

Abstract
Ischemia is a condition associated with decreased blood supply to the brain, eventually leading to death of neurons. It is associated with a diverse cascade of responses involving both degenerative and regenerative mechanisms. At the cellular level, the changes are initiated prominently in the neuronal cytoskeleton. Cofilin, a cytoskeletal actin severing protein, is known to be involved in the early stages of apoptotic cell death. Evidence supports its intervention in the progression of disease states like Alzheimer's and ischemic kidney disease. In the present study, we have hypothesized the possible involvement of cofilin in ischemia. Using PC12 cells and mouse primary cultures of cortical neurons, we investigated the potential role of cofilin in ischemia in two different in vitro ischemic models: chemical induced oxidative stress and oxygen-glucose deprivation/reperfusion (OGD/R). The expression profile studies demonstrated a decrease in phosphocofilin levels in all models of ischemia, implying stress-induced cofilin activation. Furthermore, calcineurin and slingshot 1L (SSH) phosphatases were found to be the signaling mediators of the cofilin activation. In primary cultures of cortical neurons, cofilin was found to be significantly activated after 1 h of OGD. To delineate the role of activated cofilin in ischemia, we knocked down cofilin by siRNA technique and tested the impact of cofilin silencing on neuronal viability. Cofilin siRNA-treated neurons showed a significant reduction of cofilin levels in all treatment groups (control, OGD and OGD/R). Additionally, cofilin siRNA reduced cofilin mitochondrial translocation and caspase 3 cleavage, with a concomitant increase in neuronal viability. These results strongly support the active role of cofilin in ischemia-induced neuronal degeneration and apoptosis. We believe that targeting this protein mediator has a potential for therapeutic intervention in ischemic brain injury and stroke.
Keywords: Cofilin, ischemia, Caspase 3, Oxygen glucose deprivation, reperfusion
Introduction
Ischemic stroke is a neurodegenerative disease characterized by the sudden death of neurons in the brain due to inadequate blood flow. The current drug therapy approved by the US FDA for its treatment is tissue plasminogen activator (tPA), which mainly functions to dissolve the blood clots and clear the obstruction for resumption of the blood flow to the brain. This therapy is for immediate restoration of the blood flow, but the aftermath of pathology associated with the ischemic cascade requires attention [1].
Besides neurotoxicity, calcium overload, and free radical damage, neurocytoskeletal degeneration is another important pathway involved in the progression of events following acute brain injury. The cytoskeleton, which is a dynamic structure in the cell, is composed of a filamentous network rendering homeostasis functions in the cell by maintenance of cell shape, cell movement, cell replication, apoptosis, cell differentiation, and cell signaling [2]. Actin is an essential and abundant protein of the cytoskeleton; its dynamics are regulated by actin binding proteins, among which is the ubiquitous ADF/Cofilin (AC) family of proteins. The central function of the AC family is to maintain the rapid recycling of G-actin monomers by severing filamentous actin (F-actin) into short segments and creating free barbed ends for actin elongation associated with membrane ruffling, cytokinesis and development of growth cones [3]. AC protein binding to actin is inactivated by phosphorylation at Ser 3 (LIM Kinases) and reactivated through dephosphorylation by the phosphatase slingshot or chronophin [4].
The levels of AC proteins and their binding to actin are highly dependent on the energy potentials of the cell. In conditions of ATP depletion, cofilin rods are found to be predominant in the cell as an energy conservation mechanism which, over extended periods, can eventually lead to pathological states and cell death [5]. Cellular stress-initiated formation of cofilin actin rods was found to be a key element in the progression of neurodegeneration in Alzheimer's and ischemic kidney disease [6]. Abnormal aggregates of cofilin were found in β-amyloid plaques and tau tangles [7]. During oxidative stress, cofilin is observed to translocate to mitochondria and induce the release of cytochrome C, initiating apoptotic cell death [8]. The involvement of actin cytoskeletal mediators, in particular cofilin, is not clear in ischemic conditions where ATP depletion is predominant.
In the present study, we hypothesized the possible involvement of cofilin in cytoskeletal induced neurodegenerations in ischemic conditions. To study our hypothesis, we have analyzed the expression pattern of cofilin and its role in cytoskeletal alterations using two types of cells (PC12 and primary cultures of cortical neurons) and two in vitro models of ischemia (chemical induced ischemia and OGD/R). Using PC12 cells, we tested the impact of ischemia on cofilin activation by dephosphorylation using specific and non-specific phosphatase inhibitors. In primary cultures of cortical neurons, we tested the impact of ischemia on cofilin activation status using the OGD/R model of ischemia. Additionally, we inhibited cofilin activity by using cofilin1 siRNA and then tested the impact of cofilin knockdown on neuronal survival during OGD.
