

Abstract
Nuclear factor-κB (NF-κB) plays a central role in pathogenesis of inflammation and cancer. Many phytochemicals including gamma-tocotrienol (γTE), a natural form of vitamin E, have been shown to inhibit NF-κB activation, but the underlying mechanism has not been identified. Here we show that γTE inhibited cytokine-triggered activation of NF-κB and its upstream regulator TGFβ-activated kinase-1 in murine RAW264.7 macrophages and primary bone marrow-derived macrophages. In these cells, γTE induced up-regulation of A20, an inhibitor of NF-κB. Knockout of A20 partially diminished γTE’s anti-NF-κB effect but γTE increased another NF-κB inhibitor Cezanne in A20−/− cells. In search of the reason for A20 upregulation, we found that γTE treatment increased phosphorylation of translation initiation factor 2 (eIF2α), IκBα and JNK, indicating induction of endoplasmic reticulum (ER) stress. LC-MS/MS analyses revealed that γTE modulated sphingolipids including enhancement of intracellular dihydroceramides, sphingoid bases in de novo synthesis of sphingolipid pathway. Chemical inhibition of de novo sphingolipid synthesis partially reversed γTE’s induction of A20 and anti-NF-κB effect. The importance of dihydroceramide increase is further supported by the observation that C8-dihydroceramide mimicked γTE in up-regulating A20, enhancing ER stress and attenuating TNF-triggered NF-κB activation. Our study identifies a novel anti-NF-κB mechanism where A20 is induced by stress-induced adaptive response as a result of modulation of sphingolipids, and demonstrates an immune-modulatory role of dihydrocermides.
INTRODUCTION
NF-κB is a central transcription factor that regulates immune functions and cellular survival, and therefore plays critical roles in inflammation and cancer development (1). Under resting conditions, NF-κB p50 and p65 are bound to inhibitory IκBα that sequesters inactive NF-κB complex in the cytoplasm. During inflammation, endotoxin or cytokines such as TNFα activate the assembly of receptor proximal signaling complexes containing receptor-interacting protein serine/threonine kinase (RIP) and TNF receptor-associated factors (2). This receptor assembly involves ubiquitylation and phosphorylation of RIP1 and leads to recruitment of the IκB kinase (IKK) complex to the receptor in the proximity of its upstream transforming growth factor β (TGFβ)-activated kinase 1 (TAK1). Activated TAK1 interacts with regulatory NEMO/IKKγ and stimulates the IKKs. Subsequently, activated IKKs phosphorylate IκBα, which targets IκBα for ubiquitination and proteasomal degradation. As a result, NF-κB p50 and p65 dimer is released so that they can translocate to the nucleus, where active NF-κB binds to consensus target sequences in many promoters.
Activation of NF-κB leads to up-regulation of many genes including pro-inflammatory cytokines and proteins that regulate inflammation and promote proliferation and survival of many types of cells including immune and cancer cells. To prevent excessive immune response, activation of NF-κB is tightly controlled (3). Several enzymes have been identified as negative regulators of NF-κB signaling, including CYLD (cylindromatosis), A20 and Cezanne (cellular zinc finger anti-NF-κB, a member of the A20 family) (4, 5). In particular, A20 and Cezanne, both of which are NF-κB-target proteins with ubiquitin-editing activity, are induced by NF-κB to prevent its prolonged and aberrant activation (5, 6). A20 was initially suggested to inhibit cytokine-triggered NF-κB activation via its ubiquitin-editing function (7–9). However, recent evidence indicates that the deubiquitinase activity of A20 is not required for controlling NF-κB signaling (10). Instead, A20 appears to antagonize NF-κB activation by interaction with NEMO in a non-catalytic manner to blunt activation of TAK-1 and IKKs (11).
Because of the regulatory role of NF-κB in inflammation and cancer, targeting NF-κB activation has been recognized as a potential effective strategy for preventing and treating chronic diseases. Many natural products have been shown to inhibit NF-κB in cell-based studies and animal models. For instance, γ-tocotrienol (γTE), a natural form of vitamin E rich in palm oil, has been reported to inhibit NF-κB activation in leukemia KBM-5 and other cancer cells (12) as well as macrophages (13, 14). Consistently, γTE supplementation inhibits proinflammatory cytokines in animals and human subjects (15, 16). Despite these interesting results, the molecular mechanism responsible for the anti-NF-κB effect has not been identified. Here we investigated inhibitory effects and mechanism of γTE on NF-κB in murine RAW264.7 macrophages and primary bone-marrow derived macrophages (BMDMs). Our study revealed a novel anti-NF-κB mechanism in which γTE induced up-regulation of NF-κB inhibitor A20 via altering sphingolipid metabolism and cellular stress.
MATERIALS AND METHODS
Chemicals and Reagents
γTE (>97% pure) was a gift from BASF (Germany). Recombinant mouse TNFα and IL-1β were from Sigma (St Louis, MO). Primary antibodies against phospho-IκBα, IκBα and all the secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for phosphor-JNK, JNK, A20, phosphor-p65, phosphor-eIF2α and eIF2α were from Cell Signaling Technology (Beverly, MA). Inhibitors for MEK (U0126), p38 MAPK (SB202190) and PI3K were from Calbiochem (La Jolla, CA). Myriocin from Mycelia Sterilia, C8-dihydroceramide, [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide] (MTT), and all other chemicals were from Sigma. Cell culture Media were obtained from American Type Culture Collection (ATCC) (Manassas, VA).
