
As a means to further explore the specificity of RI-STAD-2, we used a previously described fluorescence resonance energy transfer (FRET)-based PKA activity sensor, AKAR-18RBS.57 AKAR-18RBS includes a PKA-R binding helix from AKAP1858,59 and allows the reporter to detect the activity of AKAP-anchored PKA phosphorylation in real-time by monitoring changes in the YFP/CFP emission ratio in living cells. U2OS RIIΔ cells were used to measure the effects of RI-STAD-2 on RI-AKAP anchoring. The effect of displacing RI from the reporter was examined after isoproterenol (iso)-stimulated conditions (Figure 7). The RI-selective disruptor RI-STAD-2 decreased the FRET signal as compared to that in the scrambled control (scr) after elevated cAMP levels were induced. The normalized FRET ratio of scrambled-treated cells reached a maximum at 60 s and dropped gradually over 150 s. In contrast, the FRET signal of RI-STAD-2-treated cells showed a faster decline and a lower resting level. As a control, the RII-specific disruptor STAD-2 was also shown to disrupt AKAP anchoring when RII is present (Figure S7). In summary, RI-STAD-2 was shown to effectively disrupt RI anchoring at the AKAP reporter, thereby decreasing the amount of PKA in the AKAP complex in live cells.
Figure 7.

RI-STAD-2 disrupts AKAP-anchored signaling in cells. AKAP-18RBS AKAR activity was monitored, and FRET signal was measured in U2OS RIIΔ cells that were preincubated with 1 μM RI-STAD-2 (n = 26 cells) or its scrambled control for 1 h (n = 24 cells). RI-specific AKAP-18RBS AKAR activity was then monitored over 150 s after stimulation with 1 μM isoproterenol at the 40 s time point. Data was normalized relative to unstimulated basal FRET levels. RI-STAD-2 was found to reduce RI-specific AKAP anchoring in cells. Warmer colors indicate increasing phosphorylation.
Multiple strategies have been used to design cell-penetrating peptides (CPPs). One approach involves incorporation of cell-penetrating sequences such as HIV-1 TAT,60 penetratin,61 polyarginine,62 and MAP63 that allow for cargo delivery into cells. Other cell-penetrating techniques utilize chemical modifications such as stearation to further enhance cell permeability.64 Limitations of these aforementioned techniques include variability in cell penetration,65 lack of reinforcement of the peptide secondary structure, susceptibility to proteolysis,66,67 and potential mislocalization caused either by the CPP sequence or chemical moiety within the cellular environment.65 Other chemical modifications including lactam bridges were shown to stabilize the secondary structural fold of helices.68 However, peptides bearing this modification may still have limited cell permeability and susceptibility to cellular degradation.68 We contend that peptide stapling has numerous benefits. These include enhanced cell permeability, reinforcement of the secondary structural fold and resistance to proteolysis.46,49,68 Also, no additional sequence is required for conjugation, thereby reducing the overall peptide length and the possibility for off-target binding and mislocalization.
The constrained peptides reported in this study, RI-STAD-1 and RI-STAD-2, can be used to selectively disrupt the interactions between AKAPs and PKA-RI in biochemical and cell-based assays. These peptides demonstrated exquisite isoform selectivity in vitro by surface plasmon resonance and within the cellular environment. By inhibiting interactions between PKA-RI and AKAPs, one can study the direct and downstream cellular effects of localized RI-mediated signaling in a variety of normal and disease state systems. One shortcoming of this particular targeting site is that it will, in effect, nonselectively inhibit all RI-selective AKAPs that bind the canonical docking site on RI. However, so far, only a limited number of AKAPs are believed to be RI-specific. Another possible limitation is that RI-STAD peptides do not demonstrate selectivity between the RIα and RIβ isoforms. With that being said, RI-STAD peptides could be effectively used in cell lines, tissues, or subcellular compartments where the expression of a particular RI isoform predominates. For instance, although the RIα-selective anchoring protein SKIP has been shown to be localized at the mitochondria,18 key questions remain about its physiological role in both normal and disease states. Thus, dissecting the spatial and temporal dynamics of these macromolecular complexes should provide new insight into the biological role of RI isoforms. When considered with our previously reported RII-selective disruptor,42 RI-STAD-2 and STAD-2 will serve as useful probes to dissect the respective roles of AKAP-mediated type I and type II PKA signaling.