Experimental procedures
PC12 cell growth and differentiation
PC12 cells (ATCC, Manassas, VA, USA) were seeded at a population density of 0.05×106 cells onto poly-l-lysine-coated 6 cm petri dishes. After 24 h of plating the cells, growth medium (RPMI with 1% penicillin-streptomycin, 10% fetal bovine serum and 5% Horse serum) was replaced with serum free differentiating medium DMEM (Dulbecco modified Eagle medium, HyClone, Thermo scientific, West Palm Beach, FL, USA) supplemented with 1% penicillin-streptomycin, 100 ng/ml Nerve growth factor (NGF). The medium was replaced every two days with fresh differentiating medium, and the cells were cultured for a period of 7 days to allow the cells to completely transform into differentiated neuronal cells.
Primary cortical neuronal cultures
Mouse primary cultures of cortical neurons were extracted according to a protocol mentioned previously, with minor modifications [9]. Briefly, pregnant mice on gestational day 15-17 were sacrificed by CO2 overdose, and fetuses were collected in cold HBSS medium (Fisher Scientific, Hanover Park, IL, USA). The fetal brains were removed and the cortices were dissected and collected in cold HBSS. The cells were dissociated from the tissue by pipetting up and down 5-10 times. Cells were spun down at 500×g for 3 min at 18 °C, the supernatant was removed, and cells were resuspended in neurobasal medium (Invitrogen, Carlsbad, CA, USA) containing penicillin and streptomycin (50 U/ml of medium), glutamine (0.5 mM), and 2% B27 serum-free supplement (Invitrogen, Carlsbad, CA, USA). Then the cells were counted in trypan blue and plated in poly-l-lysine (0.1 mg/ml)-coated plates at a density of 2×105 neurons/well in a 24-well plate, 1×106 neurons/well in 6-well plate and 2×106 neurons/60 mm plate. Cell cultures were maintained at 37°C in 95% air and 5% CO2. At day 2 in vitro (DIV 2), neurons were treated with cytosine β-D-Arabinofuranoside (Sigma-Aldrich, St. Louis, MO, USA) at a 5 μM concentration for 24 h to curb glial cell growth.
Treatment with Stressor (tertiary Butyl hydro peroxide)
Undifferentiated PC12 cells plated at a density of 0.87×106 were subjected to different concentrations of t-Butyl hydro peroxide (t-BuOOH) such as 60 μM, 100 μM and 200 μM for a time period of 1 h. Briefly, after 24 h of plating, the medium was replaced with fresh medium and the stressor was added to the medium. The cells were then harvested and the protein expression pattern was studied using immunoblotting.
Oxygen-Glucose Deprivation (OGD) and OGD/Reperfusion (OGD/R) models of ischemia and inhibitor treatments
In this in vitro ischemia model, the growth medium of the cells was replaced with medium lacking glucose (HBSS Phenol red medium) and placed in a chamber that was rendered anaerobic by a sachet containing ascorbic acid (AnaeroGenTM, OXOID, Germany) [10]. Resazurin, an anaerobic indicator (OXOID, Germany) sensitive to changes in oxygen levels, was placed in the chamber and the lid of the chamber was tightly closed and placed in the incubator at 37°C. The complete lack of oxygen in the chamber is indicated by the change in the color of the indicator from pink to white, and the onset time for OGD was recorded. In the OGD model, cells were subjected to OGD only for 1 h, whereas for OGD/R, cells were subjected to OGD for 1 h followed by reperfusion for 24 h. Differentiated PC12 neuronal cells were cultured on coverslips in 6-well dishes at a cell density of 0.03×106 and were subjected to different treatments: inhibitor pretreatments (Sodium vanadate for 2 h, FK-506 for 1 h) and stressor treatments i.e., t-BuOOH for 1 h and OGD for 1 h in each individual set of experiments.
MTT- Cell Viability assay
Cell viability was determined using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay we used previously [11]. Briefly, cells (PC12 cells and primary cultures of cortical neurons) were incubated with MTT reagent (Promega Corporation, Madison, WI, USA) for 3 h in 5% CO2 at 37 °C. After that, the solubilizing solution/stop mix was added to dissolve formazan crystals. Viable cells were quantified by measuring the absorbance at 570 nm.
Immunocytochemistry
Differentiated PC12 neuronal cells were fixed using 4% paraformaldehyde and treated with 0.3% of Triton X-100. Primary antibodies – rabbit anti-phosphocofilin, anti-cofilin (1:100; Abcam, Cambridge, MA, USA) were incubated overnight at 4°C. Texas red labeled donkey anti-rabbit secondary IgG antibody (1:400; Jackson ImmunoResearch, West Grove, PA, USA) was used at room temperature for 1 h. Staining of F-actin in the cells was carried out using one unit of phalloidin (Invitrogen, Carlsbad, CA, USA). After rinsing with PBS, the coverslip was mounted with DAPI (Santa Cruz Biotechnology, Santa Cruz, CA, USA) over the glass slide and the gap was sealed.