Cell culture
Murine RAW264.7 macrophages from ATCC were routinely maintained in Dulbecco’s modified eagle medium (DMEM) with 10% fetal bovine serum (FBS). Confluent cells were seeded and allowed to attach overnight at 7 × 105 or 5 × 106 per well in a 24-well or a 6-well plate, respectively. γTE stock solutions were initially made in dimethyl sulfoxide (DMSO) and then diluted in 10 mg/mL fatty acid-free bovine serum albumin (BSA). In some studies, γTE in DMSO was directly diluted in cell culture media without BSA. Our unpublished data showed that similar results were observed with or without BSA. Confluent cells were incubated in DMEM containing 1% FBS and 0.05% DMSO (control) or γTE for indicated time and then stimulated by TNFα or IL-1β. Cell viability was determined by MTT assays.
Preparation of bone marrow derived macrophages (BMDMs)
BMDMs from mice were prepared according to published protocols (14, 17). The protocol on animal use was approved by the Animal Care and Use Committee at Purdue University and was strictly followed. Briefly, bone marrow was obtained by flushing femur from 7–8 week-old C57BL/6 black mice (Harlan, Indianapolis, IN). Suspension cells were cultured in 10-cm dishes in the DMEM media containing 10% FBS with 100 U/ml penicillin, 100 μg/ml streptomycin and 100 U/ml M-CSF for 5 days. Attached cells were replenished by fresh media and incubated for additional two days. Cells were harvested using non-enzymatic dissociation solution (Sigma) and characterized using flow cytometry after stained with phycoerythrin (PE)-conjugated anti-mouse F4/80/EMR1 (#FAB5580P) by Cell Lab Quanta SC–MPL flow cytometer (Beckman Coulter, Brea, CA) with excitation at 488 nm. Cells with > 95% purity were seeded in 24- or 6-well plates for subsequent studies. During experiments, cells were incubated in DMEM containing 1% FBS and 0.05% DMSO (control) or γTE for indicated time and then stimulated by TNFα.
A20−/− and A20+/+ MEFs
A20−/− and A20+/+ MEFs were prepared from the knockout and wild-type mice (18). These cells were immortalized with SV40 large T antigen and cultured in DMEM with 10% FBS, 50 μM beta mercaptoethanol and 100 U/ml penicillin-streptomycin and 2 mM L-glutamine. Cell passages < 6 were used in the study.
Quantitative PCR (qPCR)
mRNAs of A20 (TNFAIP3) and GAPDH were quantified by qPCR with SYBR green via service contract by ARQ Genetics, LLC (Bastrop, Tx). Data were obtained with samples from three independent experiments.
Western blot
Cells were lysed in a lysis buffer containing Tris-EDTA, 1% SDS, 1 mM DTT, 2 mM sodium vanadate and protease inhibitor cocktails (Sigma). Cytosolic and nuclear protein were extracted using the Pierce Kit (Pierce, Rockford, IL). Proteins (20–50 μg) were loaded on BioRad pre-cast SDS-PAGE gels. Resolved proteins were transferred onto a PVDF membrane (Millipore, Billerica, MA) and probed by antibodies. Membranes were exposed to chemiluminescent reagent (Perkin Elmer, Waltham, WA) and visualized on a Kodak film. In all the experiments, after probed by antibodies for target proteins, PVDF membranes were stripped with antibody-stripping solution from EMD Millipore Corporation (Billerica, MA) and re-immunoblotted with antibodies for internal controls such as β-actin. Results of Western blots were quantified by ImageJ.
Lipid extraction
To measure sphingolipids, total lipids were extracted as previously described (19). Briefly, cell pellets were re-suspended in chloroform/methanol/water (10:5:1; v/v/v), followed by addition of internal standards containing 0.5 nmoles of C12:0 ceramide, C17-sphingosine, C17-sphinganine, C25:0-ceramide and C12:0-sphingomyelin from Avanti Polar Lipids (Alabaster, AL). After tip sonication, samples were incubated at 48 °C overnight. The next day, 100 μl of aliquots were taken out and dried under N2 for measurement of total phospholipids (Wako Chemicals, Neuss, Germany). The rest samples were added with 75 μl of 1M KOH in methanol, sonicated for 30 min, then incubated at 37 °C for 2 h and dried in a nitrogen evaporator.
Measurement of sphingolipids using liquid chromatography tandem mass spectrometry (LC-MS/MS)
The LC-MS/MS method for sphingolipid analysis was slightly modified based on a published protocol by Merrill et al. (19). Briefly, all samples were analyzed using the Agilent 6460 triple quadrupole mass spectrometer coupled with the Agilent 1200 rapid resolution HPLC (Agilent Technologies, Santa Clara, CA) with identification of each sphingolipid by multiple reaction monitoring (MRM). The MS/MS conditions used for MRM detection of sphingolipids were the same as previously described (19). The LC column for sphingoid bases and ceramides was Agilent XDB-C18 (4.6 × 50 mm) with particle size of 1.8 μm, while the LC column for sphingomyelins was Agilent XDB-C8 (2.1 × 50 mm) with Zorbax 3.5 μm. Mobile phase A and B contains 5 mM ammonium formate and respectively has methanol-H2O-formic acid (74:25:1, v/v/v) and methanol-formic acid (99:1, v/v). Agilent mass hunter software was coupled with the LC-MS/MS for data acquisition and analysis.
Statistical Analyses
We used one-way ANOVA and the Student’s t-test to analyze the data or log transformed results. *P < 0.05 was considered significant.