Supplementary Material
ACKNOWLEDGMENTS
We kindly thank I. Georgieva, L. P. Schendowich, A. Sukhu, and S. Patel for their technical assistance. The authors would like to thank the National Institutes of Health (1K22CA154600 to E.J.K; DK54441 and DK105542 to J.D.S., F.D.S., H.H., and J.L.E.; and K99GM107355 to H.H.). In addition, we thank the European Union FP7 Health Programme (241481 AFFINOMICS to F.W.H.) and the Federal Ministry of Education and Research (funding no. 0316177F, “No Pain”, to F.W.H.). E.F. is supported by a Ph.D. fellowship from the University of Kassel as a member of the graduate program Functionomics. F.W.H. acknowledges the Center for Interdisciplinary Nanostructure Science and Technology (CINSaT) at the University of Kassel for support of this work. J.L.E. is supported by a Heart and Stroke Foundation of Canada Postdoctoral Fellowship.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Bar HP, Hechter O. Adenyl cyclase and hormone action. I. Effects of adrenocorticotropic hormone, glucagon, and epinephrine on the plasma membrane of rat fat cells. Proc. Natl. Acad. Sci. U.S.A. 1969;63:350–356. doi: 10.1073/pnas.63.2.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Carnegie GK, Smith FD, McConnachie G, Langeberg LK, Scott JD. AKAP-Lbc nucleates a protein kinase D activation scaffold. Mol. Cell. 2004;15:889–899. doi: 10.1016/j.molcel.2004.09.015. [DOI] [PubMed] [Google Scholar]
- (3).Tasken K, Aandahl EM. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev. 2004;84:137–167. doi: 10.1152/physrev.00021.2003. [DOI] [PubMed] [Google Scholar]
- (4).Taylor SS, Zhang P, Steichen JM, Keshwani MM, Kornev AP. PKA: lessons learned after twenty years. Biochim. Biophys. Acta. 2013;7:25. doi: 10.1016/j.bbapap.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Taylor SS, Ilouz R, Zhang P, Kornev AP. Assembly of allosteric macromolecular switches: lessons from PKA. Nat. Rev. Mol. Cell Biol. 2012;13:646–658. doi: 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Burns LL, Canaves JM, Pennypacker JK, Blumenthal DK, Taylor SS. Isoform specific differences in binding of a dual-specificity A-kinase anchoring protein to type I and type II regulatory subunits of PKA. Biochemistry. 2003;42:5754–5763. doi: 10.1021/bi0265729. [DOI] [PubMed] [Google Scholar]
- (7).Barradeau S, Imaizumi-Scherrer T, Weiss MC, Faust DM. Muscle-regulated expression and determinants for neuro-muscular junctional localization of the mouse RIalpha regulatory subunit of cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 2001;98:5037–5042. doi: 10.1073/pnas.081393598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Imaizumi-Scherrer T, Faust DM, Barradeau S, Hellio R, Weiss MC. Type I protein kinase a is localized to interphase microtubules and strongly associated with the mitotic spindle. Exp. Cell Res. 2001;264:250–265. doi: 10.1006/excr.2001.5164. [DOI] [PubMed] [Google Scholar]
- (9).Ilouz R, Bubis J, Wu J, Yim YY, Deal MS, Kornev AP, Ma Y, Blumenthal DK, Taylor SS. Localization and quaternary structure of the PKA RIbeta holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 2012;109:12443–12448. doi: 10.1073/pnas.1209538109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Uhler MD, McKnight GS. Expression of cDNAs for two isoforms of the catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem. 1987;262:15202–15207. [PubMed] [Google Scholar]
- (11).Amieux PS, McKnight GS. The essential role of RI alpha in the maintenance of regulated PKA activity. Ann. N.Y. Acad. Sci. 2002;968:75–95. doi: 10.1111/j.1749-6632.2002.tb04328.x. [DOI] [PubMed] [Google Scholar]
- (12).Otten AD, McKnight GS. Overexpression of the type II regulatory subunit of the cAMP-dependent protein kinase eliminates the type I holoenzyme in mouse cells. J. Biol. Chem. 1989;264:20255–20260. [PubMed] [Google Scholar]
- (13).Clegg CH, Ran W, Uhler MD, McKnight GS. A mutation in the catalytic subunit of protein kinase A prevents myristylation but does not inhibit biological activity. J. Biol. Chem. 1989;264:20140–20146. [PubMed] [Google Scholar]
