

Abstract
Inhaled corticosteroid(s) (ICS) increase community-acquired pneumonia (CAP) incidence in patients with chronic obstructive pulmonary disease (COPD) by unknown mechanisms. Apoptosis is increased in the lungs of COPD patients. Uptake of apoptotic cells (AC) (“efferocytosis”) by alveolar macrophages (AMø) reduces their ability to combat microbes, including Streptococcus pneumoniae, the most common cause of CAP in COPD patients. Having shown that ICS significantly increase AMø efferocytosis, we hypothesized that this process, termed glucocorticoid-augmented efferocytosis (GCAE), might explain the association of CAP with ICS therapy in COPD. To test this hypothesis, we studied the effects of fluticasone, AC or both on AMø of C57BL/6 mice in vitro and in an established model of pneumococcal pneumonia. Fluticasone plus AC significantly reduced TLR4-stimulated AMø IL-12 production, relative to either treatment alone, and decreased TNF-α, CCL3, CCL5 and KC, relative to AC. Mice treated with fluticasone plus AC before infection with viable pneumococci developed significantly more lung CFU at 48 h. However, none of the pretreatments altered inflammatory cell recruitment to the lungs at 48 h post-infection, and fluticasone plus AC less markedly reduced in vitro mediator production to heat-killed pneumococci. Fluticasone plus AC significantly reduced in vitro AMø killing of pneumococci, relative to other conditions, in part by delaying phagolysosome acidification without affecting production of reactive oxygen or nitrogen species. These results support GCAE as a potential explanation for the epidemiological association of ICS therapy of COPD patients with increased risk of CAP, and establish murine experimental models to dissect underlying molecular mechanisms.
Keywords: Streptococcus pneumoniae, pneumonia, bacterial, mice, inbred strain C57BL/6
INTRODUCTION
Therapy with inhaled corticosteroids (ICS) is central to chronic obstructive pulmonary disease (COPD) management, but is associated in this patient population with excess cases of community-acquired pneumonia (CAP) both in multi-center clinical trials and in analyses of administrative databases (1–10). Suggestions that this risk is balanced by reduced mortality in COPD patients admitted with CAP while using ICS (11, 12) have been contested (13–15). Defining the molecular basis of this epidemiological association could lead to more precisely personalized therapies and better outcomes in COPD, currently the third leading cause of death in the USA (16).
In COPD, as in the general population, for almost three decades the organism most commonly identified in CAP has been Streptococcus pneumoniae, also known colloquially as pneumococcus (17–19). It might seem intuitively obvious that the immunosuppressive properties of ICS should increase pneumonia frequency. However, in the sole study using a murine model, glucocorticoids (GC) alone actually reduced lung burden of pneumococcus (20). Additionally, ICS therapy is extremely prevalent in asthma, but most but not all studies have shown no similar increased risk of CAP in asthmatics (21–24). These findings suggest that additional factors may underlie the association of increased CAP risk and ICS therapy in COPD.
Apoptotic cells (AC) are increased in the lungs of COPD patients (25–29). Uptake of AC, also known as efferocytosis, is a complex, incompletely understood process that is relevant to many lung diseases (30). AC uptake by alveolar macrophages (AMø) is lower than by Mø from other organs, and is further reduced by smoking and in COPD (29, 31–37). This lung-specific suppression of AC uptake is mediated in part by interactions between the lung collectins surfactant proteins A (SP-A) & D (SP-D) and the inhibitory Mø receptor signal regulatory protein-alpha (SIRPα, CD172A) (38). The reduced basal efferocytic capacity of AMø is germane to increased CAP risk, as AC uptake induces a unique Mø activation state that favors resolution of inflammation (39), but which also blocks proinflammatory mediator production (40–42). This state differs both from classical and alternative Mø activation (43). The immunosuppressive effects of efferocytosis on host defense have been shown in vivo (39), including in a murine model of pneumococcal pneumonia (44).
We previously reported that clinically-relevant doses of the potent GC fluticasone increase in vitro AC uptake by murine AMø, via both a rapid mechanism dependent in part on down-regulation of SIRPα and by a delayed yet sustained mechanism dependent on protein synthesis (45). We term this change GC-augmented efferocytosis (GCAE). The purpose of this study was to determine whether GCAE alters host defense against pneumococcus. Early host responses to pneumococci depend crucially on AMø (46–48), yet relatively little is known about how AMø responses to pneumococcus are impacted by GC (49). Using murine AMø analyzed in vitro and an established mouse model of pneumococcal pneumonia, we demonstrate a potent detrimental effect on AMø antimicrobial function when GC therapy is used in the presence of alveolar AC.
MATERIALS AND METHODS
Mice
C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were housed under specific pathogen-free conditions in the Animal Care Facility at the VA Ann Arbor Healthcare System, which is fully accredited by the American Association for Accreditation of Laboratory Animal Care. Mice were fed standard animal chow (rodent lab chow 5008; Purina, St. Louis, MO) and chlorinated tap water ad libitum and were used for experiments between 8 and 16 weeks of age. Animal care and experimentation were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (8th edition) and were approved by the Ann Arbor VA Healthsystem Subcommittee on Animal Studies.
Pneumococcus cultivation
Streptococcus pneumoniae serotype 3 (clone 6303) stock culture was obtained from ATCC (Manassas, VA). Stocks of bacteria were immediately thawed for 30 seconds at 37°C and cultured in 5 mL Todd Hewitt (TH) broth supplemented with 0.5% yeast extract for 3 h at 37°C and 5% C02. Bacterial stock was frozen in 1 mL TH broth with 10% Glycerol (Sigma Aldrich, St. Louis, MO) and used for subsequent in vitro studies. Bacterial CFU counts were verified both by optical density (OD) at 600 nm and by quantitative culture on SRBC/tryptose agar (Fisher Scientific, Pittsburgh, PA). To generate heat-killed S. pneumoniae for use in vitro, bacteria were incubated in a water bath at 56°C for 60 min. No live bacteria were detected after plating onto agar plates.
To maintain bacterial virulence for in vivo experiments, we first passaged S. pneumoniae serotype 3 in vivo, using our established murine pneumococcal pneumonia model (50). Untreated C57BL/6 mice received a intratracheal (IT) inoculum using the surgical technique described below, at a dose (1 × 106 CFU) designed to induce bacteremia. After 24 h, mice were euthanized; spleens were harvested aseptically and processed to isolate multiple individual pneumococcal clones on blood agar plates. These clones were expanded once in TH broth, and then frozen. In all subsequent in vivo experiments, these in vivo passaged pneumococcal clones were defrosted, expanded once in TH broth and used immediately without further passage on agar plates.
Induction of thymocyte apoptosis and quantification of efferocytosis
To induce apoptosis, we treated single cell suspensions of murine thymocytes with 10 μM dexamethasone (Sigma) for 4 h at 37°C. These conditions consistently produced 50–60% Annexin+, PI− thymocytes, as we have previously shown (51). Efferocytosis was quantified using a chamber slide-based microscopic assay, as previously described (32). Data are expressed as % efferocytosis, based on the number of AMø ingesting at least one AC; and as the efferocytic index, which was generated by dividing the total number of ingested AC cells by the total number of AMø counted.
AMø isolation and culture
Murine AMø were isolated by BAL using 10–15 mL PBS containing 0.5 mM EDTA in 1 mL aliquots (45). BAL cells were plated in lymphocyte culture media (LCM) (10% FBS, 1 mM sodium pyruvate, 0.5 mM 2-Mercaptoethanol, 1 mM HEPES, 100 u/ml penicillin, 100 u/ml streptomycin, 0.292 mg/ml L-Glutamine in RPMI) for 1.5 h at 37°C and 5% CO2, and AMø were adhesion purified from this population, by discarding non-adherent cells. For in vitro stimulation studies, AMø were treated with one of four conditions: media alone; 2 μM fluticasone for 3 h; AC (at a ratio of 10 AC/AMø) for 2 h; or 2 μM fluticasone for 3 h followed by AC for 2h (Flu + AC). Without washing, LPS from E. coli K12 (InvivoGen, San Diego, CA) (1 ng/mL) or heat-killed S. pneumonia, at a multiplicity of infection (MOI) of 10 or 100 was added for an additional 24 h. Supernatants were collected and stored at −20 °C until assayed by Luminex.
Protein analysis of supernatants
We used the Luminex 200 system (Luminex Corporation, Austin, TX) running StarStation Software (Applied Cytometry, Dinnington, Sheffield, UK) according to manufacturer’s instructions to determine protein levels for TNF-α, IL-1 β, IL-6, IL-12, CCL3, CCL5 and KC (Life Technologies, Grand Island, NY).
GCAE in vivo model
For the GCAE model, mice were administered saline, fluticasone, AC, or fluticasone plus AC via the intranasal (IN) route. All mice received two IN administrations, given 4 h apart, with one of the following: saline + saline; fluticasone + saline; saline + AC, or fluticasone + AC. The dose of fluticasone varied between experiments, ranging from 100 ng to 10 μg; 1 × 107 AC per mouse was used in all experiments. To deliver the reagents, mice were anesthetized with isoflurane via the open drop method and then were held with their heads elevated. Saline, fluticasone or AC were delivered via one nostril in a volume of 30 μL. Mice were held in the upright position for an additional 60 seconds after IN administration before being returned to their cages.
In vivo pneumococcal pneumonia model
At 24 h after the last IN treatment, mice were anesthetized with an intra-peritoneal injection of ketamine/xylazine at 90 mg/kg and 10 mg/kg, respectively. The plane of anesthesia was assessed by lack of response to toe pinch and mice were positioned supine on a surgical platform elevated to a 45 degree angle. To allow visualization of the trachea, a small midline skin incision was made and the neck muscles were retracted. Using a 26 gauge needle, S. pneumoniae were injected into the trachea (20 μL PBS containing 50,000 CFU followed by 0.1 mL of air to assure deposition in the lungs). Mice were allowed to recover fully on a water-jacketed heating pad and were returned to BSL2 housing until euthanasia 48 h later by exsanguination and induction of bilateral pneumothoraces under deep anesthesia.