RESULTS
γTE inhibited cytokine-stimulated activation of NF-κB, JNK and TAK-1 in macrophages
Like previous studies (12, 14), γTE inhibited TNFα-triggered NF-κB activation in RAW 264.7 macrophages, as indicated by decreased phosphorylation and degradation of IκBα in the cytosol and diminished phosphorylation of p65 in the nucleus compared with vehicle controls (Figure 1A). The inhibitory potency by γTE appeared to be in a time-dependent fashion. Specifically, relatively short 2-h incubation with γTE did not lead to consistent inhibition of NF-κB (not shown) and longer pre-incubation resulted in stronger inhibition of phosphorylation of IκBα (comp. 16 vs. 8 h in Figure 1A). Besides RAW macrophages, γTE inhibited TNFα-stimulated phosphorylation of IκBα in bone marrow-derived macrophages (Figure 1B). In addition, γTE also attenuated TNF-triggered JNK phosphorylation, another downstream signal in response to the activation of TNF receptor (Figure 1C).
Figure 1. γTE inhibited cytokine-stimulated activation of NF-κB, JNK and TAK-1 in macrophages.

Panel A: RAW 264.7 cells were treated with γTE at 20 μM for 8 or 16 h, and then stimulated with 10 ng/ml of TNFα for 5 min. Panel B: BMDMs were pre-treated with γTE at 10 μM for 8 h, and then stimulated with 10 ng/ml of TNFα for 5 min. Panel C: RAW cells were treated with γTE for 14 h before stimulated with TNFα as described in Panel A. Panel D: RAW cells were pretreated with a TAK1 inhibitor at 1 or 5 μM (Oxozeaenol, TI1 and TI5) for 30 min, and then stimulated with 10 ng/ml of TNFα for 5 min. Panel E: RAW cells were pretreated with γTE for 16 h. Calyculin A (a Serine/Threonine phosphatase inhibitor, 50 nM) was added immediately before cells were stimulated with 10 ng/mL of IL-1β or TNFα for 5 min. In these studies, cytosolic proteins were immunoblotted for phospho-IκBα, phospho-JNK, phosphor-TAK1 and β-actin, and nuclear proteins for phosphor-p65. These data are representative results from at least three independent experiments.
It has been well-established that TNF-triggered IκBα phosphorylation is catalyzed by IKKs, which are regulated by the upstream TAK1 (2). Consistent with the key role of TAK1 in NF-κB activation, a specific inhibitor of TAK1, but not those of PKCs or PI3K (not shown), markedly inhibited TNFα-stimulated phosphorylation of IκBα in RAW macrophages (Figure 1D). Importantly, γTE treatment dampened TNFα- or IL-1β-stimulated phosphorylation of TAK1 (Figure 1E), which is in agreement with a previous study in which γTE blocked NF-κB-dependent reporter gene transcription induced by overexpression of TAK1 in A293 cells (12). These observations suggest that γTE likely targets upstream signaling that is important to the activation of TAK1 and IKKs.
γTE increased expression of A20, a negative regulator of NF-κB, in macrophages and various human cancer cell lines
Activation of TAK1 and subsequent NF-κB by TNFα requires assembly of TNF-receptor proximal adaptor proteins and involves a series of ubiquitylation and deubiquitylation events (7, 8). These ubiquitylation events can be interrupted by ubiquitin-editing enzymes such as CYLD and A20, which are recognized as endogenous NF-κB inhibitor (20–22). Alternatively, A20 is shown to block activation of TAK-1 and IKK via ubiquitin-independent mechanism by direct interaction with NEMO (11). Because γTE inhibits TAK1 phosphorylation (Figure 1E) and overexpression of CYLD or A20 blocks NF-κB activation in cells and animals (11, 20–22), we investigated whether γTE has any impact on these NF-κB negative regulators. We found that γTE treatment led to enhanced expression of A20 but had no effect on CYLD in both RAW cells and bone marrow-derived primary macrophages (Figure 2A, B and C). Enhanced A20 expression was observed as early as 4 h (not shown) and sustained during longer incubation with γTE. Since A20 has recently been shown to blunt TNF-activated JNK activation (23), the induction of A20 by γTE may also explain γTE’s suppression of TNFα-stimulated activation of JNK.
Figure 2. γTE induced A20, an inhibitor of NF-κB, in macrophages and human cancer cell lines.

Panels A and B: RAW 264.7 cells were treated with γTE at 20 or 40 μM for 16 h (A) or at 20 μM for 8 or 16 h (B). Panel C: BMDMs were treated with γTE at 10 μM for 8 h. Panel D: Subconfluent A549, PC-3 and MCF-7 cells were pre-treated with γTE (20 μM) for 14–16 h, and then stimulated with TNFα for 5 min. Panel E: Sub-confluent A549, PC-3 and MCF-7 cells were pre-treated with γTE (20 μM) with or without subsequent TNFα stimulation (10 ng/ml, 5 min). In these studies, total proteins were immunoblotted with antibodies for CYLD or A20 and β-actin, and cytosolic proteins were analyzed for phospho-IκBα. These data are representative results from at least three independent experiments.
Activation of NF-κB is believed to promote cancer cell survival and contributes to drug resistance. Since γTE has been reported to suppress NF-κB activation in cancer cells (12), we next examined whether γTE induces A20 in human A549 lung, PC-3 prostate and MCF7 breast cancer cells. We found that γTE treatment inhibited TNF-triggered phosphorylation of IκBα (Figure 2D) and resulted in induction of A20 expression in these cells (Figure 2E). Considering the well-established negative regulation of NF-κB by A20, γTE-mediated inhibition of NF-κB likely stems from its induction of A20 in these cells.