- (14).Cadd G, McKnight GS. Distinct patterns of cAMP-dependent protein kinase gene expression in mouse brain. Neuron. 1989;3:71–79. doi: 10.1016/0896-6273(89)90116-5. [DOI] [PubMed] [Google Scholar]
- (15).Caretta A, Mucignat-Caretta C. Protein kinase a in cancer. Cancers. 2011;3:913–926. doi: 10.3390/cancers3010913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Neary CL, Nesterova M, Cho YS, Cheadle C, Becker KG, Cho-Chung YS. Protein kinase A isozyme switching: eliciting differential cAMP signaling and tumor reversion. Oncogene. 2004;23:8847–8856. doi: 10.1038/sj.onc.1208165. [DOI] [PubMed] [Google Scholar]
- (17).Day ME, Gaietta GM, Sastri M, Koller A, Mackey MR, Scott JD, Perkins GA, Ellisman MH, Taylor SS. Isoform-specific targeting of PKA to multivesicular bodies. J. Cell Biol. 2011;193:347–363. doi: 10.1083/jcb.201010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Means CK, Lygren B, Langeberg LK, Jain A, Dixon RE, Vega AL, Gold MG, Petrosyan S, Taylor SS, Murphy AN, Ha T, Santana LF, Tasken K, Scott JD. An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2011;108:E1227–E1235. doi: 10.1073/pnas.1107182108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kovanich D, van der Heyden MA, Aye TT, van Veen TA, Heck AJ, Scholten A. Sphingosine kinase interacting protein is an A-kinase anchoring protein specific for type I cAMP-dependent protein kinase. ChemBioChem. 2010;11:963–971. doi: 10.1002/cbic.201000058. [DOI] [PubMed] [Google Scholar]
- (20).Esseltine JL, Scott JD. AKAP signaling complexes: pointing towards the next generation of therapeutic targets? Trends Pharmacol. Sci. 2013;34:648–655. doi: 10.1016/j.tips.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Baillie GS, Scott JD, Houslay MD. Compartmentalisation of phosphodiesterases and protein kinase A: opposites attract. FEBS Lett. 2005;579:3264–3270. doi: 10.1016/j.febslet.2005.03.089. [DOI] [PubMed] [Google Scholar]
- (22).Welch EJ, Jones BW, Scott JD. Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol. Interv. 2010;10:86–97. doi: 10.1124/mi.10.2.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).auer CW. Adenylyl cyclase-A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol. Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sanderson JL, Dell’Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist. 2011;17:321–336. doi: 10.1177/1073858410384740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- (26).Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD. Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc. Natl. Acad. Sci. U.S.A. 2008;105:12557–12562. doi: 10.1073/pnas.0805922105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hinke SA, Navedo MF, Ulman A, Whiting JL, Nygren PJ, Tian G, Jimenez-Caliani AJ, Langeberg LK, Cirulli V, Tengholm A, Dell’Acqua ML, Santana LF, Scott JD. Anchored phosphatases modulate glucose homeostasis. EMBO J. 2012;31:3991–4004. doi: 10.1038/emboj.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Burgers PP, Ma Y, Margarucci L, Mackey M, van der Heyden MA, Ellisman M, Scholten A, Taylor SS, Heck AJ. A small novel A-kinase anchoring protein (AKAP) that localizes specifically protein kinase A-regulatory subunit I (PKA-RI) to the plasma membrane. J. Biol. Chem. 2012;287:43789–43797. doi: 10.1074/jbc.M112.395970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lin RY, Moss SB, Rubin CS. Characterization of S-AKAP84, a novel developmentally regulated A kinase anchor protein of male germ cells. J. Biol. Chem. 1995;270:27804–27811. doi: 10.1074/jbc.270.46.27804. [DOI] [PubMed] [Google Scholar]
- (30).Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J. Biol. Chem. 1997;272:8057–8064. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- (31).Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. D-AKAP2, a novel protein kinase A anchoring protein with a putative RGS domain. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11184–11189. doi: 10.1073/pnas.94.21.11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).t JD. Dissection of protein kinase and phosphatase targeting interactions. Soc. Gen. Physiol. Ser. 1997;52:227–239. [PubMed] [Google Scholar]