Flow cytometry
The pulmonary vasculature was perfused via right heart injection of PBS until the effluent was clear, then the lungs were excised and mechanically disaggregated without enzyme treatments, which we have shown efficiently produces single-cell suspensions of high viability (52). After washing, lung cells were stained, fixed and run on an LSR II flow cytometer using FACSDiva software (version 6.1.3; BD Biosciences) with automatic compensation, and data were analyzed using FlowJo software (Tree Star, Ashland, OR) as previously described (45). We stained for the following antigens, using anti-murine antibodies (clone): CD1d (1B1), CD11b (M1/70), CD45 (30-F11), CD45R/B220 (RA3-6B2), CD103 (2E7), Ly6G (1A8), anti-GalCer:CD1d complex (L363) (Biolegend, San Diego, CA); CD3 (145-2C11), CD4 (GK1.5), CD11c (N418), MHC Class II (NIMR-4), NK1.1 (PK136) (eBioscience, San Diego, CA); and Ly6C (AL-21) (BD Biosciences, San Jose, CA).
We analyzed acidification of AMø phagolysosomes by measuring the pH-dependent change in fluorescence of pHrodo. AMø were plated at 40,000 AMø per well in LCM and adherence-purified for 1.5 h, then media was removed and fluticasone (2 μM) in RPMI-5 (or RPMI-5 alone) was added and plates were cultured for 22 h at 37°C in 5% CO2. Next, 400,000 AC (10:1 ratio of AC: AMø) in RPMI-5 (or RPMI-5 alone) were added and plates were incubated at 37°C in 5% CO2 for another 2 h. Then, pHrodo Zymosan Bioparticles (Life Technologies) were added according to manufacturer’s instructions and incubated at 37°C in 5% CO2 for 90 minutes. As a control, one well in each condition received media alone and not Bioparticles. Samples were stained with CD45 and analyzed by flow cytometry. To exclude the possibility of treatment-induced differences in particle ingestion, we performed a similar experiment using FITC-Zymosan Bioparticles (Life Technologies).
Bacterial killing assay
AMø were plated in two different 96-well polystyrene tissue-culture plates (Corning Incorporated, Corning, NY) at 40,000 AMø per well in LCM. One plate was designated the time 0 min (T0) control, to quantify live bacteria within AMø after phagocytosis but before killing could have time to occur; the other plate was designated the time 120 min (T120) plate, to quantify ingested bacteria remaining viable 2 h after ingestion. AMø on both plates were adherence purified for 1.5 h, then media was removed by suctioning and fluticasone (2 μM) in RPMI-5 (or RPMI-5 alone) was added and plates were cultured for 22 h at 37°C in 5% CO2. Next, 400,000 AC (10:1 ratio of AC: AMø) in RPMI-5 (or RPMI-5 alone) were added and plates were incubated at 37°C in 5% CO2 for another 2 h. Then, viable Streptococcus pneumoniae, which had been opsonized by incubation with normal rat serum (10% in Hank’s Balanced Salt Solution) for 1 h at 37°C with constant shaking, were added to all wells at 2 × 106 CFU per well. Plates were incubated for 20 min at 37°C and 5% CO2 and then centrifuged for 5 min at 250 × g. Supernatants were aspirated and discarded, then 100 μL of RPMI-5 was added to each well.
Plates were again centrifuged for 10 min at 250 × g, and at this point the two plates were treated differently. The T0 plate received 20 μL of 5% saponin per well and was incubated for 1 min at room temperature. Then, 100 μL of Todd Hewitt broth was added; the plate was covered with parafilm and stored overnight at 4°C. By contrast, the T120 plate was incubated at 37°C in 5% CO2 for 2 h before addition of saponin and then Todd Hewitt broth, followed by storage overnight at 4°C.
The following day, both the T0 and the T120 plates were incubated at 37°C and 5% CO2 for 2 h. After addition of 15 μL of 5 mg/ml 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide (MTT) (Sigma) and an additional 20 min incubation at 37°C in 5% CO2, absorbance was measured at 570 nm using a microtiter plate reader. The CFU in the T0 and T120 plates were extrapolated from a standard curve. Bacterial killing was calculated as a percentage, using the following equation: Bacterial killing (%) = ((T0 CFU − T120 CFU)/T0 CFU) × 100.
Analysis of reactive oxygen species (ROS) and reactive nitrogen species (RNS)
To detect ROS and RNS, AMø were plated in 96-well polystyrene tissue-culture plates (Corning Incorporated, Corning, NY) at 40,000 AMø per well in LCM. AMø were adherence purified for 1.5 h, then media was removed by suctioning.
To assay ROS, fluticasone (2 μM) in RPMI-5 (or RPMI-5 alone) was added and plates were cultured for 22 h at 37°C in 5% CO2. Next, 400,000 AC (10:1 ratio of AC: AMø) in RPMI-5 (or RPMI-5 alone) were added and plates were incubated at 37°C in 5% CO2 for another 2 h. Media was suctioned off and cells were washed with warm HBSS. A 50 μm solution of 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) (Life Technologies) was prepared in RPMI without phenol red and then added to each well. Cells were incubated at 37°C in 5% CO2 for 1 h. The plate was washed with HBSS, then read at an excitation of 492 nm and emission of 522 nm using an FLx800 fluorescent plate reader (BioTek, Winooski, VT). Then, viable Streptococcus pneumoniae were added at 2 × 106 CFU per well and incubated at 37°C in 5% CO2. The plate was read every 15 minutes for a total of 90 minutes and returned to the incubator after each reading.
To assay RNS, media was removed and fluticasone (2 μM) was prepared in a fluorescein amine methyl ester (FA-OMe) staining solution, as directed by the manufacturer (Cayman Chemical, Ann Arbor, MI). Wells that were not treated with fluticasone received the fluorescein amine methyl ester staining solution alone. Plates were cultured for 22 h at 37°C in 5% CO2. Next, 400,000 AC in the staining solution (or staining solution alone) were added and plates were incubated at 37°C in 5% CO2 for another 2 h. Then, viable Streptococcus pneumoniae were added at 2 × 106 CFU per well and incubated at 37°C in 5% CO2 for 2 h. Media was suctioned off and Hoechst Dye staining solution was prepared and added to each well, as directed by the manufacturer (Cayman Chemical). Plates were incubated for 10 minutes at 37°C. After a final wash, cells were analyzed using an FLx800 fluorescent plate reader. Nitric oxide staining intensity was measured with excitation and emission wavelengths of 485 and 535 nm, respectively. Cell number/density, indicated by the Hoechst Dye, was detectable at excitation and emission wavelengths of 355 and 465 nm, respectively.
Statistics
Statistical analyses were performed using GraphPad Prism 6.0.1 (GraphPad Software, Inc., La Jolla, CA) on a Macintosh Quad-Core Intel Xeon computer running OS X 10.10.3 (Apple; Cupertino, CA). To test for significant differences between groups, we used either ANOVA with appropriate post-hoc testing (Dunnett for comparison to a single control condition, Fisher LSD testing for multiple comparisons) or the analogous non-parametric Kruskal-Wallis test with appropriate post-hoc testing (Dunn for comparison to single control group, Holm-Sídák for multiple comparisons). A p value of < 0.05 was considered to indicate significance.
RESULTS
GCAE reduced production of inflammatory mediators by murine AMø
To test for suppression of host defenses by GCAE, we first studied its effect on stimulated production of inflammatory cytokines by murine AMø after in vitro pre-treatment with either fluticasone, AC, or fluticasone followed by AC. AMø activation by pneumococci depends chiefly on TLR4 recognition of pneumolysin (53), and to a lesser degree on recognition of peptidoglycan by NOD2 (53). Accordingly, we initially stimulated murine AMø in vitro via TLR4, using its prototypic agonist, purified LPS. As a read-out, we measured production of the inflammatory cytokines TNF-α, IL-6 & IL-12, which have all been shown to be essential to combat pneumococci in vivo (54–56), and the chemokines CCL3 (MIP-1α), CCL5 (RANTES) (57) and KC, a murine functional homologue of human CXCL1.
Exposure of AMø to fluticasone plus AC significantly reduced AMø elaboration of IL-12 relative to the other three conditions, and also significantly decreased secretion of TNF-α relative to AMø pretreated with AC alone or with medium alone (Fig. 1, A–C). Fluticasone plus AC treatment also led to significantly reduced secretion of CCL3 and KC (Fig. 1, D–F) in comparison to the un-pretreated AMø or those that received AC alone. Fluticasone alone strongly inhibited AMø production of all three chemokines and especially of IL-6, so that IL-12 was the only analyte that was significantly lower in AMø pretreated with fluticasone plus AC than in those pretreated with fluticasone alone, although for TNF-α, CCL5 and KC, there were non-significant trends towards lower production in response to the combined stimuli. We also measured IL-1β in these experiments, but secretion was near or below the limit of detection for all conditions (data not shown). These results imply that GCAE could reduce the ability of murine AMø to secrete inflammatory mediators crucial for recruitment of other leukocyte subsets during early pneumococcal pneumonia. Accordingly, we next set about to test that possibility in vivo.
Figure 1. GCAE reduced inflammatory cytokine production by murine AMø in response to stimulation via TLR4.

