

Abstract
The gas-phase oxidation of doubly protonated peptides is demonstrated here using ion/ion reactions with a suite of reagents derived from persulfate. Intact persulfate anion (HS2O8−), peroxymonosulfate anion (HSO5−), and sulfate radical anions (SO4−•) are all either observed directly upon negative nanoelectrospray ionization (nESI) or easily obtained via beam-type collisional activation of persulfate into the mass spectrometer. Ion/ion reactions between each of these reagents and doubly protonated peptides results in formation of a long-lived complex. Collisional activation of the complex containing a peroxymonosulfate anion results in oxygen transfer from the reagent to the peptide to generate the [M+H+O]+ species. Activation of the complex containing intact persulfate anion either results in oxygen transfer to generate the [M+H+O]+ species or abstraction of two hydrogen atoms and a proton to generate the [M-H]+ species. Activation of the complex containing sulfate radical anion results in abstraction of one hydrogen atom and a proton to form the peptide radical cation, [M]+•. This suite of reagents allows for the facile transformation of the multiply protonated peptides obtained via nESI into a variety of oxidized species capable of providing complementary information about the sequence and structure of the peptide.
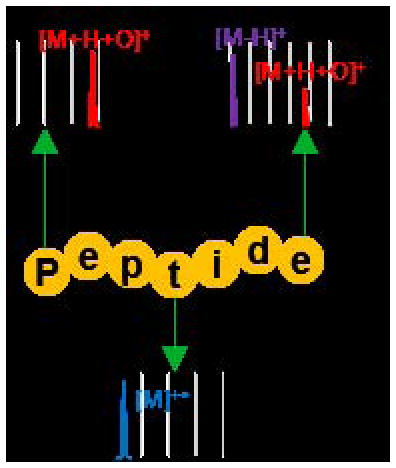
INTRODUCTION
Tandem mass spectrometry is a powerful approach for obtaining primary structural information about a bioanalyte of interest. The information obtained from a tandem mass spectrometry experiment is highly dependent on the nature of the gas-phase ions generated from the molecules of interest.1 Different ion types (e.g., protonated molecule, deprotonated molecule, metallated molecule, radical cation, radical anion, etc.) undergo different fragmentation pathways upon activation, which can yield complementary information. It is therefore useful to be able to generate different types of ions and to manipulate ion-type within the mass spectrometer. The advent of electrospray ionization (ESI) has made the generation of multiply-charged analytes straightforward.2,3 These ions are typically protonated in the positive polarity and deprotonated in the negative polarity. Gas-phase ion/ion reactions have been successful in transforming ESI-generated ions to an ion-type different from the type initially generated within a mass spectrometer.4
The reduction of charge via proton transfer,5,6 electron transfer both to and from multiply-charged analytes,7,8 and the addition or removal of metal ions9,10,11 are examples of the manipulation of ion-type in the gas-phase via ion/ion reactions. Site-selective covalent modification of peptides and proteins has also been demonstrated via ion/ion reactions. For example, N-hydroxysuccinimide (NHS) esters have been used to cross-link12,13 and covalently label14,15,16 various nucleophiles in peptide ions, 4-formyl-1,3-benzenedisulfonic acid (FBDSA) has been used to tag peptide ions via Schiff base chemistry,17,18,19 and N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide (CMC) has been used to selectively react with carboxylic acids20 in various analytes. Ion/ion reactions occur on the typical reaction time-scale of ~100 ms and can either proceed via the long-range transfer of small charged particles, e.g., protons or electrons, or through the formation of a long-lived complex.4 This gas-phase approach, particularly when implemented in a tandem mass spectrometer that enables the mass-selection of both the reagent and analyte ions, allows greater control of the extent of modification relative to solution-phase derivatization, which can complicate the mass spectrum.
Recently, the oxidation of peptides via ion/ion reactions has been described.21 Peptides containing methionine and tryptophan are selectively oxidized to [M+H+O]+ cations upon ion/ion reactions with periodate anions. The oxidation of the methionine side-chain to the sulfoxide derivative induces a signature loss of 64 Da corresponding to the ejection of methanesulfenic acid (CH3SOH).22,23,24,25,26,27,28,29,30 This signature loss can be used to determine the presence and location of a methionine residue. Additionally, strategies to oxidize protonated peptides to their radical analogs, viz., [M]+•, via ion/ion reactions have recently been described.31 The gas-phase chemistry of radical peptide ions is an area of growing interest as these species undergo different fragmentation pathways than their protonated analogs upon activation.32,33 To this end, several strategies have been developed to generate radical cations in the gas-phase to be analyzed via mass spectrometry.32,33 These strategies include collision-induced dissociation (CID) of nitrosopeptides,34,35 CID of ternary metal-ligand-peptide complexes,36,37,38,39,40,41,42 photolysis of peptides with iodinated tyrosine residues43 or iodinated electrostatic complexes,44 and free radical-initiated reactions.45,46
Here, we describe ion/ion reactions between peptide cations and three reagent anions derived from persulfate to form three oxidized species; [M+H+O]+, [M]+•, and [M-H]+. The three reagents are intact persulfate anions (HS2O8−, Supplemental Figure S-1(a)), peroxymonosulfate anions (HSO5−, Supplemental Figure S-1(b)), and sulfate radical anions (SO4−•, Supplemental Figure S-1(c)). Reactions between doubly protonated peptides and each of the three reagents results in formation of a long-lived complex. Activation of ion/ion complexes with intact persulfate anions yields oxygen addition, viz., [M+H+O]+, and hydrogen deficient species, viz., [M-H]+. Activation of analogous complexes with peroxymonosulfate anions yield the oxygen addition product, viz., [M+H+O]+. The last reagent derived from persulfate is sulfate radical anion, produced via homolytic cleavage of the persulfate peroxy-bond, which is observed upon negative nESI of an aqueous solution of sodium persulfate (Supplemental Figure S-1(d)). Activation of the complex produced via ion/ion reactions with peptide dications and sulfate radical anions yields the molecular radical peptide cation, [M]+•. This suite of reagents derived from persulfate readily and efficiently converts protonated peptides into various oxidized forms that, upon activation, may yield additional information about peptide primary structure.
EXPERIMENTAL SECTION
Materials
Methanol and glacial acetic acid were purchased from Mallinckrodt (Phillipsburg, NJ, USA). Sodium persulfate, angiotensin III, and melittin were purchased from Sigma Aldrich (St. Louis, MO, USA). Substance P and bradykinin were synthesized by Bachem (King of Prussia, PA, USA). ARAAAKA and ARAMAKA were synthesized by NeoBioLab (Cambridge, MA, USA). KGAILAGAILR and GAILAGAILR were synthesized by SynPep (Dublin, CA, USA) and GAGGMGAGGRL was synthesized by Pepnome Ltd. (Shenzhen, China). All peptide stock solutions for positive nanoelectrospray were prepared in a 49.5/49.5/1 (vol/vol/vol) solution of methanol/water/acetic acid at an initial concentration of ~1 mg/mL and diluted 100-fold prior to use. The persulfate solution was prepared in 18 MΩ purified water using a Nanopure ultrapure water system from Barnstead/Thermolyne Corp (Dubuque, IA) at a concentration of ~1 mg/mL and diluted 10-fold prior to use.