- (33).Newlon MG, Roy M, Morikis D, Hausken ZE, Coghlan V, Scott JD, Jennings PA. The molecular basis for protein kinase A anchoring revealed by solution NMR. Nat. Struct. Biol. 1999;6:222–227. doi: 10.1038/6663. [DOI] [PubMed] [Google Scholar]
- (34).Herberg FW, Maleszka A, Eide T, Vossebein L, Tasken K. Analysis of A-kinase anchoring protein (AKAP) interaction with protein kinase A (PKA) regulatory subunits: PKA isoform specificity in AKAP binding. J. Mol. Biol. 2000;298:329–339. doi: 10.1006/jmbi.2000.3662. [DOI] [PubMed] [Google Scholar]
- (35).Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, Xuong NH, Jennings PA, Taylor SS. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol. Cell. 2006;24:397–408. doi: 10.1016/j.molcel.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sarma GN, Kinderman FS, Kim C, von Daake S, Chen L, Wang BC, Taylor SS. Structure of D-AKAP2:PKA RI complex: insights into AKAP specificity and selectivity. Structure. 2010;18:155–166. doi: 10.1016/j.str.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Newlon MG, Roy M, Morikis D, Carr DW, Westphal R, Scott JD, Jennings PA. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J. 2001;20:1651–1662. doi: 10.1093/emboj/20.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Burns-Hamuro LL, Ma Y, Kammerer S, Reineke U, Self C, Cook C, Olson GL, Cantor CR, Braun A, Taylor SS. Designing isoform-specific peptide disruptors of protein kinase A localization. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4072–4077. doi: 10.1073/pnas.2628038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Carr DW, Hausken ZE, Fraser ID, Stofko-Hahn RE, Scott JD. Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J. Biol. Chem. 1992;267:13376–13382. [PubMed] [Google Scholar]
- (40).Alto NM, Soderling SH, Hoshi N, Langeberg LK, Fayos R, Jennings PA, Scott JD. Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4445–4450. doi: 10.1073/pnas.0330734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Gold MG, Lygren B, Dokurno P, Hoshi N, McConnachie G, Tasken K, Carlson CR, Scott JD, Barford D. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol. Cell. 2006;24:383–395. doi: 10.1016/j.molcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- (42).Wang Y, Ho TG, Bertinetti D, Neddermann M, Franz E, Mo GC, Schendowich LP, Sukhu A, Spelts RC, Zhang J, Herberg FW, Kennedy EJ. Isoform-selective disruption of AKAP-localized PKA using hydrocarbon stapled peptides. ACS Chem. Biol. 2014;9:635–642. doi: 10.1021/cb400900r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Carlson CR, Lygren B, Berge T, Hoshi N, Wong W, Tasken K, Scott JD. Delineation of type I protein kinase A-selective signaling events using an RI anchoring disruptor. J. Biol. Chem. 2006;281:21535–21545. doi: 10.1074/jbc.M603223200. [DOI] [PubMed] [Google Scholar]
- (44).Carr DW, Stofko-Hahn RE, Fraser ID, Bishop SM, Acott TS, Brennan RG, Scott JD. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J. Biol. Chem. 1991;266:14188–14192. [PubMed] [Google Scholar]
- (45).Torheim EA, Jarnaess E, Lygren B, Tasken K. Design of proteolytically stable RI-anchoring disruptor peptidomi-metics for in vivo studies of anchored type I protein kinase A-mediated signalling. Biochem. J. 2009;424:69–78. doi: 10.1042/BJ20090933. [DOI] [PubMed] [Google Scholar]
- (46).Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]
- (47).Schafmeister CE, Po J, Verdine GL. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000;122:5891–5892. [Google Scholar]
- (48).Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Chu Q, Moellering RE, Hilinski GJ, Kim Y-W, Grossmann TN, Yeh JTH, Verdine GL. Towards understanding cell penetration by stapled peptides. MedChemComm. 2015;6:111–119. [Google Scholar]
- (50).Burgers PP, van der Heyden MA, Kok B, Heck AJ, Scholten A. A systematic evaluation of protein kinase A-A-kinase anchoring protein interaction motifs. Biochemistry. 2014;54:11–21. doi: 10.1021/bi500721a. [DOI] [PubMed] [Google Scholar]