Adherence-purified AMø from normal C57BL/6 mice were pre-treated with media alone (none); 2 μM fluticasone for 3 h (Flu), AC (at a ratio of 10 AC/AMø) for 2 h (AC); or 2 μM fluticasone for 3 h followed by AC for 2h (Flu + AC). Next, LPS at 1 ng/mL was added to all wells for an additional 24 h. Supernatants were collected and assayed by Luminex for protein concentrations of (A) TNF-α; (B) IL-6; (C) IL-12; (D) CCL3; (E) CCL5; and (F) KC. Results are mean ± SEM of four independent experiments, each using pooled AMø from two mice. *, p < 0.05; **, p < 0.01; ***, p<0.001; ****, p<0.0001; NS, not significant by ANOVA with Fisher LSD post-hoc testing.
GCAE inhibited in vivo clearance of S. pneumoniae in a murine model
To establish a murine model of GCAE, we first needed to verify that a physiological dose of fluticasone altered murine AMø efferocytosis in vivo. Mice were pretreated with fluticasone by IN inoculation for 6 h, then received AC by the IN route. After an additional 2 h, we harvested AMø by BAL and quantified ingested AC. Results showed that fluticasone pretreatment significantly increased uptake of AC by murine AMø in vivo in a dose-dependent manner (Fig. 2).
Figure 2. Fluticasone increased in vivo uptake of AC by resident AMø.
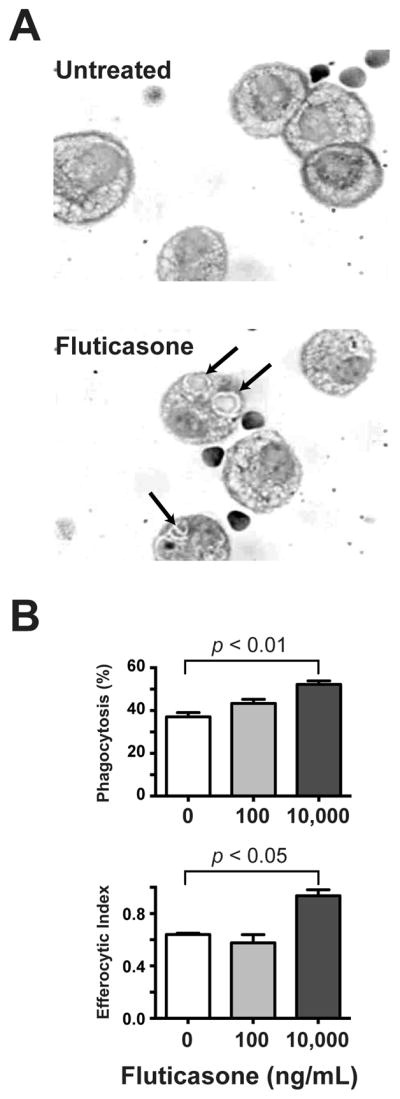
A. C57BL/6 mice received two IN administrations, of either various doses of fluticasone (100–10,000 ng/mL) or saline control, followed 6 h later by an IN administration of 1 × 107 AC. One h later, AMø were collected by BAL; cytospins were stained with H&E and ingested AC were counted under oil at 1000 × final magnification. A. Representative cytospins showing in vivo AC uptake following in vivo fluticasone treatment with either saline (top panel) or 1000 ng fluticasone. Arrows point to ingested AC. Top panel, percentage of AMø ingesting at least one AC; lower panel, efferocytic index. B. Data are mean ± SEM from three mice in a single experiment, and are representative of results of three independent experiments. Statistical testing using one-way ANOVA with Dunnett’s post-hoc testing for multiple comparisons relative to saline-only control group.
In the next set of experiments, mice were pretreated with saline, fluticasone, AC, or fluticasone followed by AC (all by IN route), and then all groups received Streptococcus pneumoniae (50,000 CFU/mouse by the IT route). After 48 h, lungs were harvested and total CFU per lung were calculated (Fig. 3A). Pretreatment by fluticasone followed by AC significantly reduced pneumococcal clearance from the lungs, relative to all other treatment groups (Fig. 3B). Thus, short-term treatment leading to GCAE in vivo had an adverse effect during bacterial pneumonia.
Figure 3. GCAE specifically reduced clearance of viable S. pneumoniae from the lungs in a murine model.

C57BL/6 mice received two IN administrations, given 4 h apart, of either saline (indicated by minus symbol), fluticasone followed by saline, saline followed by AC, or fluticasone followed by AC. All mice were infected via the IT route 24 h after the final IN treatment using 50,000 CFU S. pneumoniae serotype 3. Lungs were collected 48 h later to assay total CFU by serial dilution on blood agar plates. Data are derived from 2–3 mice per condition assayed individually in each of three independent experiments (total n = 34). A. Log lung CFU of individual mice; symbols denote mice from different experiments. B. Fold-change in lung CFU, relative to the group pre-treated twice with saline before infection, the geometric mean of which was set to 1. Data are shown as median, 25% & 75% (box) and 5%, 95% CI (whiskers), with outliers shown individually. *, p<0.5; **, p<0.01 by Kruskal-Wallis non-parametric ANOVA with Dunn’s post-hoc testing for multiple comparisons to saline-only control group.
GCAE did not alter inflammatory cell recruitment to the lungs during pneumococcal pneumonia
As a first step toward determining the mechanism by which GCAE had this adverse effect on lung host defenses, we used a separate cohort of mice to analyze inflammatory cell recruitment to the lungs during pneumonia. Mice were treated exactly as in the previous experiment, but at 48 h, lungs were harvested without previous bronchoalveolar lavage and total cells were stained for flow cytometric analysis.
All cell populations were initially gated on viable CD45+ cells. We defined AMø as high autofluorescent, CD11c+ cells that were negative for CD3, CD19 & Ly6G. Neutrophils were identified as high side scatter cells abundantly expressing Ly6G. We defined dendritic cells (DC) as low autofluorescent, MHC II+, CD11cdim cells that were negative for CD3, CD19 and Ly6G; two DC subsets were distinguished, CD11b+ CD103− (which we and others have shown are located predominately in parenchymal lung interstitium) and CD11b− CD103+ (which have been shown to reside largely in airways) (58, 59). Similarly, exudate Mø were identified as low autofluorescent, CD11bhigh cells, among which we distinguished two subsets by their expression of Ly6C. We also identified CD4+ T cells, which have recently been recognized to contribute importantly and acutely to host defense during pneumococcal pneumonia (60, 61). Finally, we searched for NK cells and NKT cells, using staining for NK1.1, CD1d, CD3 and anti-GalCer.
There were no significant differences between the four in vivo treatments for any of these inflammatory cell populations at this time post-infection, whether expressed as absolute number of cells per mouse lung (Fig. 4) or as percentage of each cell type among all CD45+ lung cells (data not shown). Although we identified lung NK cells (NK1.1+, CD3−, CD1d−, anti-GalCer−), we found no evidence of classical (type I) NKT cells (NK1.1+, CD1d+ anti-GalCer+) or non-classical (type II) NKT cells (CD1d+ anti-GalCer−). Interestingly, for all cell types except the CD11b−, CD103+ DC subset, there was a non-significant trend towards higher absolute numbers in the fluticasone-treated group (Fig. 4). The reason for this trend is not apparent. Thus, the observed significant differences in lung pneumococcal CFU associated with GCAE (Fig. 3) could not be attributed to disparity in numbers or relative composition of lung inflammatory cells.
Figure 4. GCAE did not alter inflammatory cell recruitment to the lungs during pneumococcal pneumonia.

Mice were pre-treated by the IN route and infected by the IT route with 50,000 CFU/mouse S. pneumoniae serotype 3, exactly as described in the Legend to Figure 3, except that 48 h post-infection, lungs were harvested and processed individually for flow cytometry. Hematopoietic cells were gated using light-scatter parameters and CD45 staining as described in the Results. Data are expressed as the absolute number of cells per lung for each cell type on the vertical axis (note differences in scales), as mean ± SEM of 2–3 mice per condition assayed individually in each of two independent experiments (total n = 21). There were no statistically significant differences between treatment groups by ANOVA with Fisher’s LSD post-hoc testing.
GCAE minimally decreases production of cytokine and chemokine murine AMø in response to heat-killed pneumococcus
This unanticipated result for inflammatory cell accumulation led us to repeat our initial in vitro stimulation of murine AMø pre-treated with fluticasone, AC or both, but this time using heat-killed pneumococci to induce inflammatory mediator production. Results were strikingly different than with LPS, as the combined treatment induced a significant decrease, relative to the other three conditions, only in IL-6 production (Fig. 5). At this ratio of heat-killed pneumococci to AMø (MOI 100), the combined stimulus of fluticasone followed by AC significantly decreased production of TNF-α and CCL3, relative to no pretreatment or AC alone, but the change was not statistically significant for fluticasone pretreatment alone. Interestingly, there were no differences between groups in response to heat-killed organisms for IL-12 or KC, which had shown the greatest difference in response to LPS.
Figure 5. GCAE reduced inflammatory cytokine production by murine AMø in response to stimulation by heat-killed pneumococci.

Adherence-purified murine AMø from C57BL/6 mice were pre-treated with media alone (none); 2 μM fluticasone for 22 h (Flu), AC (at a ratio of 10 AC/AMø) for 2 h (AC); or 2 μM fluticasone for 22 h followed by AC for 2h (Flu + AC). All conditions were incubated for and additional 24 h with heat-killed S. pneumoniae serotype 3 at a multiplicity of infection (MOI) of 100 (except for CCL3, for which MOI = 10). Supernatants were collected and assayed by Luminex for protein concentrations of TNF-α, IL-6, IL-12 (top row) and CCL3, CCL5 and KC (bottom row). Results are mean ± SEM of three independent experiments, each using pooled AMø from two mice. *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant by ANOVA with Fisher LSD post-hoc testing.