Mass Spectrometry
All experiments were performed on a QTRAP 4000 hybrid triple quadrupole/linear ion trap mass spectrometer (AB Sciex, Concord, ON, Canada), previously modified for ion/ion reactions.47 Multiply protonated peptides were isolated in the Q1-mass filter and injected into the q2 reaction cell followed by singly charged reagent anions via alternately pulsed nano-electrospray (nESI).48 Both the intact persulfate anion (HS2O8−) and sulfate radical anion species (SO4−•) were present upon negative nESI of aqueous persulfate and were isolated in Q1 prior to injection into the q2 reaction cell. Peroxymonosulfate anion (HSO5−), however, is a fragment derived from intact persulfate and was obtained through beam-type CID into the instrument via energetic interface conditions prior to isolation and injection into q2. The peptide cations and reagent anions were allowed to react for a mutual storage reaction time of 20 ms – 1000 ms. The ion/ion reaction products were then transferred to Q3, where the complex was subjected to further characterization via MSn and mass analysis using mass-selective axial ejection (MSAE).49
Calculations
Density function theory calculations have been carried out using the Gaussian 09 package.50 Structural optimizations and energy calculations were performed with unrestricted B3LYP at the 6-31G(d) basis set for HSO4−, SO4−•, CH3COOH, and CH3COO•. The bond dissociation energy (BDE) for H-SO4− was calculated by the isodesmic reaction method51 using CH3COO-H as the reference molecule, the BDE of which was previously determined experimentally.52 In detail, BDE(H-SO4−) was calculated using the following equation.
The value was then compared with amino acid BDEs reported by the Julian group calculated via the same method.53
RESULTS AND DISCUSSION
Ion/Ion Reactions with the Suite of Reagents Derived from Persulfate
The nESI spectrum of an aqueous solution of sodium persulfate is shown in Supplemental Figure S-1(d). The base peak is the sodiated persulfate anion, though protonated persulfate is also present in a high enough abundance for ion/ion reactions. While the sulfate radical anion is observed at the same mass-to-charge ratio as the persulfate dianion, the majority of this peak is the singly-charged radical species based on the isotopic distribution and lack of anionic products observed during charge-inversion experiments (e.g., the reaction of singly protonated peptides with dianions resulting in final negative charge overall). The doubly charged species cannot be separated from the sulfate radical anion species of interest on the basis of mass-to-charge ratio alone. However, the only deleterious impact from the presence of a small persulfate dianion population in the sulfate radical anion population in reactions with doubly charged peptide cations is a slight decrease in overall peptide ion signal due to neutralization of the peptide by the dianion. While some peroxymonosulfate anion is present in the original nESI spectrum acquired using a nozzle-skimmer voltage difference of 25 V (Supplemental Figure S-1(d)), the abundance is significantly increased upon use of a nozzle-skimmer voltage difference of 75 V (Supplemental Figure S-1(e)). A separate MS/MS experiment indicated that CID of the singly charged persulfate anion results predominantly in formation of the peroxymonosulfate anion (data not shown).
Ion/ion reactions between doubly protonated peptides and each of the reagent anions derived from negative nESI of persulfate results in the formation of a long-lived complex. Activation of this complex can either result in proton transfer from the peptide to the reagent to generate the charge-reduced [M+H]+ species or undergo one of the oxidative pathways outlined in Scheme 1. The first two pathways (red arrows in Scheme 1) result in oxygen transfer to the peptide to form the [M+H+O]+ species. Pathway A occurs via H2SO4 loss from either CID of complexes with peroxymonosulfate anions or MS3 of the SO3 loss from complexes with persulfate anions. Pathway B occurs via H2SO4 loss followed by SO3 loss from complexes with persulfate anions. While Pathway B also generates an oxidized species of the form [M+H+O]+, the structure of the final product is not necessarily the same as the one obtained via Pathway A, as they proceed through different intermediates. Loss of two sulfuric acid moieties from the ion/ion complex with persulfate also occurs and results in the hydrogen-deficient [M-H]+ species via Pathway C (blue arrow in Scheme 1). The formation of the peptide radical cation, viz., [M]+•, takes place via Pathway D (orange arrow in Scheme 1), which involves doubly protonated peptide in reaction with the sulfate radical anion and subsequent CID of the complex to lose H2SO4. To demonstrate each of these pathways, doubly protonated substance P (RPKPQQFFGLM) was reacted with each of the reagents derived from persulfate (HS2O8−, HSO5−, and SO4−•) and the resulting ion/ion complexes were subjected to CID (Figure 1). Activation of the ion/ion complex between substance P and peroxymonosulfate anion proceeds exclusively via Pathway A to yield the [M+H+O]+ species (Figure 1(a)). Activation of the ion/ion complex between substance P and persulfate anion yields loss of SO3 and loss of H2SO4 as well as formation of the oxidized [M+H+O]+ and [M-H]+ species (Figure 1(b)). The [M+H+O]+ species is produced via a combination of Pathways A and B whereas the [M-H]+ species is produced via Pathway C. Pathway D is illustrated in Figure 1(c) via CID of the complex produced upon ion/ion reaction of doubly protonated substance P with sulfate radical anion, which undergoes loss of H2SO4 to yield the [M]+• cation. Each of the reagents derived from persulfate are discussed in detail below.
Scheme 1.

Summary of oxidative pathways available to a doubly protonated peptide upon ion/ion reaction with each of the reagents derived from persulfate and subsequent MSn (CID indicated by Δ) of the complexes. Black arrows indicate an ion/ion reaction, the orange arrow indicates the pathway to create a radical peptide cation, the blue arrow indicates the pathway to form the hydrogen-deficient [M-H]+ species, red arrows indicate oxygen transfer pathways to form the [M+H+O]+ species with the dashed arrow indicating a pathway that results in a structure different than that obtained via solid-arrow pathways, and the purple arrow indicates the generation of a species capable of proceeding via either the oxygen transfer or the hydrogen abstraction pathway.
Figure 1.

Product ion spectra derived from CID of complexes produced via ion/ion reactions between doubly protonated substance P (RPKPQQFFGLM) and (a) HSO5−, (b), HS2O8−, and (c) SO4−•. The lightning bolt (
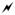 ) indicates species subjected to CID.
) indicates species subjected to CID.