Subcellular Fractionation and Western blotting
To obtain subcellular fractions, we used the protocol developed by Dimauro et al. [12], with some modifications. Briefly, primary cultures of cortical neurons were harvested using ice-cold lysis buffer (250 mM sucrose, 1 mM EDTA, 1 mM EGTA, 1.5 MgCl2, 10 mM KCl, 20 mM HEPES (PH 7.5), 1 mM dithiothreitol (DTT), 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 50 mM NaF, 10 mM Na Vanadate, 20 mM Na pyrophosphate and protease inhibitor cocktail (Thermo scientific). Cell homogenates were kept on ice for 15 min and then centrifuged at 1000 × g for 15 min to sediment nuclear pellets. The resultant supernatant was collected and recentrifuged at 11000 × g for 20 min to sediment mitochondrial pellets. Nuclear pellets were resuspended in NET buffer (20 mM HEPES PH 7.9, 20% glycerol, 0.5 M NaCl, 1.5 mM MgCl2, 1% Triton-X-100, 1 Mm DTT, 0.1 mM PMSF, 50 mM NaF, 10 mM Na Vanadate, 20 mM Na pyrophosphate and protease inhibitor cocktail (Thermo Scientific)), whereas mitochondrial pellets were suspended in SOL buffer (50 mM Tris-HCl pH 6.8, 1 mM EDTA, 0.5% Triton-X-100, protease and phosphatase inhibitors). Both nuclear and mitochondrial suspensions were kept on ice for 30 min with intermittent shaking and then centrifuged at 9000 × g for 30 min to obtain nuclear and mitochondrial fractions, respectively.
Protein concentrations were determined using Bradford reagent (Bio-Rad Laboratories, CA, USA), and samples were analyzed by loading equivalent amounts of protein (2-25 μg) onto 10-15% SDS-polyacrylamide gels. Proteins separated on the gels were transferred onto a pretreated PVDF membrane and were blocked with 3% BSA for 1 h to prevent nonspecific binding. The membrane was then incubated overnight at 4°C with the following primary antibodies: rabbit anti-phosphocofilin and anti-cofilin (1:1000; Abcam, Cambridge, MA, USA), rabbit anti-actin (1:2000; Sigma Aldrich), rabbit anti-α-tubulin (1:2000; Cell Signaling, Danvers, MA, USA), rabbit anti-histone H3 (1:2000; Fisher), mice anti-COX IV (1:1000; Cell Signaling), rabbit anti-GAPDH (1:2000; Fisher) and rabbit anti-caspase 3 (specific for activated (cleaved) form of caspase 3, Cell Signaling). Following the incubation, the blots were washed and incubated with HRP (Horseradish peroxidase) conjugated goat anti-rabbit or anti-mouse secondary antibody (1:6000; Jackson ImmunoResearch) for 1 h at room temperature. Actin, α-Tubulin, and GAPDH were used as a loading control for cytosolic proteins, Histone H3 for nuclear proteins, and COX IV for mitochondrial proteins. The images were analyzed using Adobe Photoshop and Image J software.
Cofilin siRNA Neuronal Transfection
Cofilin1 siRNA with sequence 5′AGGAGAUCCUGGUAGGAGAtt 3′ was ordered from Ambion (Life Technologies, Carlsbad, CA, USA). At DIV 4, neurons were transfected with cofilin1 siRNA at different concentrations for 72 h using the SilenceMag transfection reagent (OZ Biosciences) and following the manufacturer's instructions. Briefly, in one set of microtubes, siRNA was diluted at different concentrations with OPTI-MEM serum reduced medium (Life Technology). In another set of microtubes, SilenceMag transfection reagent was added at different amounts depending on the final concentrations of siRNA required. Then, diluted siRNA solution was added to the SilenceMag tube and mixed immediately 4-5 times by vigorous pipetting followed by incubation for 20 min at room temperature. The formed complexes (siRNA and SilenceMag) were added drop by drop directly onto the cells, and the cell culture plate was placed on the magnetic plate for 15 min in the incubator. After that, the magnetic plate was removed and the cell culture plate was kept in the incubator. In OGD experiments, neurons were transfected for 72 h (DIV 7) and then subjected to 1 h of OGD, whereas in OGD/R experiments, neurons were subjected to 48 h transfection (DIV 6) followed by 1 h OGD and then 24 h of reperfusion. At the end, all neurons were tested at DIV 7, where some of them were harvested for Western blotting and the rest were tested for viability using MTT assay.
Statistical analysis
The experimental results are expressed as the mean ± SEM and are accompanied by the number of observations (independent preparations of cultured cells). ANOVA test was used to compare the control group to different treatment groups. Additionally, student's unpaired t-test was used to determine significant differences between the control group (scrambled siRNA) and the cofilin1 siRNA treatment group. A value of P < 0.05 was considered to be statistically significant.