We considered that the ratio of organisms to AMø, the MOI, might be too great to see an effect of GCAE, so in one experiment performed in parallel we analyzed an MOI of 10. However, at that lower MOI, the only analyte detected in any condition was CCL-3 (data not shown). Collectively, these findings are consistent with the lack of differences in inflammatory cell recruitment in vivo, and imply that the effect of GCAE varies with the stimulus, being much stronger with stimulation of TLR4 than with the heat-killed organism. Nevertheless, these results do not provide an explanation for the significant differences observed in lung CFU.
GCAE significantly reduced in vitro killing of pneumococcus by murine AMø
To test whether bacterial killing by AMø was inhibited by GCAE and could contribute to the observed increase in bacterial burden in vivo, we performed an in vitro bacterial killing assay. AMø from normal C57BL/6 mice were adherence-purified before pre-treatment with either saline, fluticasone, AC, or fluticasone followed by AC. Pre-treatment with fluticasone plus AC significantly reduced AMø-mediated bacterial killing, relative to each of the three other conditions (Fig. 6A). Fluticasone pre-treatment also resulted in a modest decrease in bacterial killing, which was significantly different from the saline control. Hence, an additive reduction in AMø killing of pneumococcus is one means by which GCAE impairs host defense against pneumonia. Numbers of CFU at T0 were significantly increased in AMø pretreated with fluticasone plus AC, relative to the untreated and AC only groups (Fig. 6B). This result implies that the observed GCAE-induced deficit in killing by T120 mins did not result from reduced initial phagocytosis of pneumococcus.
Figure 6. GCAE significantly inhibited in vitro killing of S. pneumoniae by murine AMø.

Adherence-purified AMø from normal C57BL/6 mice were treated with one of four regimens: saline (indicated by minus sign) alone twice; 2 μM fluticasone for 22 h followed by saline for 2 h; saline for 22 h followed by AC for 2 h; or 2 μM fluticasone for 22 h followed by AC for 2 h. AC were added at a ratio of 10:1 relative to AMø. Next, viable pneumococci (2 × 106 CFU) were added, and then bacterial killing was assayed as described in Material & Methods. Data are expressed as (A) the percentage of bacterial killing at T120 mins; and (B) lung CFU (in millions) at T0 mins; both are derived from four independent experiments each containing at least three mice per group. Results are depicted as a box & whiskers plot, indicating median, 25th & 75th percentiles and minimum, maximum values; *, p < 0.05; **, p < 0.01, by one-way ANOVA with Holm-Sídák post-hoc testing for multiple comparisons.
An effect of AC on in vitro killing of K. pneumoniae by rat AMø was previously shown in experiments using opsonization by specific IgG (44). Accordingly, we investigated whether the GCAE-induced decrement in killing of pneumococcus by murine AMø that we showed using complement-opsonized bacteria would also be seen with specific IgG opsonization. We compared killing of pneumococci opsonized by the two methods by untreated murine AMø or AMø treated in vitro with AC plus fluticasone. Results showed a similar reduction in bacterial killing by GCAE regardless of the method of opsonization (Supplemental Figure 1), although the difference from untreated AMø did not attain statistical significance in this single experiment.
GCAE and fluticasone impaired AMø killing of S. pneumoniae in part by reducing phagolysosome acidification
We performed several types of experiments to begin to define the possible mechanisms by which GCAE impairs in vitro killing of S. pneumoniae by murine AMø. Bacterial killing by murine Mø often involves production of ROS by the NADPH oxidase complex and of RNS by inducible nitric oxide. However, neither exposure of murine AMø to fluticasone, AC or both significantly affected ROS production in response to ingestion of viable pneumococci (Supplemental Fig. 2A). Similarly, none of these treatments impacted production of RNS (Supplemental Fig. 2B).
Finally, we examined the effect of GCAE on phagolysosome acidification. We assayed AMø phagolysosome acidification by quantifying the change in fluorescence of the pH-sensitive dye pHrodo using flow cytometry. Pretreatment with either fluticasone or fluticasone followed by AC significantly impaired phagosome acidification in vitro (Fig. 7A, B). To exclude the possibility that this difference was artifactual, and instead reflected treatment-induced differences in particle ingestion, we performed a similar experiment using FITC-Zymosan Bioparticles. That control experiment showed that the combination of fluticasone followed by AC slightly but significantly increased AMø ingestion, which was avid for all treatments (Fig. 7C).
Figure 7. Fluticasone alone and GCAE significantly inhibited phagolysosome acidification.

Adherence-purified AMø from normal C57BL/6 mice were treated with one of four regimens: saline alone twice; 2 μM fluticasone for 22 h followed by saline for 2 h; saline for 22 h followed by AC for 2 h; or 2 μM fluticasone for 22 h followed by AC for 2 h. Next, either pHrodo Bioparticles (A, B) or FITC Zymosan Bioparticles (C) were added and plates were cultured for an additional 90 minutes. Finally, AM were washed, harvested and analyzed by flow cytometry. Corrected MFI was calculated individually for each condition by subtracting the MFI of the well without added particles from the well with added particles in the same experiment. (A) Representative staining for pHrodo Bioparticles (FITC+) for each condition. (B) Aggregated corrected MFI for pHrodo. Results are mean ± SEM of three independent experiments (4–5 mice per experiment); each symbol represents an individual well. (C) Aggregated corrected MFI for FITC-Zymosan; note compressed range. Results are mean ± SEM from one experiment using pooled AMø from five mice; each symbol represents an individual well. *, p<0.05; **, p < 0.01, by one-way ANOVA with Holm-Sídák post-hoc testing for multiple comparisons; NS, not significant.
DISCUSSION
The results of this study demonstrate that by the process we term GCAE, the combination of the potent GC fluticasone and subsequent AC exposure impairs the ability of resident AMø to defend against pneumococcus more greatly than either stimulus alone. GC pre-treatment in vitro significantly reduced pro-inflammatory production by murine AMø in response to TLR4-stimulation, although the effect was less marked using heat-killed pneumococci under the conditions tested. The combination of steroid pre-treatment and intra-alveolar AC led to significantly greater lung bacterial burdens in a murine model of pneumococcal pneumonia, without altering inflammatory cell recruitment to the lungs. GCAE also decreased in vitro killing of pneumococci by murine AMø, but not their uptake, in association with a decrease in phagolysosome acidification. These data provide one mechanism for the increased susceptibility to CAP when COPD patients are treated with potent ICS therapy.
Streptococcus pneumoniae is a near-obligate human pathogen that causes more deaths globally than any other organism, principally due to infections in children under the age of five years in the developing world (62). However, pneumococcal pneumonia also continues to be a very significant health problem in industrialized nations, despite vaccines that have efficacy even in elderly patients with COPD (63). Moreover, the lethality of pneumococcal pneumonia remains considerable even when appropriate antibiotic therapy is initiated promptly (64). Providing appropriate antibiotic therapy against pneumococcus is becoming more difficult, as high-grade resistance to multiple antibiotics is already prevalent in many regions, especially Asia. Although antibiotic stewardship may delay the global spread of resistance, the profound ability of pneumococcus to undergo DNA transformation virtually assures that the trend will continue. Hence, investigating the immunological basis of susceptibility to pneumococcal pneumonia will remain important.
Our studies employed mice, a well-accepted model of human pneumococcal pneumonia, provided appropriate attention is given to pneumococcal serotype, murine strain and anesthesia method (46, 65, 66). We used the encapsulated serotype 3, which in humans remains associated both with common nasopharyngeal carriage and frequent pneumonia with relatively high mortality (67–69), perhaps recently in part due to its lower immunogenicity relative to other strains in polyvalent vaccines (70). We (50, 71, 72) and others (44, 73–85) have used variations of this murine model extensively in wild-type and transgenic mice to define the molecular mechanisms of lung host defense against pneumococcus. Together with established murine models mimicking the pathogenic changes of COPD, either employing or independent of cigarette smoke-exposure (86, 87), the tools are now available to determine whether the beneficial effects of ICS can be dissociated from this and other adverse effects.
The demonstration that GCAE was associated with both significantly greater lung bacterial burdens and reduced killing in vitro is important because resident AMø are so crucial to defend against pneumococcal pneumonia (88). Part of their role depends on killing pneumococci, which for AMø occurs almost entirely intracellularly. AMø require opsonization to ingest encapsulated strains, which are associated with most episodes of CAP in humans. Accordingly, we used the encapsulated serotype 3 and performed serum opsonization in all experiments. Ingestion depends on several Mø receptors, including FcγR and the scavenger receptors SR-A and MARCO (89) and (primarily for unopsonized pneumococci) mannose receptor. Our data indicate GCAE and fluticasone alone increased ingestion of pneumococcus by murine AMø in vitro, which literature searching leads us to conclude is a novel finding. The reason for this increase is uncertain. Our previous demonstration that fluticasone induced downregulation by murine AMø of the inhibitory receptor SIRPα (45) together with the finding that alveolar lining fluid [which contain surfactant proteins (SP−) A and SP-D, the collectin ligands for SIRPα] impaired phagocytosis by murine AMø (90) provide a possible explanation. However, increased phagocytosis of pneumococcus was induced in human neutrophils by both SP-A and SP-D (91), and in murine AMø by SP-A (via surface localization of SR-A) but not by SP-D (92). Hence, further study will be needed to establish that mechanism. Importantly, the calculation of percentage bacterial killing takes into account difference in bacterial uptake, so this effect does not explain the GCAE-induced killing defect we show.