Ion/Ion Reactions with Peroxymonosulfate (HSO5−) Anions to Form [M+H+O]+ Species
Ion/ion reactions with peroxymonosulfate anions result in oxidation of a peptide via oxygen transfer from the reagent to the peptide. While the selective oxidation of methionine and tryptophan side-chains via ion/ion reactions with periodate has recently been described,21 peroxymonosulfate appears to be a stronger oxidizing reagent as peptides both containing and lacking methionine and tryptophan residues are oxidized. This is demonstrated in Figure 2 with ARAAAKA, a peptide that does not contain methionine or tryptophan residues and, as such, is not susceptible to oxidation by periodate. The ion/ion reaction between doubly protonated ARAAAKA and peroxymonosulfate results in both proton transfer from the peptide to the anionic reagent resulting in the charge-reduced [M+H]+ species and complex formation to produce the [M+2H+HSO5−]+ species (Figure 2(a)). Activation of the complex either results in generation of the charge-reduced species via proton transfer or formation of the oxidized [M+H+O]+ species via Pathway A (Figure 2(b)). Note that the extent of oxidation in this case is much lower than for substance P (Figure 1(a)), which has a methionine residue at the C-terminus. A proposed mechanism for peroxymonosulfate oxidation is outlined in Scheme 2, which is analogous to the accepted mechanism for oxygen transfer from alkyl hydrogen peroxides.54 The peroxymonosulfate anion first abstracts a proton from the peptide. One of the nitrogen atoms on the arginine side-chain then initiates a nucleophilic attack on the distal peroxy oxygen atom. Rearrangement to lose neutral H2SO4 occurs and yields a charge-separated [M+H+O]+ species. Further rearrangement to eject NH2OH (33 Da) can proceed via abstraction of a proton by the negatively charged oxygen atom to yield the final species shown in Scheme 2. While arginine was chosen to illustrate the proposed mechanism in Scheme 2, other sites on the peptide are capable of being oxidized via similar mechanisms as well (Supplemental Figure S-2).
Figure 2.
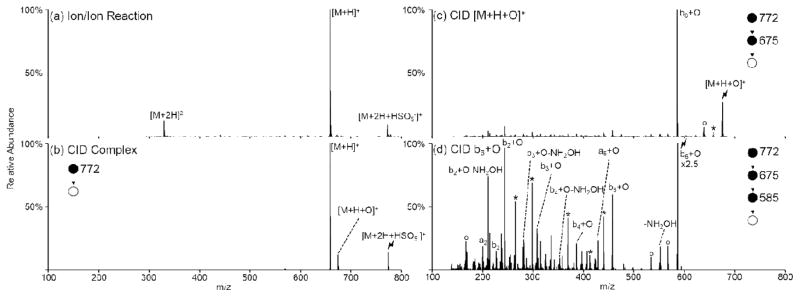
Oxidation of ARAAAKA via ion/ion reaction with peroxymonosulfate. (a) Ion/ion reaction between doubly protonated ARAAAKA and peroxymonosulfate anion, (b) activation of the ion/ion complex, (c) activation of the [M+H+O]+ oxidized species, and (d) further activation of the oxidized b6 ion to further provide sequence information. Asterisks (*) indicate ammonia losses, degree signs (°) indicate water losses, and the lightning bolt (
) indicates species subjected to CID.
Scheme 2.
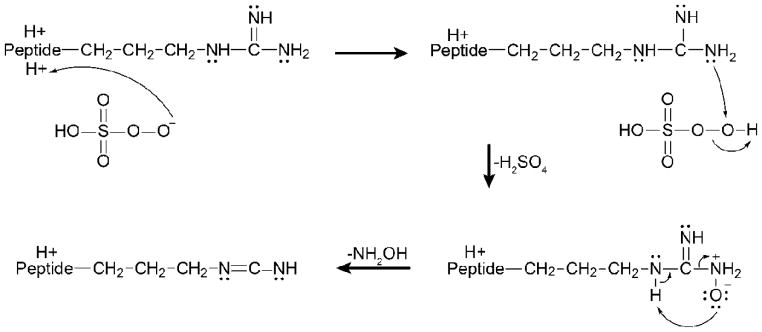
Mechanism of [M+H+O]+ formation via ion/ion reaction between peroxymonosulfate anion and an arginine-containing peptide and NH2OH loss from oxidized arginine side-chain.
Activation of the oxidized species results in formation of an abundant b6+O ion (Figure 2(c)), which is produced via cleavage C-terminal to the lysine residue. This spectrum is similar to CID of the unmodified peptide in which the b6 ion is the dominant fragment observed (Supplemental Figure S-3). Further activation of the b6+O ion was performed to gather more information about the site of oxidation, as shown in Figure 2(d). There are 33 Da losses present throughout the product spectrum likely corresponding to losses of NH2OH from oxidized lysine or arginine side-chains (Scheme 2). Additionally, while a series of oxidized b-ions are observed, the presence of both the oxidized and the non-oxidized b2-ion indicate the likelihood of multiple sites within the peptide being oxidized upon ion/ion reaction with peroxymonosulfate anions.
While activation of the ion/ion complex between doubly protonated ARAAAKA and peroxymonosulfate anion resulted in mainly proton transfer to produce the charge-reduced species with oxidation of the peptide observed as a minor pathway (Figure 2(b)), activation of the analogous complex with ARAMAKA exclusively results in oxygen atom transfer (Figure 3(a)). Additionally, activation of the oxidized ARAMAKA species, [M+H+O]+, yields dominant 64 Da losses corresponding to ejection of CH3SOH from the oxidized methionine side-chain (Figure 3(b)).22–30 While the presence of a methionine or tryptophan residue is not necessary for oxidation to occur, peptides containing these residues demonstrate increased complex formation and oxidation efficiencies and undergo preferential oxidation of the methionine or tryptophan side-chain. The peroxymonosulfate anion oxidation chemistry is also demonstrated with bradykinin, a widely studied 9-residue peptide (RPPGFSPFR). The efficiency of oxidation with bradykinin is ~10%, which is similar to that observed with ARAAAKA (compare Figure 3(c) with Figure 2(b)). Additionally, similarly to ARAAAKA, the presence of both oxidized and non-oxidized y8 ions upon CID of the [M+H+O]+ species indicates the presence of multiple oxidation sites in the peptide (Figure 3(d)). Spectra illustrating the reactions between HSO5− and the [M+2H]2+ species of ARAMAKA and bradykinin are shown in Supplemental Figure S-4.
Figure 3.
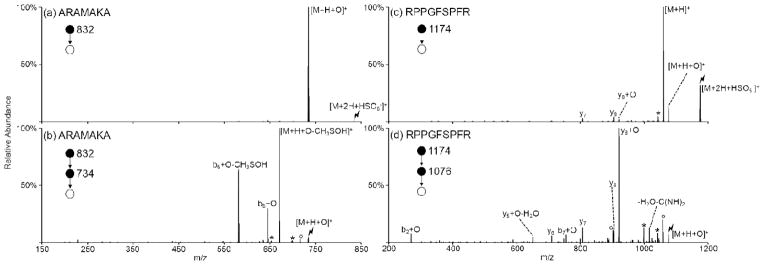
Activation of the ion/ion complex produced via the ion/ion reaction between peroxymonosulfate anions and doubly protonated (a) ARAMAKA and (c) bradykinin (RPPGFSPFR). Further CID of the [M+H+O]+ species produced for (b) ARAMAKA and (d) bradykinin. Asterisks (*) indicate ammonia losses, degree signs (°) indicate water losses, and the lightning bolt (
) indicates species subjected to CID.