Results
Effect of cofilin against t-BuOOH-induced oxidative stress in PC12 cells
In our first study, we investigated changes in the expression levels of the inactive form of cofilin (phosphocofilin) in cultured PC12 cells in response to t-butyl hydro peroxide (t-BuOOH)-induced oxidative stress. The rationale for using t-BuOOH in our first series of experiments was to evaluate cofilin expression changes during oxidative stress, which is the main component of ischemic cascade and results in ATP depletion and eventual cell death by inducing mitochondrial permeability transition, reactive oxygen species (ROS), mitochondrial NAD(P)H oxidation, increased mitochondrial free Ca2+, intramitochondrial generation of ROS and mitochondrial depolarization and inner membrane permeabilization [13]. PC12 cells were exposed for 1 h to different concentrations of t-BuOOH 60μM, 100μM and 200μM. As cofilin is regulated by phosphorylation to an inactive form, phosphocofilin, the levels of cofilin were studied and interpreted in terms of changes in the phosphocofilin expression. The expression of phosphocofilin was found to be significantly decreased with exposure to higher concentrations of t-BuOOH (Fig.1A-B). Total cofilin expression was unchanged in all the treatment groups. The expression pattern of the ratio of phosphocofilin to total cofilin was plotted against the treatment groups. The results suggest that cofilin plays an important role in conditions associated with oxidative stress.
Figure 1. Oxidative stress and OGD-induced changes in phosphocofilin and cofilin expression.
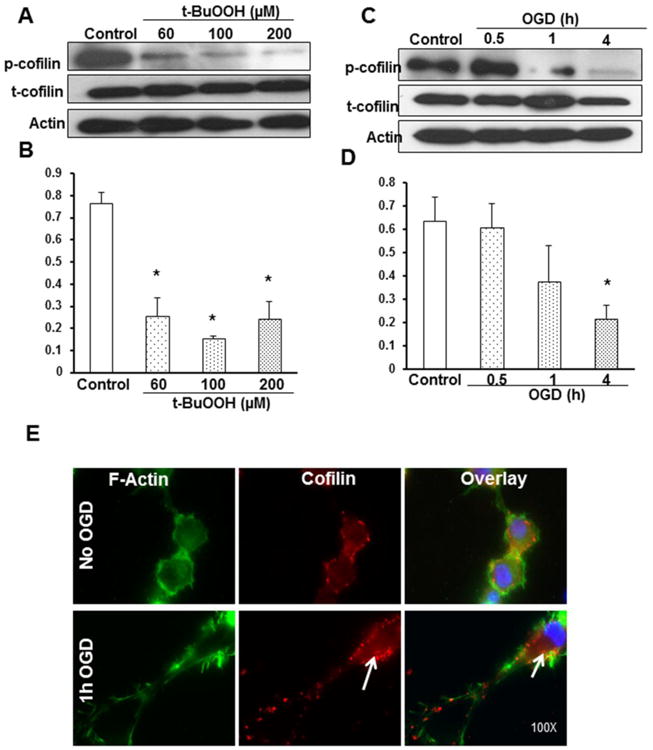
A PC12 cells were subjected to treatment with 60, 100 and 200μM concentrations of t-BuOOH for 1 h, and the protein expression levels were evaluated. Representative blot shows phosphocofilin expression, which was found to be significantly decreased with increasing concentration of t-BuOOH. C. Representative blot shows phosphocofilin, total cofilin and actin expression pattern at 30 min, 1 and 4 h of OGD using differentiated PC12 neuronal cells. Time course study demonstrated the dephosphorylation of phosphocofilin occurring with increasing periods of stress. The decline in the phosphorylated form initiated within 1 h of OGD, followed by a complete decline within 4 h of OGD. B and D. Graphs represent the densitometric analysis values normalized with actin and the ratio of phosphocofilin to total cofilin levels plotted against treatment groups. E. Representative immunofluorescence pictures show that cofilin (stained red) is co-localized with the F-actin filaments, predominantly in the cell periphery and cytoplasm of differentiated PC12 neuronal cells. After OGD, most of the actin filaments were rigid and degraded. The areas with white arrows show cofilin prevalence in aggregated actin filaments. Scale bar is 10 μm. Experiments were performed thrice with separate batches of cells, and the graphs are the cumulative results of three different experiments. Data are expressed as mean±SEM, where p<0.05 was considered significant. *vs control
Oxygen glucose deprivation (OGD) induced changes in phosphocofilin expression levels
After initial success with PC12 cells and oxidative stress, we further investigated the role of cofilin in differentiated PC12 cells in a more physiologically relevant environment of ischemic conditions, i.e., the in vitro model of oxygen glucose deprivation (OGD). Differentiated PC12 neuronal cells were subjected to different durations of OGD (0.5-4 h), and the protein expression pattern was studied. A gradient decrease in the phosphocofilin expression was observed with increasing periods of OGD. There were no discernible changes in the total cofilin levels, clearly indicating that the dephosphorylation is an activation process in hypoglycemic and hypoxic stress conditions, not a cellular degradation mechanism. Phosphocofilin dephosphorylation was initially unchanged at 30 min of OGD, followed by a steep decline with 1 h and highly significant dephosphorylation at 4 h of OGD (Fig. 1C-D). The results provide further proof of concept for the involvement of cofilin in ischemic injury. To study morphological changes in cofilin expression, we used fluorescence microscopy and observed that phosphocofilin was localized in the nucleus. With the help of fluorescein isothiocyanate (FITC) staining, cofilin was found prominently co-localized in the F-actin filaments and expressed in the cell periphery, cytoplasm and neurite extensions (Fig. 1E).