Unlike the better studied neutrophil, which eliminate pneumococci using multiple extracellular and intracellular mechanisms, precisely which elements are essential for AMø to kill ingested pneumococci remains incompletely defined. Our finding that neither GCAE nor fluticasone alone affected ROS production extends results of Marriott and colleagues, who used gp91phox−/− mice to show that ROS production was dispensable in a subclinical pneumonia model (93). Glucocorticoids do reduce phagocyte generation of superoxide at least in part via reduced eicosanoid signaling (94), but that effect is seen at micromolar concentrations irrelevant to GCAE, which occurs at nanomolar concentration identical to those achieved clinically during ICS therapy. Further investigation is needed to define the roles of ROS and RNS in AMø killing of pneumococcus, but our results show that changes in these mediators cannot explain the defect induced by GCAE.
To our knowledge, corticosteroids have not previously been shown in any mammalian phagocyte to affect phagolysosome acidification, a particularly crucial step in the killing process (95). An acidic intraluminal pH is necessary for optimal activity of cathepsins, which contribute to pneumococcal clearance in murine models both by inducing AMø apoptosis linked to bacterial killing (83), and at least in neutrophils, by direct anti-bacterial activity (96). Additionally, acidification of the maturing phagolysosome is important to counteract three potentially detrimental effects of the oxidative burst that would otherwise increase pH: (a) consumption of protons as superoxide undergoes dismutation to hydrogen peroxide; (b) leakage of H+ into the cytoplasm due to membrane oxidation; and (c) impaired recruitment to the phagosome of V-ATPase, which is essential to complete acidification (97). Because the effect on acidification was equivalent in AMø treated with fluticasone without or with subsequent AC, this mechanism cannot explain the even greater reduction in killing induced by GCAE, relative to fluticasone alone. Considerable additional investigation will be required to define the molecular basis for this GCAE-induced defect, but these results advance the field by excluding defective ROS or RNS generation.
In experimental models, AMø also contribute to defense against pneumococcal pneumonia by production of cytokines and chemokines, which are particularly crucial to activate and recruit other cell types in response to larger inocula (98). The disparity in results of stimulating AMø in vitro with purified LPS versus with heat-killed pneumococci does not necessarily detract from the relevance of GCAE shown by our lung CFU and in vitro killing data because neither experiment fully simulates the indirect contribution of AMø to host defense during pneumococcal pneumonia. For example, production of CCL5 by murine Mø in response to pneumococcus requires pneumolysin-dependent escape from phagolysosomes that likely does not occur efficiently using heat-killed organisms (99, 100). However, the absence of any significant difference between treatment groups in recruitment of inflammatory cell subsets implies that in vivo, either AMø production of recruitment signals was not essential under the conditions tested, or that other factors compensated. We suspect that the disparity in our results regarding neutrophil recruitment from the decreases seen by Medeiros and colleagues in response to AC alone (44) relate to differences from our experiments in pneumococcal inoculum (106 CFU, versus 5 × 104 CFU in this study) (98). Moreover, it is likely that GCAE-induced failure of early pneumococcal eradication in our study led to compensatory changes by other cell populations that culminated in increased neutrophil recruitment to the lungs by the time-point we studied.
In adults, COPD is one of the strongest risk factors for development of CAP. COPD is also a pervasive and rapidly increasing problem worldwide, due to the juxtaposition in much of the developing world of rapidly increasing cigarette consumption and air pollution, particularly by indoor use of biomass fuel or coal. Although COPD is also increasing as a directly attributable cause of death in industrialized nations, its true burden is likely vastly underestimated. COPD is under-diagnosed, especially in women (101). Hence, defining precisely why there is an association between ICS therapy, which clearly has beneficial effects in COPD, and potentially lethal hospitalizations for CAP should be of significant public health interest. Our data imply that the risk of GCAE contributing to CAP might be greatest in those with emphysema, which among the several pathological processes contributing to COPD phenotypes is the one most closely associated with excessive apoptosis of parenchymal lung cells. To date, none of the studies on the association of CAP and ICS usage have included the imaging data necessary to address that possibility. Indirect support for that possibility comes from the recent identification of the emphysematous subset as being at elevated risk for acute exacerbations of COPD (102), a largely infectious syndrome that blends clinically with CAP.
Based on our in vitro findings that budesonide also induced GCAE in murine AMø (45), we suspect that the effect we have shown here using fluticasone is a class-effect of all potent ICS medications. However, we recognize that there is controversy on this important clinical point. A retrospective observational trial using administrative health data found lower pneumonia event rates in COPD patients using budesonide compared to fluticasone (10). An individual-subject data meta-analysis relying heavily on adverse event reporting and including studies of relatively short duration found no difference in incidence of CAP from placebo among users of budesonide (103). However, this conclusion has also been contested (104, 105). Verifying whether specific ICS agents have different risks of infection or other adverse effects is a highly significant question, but one which will likely require large clinical trials or very careful epidemiological approaches. Importantly for our hypothesis that GCAE might explain the increased risk of CAP among COPD patients using ICS therapy, asthma is not associated with apoptosis of alveolar epithelial and endothelial cells that can interact with AMø.
In summary, we have extended our previous identification of GCAE as a property of resident tissue Mø (45) (i.e., not only a process relevant during maturation of blood monocytes) by showing its impact on a clinically relevant bacterial pathogen in vitro and in vivo. Our results support GCAE as a potential explanation for the epidemiological association between ICS therapy of COPD patients and an increased risk of CAP, although further support for that possibility is required, including studies using human AMø that we have underway. These findings also establish murine in vitro and in vivo experimental models to dissect the underlying molecular mechanisms by which GCAE impacts lung host defense.
Supplementary Material
Acknowledgments
The authors thank Drs. David M. Aronoff, Gary B. Huffnagle, Bethany B. Moore, Theodore J. Standiford, Joel A. Swanson and Debra A. Thompson for helpful discussion and suggestions; Zarinah Aquil, Donna Boyer, Mary Freer, Tameka Lewis, Cat Meyer, Joyce O’Brien and Melina White for administrative support; and Sherry Wagar and the staff of the VA Ann Arbor Animal Care Facility for expert and compassionate veterinary support.
Sources of support: Merit Review Awards I01 CX000911 from the Clinical Research & Development Services (JLC) and I01 BX001389 from the Biomedical Laboratory Sciences Research & Development Service (CMF), Department of Veterans Affairs; U01 HL098961 (JLC), T32 HL07749 (JPB) and T32 AI007413 (ALM, SHT) from the USPHS; Flight Attendants Medical Research Institute Award CIA-103071 (PM); and a Rackham Pre-doctoral Fellowship from the University of Michigan (ALM).
LIST OF ABBREVIATIONS USED
- AC
apoptotic cell(s)
- AMø
alveolar macrophages(s)
- BAL
bronchoalveolar lavage
- CAP
community acquired pneumonia
- COPD
chronic obstructive pulmonary disease
- GC
glucocorticoid
- GCAE
glucocorticoid-augmented efferocytosis
- ICS
inhaled corticosteroid(s)
- IN
intranasal
- IT
intratracheal
- TH
Todd Hewitt (broth)
References
- 1.Calverley PM, Anderson JA, Celli B, Ferguson GT, Jenkins C, Jones PW, Yates JC, Vestbo J. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356:775–789. doi: 10.1056/NEJMoa063070. [DOI] [PubMed] [Google Scholar]
- 2.Wedzicha JA, Calverley PM, Seemungal TA, Hagan G, Ansari Z, Stockley RA. The prevention of chronic obstructive pulmonary disease exacerbations by salmeterol/fluticasone propionate or tiotropium bromide. Am J Respir Crit Care Med. 2008;177:19–26. doi: 10.1164/rccm.200707-973OC. [DOI] [PubMed] [Google Scholar]
- 3.Crim C, Calverley PM, Anderson JA, Celli B, Ferguson GT, Jenkins C, Jones PW, Willits LR, Yates JC, Vestbo J. Pneumonia risk in COPD patients receiving inhaled corticosteroids alone or in combination: TORCH study results. Eur Respir J. 2009;34:641–647. doi: 10.1183/09031936.00193908. [DOI] [PubMed] [Google Scholar]