Ion/Ion Reactions with Intact Persulfate (HS2O8−) Anions to Form [M+H+O]+ and [M-H]+ Species
Intact persulfate is capable of oxidation via oxygen atom transfer to form [M+H+O]+ as well as hydrogen abstraction to form the unique [M-H]+ species upon ion/ion reactions with doubly protonated peptides. Oxygen atom transfer is capable of proceeding via SO3 loss to form [M+2H+HSO5−]+, which is identical to the complex obtained via ion/ion reactions directly with peroxymonosulfate anions, followed by H2SO4 loss (Pathway A). Oxygen atom transfer may also proceed via initial loss of H2SO4 from the complex to form [M+H+SO4]+ followed by SO3 loss (Pathway B). These processes can result in oxidation at different sites in the peptides, as shown below. Additionally, loss of two sulfuric acid moieties from the complex can occur to form a species of the nominal form [M-H]+ (Pathway C).
These three oxidation pathways are demonstrated in Figure 4 with the peptide ARAMAKA. The ion/ion reaction between doubly protonated ARAMAKA and intact persulfate anions is shown in Figure 4(a). Both proton transfer to form the charge-reduced [M+H]+ species and complex formation occur upon ion/ion reaction. Some fragmentation of the complexes due to energetic transfer conditions from the collision quadrupole to Q3 is also observed, as illustrated by the losses of SO3, H2SO4, and H2SO5 as well as the formation of the [M+H+O]+ and [M-H]+ species. Collisional activation of the ion/ion complex, [M+2H+HS2O8−]+, yields product ions from losses of SO3, H2SO4, and H2SO5, each of which is discussed below. Additionally, formation of both oxidized species is dominant, though it is unclear what percentage of these species is consecutive from the SO3 and/or H2SO4 losses versus directly from the complex itself. Activation of the product ion from SO3 loss exclusively yields the [M+H+O]+ species via Pathway A for ARAMAKA (Figure 4(c)) though proton transfer may also be observed for some peptides. Activation of the H2SO4 loss yields both an SO3 loss to produce the [M+H+O]+ species and another H2SO4 loss to produce the [M-H]+ species (Figure 4(d)). Losses of 30 Da neutral species, corresponding to CH2O loss, from the complex, [M+H+O]+, and b6+O species are also observed. This loss is likely to arise from oxidation of the terminal carbon on the methionine side-chain via a mechanism proposed by Froelich and Reid.55 Activation of the H2SO5 loss exclusively undergoes ejection of neutral SO3 to form the charge-reduced [M+H]+ species (Supplemental Figure S-5).
Figure 4.
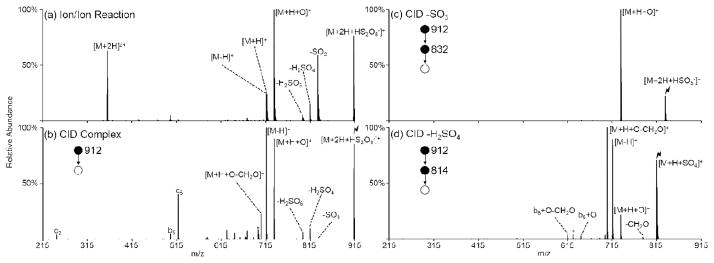
Reaction between doubly protonated ARAMAKA and intact persulfate. (a) Ion/ion reaction between [ARAMAKA+2H]2+ and HS2O8−, (b) activation of the ion/ion complex, (c) activation of the SO3 loss from the complex, and (d) activation of the H2SO4 loss from the complex. Asterisks (*) indicate ammonia losses, degree signs (°) indicate water losses, and the lightning bolt (
) indicates species subjected to CID.
While a nominal [M+H+O]+ species is observed from both the SO3 and the H2SO4 losses, the oxidation does not necessarily occur at the same site(s), as demonstrated in Supplemental Figure S-6 in which the [M+H+O]+ products from both pathways with ARAMAKA are subjected to CID. Activation of the [M+H+O]+ species produced via initial SO3 loss yields dominant CH3SOH losses from the oxidized parent and b6 ions indicating exclusive oxidation of the sulfur atom in the methionine side-chain (Figure S-6(a)). This spectrum is identical to that obtained from activation of the [M+H+O]+ species produced via activation of the ion/ion complex between doubly protonated ARAMAKA and peroxymonosulfate anions (see Figure 3(b)). While activation of the [M+H+O]+ species produced via initial H2SO4 loss (Pathway B, Figure S-6(b)) produces many of the same peaks observed in the initial SO3 loss (Pathway A, Figure S-6(a)) spectrum, there are some key differences. The 64 Da loss corresponding to CH3SOH loss from the sulfoxide derivative of the methionine side-chain is observed at a significantly lesser relative abundance for the initial H2SO4 loss pathway. This indicates that, while oxygen transfer to the sulfur atom is possible via this mechanism, there are likely other sites oxidized via this pathway as well. Furthermore, CH2O loss is observed from initial H2SO4 loss (Figure S-6(b)) but not initial SO3 loss (Figure S-6(a)). As discussed above, this loss indicates that the [M+H+O]+ ion produced via initial H2SO4 loss contains some population oxidized at the terminal carbon on the methionine side-chain. Lastly, small signals corresponding to non-oxidized y4 and y5 ions in Figure S-6(b) but not in Figure S-6(a) suggest that some of the oxidation occurs on a site in the peptide other than the methionine residue upon oxidation via Pathway B.
Scheme 3 depicts the proposed mechanisms for oxidation of methionine via ion/ion reactions with intact persulfate anions. Scheme 3(a) illustrates oxidation of the sulfur atom while Scheme 3(b) depicts oxidation of the terminal carbon atom in the methionine side-chain via initial H2SO4 loss from an ion/ion complex with intact persulfate. Oxidation of the sulfur atom proceeds via initial proton transfer from the peptide to the reagent followed by nucleophilic attack by the sulfur atom on one of the oxygen atoms in the peroxy bond, resulting in loss of H2SO4. Further rearrangement occurs to eject neutral SO3 and form a double bond between the sulfur and oxygen atoms (Scheme 3(a)). Oxidation of the terminal carbon atom also undergoes proton transfer from the peptide to the reagent, but is instead followed by abstraction of a hydrogen atom to cleave the peroxy bond, eject neutral H2SO4, and form a bond between the β-peroxy oxygen atom and the terminal carbon atom. The oxidizing oxygen then abstracts a proton, initiating SO3 loss (Scheme 3(b)). This species can then lose CH2O via a mechanism proposed by Froelich and Reid to yield a homocysteine functionality.55 Both mechanisms illustrated in Scheme 3(a) and (b) proceed via initial H2SO4 loss (Pathway B). However, activation of a persulfate-containing ion/ion complex can also generate the [M+H+O]+ species via initial SO3 loss (Pathway A) as depicted in Scheme 3(c). Initial SO3 loss results in a species identical to the complex observed directly from ion/ion reactions between doubly protonated peptides and peroxymonosulfate anions. After SO3 loss, proton transfer from the peptide to the reagent anion occurs. This is followed by attack by the sulfur atom on the distal oxygen atom with nucleophilic displacement of the peroxy oxygen and simultaneous hydrogen migration as described by Bach, et al., for general reactions between nucleophiles and peroxides.54 The specific reaction between HSO5− and thioesters has also been previously investigated.56
Scheme 3.