Effect of nonspecific tyrosine phosphatase inhibitor (Na3VO4) on the phosphocofilin expression and cell survival against oxygen glucose deprivation (OGD)
Sodium orthovanadate (Na3VO4), a nonspecific tyrosine phosphatase inhibitor, was employed to target the broad range of phosphatases, based on the hypothesis about their involvement in the activation of cofilin, which led us to study the outcomes of cofilin activation and inhibition. Cells were pre-incubated with 1mM Na3VO4 for 2 h and then subjected to OGD for 1 h duration. Western blot analysis indicated significant restoration of phosphocofilin levels with inhibitor pretreated cells during OGD, when compared to untreated cells (Fig. 2A-B). Inhibitor pretreated neurons did not exhibit any cell death demarcations, suggesting that the inhibitor by itself, comparable to the control, does not have a deleterious effect on cell viability. Inhibitor pretreatment for 2 h followed by OGD exposure showed significant restoration of cell viability against OGD-induced stress, suggesting the importance of phosphocofilin expression on cell viability (Fig 2C). To further our understanding on the morphological changes in phosphor- and total cofilin during OGD and with inhibitor treatment, immunofluorescence analysis was used. Immunofluorescence analysis revealed that within 1 h of OGD, the levels of phosphocofilin in the nucleus were found to be decreased, but the inhibitor pretreated group displayed a restored level, which was consistent with the Western blotting data. Ischemic stress-induced destruction of F-actin filaments was depicted from decreased FITC staining. Upon close observation of the morphology of the filaments, rigidity and intense staining at the filament ends was noticed, suggesting the aggregation of severed actin filaments. Cofilin was found to be localized to these aggregated filaments. This supports our hypothesis that cofilin enhances actin severing activity in stress conditions (Fig. 2D).
Figure 2. Phosphocofilin and cofilin protein expression and cell viability were restored in differentiated PC12 neuronal cells pretreated with inhibitors.
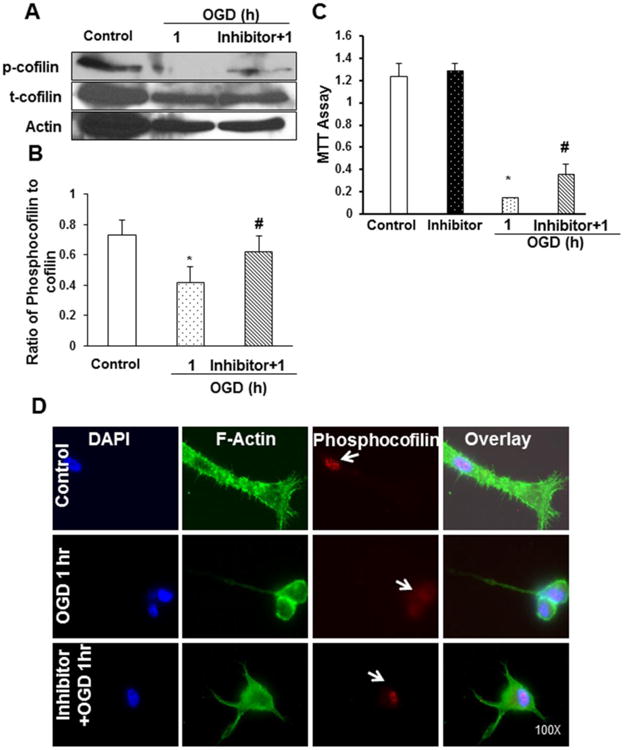
Differentiated PC12 cells were pretreated with non-specific phosphatase inhibitor (1mM Na3VO4) for 2 h and then subjected to 1 h of OGD. A. Representative blot shows a decrease in phosphocofilin expression with OGD and a remarkable restoration of its levels with inhibitor pretreatment. Actin was used as a loading control for densitometric analysis. C. Differentiated PC12 neuronal cells were assayed for cell viability during OGD conditions. The assay results display a decreased percentage of metabolically active cells during OGD. The inhibitor pretreatment was able to restore cell viability when compared to OGD only. The values were expressed as a percentage of control, and data were expressed as mean±SEM of three independent experiments. B. Graphs represent the cumulative data of three experiments conducted on separate batches of cells. D. Representative fluorescence pictures show nucleus stained with DAPI (blue color), F-actin filaments stained with phalloidin (green), and phosphocofilin stained with Texas red (red). The top panel shows the control followed by OGD and inhibitor-pretreated differentiated PC12 neuronal cells in the middle and lower panels. The right column shows the merged overlay of all figures. The fluorescence imaging clearly illustrates that the inhibitor pretreatment restored the phosphocofilin levels in the nucleus, which were decreased with OGD. Scale bar is 10 μm. Data are expressed as mean±SEM, where p<0.05 was considered significant. *vs control; #vs 1 h of OGD.