γTE’s inhibition of NK-κB was partially diminished in A20−/− cells and γTE induced Cezanne in the absence of A20
To further examine the role of A20 induction in γTE’s anti-NF-κB effect, we used A20−/− MEFs (mouse embryonic fibroblasts). Like observations in macrophages, γTE treatment induced A20 expression in A20+/+ MEFs, whereas A20 was not detectable in A20−/− cells (Figure 3A). Consistent with anti- NF-κB role of A20, TNFα-induced phosphorylation of IκBα was more pronounced in A20−/− cells than that in wild-type cells (Figure 3B, comp. lanes of 3 vs. 4). The suppressive effect of γTE on phosphorylation of IκBα was significantly, though partially, reversed in A20−/− cells compared with A20+/+ cells (Figure 3B, comp. lanes 7 vs. 8). These results, together with known functions of A20 support the idea that A20 induction plays a key role in γTE’s inhibitory effect on NF-κB. On the other hand, we observed that γTE still inhibited NF-κB in the absence of A20, suggesting that other mechanisms are involved in antagonizing NF-κB especially under A20 knockout condition. Consistent with this hypothesis, γTE treatment increased another NF-κB inhibitor Cezanne in A20−/− MEFs (Figure 3C), but had no effect on CYLD (not shown). Interestingly, Cezanne was not induced by γTE in A20+/+ MEFs (Figure 3C).
Figure 3. γTE’s anti-NF-κB effect in A20+/+ and A20−/− MEFs.

Panel A: A20+/+ and A20−/− MEFs were treated with γTE (10 or 20 μM) for 8 h, and A20 expression was detected by Western blots. Results from A20+/+ cells were quantified by ImageJ. Panel B: After pre-incubated with γTE (10 μM) for 8 h, cells were stimulated by TNFα (10 ng/mg) for 5 min and then collected for probing IκBα (cytosol) by Western blot. ANOVA was performed to analyze statistical differences among different groups. Quantitative results with no common letter differ (P < 0.05, Mean ± SD, n = 4–5). Panel C. A20+/+ and A20−/− MEFs were treated with γTE (10 μM) for 8 h, and Cezanne was analyzed by Western blot that was quantified by ImageJ. For Panels A and C, Student’s t-tests were used to analyze Western blot data. *P < 0.05 indicates significant difference between control and γTE group (Mean ± SEM, n =4–6).
γTE treatment increased A20 mRNA and phosphorylation of IκBα, JNK and translation initiation factor 2 (eIF2α)
In search of the mechanism underlying A20 induction, we found that γTE enhanced A20 mRNA, indicating that A20 was up-regulated at the transcriptional level (Figure 4A). Since A20 expression is known to be regulated by NF-κB (24), we investigated whether γTE has any effect on the basal activity of NF-κB. To this end, γTE treatment mildly increased basal IκBα phosphorylation (Figure 4B). As previously observed in other types of cells (25, 26), γTE enhanced phosphorylation of JNK in macrophages (Figure 4B). In addition, γTE increased phosphorylation of eIF2α, a marker of endoplasmic reticulum (ER) stress (Figure 4C). These data indicate that γTE treatment induced moderate stress to the cells, although no obvious changes in cell morphology or viability were observed in confluent macrophages based on microscopic examination and the MTT assays (data not shown). Because phosphorylation of eIF2α has been demonstrated to be sufficient to activate NF-κB (27) and A20 expression is regulated by NF-κB (24), we reason that γTE-induced basal activation of NF-κB and ER stress is responsible for A20 up-regulation.
Figure 4. γTE treatment increased A20 mRNA and basal IκBα phosphorylation and induced ER stress as indicated by phosphorylation of JNK and eIF2α.

Panel A: RAW macrophages were incubated with γTE (20 μM) for 14 h and collected for measurement of mRNA by qPCR. Panel B: RAW 264.7 cells or BMDMs were treated with γTE at 20 or 10 μM for 16 or 8 h, respectively. Panel C. RAW 264.7 cells were incubated with γTE at 20 μM for 8 h. Cytosolic proteins (for IκBα and JNK) or total proteins (for eIF2α) were analyzed by immunoblots. *P < 0.05 indicates significant difference between control and γTE treated cells (Mean ± SEM, n = or > 3).
γTE modulated sphingolipid metabolism in macrophages
We have previously demonstrated that vitamin E forms including γTE induced JNK phosphorylation and ER stress in MCF7 cells and inhibited prostate cancer cell growth by modulation of de novo synthesis of sphingolipids pathway (Figure 5A) (26, 28, 29). Because γTE appeared to induce cellular stress in the present study, we examined whether it has any effect on sphingolipids in macrophages by a sphingolipodomic approach using LC-MS/MS (26). Consistent with a previous study (30), C16:0-, C24:0- and C24:1-dihydroceramide (dhCer) or ceramide (Cer) were the predominant (dihydro)ceramides in macrophages (Table S1). Compared with controls, γTE treatment significantly enhanced individual and total dhCer (Figure 5B, Table S1). In contrast, γTE treatment resulted in decreased total and specific ceramides at 8 h, but increased C24:0-Cer but had no impact on total or other ceramides at 16 h (Figure 5B, Table S1). Prolonged incubation with γTE for 16 h but not 8 h decreased total sphingomeylin (SM) (Figure 5, Table S1).
Figure 5. γTE modulated sphingolipid metabolism.