Mechanism of reaction for methionine residues and HS2O8− to yield oxidation of (a) the sulfur atom, and (b) the carbon atom, via initial H2SO4 loss and (c) oxidation of the sulfur atom via initial SO3 loss.
The [M-H]+ species, which was observed to be generated as a major product only with the persulfate anion, can nominally correspond to the loss of two sulfuric acid moieties from the complex, viz., [M+2H+HS2O8−-2H2SO4]+, or water loss from the oxygen transfer species, viz., [M+H+O-H2O]+. To investigate which structure should be assigned to this unique species, the CID spectrum of the [M-H]+ species generated directly from the complex was compared to that of the [M+H+O-H2O]+ species (Supplemental Figure S-7). The b6(-2) ion is the base peak in both spectra, similar to the spectrum corresponding to CID of the unmodified peptide in which the b6 is the base peak (data not shown). Parentheses indicate that the mass of the reported ion is 2 Da less than expected for an unmodified peptide. The 2 Da mass difference can either come from loss of water from an oxygen transfer species or abstraction of two hydrogen atoms. Various b and b(-2) ions are observed throughout both spectra. However, there are 64 Da losses (CH3SOH) in the [M+H+O-H2O]+ product spectrum (Figure S-7(b)) that are not observed in the [M-H]+ product spectrum (Figure S-7(a)). These CH3SOH losses indicate that there is some population of peptides with oxidized sulfur atoms on the methionine side-chain that have lost water from elsewhere in the peptide. The absence of these losses in the product spectrum of the [M-H]+ ion indicate that no sulfur oxidation is present. Furthermore, small signals due to c2, c3, and c5 ions are observed upon CID of [M-H]+ (Figure S-7(a)) but not [M+H+O-H2O]+ and a small y5 ion is observed upon CID for [M+H+O-H2O]+ (Figure S-7(b)) but not [M-H]+. These differences suggest that very little, if any, of the [M-H]+ ion population results from water loss from the oxidized species and is likely produced by hydrogen abstraction via loss of two neutral H2SO4 moieties. It is also worth noting that the abundance of water loss from CID of the [M+H+O]+ species is low (see Supplemental Figure S-6) compared to the abundance of the [M-H]+ observed from activation of the complex (Figure 4(b)), further supporting the hydrogen abstraction pathway. Oxidation of the non-arginine containing peptide KAKAKAA via ion/ion reaction with intact persulfate anion is shown in Supplemental Figure S-2.
We have found that the formation of the [M-H]+ ion from the [M+2H]2+ ion via reaction with the persulfate anion requires the presence of an unprotonated basic site. For instance, doubly protonated GAILAGAILR forms an ion/ion complex upon reaction with persulfate anion, but activation of this complex leads predominantly to [M+H]+ (i.e., proton transfer from the peptide to the reagent anion) and, to a lesser degree, SO3 loss (Supplemental Figure S-8(a)). Activation of the reaction complex between persulfate and doubly protonated KGAILAGAILR, on the other hand, yields the [M-H]+ ion as the base peak along with less abundant formation of the [M+H+O]+ ion as well as losses of SO3, H2SO4, and H2SO5 (Figure S-8(b)). Furthermore, triply protonated KGAILAGAILR primarily leads to proton transfer and a minor loss of SO3 upon activation of the ion/ion complex with persulfate anion (Figure S-8(c)). We also note that species containing easily oxidized residues undergo oxygen transfer in the absence of an unprotonated basic residue to form the [M+H+O]+ species, as illustrated with the doubly protonated peptide GAGGMGAGGRL in Supplemental Figure S-9, but not the [M-H]+ ion. These trends indicate that the chemistry proceeding via the initial H2SO4 loss pathway requires an unprotonated basic site to occur. Potential mechanisms for the generation of the [M-H]+ species for each of the three unprotonated basic amino acids are proposed in Scheme 4. The peroxy bond in persulfate is labile (BDE reported to be 33.5 kcal/mol)57 and is capable of homolytic cleavage to form two radical species. The proposed reaction proceeds via formation of a 6-membered ring upon which radical rearrangement occurs to abstract two hydrogen atoms from adjacent atoms. Additionally, the anionic site on the reagent takes a proton from another basic site along the peptide. This results in ejection of two sulfuric acid moieties and the creation of a double bond between adjacent atoms in the unprotonated amino acid side-chain, or in the case of histidine, between the α– and β–carbons. The hydrogen atoms abstracted in Scheme 4 were chosen because they had the lowest local bond dissociation energies, as reported by Benjamin and Julian.53 While the mechanism proposed in Scheme 4 is one possibility for how the reaction between basic sites and intact persulfate proceeds, there are other possible mechanisms, one of which is presented in Supplemental Scheme S-1.
Scheme 4.

Mechanisms to form [M-H]+ species upon reaction between intact persulfate anion and (a) lysine, (b) arginine, and (c) histidine.
Ion/Ion Reactions with Sulfate Radical Anion (SO4−•) to Form Peptide Radical Cations ([M]+•)
The creation of gas-phase peptide radical cations is an area of growing interest as the activation of these radical species yields information complementary to that obtained via CID of the protonated species. Strategies to generate radical cations and examination of the fragmentation pathways they undergo have recently been reviewed.58,33,59 Several strategies for the creation of radical cations via gas-phase ion/ion reactions have also recently been described.31 Here, we demonstrate the generation of peptide radical cations in the gas-phase via ion/ion reactions with the sulfate radical anion. The reaction proceeds via a long-lived complex of the form [M+2H+SO4−•]+, which undergoes facile loss of sulfuric acid to efficiently yield the peptide radical cation of interest, viz., [M]+•.
The generation of the molecular radical cation from doubly protonated angiotensin III (RVYIHPF) via ion/ion reactions with sulfate radical anion is shown in Figure 5. Activation of the ion/ion complex, [M+2H+SO4−•]+, yields a dominant sulfuric acid loss to produce the peptide radical cation (Figure 5(a)). Further activation of the [M]+• yields neutral losses as well as cleavages along the peptide backbone to form the a3 through a5-ions and the z4•-ion (Figure 5(b)). The dominant neutral losses observed are CO2 from the C-terminus and p-quinomethide (106 Da) from the tyrosine side-chain. This spectrum agrees well with the SID spectrum of the angiotensin III radical cation produced by Yang et al. via in-source fragmentation of [CoIII(salen)M]+ and [CuII(terpy)M]+ complexes.60 Activation of the protonated peptide is shown in Figure 5(c) for comparison and is dominated by water and ammonia losses, although a-, b-, and y-type ions are also observed.