Calcineurin dephosphorylates cofilin during OGD
Calcineurin is a calcium-dependent serine threonine phosphatase involved in dephosphorylation and activation of various proteins and enzymes. In particular, Ca2+-induced cofilin dephosphorylation is shown to be mediated by calcineurin-dependent activation of slingshot-1L (SSH1L) [14]. As calcium levels are elevated during OGD, we present the hypothesis for the involvement of calcineurin in the activation of slingshot-1L (SSH1L) phosphatase and cofilin during stress. Differentiated PC12 neuronal cells were pretreated for 1 h with different concentrations (50, 100 and 500 nM) of calcineurin inhibitor (FK-506) and then subjected to OGD for another 1 h. The decrease in phosphocofilin levels observed during OGD was restored with 100 nM and 500 nM of calcineurin inhibitor, illustrating clearly the role of SSH1L phosphatase and calcineurin in phosphocofilin dephosphorylation (Fig. 3A-B).
Figure 3. Calcineurin inhibitor dephosphorylates cofilin during OGD.
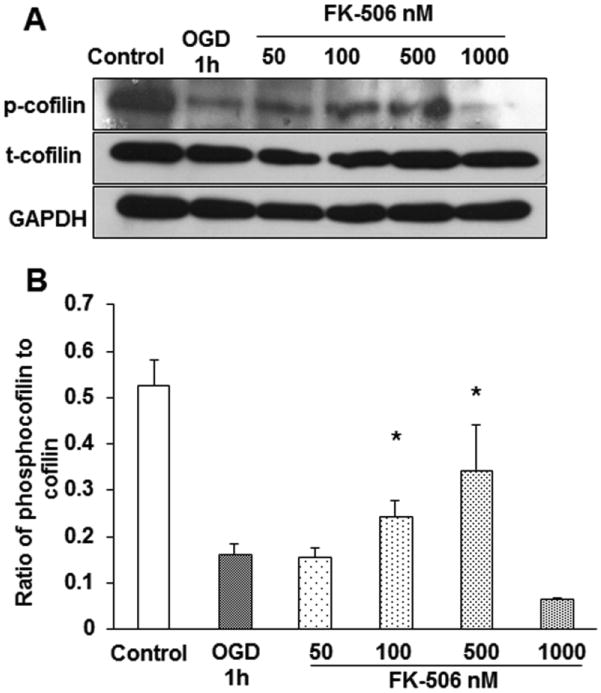
Differentiated PC12 neuronal cells were pretreated with different concentrations of calcineurin inhibitor (FK-506) for 1 h and then subjected to 1 h of OGD. A. The decreased levels of phosphocofilin in OGD were found to be restored with 100 and 500 nM concentrations of the inhibitor. B. The bar graph represents the cumulative result of three experiments; densitometric values of the bands were calculated using Image J software. The ratio of phosphocofilin to cofilin levels were plotted against the different treatment groups. The data was expressed as mean±SEM, where p<0.05 was considered significant. *vs 1 h of OGD.
Cofilin is upregulated in cytosolic fraction and downregulated in nuclear fraction in primary cultures of cortical neurons subjected to OGD
We demonstrated that cofilin is activated during OGD and this activation is involved in differentiated PC12 cell death and degeneration. Accordingly, we explored the expression profile of cofilin in mouse primary cultures of cortical neurons subjected to OGD and OGD/R before starting the cofilin silencing experiment. This experiment involved three sets of cultures wherein the first set was the control (no OGD and OGD/R); the second set was subjected to 1 h OGD at DIV 7 and then harvested directly for Western blot analysis (OGD); and the third set was subjected to 1 h OGD at DIV 6 and then 24 h reperfusion before harvesting (OGD/R). Cytosolic fraction analysis showed significant upregulation of cofilin in OGD-treated neurons compared to control neurons (Fig. 4A-B). Surprisingly, nuclear fraction analysis showed highly significant downregulation of cofilin (Fig. 4C-D). Furthermore, cofilin levels gradually went back to normal in cytosolic and nuclear fractions of neurons subjected to 24 h reperfusion.
Figure 4. Cofilin expression levels in the cytosolic and nuclear fractions after OGD and OGD/R.