Panel A: De novo synthesis of sphingolipids. R = Acyl group with C16 - C26. Panel B: RAW 264.7 cells were treated with γTE at 20 μM for 8 or 16 h. Sphingolipids including total dihydroceramides (dhCers), ceramides (Cers) and sphingomyelins (SMs) were analyzed by LC-MS/MS and expressed as Mean ± SD (n = 4 or 5). *P < 0.05 indicates significant difference between control and γTE treated cells.
Modulation of sphingolipid metabolism by γTE plays a significant role in its induction of A20 and inhibition of NF-κB
Modulation of sphingolipids including increase of dhCer has been shown to cause ER stress (31). To examine the role of sphingolipid modulation in γTE-induced effects, we pharmacologically inhibited de novo sphingolipid biosynthesis with myriocin, a specific inhibitor of serine palmitoyl-CoA transferase that catalyzes the rate-limiting reaction in de novo synthesis of sphingolipids (Figure 5A). Myriocin blocked γTE-caused increase of dhCer (Figure 6A) and partially reversed γTE-induced upregulation of A20 and phosphorylation of JNK (Figure 6B). More importantly, myriocin counteracted γTE’s-inhibition of NF-κB activation (figure 6C).
Figure 6. Myriocin reversed γTE-caused increase of dhCer, induction of A20 and subsequent inhibition of NF-κB.

Panels A and B: RAW264.7 cells were incubated with γTE (20 μM) for 8 h (A) or 14 h (B) in the presence or absence of 6 μM myriocin (M). Total amounts of dhCers were measured by LC-MS/MS (A). Cytosolic proteins (for p-JNK) or whole proteins (for A20) were analyzed by Western blot that was quantified by ImageJ (B). Panel C: RAW cells were incubated with γTE (20 μM) for 14 h with or w/o 6 μM myriocin (M) and then were stimulated with TNFα for 5 min. Cytosolic proteins were used for monitoring IκBα phosphorylation. **P < 0.01 indicates significant difference between γTE and γTE plus myriocin (γTE+M) treated cells (Mean ± SEM, n = 3–4).
To further investigate the role of increased dhCer in induction of cell stress and A20 as well as NF-κB activation in macrophages, we incubated RAW cells with C8-dhCer. Similar to γTE, C8-dhCer treatment did not change cell morphology but led to increased phosphorylation of JNK and IκBα as well as the ER stress marker eIF2α (Figure 7). Moreover, like γTE, dhCer induced A20 expression (Figure 7A) and attenuated TNFα-caused phosphorylation of IκBα (Figure 7B). These results, together with myriocin’s counteraction of γTE’s anti-NF-κB, indicate that modulation of de novo synthesis of sphingolipids, especially accumulation of dhCer, plays a significant role in γTE-mediated anti-NF-κB effects.
Figure 7. C8-dhCer induced phosphorylation of JNK, IκBα, eIF2α and inhibited TNFα-stimulated NF-κB activation.
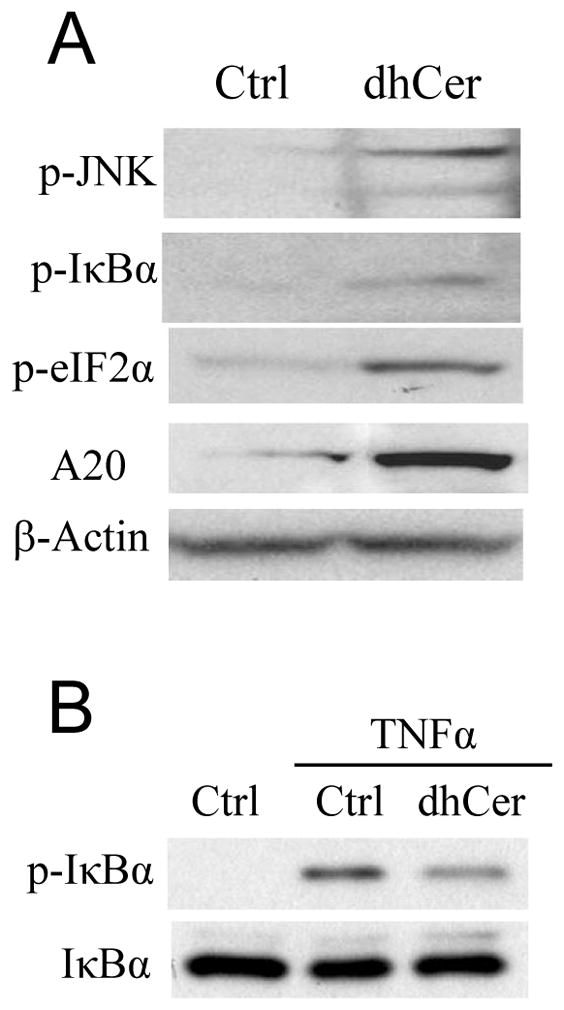
Panel A: RAW264.7 cells were treated with C8-dhCer (10 μM) for 16 h. Panel B: RAW cells were treated with C8-dhCer for 16 h and stimulated with TNFα (10 ng/mL) for 5 min. Cytosolic proteins (for p-JNK, p-IκBα, IκBα) or whole proteins (for A20 and p-eIF2α) were analyzed by Western blot.