- (51).Scott JD, Dessauer CW, Tasken K. Creating order from chaos: cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 2013;53:187–210. doi: 10.1146/annurev-pharmtox-011112-140204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Diviani D, Langeberg LK, Doxsey SJ, Scott JD. Pericentrin anchors protein kinase A at the centrosome through a newly identified RII-binding domain. Curr. Biol. 2000;10:417–420. doi: 10.1016/s0960-9822(00)00422-x. [DOI] [PubMed] [Google Scholar]
- (53).Lim CJ, Han J, Yousefi N, Ma Y, Amieux PS, McKnight GS, Taylor SS, Ginsberg MH. Alpha4 integrins are type I cAMP-dependent protein kinase-anchoring proteins. Nat. Cell Biol. 2007;9:415–421. doi: 10.1038/ncb1561. [DOI] [PubMed] [Google Scholar]
- (54).Jarnaess E, Ruppelt A, Stokka AJ, Lygren B, Scott JD, Tasken K. Dual specificity A-kinase anchoring proteins (AKAPs) contain an additional binding region that enhances targeting of protein kinase A type I. J. Biol. Chem. 2008;283:33708–33718. doi: 10.1074/jbc.M804807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Burton KA, Johnson BD, Hausken ZE, Westenbroek RE, Idzerda RL, Scheuer T, Scott JD, Catterall WA, McKnight GS. Type II regulatory subunits are not required for the anchoring-dependent modulation of Ca2+ channel activity by cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11067–11072. doi: 10.1073/pnas.94.20.11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Vijayaraghavan S, Goueli SA, Davey MP, Carr DW. Protein kinase A-anchoring inhibitor peptides arrest mammalian sperm motility. J. Biol. Chem. 1997;272:4747–4752. doi: 10.1074/jbc.272.8.4747. [DOI] [PubMed] [Google Scholar]
- (57).Smith FD, Reichow SL, Esseltine JL, Shi D, Langeberg LK, Scott JD, Gonen T. Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. eLife. 2013;2:e01319. doi: 10.7554/eLife.01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Fraser ID, Tavalin SJ, Lester LB, Langeberg LK, Westphal AM, Dean RA, Marrion NV, Scott JD. A novel lipid-anchored A-kinase anchoring protein facilitates cAMP-responsive membrane events. EMBO J. 1998;17:2261–2272. doi: 10.1093/emboj/17.8.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Gray PC, Johnson BD, Westenbroek RE, Hays LG, Yates JR, III, Scheuer T, Catterall WA, Murphy BJ. Primary structure and function of an A kinase anchoring protein associated with calcium channels. Neuron. 1998;20:1017–1026. doi: 10.1016/s0896-6273(00)80482-1. [DOI] [PubMed] [Google Scholar]
- (60).Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- (61).Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- (62).Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- (63).Pooga M, Hallbrink M, Zorko M, Langel U. Cell penetration by transportan. FASEB J. 1998;12:67–77. doi: 10.1096/fasebj.12.1.67. [DOI] [PubMed] [Google Scholar]
- (64).Futaki S, Ohashi W, Suzuki T, Niwa M, Tanaka S, Ueda K, Harashima H, Sugiura Y. Stearylated arginine-rich peptides: a new class of transfection systems. Bioconjugate Chem. 2001;12:1005–1011. doi: 10.1021/bc015508l. [DOI] [PubMed] [Google Scholar]
- (65).Zaro JL, Vekich JE, Tran T, Shen WC. Nuclear localization of cell-penetrating peptides is dependent on endocytosis rather than cytosolic delivery in CHO cells. Mol. Pharmaceutics. 2009;6:337–344. doi: 10.1021/mp800239p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Tikhonov I, Ruckwardt TJ, Berg S, Hatfield GS, David Pauza C. Furin cleavage of the HIV-1 Tat protein. FEBS Lett. 2004;565:89–92. doi: 10.1016/j.febslet.2004.03.079. [DOI] [PubMed] [Google Scholar]
- (67).Trehin R, Nielsen HM, Jahnke HG, Krauss U, Beck-Sickinger AG, Merkle HP. Metabolic cleavage of cell-penetrating peptides in contact with epithelial models: human calcitonin (hCT)-derived peptides, Tat(47-57) and penetratin(43-58) Biochem. J. 2004;382:945–956. doi: 10.1042/BJ20040238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules. Nat. Chem. 2013;5:161–173. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.