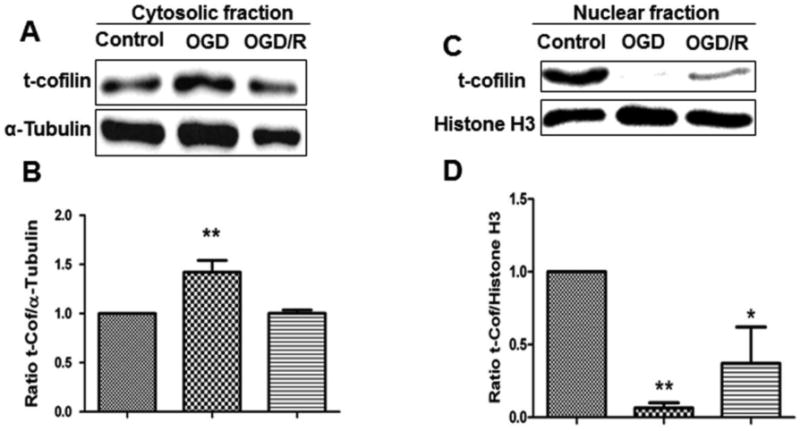
One set of mouse primary cultures of cortical neurons was subjected to 1 h of OGD and the other to 1 h of OGD and 24 h of reperfusion. Cytosolic and nuclear fractions of neurons were analyzed to check for cofilin expression levels by comparing them to control neurons (no OGD or OGD/R). A and C. Representative blots show increased cofilin expression after 1 h of OGD in the cytosolic fraction and decreased in the nuclear fraction. During the 24 h of reperfusion period, cofilin goes back gradually to normal levels in both fractions. B and D. Graphs represent the densitometric analysis of cofilin normalized with α-tubulin in the cytosolic fractions and Histone H3 in the nuclear fractions as loading control. Data are represented as mean±SEM of three independent experiments where p<0.05 was considered significant. *vs control.
Cofilin knockdown prevented neuronal death during OGD and OGD/R
Our results in the previous experiments showed that cofilin is highly activated during stressful conditions like oxidative stress and OGD. Therefore, our next hypothesis is that knockdown of cofilin can reduce the impact of its activation during ischemia and enhance neuronal survival. Primary cultures of cortical neurons were transfected with cofilin1 siRNA at DIV 4 using the SilenceMag transfection reagent. In this experiment, three sets of neurons were selected. The first set was transfected with cofilin1 siRNA for 72 h without OGD (control); the second set was transfected with siRNA as in the first, but was subjected to 1 h OGD (OGD); and the third set was transfected with siRNA for 48 h followed by 1 h OGD and 24 h reperfusion (OGD/R). All cofilin1 transfected neurons were compared with scrambled siRNA-treated neurons. Figures 5A-B show the highly significant reduction of cofilin levels in neurons treated with cofilin1 siRNA 50 nM compared to the scrambled siRNA-treated group. We used higher concentrations of cofilin1 siRNA (100 and 200 nM), but we did not observe a further decrease in cofilin levels (data not shown). In neurons subjected to 1 h OGD (Fig. 5C-D), the reduction in the cofilin level at 50 nM siRNA was less than that observed in control neurons (cofilin level was reduced to about 67%). In OGD/R subjected neurons, cofilin1 siRNA at 50 nM reduced the cofilin level to about 60% compared to scrambled siRNA-treated neurons (Fig.5E-F).
Figure 5. Cofilin expression levels in cofilin knock-down primary cultures of cortical neurons.
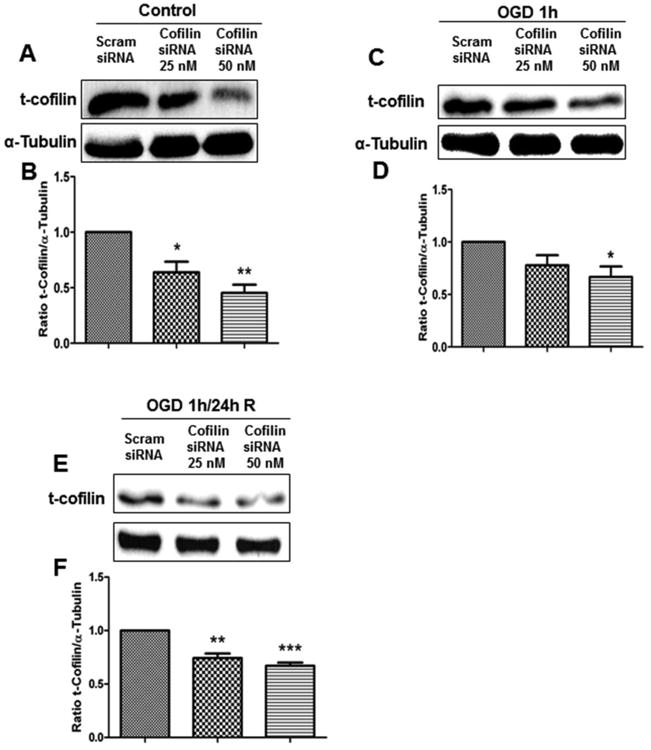
Mouse primary cultures of cortical neurons at DIV 4 were transfected with cofilin1 siRNA (25 and 50 nM) for 72 h and then harvested for Western blot analysis (control). OGD-treated primary cultures of cortical neurons were subjected to 1 h of OGD 72 h posttransfection and then harvested for immunoblotting (OGD). OGD/R-treated primary cultures of cortical neurons were subjected to 48 h transfection, followed by 1 h of OGD and 24 h of reperfusion, and were harvested for immunoblotting (OGD/R). A, C and E. Representative blots show a decrease in cofilin expression levels in cofilin knock-down in all treatment paradigms (control, OGD and OGD/R). B, D and F. Graphs represent densitometric analysis of cofilin normalized to α-tubulin as a loading control. Data are expressed as mean±SEM of three independent experiments, where p<0.05 was considered significant. *vs control (scrambled siRNA).