DISCUSSION
We have identified a novel anti-NF-κB mechanism in which modulation of sphingolipids leads to cellular stress and upregulation of A20, a well-established NF-κB negative regulator (Figure 8). The importance of A20 in anti-NF-κB action was verified by the observation that NF-κB activation was enhanced in the absence of A20 and compared with A20+/+ cells, γTE showed diminished inhibitory effect on NF-κB in A20−/− cells. A causal role of sphingolipid modulation in γTE’s anti-NF-κB effect and induction of A20 and stress is supported by the following lines of evidence. γTE treatment led to significant increase of intracellular dihydroceramides, which are key sphingoid bases in de novo synthesis of sphingolipids and known to cause ER stress (31, 32). Exogenous addition of C8-dihydroceramide induced similar cellular changes to those observed in γTE-treated cells including upregulation of A20, increase of ER stress and suppression of TNFα-induced IκBα phosphorylation. Moreover, myriocin, which blocks de novo synthesis of sphingolipids and thus prevents accumulation of dhCer counteracted γTE-caused anti-NF-κB effect and induction of A20. Although γTE was previously suggested to have anti-inflammatory effects by suppressing the proteasome activity (15), there is no evidence indicating that γTE inhibits the proteasome activity in the present study. This is because in contrast to proteasome inhibitors shown to increase TNFα-induced phosphorylation of IκBα by blocking its proteasome-mediated degradation (33), γTE inhibited TNFα-triggered phosphorylation of IκBα and upstream regulator TAK-1.
Figure 8. Proposed mechanism underlying γTE’s anti-NF-κB action.

γTE modulates sphingolipid metabolism including increase of intracellular dihydroceramides. This leads to ER stress, which results in activation of basal NF-κB and subsequent A20 upregulation. Increased expression of A20 is responsible for suppression of TNFα-stimulated activation of JNK, TAK1 and NF-κB.
The induction of A20 explains γTE’s inhibition of TNFα-stimulated activation of TAK-1, JNK and NF-κB and has important biological implications. Although A20 was initially proposed to inhibit NF-κB via its ubiquitin-editing activity, recent studies have shown that A20 blocked NF-κB in a non-catalytic manner (10) via direct association with NEMO to inhibit TAK-1 and IKKs (11). Besides its mechanistic action, A20 as a key regulator of immunopathology has also been firmly established in vivo. For instance, A20−/− mice developed severe inflammation and hypersensitivity to LPS or TNFα, and died prematurely as a result of unrestrained inflammation compared with wild-type mice (18, 34). Mice lacking A20 in dendritic cells spontaneously developed colitis (35). Besides NF-κB, A20 has also been shown to antagonize TNF-induced JNK signaling (36, 37) by targeting apoptosis signal-regulating kinase1 (ASK1) and mediating ASK1 degradation (23). Consistently, ectopic expression of A20 suppressed TNF-induced activation of NF-κB and JNK (11, 23). In agreement with these studies, deficiency in A20 is associated with inflammatory diseases in humans such as Crohn’s disease, arthritis and autoimmune type I diabetes (9). In addition, A20 has been recognized as a tumor suppressor in lymphomas as A20 was inactivated in many lymphomas and reintroduction of A20 promoted apoptosis and growth arrest of cancer cells (4). Given that NF-κB target genes regulate cell proliferation, cytokines and survival, the induction of A20 and consequent inhibition of NF-κB by γTE likely contributes to its antiproliferative, proapoptotic, anti-inflammatory and immunomodulatory effects (12, 15, 16, 28).
We demonstrate a novel immunomodulatory role of dihydroceramide and propose links among sphingolipid metabolism, ER stress and anti-NF-κB activities. Although dihydroceramides used to be considered inert precursors of ceramides, recent evidence indicates that they are bioactive (38, 39). For instance, dihydroceramides have been shown to induce growth arrest in cancer cells (28, 40) and induce ER stress and autophagy (31, 32). In the present study, exogenous addition of C8-dihydroceramide blocked TNF-stimulated NF-κB activation, up-regulated A20 and appeared to cause ER stress in macrophages as indicated by enhanced phosphorylation of eIF2α. Interestingly, ER stress related eIF2α phosphorylation has been shown to be necessary and sufficient to activation of NF-κB (27). Consistently, both γTE and C8-dihydroceramide moderately elevated basal levels of IκBα phosphorylation. Since A20 is a NF-κB target gene (24), the basal activation of NF-κB is likely responsible for the upregulation of A20. This is further supported by the fact that ER stress inducers like thapsigargin, ionophore A23187 and tunicamycin have been shown to enhance A20 and blunt cytokine-induced NF-κB activation (41). It is noteworthy that besides γTE, some other bioactive phytochemicals may have similar anti-inflammatory mechanism. For instance, we found that curcumin inhibited NF-κB, modulated sphingolipids and up-regulated A20 in macrophages (Wang Y and Jiang Q, unpublished data).
Despite the unambiguous role of A20 in inhibition of NF-κB, γTE’s anti-NF-κB action was not exclusively dependent upon A20 because γTE diminished IκBα phosphorylation even in A20−/− cells. In this regard, we found that γTE induced another inhibitor Cezanne in the absence of A20. Cezanne is an A20 family member and is also regulated by NF-κB (5). Previously, we have shown that γTE blocked IL-13-stimulated JAK-STAT6 activation by upregulation of PAR-4 (prostate apoptosis response-4) that interacts with atypical PKC to limit STAT6 signaling in lung epithelial cells (42). Interestingly, PAR-4 is known to be up-regulated by ER stress (43). Therefore, we propose that upregulation of A20, Cezanne and PAR-4 by γTE (and possibly other phytochemicals) in response to ER stress via sphingolipid modulation represents a stress-adaptive mechanism that prepares cells for antagonizing pro-inflammatory insults. This theory should be further verified by investigation of anti-NF-κB mechanisms by other phytochemicals in various cell types.