Figure 5.

Product ion spectra derived from CID of (a) the ion/ion complex between angiotensin III (RVYIHPF) and sulfate radical anion, (b) the radical peptide cation of angiotensin III, and (c) protonated angiotensin III. Asterisks (*) indicate ammonia losses, degree signs (°) indicate water losses, and the lightning bolt (
) indicates species subjected to CID. Loss of 106 Da indicates loss of p-quinomethide (C7H6O) from the tyrosine side-chain.
Calculations were performed to determine the bond dissociation energy (BDE) of H-SO4− and compared to the values previously reported by the Julian group for abstraction of each of the hydrogen atoms on all of the amino acids.53 The calculated BDE for H-SO4− is 454 kJ/mol with an expected error of ~10 kJ/mol.53 This value is higher than most of the values reported for abstracting H-atoms from amino acids with the exception of aromatic carbon radical centers which, in general, are the least stable radical centers. Notably, the H-SO4− BDE is higher than those reported for abstraction of hydrogen atoms attached to alpha- (325.5 – 372.3 kJ/mol) and beta- (348.2 – 428.5 kJ/mol) carbons, indicating that the sulfate radical anion can theoretically abstract a hydrogen atom from any peptide upon reaction.53 Ion/ion reactions with sulfate radical anions are a robust and efficient method of generating peptide radical cations from their protonated dicationic counterparts in the gas-phase.
Peptide cations with multiple radical sites can be generated via sequential ion/ion reactions between multiply protonated peptides and sulfate radical anions. Multiply protonated peptides of the form [M+nH]n+ can, in principle, undergo up to (n-1) additions of sulfate radical anion before neutralization. The [M+5H]5+ species of melittin, a large 26-residue peptide, was subjected to ion/ion reactions with sulfate radical anions. Addition of up to three sulfate radical anions was observed, as shown in Figure 6(a). (Addition of a fourth sulfate radical anion is possible prior to neutralization but is not observed as it is beyond the upper mass-to-charge limit of the instrument.) Activation of the triply adducted species yields sequential losses of sulfuric acid to produce a species of the nominal form [M-H]2+••• (Figure 6(b)). Further MSn on this peak is dominated by neutral losses with smaller peaks corresponding to both charge-directed (b-, y-type ions) and radical-directed (z- and c-type ions) backbone cleavages (Figure 6(c)). This example also demonstrates the facile ability to generate radical cations from relatively large polypeptide systems via ion/ion reactions with the sulfate radical anion.
Figure 6.

Generation of multiple radical sites in melittin. (a) Ion/ion reaction between sulfate radical anion and melittin [M+5H]5+. (b) CID of the triply adducted species. (c) CID of the third H2SO4 loss from the triply adducted species, viz., [M-H]2+•••. Asterisks (*) indicate ammonia losses, degree signs (°) indicate water losses, and the lightning bolt (
) indicates species subjected to CID. Green circles indicate H2SO4 loss.
CONCLUSIONS
The generation of various oxidized species, including [M-H]+, [M+H+O]+, and [M]+• peptide cations, from multiply-protonated peptides has been demonstrated using ion/ion reactions with a novel suite of reagents derived from persulfate. Peroxymonosulfate anion, HSO5−, results in oxygen transfer from the reagent to the peptide to yield the [M+H+O]+ cation upon loss of SO3. The intact persulfate anion, HS2O8−, can transfer oxygen from the reagent to the peptide via two distinct pathways to yield the [M+H+O]+ cation or it can abstract two hydrogen atoms from unprotonated basic side-chains to yield the [M-H]+ cation upon ejection of two neutral sulfuric acid molecules. The latter reaction represents a process without precedent in gas-phase ion/ion chemistry. The sulfate radical anion, SO4−•, abstracts a hydrogen atom from the peptide to generate the peptide radical cation, [M]+•, upon loss of one neutral sulfuric acid molecule. These three reagents are either observed directly upon negative nESI of an aqueous solution of persulfate under gentle interface conditions or produced via CID of the reagent under harsher interface conditions thereby providing multiple reagent options within one sample. These results demonstrate that persulfate can provide a unique array of reagent anions for the transformation of multiply protonated peptide ions to other ion-types.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health under Grant GM 45372. Graduate student support for A.L.P. provided by an Emerson Kampen Fellowship.
References
- 1.McLuckey SA, Mentinova M. Ion/Neutral, Ion/Electron, Ion/Photon, and Ion/Ion Interactions in Tandem Mass Spectrometry: Do we need them all? Are they enough? J Am Soc Mass Spectrom. 2011;22:3–12. doi: 10.1007/s13361-010-0004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 3.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray Ionization-Principles and Practice. Mass Spectrom Rev. 1990;9:37–70. [Google Scholar]
- 4.Prentice BM, McLuckey SA. Gas-Phase Ion/Ion Reactions of Peptides and Proteins: Acid/Base, Redox, and Covalent Chemistries. Chem Commun. 2013;49:947–965. doi: 10.1039/c2cc36577d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLuckey SA, Stephenson JL., Jr Ion/Ion Chemistry of High-Mass Multiply Charged Ions. Mass Spectrom Rev. 1998;17:369–407. doi: 10.1002/(SICI)1098-2787(1998)17:6<369::AID-MAS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 6.Scalf M, Westphall MS, Krause J, Kaufman SL, Smith LM. Controlling Charge States of Large Ions. Science. 1999;283:194–197. doi: 10.1126/science.283.5399.194. [DOI] [PubMed] [Google Scholar]