- 4.Singanayagam A, Chalmers JD, Hill AT. Inhaled corticosteroids and risk of pneumonia: evidence for and against the proposed association. Q J Med. 2010;103:379–385. doi: 10.1093/qjmed/hcq023. [DOI] [PubMed] [Google Scholar]
- 5.Joo MJ, Au DH, Fitzgibbon ML, Lee TA. Inhaled corticosteroids and risk of pneumonia in newly diagnosed COPD. Respir Med. 2010;104:246–252. doi: 10.1016/j.rmed.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Calverley PM, Stockley RA, Seemungal TA, Hagan G, Willits LR, Riley JH, Wedzicha JA Investigating New Standards for Prophylaxis in Reduction of Exacerbations (INSPIRE) Investigators. Reported pneumonia in patients With COPD: Findings from the INSPIRE study. Chest. 2011;139:505–512. doi: 10.1378/chest.09-2992. [DOI] [PubMed] [Google Scholar]
- 7.Eurich DT, Lee C, Marrie TJ, Majumdar SR. Inhaled corticosteroids and risk of recurrent pneumonia: population based nested case control study. Clin Infect Dis. 2013;57:1138–1144. doi: 10.1093/cid/cit472. [DOI] [PubMed] [Google Scholar]
- 8.Yawn BP, Li Y, Tian H, Zhang J, Arcona S, Kahler KH. Inhaled corticosteroid use in patients with chronic obstructive pulmonary disease and the risk of pneumonia: a retrospective claims data analysis. Int J COPD. 2013;8:295–304. doi: 10.2147/COPD.S42366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang IA, Clarke MS, Sim EH, Fong KM. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2012;7:CD002991. doi: 10.1002/14651858.CD002991.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janson C, Larsson K, Lisspers KH, Stallberg B, Stratelis G, Goike H, Jorgensen L, Johansson G. Pneumonia and pneumonia related mortality in patients with COPD treated with fixed combinations of inhaled corticosteroid and long acting beta2 agonist: observational matched cohort study (PATHOS) BMJ. 2013;346:f3306. doi: 10.1136/bmj.f3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malo de Molina R, Mortensen EM, Restrepo MI, Copeland LA, Pugh MJ, Anzueto A. Inhaled corticosteroid use is associated with lower mortality for subjects with COPD and hospitalised with pneumonia. Eur Respir J. 2010;36:751–757. doi: 10.1183/09031936.00077509. [DOI] [PubMed] [Google Scholar]
- 12.Chen D, Restrepo MI, Fine MJ, Pugh MJ, Anzueto A, Metersky ML, Nakashima B, Good C, Mortensen EM. Observational study of inhaled corticosteroids on outcomes for COPD patients with pneumonia. Am J Respir Crit Care Med. 2011;184:312–316. doi: 10.1164/rccm.201012-2070OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drummond MB, Dasenbrook EC, Pitz MW, Murphy DJ, Fan E. Inhaled corticosteroids in patients with stable chronic obstructive pulmonary disease. JAMA. 2008;300:2407–2416. doi: 10.1001/jama.2008.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singanayagam A, Chalmers JD, Akram AR, Hill AT. Impact of inhaled corticosteroid use on outcome in COPD patients admitted with pneumonia. Eur Respir J. 2011;38:36–41. doi: 10.1183/09031936.00077010. [DOI] [PubMed] [Google Scholar]
- 15.Loke YK, Kwok CS, Wong JM, Sankaran P, Myint PK. Chronic obstructive pulmonary disease and mortality from pneumonia: meta-analysis. Int J Clin Pract. 2013;67:477–487. doi: 10.1111/ijcp.12120. [DOI] [PubMed] [Google Scholar]
- 16.Minino Arialdi M, Kenneth JX, Kochanek D. Deaths: Preliminary Data for 2008. National Vital Statistics Reports. 2010;59:1–72. [PubMed] [Google Scholar]
- 17.Torres A, Dorca J, Zalacain R, Bello S, El-Ebiary M, Molinos L, Arevalo M, Blanquer J, Celis R, Iriberri M, Prats E, Fernandez R, Irigaray R, Serra J. Community-acquired pneumonia in chronic obstructive pulmonary disease: a Spanish multicenter study. Am J Respir Crit Care Med. 1996;154:1456–1461. doi: 10.1164/ajrccm.154.5.8912764. [DOI] [PubMed] [Google Scholar]
- 18.Reissig A, Mempel C, Schumacher U, Copetti R, Gross F, Aliberti S. Microbiological diagnosis and antibiotic therapy in patients with community-acquired pneumonia and acute COPD exacerbation in daily clinical practice: comparison to current guidelines. Lung. 2013;191:239–246. doi: 10.1007/s00408-013-9460-x. [DOI] [PubMed] [Google Scholar]
- 19.Torres A, Blasi F, Peetermans WE, Viegi G, Welte T. The aetiology and antibiotic management of community-acquired pneumonia in adults in Europe: a literature review. Eur J Clin Microbiol Infect Dis. 2014;33:1065–1079. doi: 10.1007/s10096-014-2067-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barbier M, Agusti A, Alberti S. Fluticasone propionate reduces bacterial airway epithelial invasion. Eur Respir J. 2008;32:1283–1288. doi: 10.1183/09031936.00020608. [DOI] [PubMed] [Google Scholar]
- 21.To M, To Y, Yamada H, Ogawa C, Otomo M, Suzuki N, Sano Y. Influence of inhaled corticosteroids on community-acquired pneumonia in patients with bronchial asthma. Intern Med. 2004;43:674–678. doi: 10.2169/internalmedicine.43.674. [DOI] [PubMed] [Google Scholar]
- 22.O’Byrne PM, Pedersen S, Carlsson LG, Radner F, Thoren A, Peterson S, Ernst P, Suissa S. Risks of pneumonia in patients with asthma taking inhaled corticosteroids. Am J Respir Crit Care Med. 2011;183:589–595. doi: 10.1164/rccm.201005-0694OC. [DOI] [PubMed] [Google Scholar]
- 23.McKeever T, Harrison TW, Hubbard R, Shaw D. Inhaled corticosteroids and the risk of pneumonia in people with asthma: a case-control study. Chest. 2013;144:1788–1794. doi: 10.1378/chest.13-0871. [DOI] [PubMed] [Google Scholar]
- 24.Festic E, Bansal V, Gajic O, Lee AS. Prehospital use of inhaled corticosteroids and point prevalence of pneumonia at the time of hospital admission: secondary analysis of a multicenter cohort study. Mayo Clin Proc. 2014;89:154–162. doi: 10.1016/j.mayocp.2013.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majo J, Ghezzo H, Cosio MG. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir J. 2001;17:946–953. doi: 10.1183/09031936.01.17509460. [DOI] [PubMed] [Google Scholar]
- 26.Yokohori N, Aoshiba K, Nagai A. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest. 2004;125:626–632. doi: 10.1378/chest.125.2.626. [DOI] [PubMed] [Google Scholar]
- 27.Calabrese F, Giacometti C, Beghe B, Rea F, Loy M, Zuin R, Marulli G, Baraldo S, Saetta M, Valente M. Marked alveolar apoptosis/proliferation imbalance in end-stage emphysema. Respir Res. 2005;6:14. doi: 10.1186/1465-9921-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imai K, Mercer BA, Schulman LL, Sonett JR, D’Armiento JM. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J. 2005;25:250–258. doi: 10.1183/09031936.05.00023704. [DOI] [PubMed] [Google Scholar]
- 29.Hodge S, Hodge G, Holmes M, Reynolds PN. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur Respir J. 2005;25:447–454. doi: 10.1183/09031936.05.00077604. [DOI] [PubMed] [Google Scholar]
- 30.McCubbrey AL, Curtis JL. Efferocytosis and lung disease. Chest. 2013;143:1750–1757. doi: 10.1378/chest.12-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newman SL, Henson JE, Henson PM. Phagocytosis of senescent neutrophils by human monocyte-derived macrophages and rabbit inflammatory macrophages. J Exp Med. 1982;156:430–442. doi: 10.1084/jem.156.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu B, Sonstein J, Christensen PJ, Punturieri A, Curtis JL. Deficient in vitro and in vivo phagocytosis of apoptotic T cells by resident murine alveolar macrophages. J Immunol. 2000;165:2124–2133. doi: 10.4049/jimmunol.165.4.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81:289–296. doi: 10.1046/j.1440-1711.2003.t01-1-01170.x. [DOI] [PubMed] [Google Scholar]
- 34.Hodge S, Hodge G, Brozyna S, Jersmann H, Holmes M, Reynolds PN. Azithromycin increases phagocytosis of apoptotic bronchial epithelial cells by alveolar macrophages. Eur Respir J. 2006;28:486–495. doi: 10.1183/09031936.06.00001506. [DOI] [PubMed] [Google Scholar]
- 35.Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds PN. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2007;37:748–755. doi: 10.1165/rcmb.2007-0025OC. [DOI] [PubMed] [Google Scholar]
- 36.Richens TR, Linderman DJ, Horstmann SA, Lambert C, Xiao YQ, Keith RL, Boe DM, Morimoto K, Bowler RP, Day BJ, Janssen WJ, Henson PM, Vandivier RW. Cigarette smoke impairs clearance of apoptotic cells through oxidant-dependent activation of RhoA. Am J Respir Crit Care Med. 2009;179:1011–1021. doi: 10.1164/rccm.200807-1148OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrusca DN, Gu Y, Adamowicz JJ, Rush NI, Hubbard WC, Smith PA, Berdyshev EV, Birukov KG, Lee CH, Tuder RM, Twigg HL, 3rd, Vandivier RW, Petrache I. Sphingolipid-mediated inhibition of apoptotic cell clearance by alveolar macrophages. J Biol Chem. 2010;285:40322–40332. doi: 10.1074/jbc.M110.137604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janssen WJ, McPhillips KA, Dickinson MG, Linderman DJ, Morimoto K, Xiao YQ, Oldham KM, Vandivier RW, Henson PM, Gardai SJ. Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am J Respir Crit Care Med. 2008;178:158–167. doi: 10.1164/rccm.200711-1661OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Birge RB, Ucker DS. Innate apoptotic immunity: the calming touch of death. Cell Death Differ. 2008;15:1096–1102. doi: 10.1038/cdd.2008.58. [DOI] [PubMed] [Google Scholar]