In addition to the present study in macrophages, γTE’s modulatory effect on sphingolipid metabolism has previously been observed in other types of cells. γTE induced accumulation of dhCer and dihydrosphingosine in prostate cancer cells where sphingolipid modulation appeared to play a key role in induction of apoptosis and autophagy (28, 29). γTE has been shown to cause ER stress and JNK phosphorylation by potentiating de novo synthesis of sphingolipids (26). Our unpublished results indicate that the mechanism underlying sphingolipid modulation by γTE appears to be rooted in its direct inhibition of dihydroceramide desaturase (DEGS1), the enzyme converting dihyceramides to ceramides (Jang Y and Jiang Q, manuscript in preparation). On the other hand, prolonged incubation of γTE likely induces other changes besides increase of dhCer. In this study, γTE treatment for 16 h resulted in decrease of sphingomyelin but increase of C24:0-Cer (Supplemental Materials). These changes may intensify inhibition of NF-κB, which should be further characterized.
Supplementary Material
Acknowledgments
The authors would like to thank Amber S Jannasch for helps with LC-MS/MS analysis of sphingolipids.
This work was in part supported by grants R21CA152588 and R01AT006882 (QJ) from National Institutes of Health. This project was also partially supported by NIH grant P30CA023168.
ABBREVIATION
- γTE
gamma-tocotrienol
- TAK1
transforming growth factor β–activated kinase 1
- eIF2α
translation initiation factor 2α
- JNK
c-jun N-terminal kinase
- IKKs
IκB kinases
- NF-κB
Nuclear factor-κB
- BMDM
bone marrow-derived macrophage
- ER stress
endoplasmic reticulum stress
- dhCer
dihydroceramide
- Cer
ceramide
- SM
sphingomyelin
References
- 1.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nature immunology. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 2.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nature immunology. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 3.Wertz IE, Dixit VM. Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harbor perspectives in biology. 2010;2:a003350. doi: 10.1101/cshperspect.a003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hymowitz SG, Wertz IE. A20: from ubiquitin editing to tumour suppression. Nature reviews. Cancer. 2010;10:332–341. doi: 10.1038/nrc2775. [DOI] [PubMed] [Google Scholar]
- 5.Ruland J. Return to homeostasis: downregulation of NF-kappaB responses. Nature immunology. 2011;12:709–714. doi: 10.1038/ni.2055. [DOI] [PubMed] [Google Scholar]
- 6.Jaattela M, Mouritzen H, Elling F, Bastholm L. A20 zinc finger protein inhibits TNF and IL-1 signaling. J Immunol. 1996;156:1166–1173. [PubMed] [Google Scholar]
- 7.Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, Komuves L, French DM, Ferrando RE, Lam C, Compaan D, Yu C, Bosanac I, Hymowitz SG, Kelley RF, Dixit VM. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 8.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 9.Vereecke L, Beyaert R, van Loo G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends in immunology. 2009;30:383–391. doi: 10.1016/j.it.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 10.De A, Dainichi T, Rathinam CV, Ghosh S. The deubiquitinase activity of A20 is dispensable for NF-kappaB signaling. EMBO reports. 2014;15:775–783. doi: 10.15252/embr.201338305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skaug B, Chen J, Du F, He J, Ma A, Chen ZJ. Direct, noncatalytic mechanism of IKK inhibition by A20. Molecular cell. 2011;44:559–571. doi: 10.1016/j.molcel.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahn KS, Sethi G, Krishnan K, Aggarwal BB. Gamma-tocotrienol inhibits nuclear factor-kappaB signaling pathway through inhibition of receptor-interacting protein and TAK1 leading to suppression of antiapoptotic gene products and potentiation of apoptosis. J Biol Chem. 2007;282:809–820. doi: 10.1074/jbc.M610028200. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Q. Natural forms of vitamin E: metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free radical biology & medicine. 2014;72:76–90. doi: 10.1016/j.freeradbiomed.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Jiang Q. gamma-Tocotrienol inhibits lipopolysaccharide-induced interlukin-6 and granulocyte colony-stimulating factor by suppressing C/EBPbeta and NF-kappaB in macrophages. The Journal of nutritional biochemistry. 2013;24:1146–1152. doi: 10.1016/j.jnutbio.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qureshi AA, Reis JC, Papasian CJ, Morrison DC, Qureshi N. Tocotrienols inhibit lipopolysaccharide-induced pro-inflammatory cytokines in macrophages of female mice. Lipids in health and disease. 2010;9:143. doi: 10.1186/1476-511X-9-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahalingam D, Radhakrishnan AK, Amom Z, Ibrahim N, Nesaretnam K. Effects of supplementation with tocotrienol-rich fraction on immune response to tetanus toxoid immunization in normal healthy volunteers. European journal of clinical nutrition. 2011;65:63–69. doi: 10.1038/ejcn.2010.184. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14(Unit 14):11. doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merrill AH, Jr, Sullards MC, Allegood JC, Kelly S, Wang E. Sphingolipidomics: high-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods. 2005;36:207–224. doi: 10.1016/j.ymeth.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 20.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 22.Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327:1135–1139. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Won M, Park KA, Byun HS, Sohn KC, Kim YR, Jeon J, Hong JH, Park J, Seok JH, Kim JM, Yoon WH, Jang IS, Shen HM, Liu ZG, Hur GM. Novel anti-apoptotic mechanism of A20 through targeting ASK1 to suppress TNF-induced JNK activation. Cell death and differentiation. 2010;17:1830–1841. doi: 10.1038/cdd.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. The Journal of biological chemistry. 1992;267:17971–17976. [PubMed] [Google Scholar]