- 7.Good DM, Wirtala M, McAlister GC, Coon JJ. Performance Characteristics of Electron Transfer Dissociation Mass Spectrometry. Mol Cell Proteomics. 2007;6:1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Sohn CH, Chung CK, Yin S, Ramachandran P, Loo JA, Beauchamp JL. Probing the Mechanism of Electron Capture and Electron Transfer Dissociation Using Tags with Variable Electron Affinity. J Am Chem Soc. 2009;131:5444–5459. doi: 10.1021/ja806534r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newton KA, Amunugama R, McLuckey SA. Gas-Phase Ion/Ion Reactions of Multiply Protonated Polypeptides with Metal Containing Anions. J Phys Chem A. 2005;109:3608–3616. doi: 10.1021/jp04416i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Payne AH, Glish GL. Gas-phase ion/ion interactions between peptides or proteins and iron ions in a quadrupole ion trap. Int J Mass Spectrom. 2001;204:47–54. [Google Scholar]
- 11.Luongo CA, Bu J, Burke NL, Gilbert JD, Prentice BM, Cummings S, Reed CA, McLuckey SA. Selective Removal of Alkali Metal Cations from Multiply-Charged Ions via Gas-Phase Ion/Ion Reactions using Weakly Coordinating Anions. J Am Soc Mass Spectrom. 2015;26:404–415. doi: 10.1007/s13361-014-1052-3. [DOI] [PubMed] [Google Scholar]
- 12.Mentinova M, McLuckey SA. Intra- and Inter-Molecular Cross-Linking of Peptide Ions in the Gas-Phase: Reagents and Conditions. J Am Soc Mass Spectrom. 2011;22:912–921. doi: 10.1007/s13361-011-0103-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Webb IK, Mentinova M, McGee WM, McLuckey SA. Gas-Phase Intramolecular Protein Crosslinking via Ion/Ion Reactions: Ubiquitin and a Homobifunctional sulfo-NHS Ester. J Am Soc Mass Spectrom. 2013;24:733–743. doi: 10.1007/s13361-013-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mentinova M, McLuckey SA. Covalent Modification of Gaseous Peptide Ions with N-Hydroxysuccinimide Ester Reagent Ions. J Am Chem Soc. 2010;132:18248–18257. doi: 10.1021/ja107286p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mentinova M, Barefoot NZ, McLuckey SA. Solution versus Gas-Phase Modification of Peptide Cations with NHS-Ester Reagents. J Am Soc Mass Spectrom. 2012;23:282–289. doi: 10.1007/s13361-011-0291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGee WM, Mentinova M, McLuckey SA. Gas-Phase Conjugation to Arginine Residues in Polypeptide Ions via N-Hydroxysuccinimide Ester-based Reagent Ions. J Am Chem Soc. 2012;134:11412–11414. doi: 10.1021/ja304778j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han H, McLuckey SA. Selective Covalent Bond Formation in Polypeptide Ions via Gas-Phase Ion/Ion Reaction Chemistry. J Am Chem Soc. 2009;131:12884–12885. doi: 10.1021/ja904812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hassell KM, Stutzman JR, McLuckey SA. Gas Phase Bio-conjugation of Peptides via Ion/Ion Charge Inversion: Schiff Base Formation on the Conversion of Cations to Anions. Anal Chem. 2010;82:1594–1597. doi: 10.1021/ac902732v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stutzman JR, McLuckey SA. Ion/Ion Reactions of MALDI-Derived Peptide Ions: Increased Sequence Coverage via Covalent and Electrostatic Modification upon Charge Inversion. Anal Chem. 2012;84:10679–10685. doi: 10.1021/ac302374p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prentice BM, Gilbert JD, Stutzman JR, Forrest WP, McLuckey SA. Gas-Phase Reactivity of Carboxylic Acid Functional Groups with Carbodiimides. J Am Soc Mass Spectrom. 2013;24:30–37. doi: 10.1007/s13361-012-0506-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pilo AL, McLuckey SA. Oxidation of Methionine Residues in Polypeptide Ions Via Gas-Phase Ion/Ion Chemistry. J Am Soc Mass Spectrom. 2014;25:1049–1057. doi: 10.1007/s13361-014-0861-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schey KL, Finley EL. Identification of Peptide Oxidation by Tandem Mass Spectrometry. Acc Chem Res. 2000;33:299–306. doi: 10.1021/ar9800744. [DOI] [PubMed] [Google Scholar]
- 23.Lagerwerf FM, van de Weert M, Heerma W, Haverkamp J. Oxidation of Oxidized Methionine Peptides. Rapid Commun Mass Spectrom. 1996;10:1905–1910. doi: 10.1002/(SICI)1097-0231(199612)10:15<1905::AID-RCM755>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 24.Reid GE, Roberts KD, Kapp EA, Simpson RJ. Statistical and Mechanistic Approaches to Understanding the Gas-Phase Fragmentation Behavior of Methionine Sulfoxide Containing Peptides. J Proteome Res. 2004;3:751–759. doi: 10.1021/pr0499646. [DOI] [PubMed] [Google Scholar]
- 25.Qin J, Chait BT. Identification and characterization of posttranslational modifications of proteins by MALDI ion trap mass spectrometry. Anal Chem. 1997;69:3995–4001. doi: 10.1021/ac970489n. [DOI] [PubMed] [Google Scholar]
- 26.Jiang XY, Smith JB, Abraham EC. Identification of a MS-MS fragment diagnostic for methionine sulfoxide. J Mass Spectrom. 1996;31:1309–1310. [Google Scholar]
- 27.Tureček F, Drinkwater DE, McLafferty FW. Gas-Phase Chemistry of CH3SOH, CH2+SHOH, CH3SO•, and •CH2SOH by Neutralization-Reionization Mass Spectrometry. J Am Chem Soc. 1989;111:7696. [Google Scholar]
- 28.O’Hair RAJ, Reid GE. Neighboring group versus cis-elimination mechanisms for side chain loss from protonated methionine, methionine sulfoxide and their peptides. Eur J Mass Spectrom. 1999;5:325–334. [Google Scholar]
- 29.Amunugama M, Roberts KD, Reid GE. Mechanisms for the Selective Gas-phase Fragmentation Reactions of Methionine Side Chain Fixed Charge Sulfonium Ion Containing Peptides. J Am Soc Mass Spectrom. 2006;17:1631–1642. doi: 10.1016/j.jasms.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 30.Lioe H, Laskin J, Reid GE, O’Hair RAJ. Energetics and Dynamics of the Fragmentation Reactions of Protonated Peptides containing Methionine Sulfoxide or Aspartic Acid via Energy- and Time-Resolved Surface Induced Dissociation. J Phys Chem A. 2007;111:10580–10588. doi: 10.1021/jp073040z. [DOI] [PubMed] [Google Scholar]
- 31.Gilbert JD, Fisher CM, Bu J, Prentice BM, Redwine JG, McLuckey SA. Strategies for generating peptide radical cations via ion/ion reactions. J Mass Spectrom. 2014;50:418–426. doi: 10.1002/jms.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tureček F. Peptide Radicals and Cation Radicals in the Gas Phase. Chem Rev. 2013;113:6691–6733. doi: 10.1021/cr400043s. [DOI] [PubMed] [Google Scholar]