- 40.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-b, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cvetanovic M, Mitchell JE, Patel V, Avner BS, Su Y, van der Saag PT, Witte PL, Fiore S, Levine JS, Ucker DS. Specific recognition of apoptotic cells reveals a ubiquitous and unconventional innate immunity. J Biol Chem. 2006;281:20055–20067. doi: 10.1074/jbc.M603920200. [DOI] [PubMed] [Google Scholar]
- 42.Lucas M, Stuart LM, Zhang A, Hodivala-Dilke K, Febbraio M, Silverstein R, Savill J, Lacy-Hulbert A. Requirements for apoptotic cell contact in regulation of macrophage responses. J Immunol. 2006;177:4047–4054. doi: 10.4049/jimmunol.177.6.4047. [DOI] [PubMed] [Google Scholar]
- 43.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medeiros AI, Serezani CH, Lee SP, Peters-Golden M. Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J Exp Med. 2009;206:61–68. doi: 10.1084/jem.20082058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCubbrey AL, Sonstein J, Ames TM, Freeman CM, Curtis JL. Glucocorticoids relieve collectin-driven suppression of apoptotic cell uptake in murine alveolar macrophages through downregulation of SIRPalpha. J Immunol. 2012;189:112–119. doi: 10.4049/jimmunol.1200984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jonsson S, Musher DM, Chapman A, Goree A, Lawrence EC. Phagocytosis and killing of common bacterial pathogens of the lung by human alveolar macrophages. J Infect Dis. 1985;152:004–013. doi: 10.1093/infdis/152.1.4. [DOI] [PubMed] [Google Scholar]
- 47.Franke-Ullmann G, Pfortner C, Walter P, Steinmuller C, Lohmann-Matthes ML, Kobzik L. Characterization of murine lung interstitial macrophages in comparison with alveolar macrophages in vitro. J Immunol. 1996;157:3097–3104. [PubMed] [Google Scholar]
- 48.Koppe U, Suttorp N, Opitz B. Recognition of Streptococcus pneumoniae by the innate immune system. Cell Microbiol. 2012;14:460–466. doi: 10.1111/j.1462-5822.2011.01746.x. [DOI] [PubMed] [Google Scholar]
- 49.Taylor AE, Finney-Hayward TK, Quint JK, Thomas CM, Tudhope SJ, Wedzicha JA, Barnes PJ, Donnelly LE. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J. 2010;35:1039–1047. doi: 10.1183/09031936.00036709. [DOI] [PubMed] [Google Scholar]
- 50.Phipps JC, Aronoff DM, Curtis JL, Goel D, O’Brien E, Mancuso P. Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement-mediated phagocytosis of Streptococcus pneumoniae. Infect Immun. 2010;78:1214–1220. doi: 10.1128/IAI.00963-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu B, Punturieri A, Todt J, Sonstein J, Polak T, Curtis JL. Recognition and phagocytosis of apoptotic T cells by resident murine tissue macrophages require multiple signal transduction events. J Leukoc Biol. 2002;71:881–889. [PMC free article] [PubMed] [Google Scholar]
- 52.Freeman CM, Curtis JL, Chensue SW. CC chemokine receptor 5 and CXC chemokine receptor 6 expression by lung CD8+ cells correlates with chronic obstructive pulmonary disease severity. Am J Pathol. 2007;171:767–776. doi: 10.2353/ajpath.2007.061177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A. 2003;100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takashima K, Tateda K, Matsumoto T, Iizawa Y, Nakao M, Yamaguchi K. Role of tumor necrosis factor alpha in pathogenesis of pneumococcal pneumonia in mice. Infect Immun. 1997;65:257–260. doi: 10.1128/iai.65.1.257-260.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van der Poll T, Keogh CV, Guirao X, Buurman WA, Kopf M, Lowry SF. Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J Infect Dis. 1997;176:439–444. doi: 10.1086/514062. [DOI] [PubMed] [Google Scholar]
- 56.Sun K, Salmon SL, Lotz SA, Metzger DW. Interleukin-12 promotes gamma interferon-dependent neutrophil recruitment in the lung and improves protection against respiratory Streptococcus pneumoniae infection. Infect Immun. 2007;75:1196–1202. doi: 10.1128/IAI.01403-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palaniappan R, Singh S, Singh UP, Singh R, Ades EW, Briles DE, Hollingshead SK, Royal W, 3rd, Sampson JS, Stiles JK, Taub DD, Lillard JW., Jr CCL5 modulates pneumococcal immunity and carriage. J Immunol. 2006;176:2346–2356. doi: 10.4049/jimmunol.176.4.2346. [DOI] [PubMed] [Google Scholar]
- 58.Sung SS, Fu SM, Rose CE, Jr, Gaskin F, Ju ST, Beaty SR. A major lung CD103 ({alpha}E)-beta7 integrin-positive epithelial dendritic cellpopulation expressing langerin and tight junction proteins. J Immunol. 2006;176:2161–2172. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 59.Stolberg VR, Martin B, Mancuso P, Olszewski MA, Freeman CM, Curtis JL, Chensue SW. Role of CC Chemokine Receptor 4 in natural killer cell activation during acute cigarette smoke exposure. Am J Pathol. 2014;184:454–463. doi: 10.1016/j.ajpath.2013.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marques JM, Rial A, Munoz N, Pellay FX, Van Maele L, Leger H, Camou T, Sirard JC, Benecke A, Chabalgoity JA. Protection against Streptococcus pneumoniae serotype 1 acute infection shows a signature of Th17- and IFN-gamma-mediated immunity. Immunobiology. 2012;217:420–429. doi: 10.1016/j.imbio.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 61.Paats MS, Bergen IM, Hanselaar WE, van Zoelen EC, Verbrugh HA, Hoogsteden HC, van den Blink B, Hendriks RW, van der Eerden MM. T helper 17 cells are involved in the local and systemic inflammatory response in community-acquired pneumonia. Thorax. 2013;68:468–474. doi: 10.1136/thoraxjnl-2012-202168. [DOI] [PubMed] [Google Scholar]
- 62.Henriques-Normark B, Tuomanen EI. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med. 2013:3. doi: 10.1101/cshperspect.a010215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dransfield MT, Harnden S, Burton RL, Albert RK, Bailey WC, Casaburi R, Connett J, Cooper JA, Criner GJ, Curtis JL, Han MK, Make B, Marchetti N, Martinez FJ, McEvoy C, Nahm MH, Niewoehner DE, Porszasz J, Reilly J, Scanlon PD, Scharf SM, Sciurba FC, Washko GR, Woodruff PG, Lazarus SC. Long-term comparative immunogenicity of protein conjugate and free polysaccharide pneumococcal vaccines in chronic obstructive pulmonary disease. Clin Infect Dis. 2012;55:e35–44. doi: 10.1093/cid/cis513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mongardon N, Max A, Bougle A, Pene F, Lemiale V, Charpentier J, Cariou A, Chiche JD, Bedos JP, Mira JP. Epidemiology and outcome of severe pneumococcal pneumonia admitted to intensive care unit: a multicenter study. Crit Care. 2012;16:R155. doi: 10.1186/cc11471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mizrachi-Nebenzahl Y, Lifshitz S, Teitelbaum R, Novick S, Levi A, Benharroch D, Ling E, Dagan R. Differential activation of the immune system by virulent Streptococcus pneumoniae strains determines recovery or death of the host. Clin Exp Immunol. 2003;134:23–31. doi: 10.1046/j.1365-2249.2003.02261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chiavolini D, Pozzi G, Ricci S. Animal models of Streptococcus pneumoniae disease. Clin Microbiol Rev. 2008;21:666–685. doi: 10.1128/CMR.00012-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Finland M. Significance of specific pneumococcus types in disease, including types IV to XXXII (Cooper) Ann Intern Med. 1937;10:1531–1543. [Google Scholar]
- 68.Austrian R, Gold J. Pneumococcal bactermia with especial reference to bacteremic pneumococcal pneumonia. Ann Intern Med. 1964;60:759–776. doi: 10.7326/0003-4819-60-5-759. [DOI] [PubMed] [Google Scholar]
- 69.Weinberger DM, Harboe ZB, Sanders EA, Ndiritu M, Klugman KP, Ruckinger S, Dagan R, Adegbola R, Cutts F, Johnson HL, O’Brien KL, Scott JA, Lipsitch M. Association of serotype with risk of death due to pneumococcal pneumonia: a meta-analysis. Clin Infect Dis. 2010;51:692–699. doi: 10.1086/655828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Madhi F, Godot C, Bidet P, Bahuaud M, Epaud R, Cohen R. Serotype 3 pneumococcal pleural empyema in an immunocompetent child after 13-valent pneumococcal conjugate vaccine. Pediatr Infect Dis J. 2014;33:545–546. doi: 10.1097/INF.0000000000000252. [DOI] [PubMed] [Google Scholar]
- 71.Mancuso P, Huffnagle GB, Olszewski MA, Phipps J, Peters-Golden M. Leptin corrects host defense defects after acute starvation in murine pneumococcal pneumonia. Am J Respir Crit Care Med. 2006;173:212–218. doi: 10.1164/rccm.200506-909OC. [DOI] [PubMed] [Google Scholar]
- 72.Hsu A, Aronoff DM, Phipps J, Goel D, Mancuso P. Leptin improves pulmonary bacterial clearance and survival in ob/ob mice during pneumococcal pneumonia. Clin Exp Immunol. 2007;150:332–339. doi: 10.1111/j.1365-2249.2007.03491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amory-Rivier CF, Mohler J, Bedos JP, Azoulay-Dupuis E, Henin D, Muffat-Joly M, Carbon C, Moine P. Nuclear factor-kappaB activation in mouse lung lavage cells in response to Streptococcus pneumoniae pulmonary infection. Crit Care Med. 2000;28:3249–3256. doi: 10.1097/00003246-200009000-00021. [DOI] [PubMed] [Google Scholar]