- 25.Park SK, Sanders BG, Kline K. Tocotrienols induce apoptosis in breast cancer cell lines via an endoplasmic reticulum stress-dependent increase in extrinsic death receptor signaling. Breast cancer research and treatment. 2010;124:361–375. doi: 10.1007/s10549-010-0786-2. [DOI] [PubMed] [Google Scholar]
- 26.Gopalan AYW, Jiang Q, Jang Y, Sanders BG, Kline K. Involvement of de novo ceramide synthesis in gamma-tocopherol and gamma-tocotrienol-induced apoptosis in human breast cancer cells. Mol Nutr Food Res. 2012;56:1803–1811. doi: 10.1002/mnfr.201200350. [DOI] [PubMed] [Google Scholar]
- 27.Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Molecular and cellular biology. 2004;24:10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang Q, Rao X, Kim CY, Freiser H, Zhang Q, Jiang Z, Li G. Gamma-tocotrienol induces apoptosis and autophagy in prostate cancer cells by increasing intracellular dihydrosphingosine and dihydroceramide. International journal of cancer. Journal international du cancer. 2012;130:685–693. doi: 10.1002/ijc.26054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Q, Wong J, Fyrst H, Saba JD, Ames BN. {gamma}-Tocopherol or combinations of vitamin E forms induce cell death in human prostate cancer cells by interrupting sphingolipid synthesis. Proc Natl Acad Sci U S A. 2004;101:17825–17830. doi: 10.1073/pnas.0408340102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sims K, Haynes CA, Kelly S, Allegood JC, Wang E, Momin A, Leipelt M, Reichart D, Glass CK, Sullards MC, Merrill AH., Jr Kdo2-lipid A, a TLR4-specific agonist, induces de novo sphingolipid biosynthesis in RAW264.7 macrophages, which is essential for induction of autophagy. J Biol Chem. 2010;285:38568–38579. doi: 10.1074/jbc.M110.170621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gagliostro V, Casas J, Caretti A, Abad JL, Tagliavacca L, Ghidoni R, Fabrias G, Signorelli P. Dihydroceramide delays cell cycle G1/S transition via activation of ER stress and induction of autophagy. The international journal of biochemistry & cell biology. 2012;44:2135–2143. doi: 10.1016/j.biocel.2012.08.025. [DOI] [PubMed] [Google Scholar]
- 32.Fabrias G, Munoz-Olaya J, Cingolani F, Signorelli P, Casas J, Gagliostro V, Ghidoni R. Dihydroceramide desaturase and dihydrosphingolipids: debutant players in the sphingolipid arena. Progress in lipid research. 2012;51:82–94. doi: 10.1016/j.plipres.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Traenckner EB, Wilk S, Baeuerle PA. A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of I kappa B-alpha that is still bound to NF-kappa B. The EMBO journal. 1994;13:5433–5441. doi: 10.1002/j.1460-2075.1994.tb06878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, McNally E, Pickart C, Ma A. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 35.Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S, Barrera J, Huang EJ, Hou B, Malynn BA, Reizis B, DeFranco A, Criswell LA, Nakamura MC, Ma A. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nature immunology. 2011;12:1184–1193. doi: 10.1038/ni.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lademann U, Kallunki T, Jaattela M. A20 zinc finger protein inhibits TNF-induced apoptosis and stress response early in the signaling cascades and independently of binding to TRAF2 or 14-3-3 proteins. Cell death and differentiation. 2001;8:265–272. doi: 10.1038/sj.cdd.4400805. [DOI] [PubMed] [Google Scholar]
- 37.Papa S, Zazzeroni F, Pham CG, Bubici C, Franzoso G. Linking JNK signaling to NF-kappaB: a key to survival. Journal of cell science. 2004;117:5197–5208. doi: 10.1242/jcs.01483. [DOI] [PubMed] [Google Scholar]
- 38.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nature reviews. Molecular cell biology. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez-Cuenca S, Barbarroja N, Vidal-Puig A. Dihydroceramide desaturase 1, the gatekeeper of ceramide induced lipotoxicity. Biochimica et biophysica acta. 2014 doi: 10.1016/j.bbalip.2014.09.021. [DOI] [PubMed] [Google Scholar]
- 40.Kraveka JM, Li L, Szulc ZM, Bielawski J, Ogretmen B, Hannun YA, Obeid LM, Bielawska A. Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. The Journal of biological chemistry. 2007;282:16718–16728. doi: 10.1074/jbc.M700647200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayakawa K, Hiramatsu N, Okamura M, Yamazaki H, Nakajima S, Yao J, Paton AW, Paton JC, Kitamura M. Acquisition of anergy to proinflammatory cytokines in nonimmune cells through endoplasmic reticulum stress response: a mechanism for subsidence of inflammation. J Immunol. 2009;182:1182–1191. doi: 10.4049/jimmunol.182.2.1182. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Moreland M, Wagner JG, Ames BN, Illek B, Peden DB, Jiang Q. Vitamin E forms inhibit IL-13/STAT6-induced eotaxin-3 secretion by up-regulation of PAR4, an endogenous inhibitor of atypical PKC in human lung epithelial cells. The Journal of nutritional biochemistry. 2012;23:602–608. doi: 10.1016/j.jnutbio.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell. 2009;138:377–388. doi: 10.1016/j.cell.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.