- 33.Oh HB, Moon B. Radical-Driven Peptide Backbone Dissociation Tandem Mass Spectrometry. Mass Spectrom Rev. 2014;9999:1–17. doi: 10.1002/mas.21426. [DOI] [PubMed] [Google Scholar]
- 34.Knudsen ER, Julian RR. Fragmentation chemistry observed in hydrogen deficient radical peptides generated from N-nitrosotryptophan residues. Int J Mass Spectrom. 2010;294:83–87. [Google Scholar]
- 35.Hao G, Gross SS. Electrospray Tandem Mass Spectrometry Analysis of S- and N-Nitrosopeptides: Facile Loss of NO and Radical-Induced Fragmentation. J Am Soc Mass Spectrom. 2006;17:1725–1730. doi: 10.1016/j.jasms.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 36.Chu IK, Rodriguez CF, Lau TC, Hopkinson AC, Siu KWM. Molecular radical cations of oligopeptides. J Phys Chem B. 2000;104:3393–3397. [Google Scholar]
- 37.Chu IK, Zhao J, Xu M, Siu SO, Hopkinson AC, Siu KWM. Are the radical centeres in peptide radical cations mobile? The generation, tautomerism, and dissociation of isomeric α-carbon-centered triglycine radical cations in the gas phase. J Am Chem Soc. 2008;130:7862–7872. doi: 10.1021/ja801108j. [DOI] [PubMed] [Google Scholar]
- 38.Chu IK, Rodriguez CF, Lau TC, Hopkinson AC, Siu KWM. Formation of peptide radical ions through dissociative electron transfer in ternary metal-ligand-peptide complexes. Eur J Mass Spectrom. 2011;17:543–556. doi: 10.1255/ejms.1156. [DOI] [PubMed] [Google Scholar]
- 39.Bagheri-Madji E, Ke YY, Orlova G, Chu IK, Hopkinson AC, Siu KWM. Copper-mediated peptide radical ions in the gas-phase. J Phys Chem B. 2004;108:11170–11181. [Google Scholar]
- 40.Barlow CK, McFadyen WD, O’Hair RAJ. Formation of cationic peptide radicals by gas-phase redox reactions with trivalent chromium, manganese, iron, and cobalt complexes. J Am Chem Soc. 2005;127:6109–6115. doi: 10.1021/ja043088f. [DOI] [PubMed] [Google Scholar]
- 41.Barlow CK, Moran D, Radom L, McFadyen WD, O’Hair RAJ. Metal-mediated formation of gas-phase amino acid radical cations. J Phys Chem A. 2006;110:8304–8315. doi: 10.1021/jp056471v. [DOI] [PubMed] [Google Scholar]
- 42.Laskin J, Yang Z, Chu IK. Energetics and dynamics of electron transfer and proton transfer in dissociation of MetalIII(salen)-peptide complexes in the gas phase. 2008;130:3218–3230. doi: 10.1021/ja077690s. [DOI] [PubMed] [Google Scholar]
- 43.Ly T, Julian RR. Residue-Specific Radical-Directed Dissociation of Whole Proteins in the Gas Phase. J Am Chem Soc. 2008;130:351–358. doi: 10.1021/ja076535a. [DOI] [PubMed] [Google Scholar]
- 44.Sun QY, Nelson H, Ly T, Stoltz BM, Julian RR. Side chain chemistry mediates backbone fragmentation in hydrogen deficient peptide radicals. J Proteome Res. 2009;8:958–966. doi: 10.1021/pr800592t. [DOI] [PubMed] [Google Scholar]
- 45.Masterson DS, Yin H, Chacon A, Hachey DL, Norris JL, Porter NA. Lysine peroxycarbamates: Free radical-promoted peptide cleavage. J Am Chem Soc. 2004;126:720–721. doi: 10.1021/ja038615u. [DOI] [PubMed] [Google Scholar]
- 46.Hodyss R, Cox HA, Beauchamp JL. Bioconjugates for tunable peptide fragmentation: Free radical initiated peptide sequencing (FRIPS) J Am Chem Soc. 2005;127:12436–12437. doi: 10.1021/ja052042z. [DOI] [PubMed] [Google Scholar]
- 47.Xia Y, Wu J, Londry FA, Hager JW, McLuckey SA. Mutual Storage Mode Ion/Ion Reactions in Hybrid Linear Ion Trap. J Am Soc Mass Spectrom. 2005;16:71–81. doi: 10.1016/j.jasms.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 48.Liang W, Xia Y, McLuckey SA. Alternately pulsed nano-electrospray ionization/atmospheric pressure chemical ionization for ion/ion reactions in an electrodynamic ion trap. Anal Chem. 2006;78:3208–3212. doi: 10.1021/ac052288m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Londry FA, Hager JW. Mass selective axial ion ejection from a linear quadrupole ion trap. J Am Chem Soc. 2003;14:1130–1147. doi: 10.1016/S1044-0305(03)00446-X. [DOI] [PubMed] [Google Scholar]
- 50.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam MJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision D.01. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 51.Hehre WJ, Ditchfield R, Radom L, Pople JA. Molecular orbital theory of the electronic structure of organic compounds. V. Molecular theory of bond separation. J Am Chem Soc. 1970;92:4796–4801. [Google Scholar]
- 52.Blanksby SJ, Ellison GB. Bond Dissociation Energies of Organic Molecules. Acc Chem Res. 2003;36:255–263. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- 53.Benjamin NM, Ryan RR. Dissociation energies of X–H bonds in amino acids. Phys Chem Chem Phys. 2012;14:3148–3154. doi: 10.1039/c2cp23443b. [DOI] [PubMed] [Google Scholar]
- 54.Bach RD, Owensby AL, Gonzalez C, Bernhard Schlegel H, McDouall JJW. Nature of the Transition Structure for Oxygen Atom Transfer from a Hydroperoxide. Theoretical Comparison between Water Oxide and Ammonia Oxide. J Am Chem Soc. 1991;113:6001–6011. [Google Scholar]
- 55.Froelich JM, Reid GE. Mechanisms for the Proton Mobility-Dependent Gas-Phase Fragmentation Reactions of S-alkyl Cysteine Sulfoxide-Containing Peptide Ions. J Am Soc Mass Spectrom. 2007;18:1690–1705. doi: 10.1016/j.jasms.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 56.Bunton CA, Foroudian HJ, Kumar A. Sulfide Oxidation and Oxidative Hydrolysis of Thioesters by Peroxymonosulfate Ion. J Chem Soc Perkin Trans 2. 1995;1:33–39. [Google Scholar]
- 57.Kolthoff IM, Miller IK. The Chemistry of Persulfate. I. The Kinetics and Mechanism of the Decomposition of the Persulfate Ion in Aqueous Medium. J Am Chem Soc. 1951;73:3055–3059. [Google Scholar]
- 58.Tureček F. Peptide Radicals and Cation Radicals in the Gas Phase. Chem Rev. 2013;113:6691–6733. doi: 10.1021/cr400043s. [DOI] [PubMed] [Google Scholar]
- 59.Hopkinson AC. Radical Cations of Amino Acids and Peptides: Structures and Stabilities. Mass Spectro Rev. 2007;28:655–671. doi: 10.1002/mas.20229. [DOI] [PubMed] [Google Scholar]
- 60.Yang Z, Lam C, Chu IK, Laskin J. The Effect of the Secondary Structure on Dissociation of Peptide Radical Cations: Fragmentation of Angiotensin III and Its Analogues. J Phys Chem B. 2008;112:12468–12478. doi: 10.1021/jp805226x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.