- 74.Dockrell DH, Lee M, Lynch DH, Read RC. Immune-mediated phagocytosis and killing of Streptococcus pneumoniae are associated with direct and bystander macrophage apoptosis. J Infect Dis. 2001;184:713–722. doi: 10.1086/323084. [DOI] [PubMed] [Google Scholar]
- 75.Dockrell DH, Marriott HM, Prince LR, Ridger VC, Ince PG, Hellewell PG, Whyte MK. Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J Immunol. 2003;171:5380–5388. doi: 10.4049/jimmunol.171.10.5380. [DOI] [PubMed] [Google Scholar]
- 76.Marriott HM, Ali F, Read RC, Mitchell TJ, Whyte MK, Dockrell DH. Nitric oxide levels regulate macrophage commitment to apoptosis or necrosis during pneumococcal infection. FASEB J. 2004;8:1126–1128. doi: 10.1096/fj.03-1450fje. [DOI] [PubMed] [Google Scholar]
- 77.Matute-Bello G, Liles WC, Frevert CW, Dhanireddy S, Ballman K, Wong V, Green RR, Song HY, Witcher DR, Jakubowski JA, Martin TR. Blockade of the Fas/FasL system improves pneumococcal clearance from the lungs without preventing dissemination of bacteria to the spleen. J Infect Dis. 2005;191:596–606. doi: 10.1086/427261. [DOI] [PubMed] [Google Scholar]
- 78.Marriott HM, Hellewell PG, Cross SS, Ince PG, Whyte MK, Dockrell DH. Decreased alveolar macrophage apoptosis is associated with increased pulmonary inflammation in a murine model of pneumococcal pneumonia. J Immunol. 2006;177:6480–6488. doi: 10.4049/jimmunol.177.9.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Winter C, Taut K, Langer F, Mack M, Briles DE, Paton JC, Maus R, Srivastava M, Welte T, Maus UA. FMS-like tyrosine kinase 3 ligand aggravates the lung inflammatory response to Streptococcus pneumoniae infection in mice: role of dendritic cells. J Immunol. 2007;179:3099–3108. doi: 10.4049/jimmunol.179.5.3099. [DOI] [PubMed] [Google Scholar]
- 80.Taut K, Winter C, Briles DE, Paton JC, Christman JW, Maus R, Baumann R, Welte T, Maus UA. Macrophage turnover kinetics in the lungs of mice infected with Streptococcus pneumoniae. Am J Respir Cell Mol Biol. 2008;38:105–113. doi: 10.1165/rcmb.2007-0132OC. [DOI] [PubMed] [Google Scholar]
- 81.Herbold W, Maus R, Hahn I, Ding N, Srivastava M, Christman JW, Mack M, Reutershan J, Briles DE, Paton JC, Winter C, Welte T, Maus UA. Importance of CXC chemokine receptor 2 in alveolar neutrophil and exudate macrophage recruitment in response to pneumococcal lung infection. Infect Immun. 2010;78:2620–2630. doi: 10.1128/IAI.01169-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schabbauer G, Matt U, Gunzl P, Warszawska J, Furtner T, Hainzl E, Elbau I, Mesteri I, Doninger B, Binder BR, Knapp S. Myeloid PTEN promotes inflammation but impairs bactericidal activities during murine pneumococcal pneumonia. J Immunol. 2010;185:468–476. doi: 10.4049/jimmunol.0902221. [DOI] [PubMed] [Google Scholar]
- 83.Bewley MA, Marriott HM, Tulone C, Francis SE, Mitchell TJ, Read RC, Chain B, Kroemer G, Whyte MK, Dockrell DH. A cardinal role for cathepsin d in co-ordinating the host-mediated apoptosis of macrophages and killing of pneumococci. PLoS Pathog. 2011;7:e1001262. doi: 10.1371/journal.ppat.1001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brumshagen C, Maus R, Bischof A, Ueberberg B, Bohling J, Osterholzer JJ, Ogunniyi AD, Paton JC, Welte T, Maus UA. FMS-like tyrosine kinase 3 ligand treatment of mice aggravates acute lung injury in response to Streptococcus pneumoniae: role of pneumolysin. Infect Immun. 2012;80:4281–4290. doi: 10.1128/IAI.00854-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Steinwede K, Henken S, Bohling J, Maus R, Ueberberg B, Brumshagen C, Brincks EL, Griffith TS, Welte T, Maus UA. TNF-related apoptosis-inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. J Exp Med. 2012;209:1937–1952. doi: 10.1084/jem.20120983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tokairin Y, Shibata Y, Sata M, Abe S, Takabatake N, Igarashi A, Ishikawa T, Inoue S, Kubota I. Enhanced immediate inflammatory response to Streptococcus pneumoniae in the lungs of mice with pulmonary emphysema. Respirology. 2008;13:324–332. doi: 10.1111/j.1440-1843.2007.01229.x. [DOI] [PubMed] [Google Scholar]
- 87.Sajjan U, Ganesan S, Comstock AT, Shim J, Wang Q, Nagarkar DR, Zhao Y, Goldsmith AM, Sonstein J, Linn MJ, Curtis JL, Hershenson MB. Elastase- and LPS-exposed mice display altered responses to rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. 2009;297:L931–944. doi: 10.1152/ajplung.00150.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Aberdein JD, Cole J, Bewley MA, Marriott HM, Dockrell DH. Alveolar macrophages in pulmonary host defence the unrecognized role of apoptosis as a mechanism of intracellular bacterial killing. Clin Exp Immunol. 2013;174:193–202. doi: 10.1111/cei.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Paterson GK, Orihuela CJ. Pneumococci: immunology of the innate host response. Respirology. 2010;15:1057–1063. doi: 10.1111/j.1440-1843.2010.01814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nibbering PH, van den Barselaar MT, van de Gevel JS, Leijh PC, van Furth R. Deficient intracellular killing of bacteria by murine alveolar macrophages. Am J Respir Cell Mol Biol. 1989;1:417–422. doi: 10.1165/ajrcmb/1.5.417. [DOI] [PubMed] [Google Scholar]
- 91.Hartshorn KL, Crouch E, White MR, Colamussi ML, Kakkanatt A, Tauber B, Shepherd V, Sastry KN. Pulmonary surfactant proteins A and D enhance neutrophil uptake of bacteria. Am J Physiol. 1998;274:L958–969. doi: 10.1152/ajplung.1998.274.6.L958. [DOI] [PubMed] [Google Scholar]
- 92.Kuronuma K, Sano H, Kato K, Kudo K, Hyakushima N, Yokota S, Takahashi H, Fujii N, Suzuki H, Kodama T, Abe S, Kuroki Y. Pulmonary surfactant protein A augments the phagocytosis of Streptococcus pneumoniae by alveolar macrophages through a casein kinase 2-dependent increase of cell surface localization of scavenger receptor A. J Biol Chem. 2004;279:21421–21430. doi: 10.1074/jbc.M312490200. [DOI] [PubMed] [Google Scholar]
- 93.Marriott HM, Hellewell PG, Whyte MK, Dockrell DH. Contrasting roles for reactive oxygen species and nitric oxide in the innate response to pulmonary infection with Streptococcus pneumoniae. Vaccine. 2007;25:2485–2490. doi: 10.1016/j.vaccine.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peters-Golden M, Thebert P. Inhibition by methylprednisolone of zymosan-induced leukotriene synthesis in alveolar macrophages. Am Rev Respir Dis. 1987;135:1020–1026. doi: 10.1164/arrd.1987.135.5.1020. [DOI] [PubMed] [Google Scholar]
- 95.Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9:781–795. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Standish AJ, Weiser JN. Human neutrophils kill Streptococcus pneumoniae via serine proteases. J Immunol. 2009;183:2602–2609. doi: 10.4049/jimmunol.0900688. [DOI] [PubMed] [Google Scholar]
- 97.Canton J. Phagosome maturation in polarized macrophages. J Leukoc Biol. 2014;96:729–738. doi: 10.1189/jlb.1MR0114-021R. [DOI] [PubMed] [Google Scholar]
- 98.Marriott HM, Dockrell DH. The role of the macrophage in lung disease mediated by bacteria. Exp Lung Res. 2007;33:493–505. doi: 10.1080/01902140701756562. [DOI] [PubMed] [Google Scholar]
- 99.Parker D, Martin FJ, Soong G, Harfenist BS, Aguilar JL, Ratner AJ, Fitzgerald KA, Schindler C, Prince A. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. MBio. 2011;2:e00016–00011. doi: 10.1128/mBio.00016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koppe U, Hogner K, Doehn JM, Muller HC, Witzenrath M, Gutbier B, Bauer S, Pribyl T, Hammerschmidt S, Lohmeyer J, Suttorp N, Herold S, Opitz B. Streptococcus pneumoniae stimulates a STING- and IFN regulatory factor 3-dependent type I IFN production in macrophages, which regulates RANTES production in macrophages, cocultured alveolar epithelial cells, and mouse lungs. J Immunol. 2012;188:811–817. doi: 10.4049/jimmunol.1004143. [DOI] [PubMed] [Google Scholar]
- 101.Han MK, Postma D, Mannino DM, Giardino ND, Buist S, Curtis JL, Martinez FJ. Gender and chronic obstructive pulmonary disease: why it matters. Am J Respir Crit Care Med. 2007;176:1179–1184. doi: 10.1164/rccm.200704-553CC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Han MK, Kazerooni EA, Lynch DA, Liu LX, Murray S, Curtis JL, Criner GJ, Kim V, Bowler RP, Hanania NA, Anzueto AR, Make BJ, Hokanson JE, Crapo JD, Silverman EK, Martinez FJ, Washko GR. Chronic obstructive pulmonary disease exacerbations in the COPDGene study: associated radiologic phenotypes. Radiology. 2011;261:274–282. doi: 10.1148/radiol.11110173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sin DD, Tashkin D, Zhang X, Radner F, Sjobring U, Thoren A, Calverley PM, Rennard SI. Budesonide and the risk of pneumonia: a meta-analysis of individual patient data. Lancet. 2009;374:712–719. doi: 10.1016/S0140-6736(09)61250-2. [DOI] [PubMed] [Google Scholar]
- 104.Cates C. Inhaled corticosteroids in COPD: quantifying risks and benefits. Thorax. 2013;68:499–500. doi: 10.1136/thoraxjnl-2012-202959. [DOI] [PubMed] [Google Scholar]
- 105.Nannini LJ, Lasserson TJ, Poole P. Combined corticosteroid and long-acting beta(2)-agonist in one inhaler versus long-acting beta(2)-agonists for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2012;9:CD006829. doi: 10.1002/14651858.CD006829.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.