The increased neuronal viability in cofilin knock-down neurons is a result of reduction in cofilin mitochondrial translocation and caspase 3 cleavage
After successful knockdown of cofilin in the previous experiments, we tested the impact of this knockdown on neuronal viability using the MTT viability assay. Cofilin1 siRNA transfected neurons showed a significant increase in neuronal viability compared to scrambled siRNA-transfected cells (Fig. 6A). Cofilin siRNA at 50 nM increased neuronal viability from 25% to about 60%. Such an increase in neuronal viability encouraged us to look for signaling pathways and mediators involved in improving neuronal viability in cofilin siRNA-treated neurons. Chua et al. [15] and Posadas et al. [16] showed that cofilin translocation to mitochondria is an important step in the initiation of apoptosis. Accordingly, we tested the localization of cofilin in mitochondrial fractions of neurons transfected with cofilin1 siRNA and subjected to OGD/R. Our results demonstrate that cofilin translocation into mitochondria is significantly reduced in cofilin-silenced neurons (Fig. 6B-C) as compared to scrambled siRNA-treated neurons. It was also shown [16] that cofilin activation during excitotoxicity mediates Bax transclocation to mitochondria, cytochrome C release and caspase 3 cleavage. Therefore, we tested the impact of cofilin silencing on caspase 3 cleavage in neurons that had undergone OGD/R. It was observed that caspase 3 cleavage is significantly reduced in cofilin1 siRNA-transfected neurons compared to control neurons (Fig. 6D-E).
Figure 6. Cofilin knock-down increases neuronal viability and reduces cofilin mitochondrial translocation and caspase 3 cleavage.
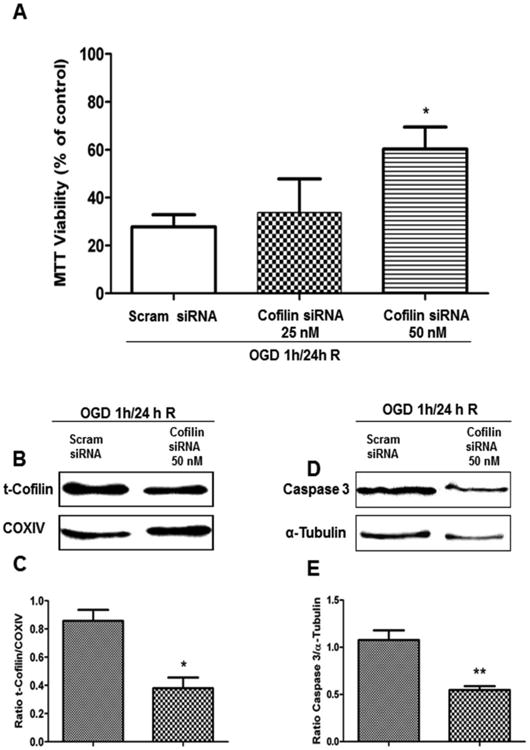
Primary cultures of cortical neurons at DIV 4 were transfected with cofilin1 siRNA for 48 h and then subjected to 1 h of OGD and 24 h of reperfusion. At DIV 7, neurons were harvested and analyzed for MTT assay and also fractionated to get cytosolic and mitochondrial fractions. A. Cofilin knock-down with siRNA (25 and 50nM) and subjecting to OGD/R rescued neuronal death. B. Representative blot shows a decrease in mitochondrial cofilin levels in cofilin knock-down neurons as compared to control. D. Representative blot of cytosolic fractions shows a decrease in cleaved caspase 3 in cofilin knock-down neurons as compared to the control. C and E. Graphs show densitometric analysis of cofilin normalized to α-tubulin in cytosolic fraction and COXIV in mitochondrial fraction used as a loading control. Data are expressed as mean±SEM of three independent experiments, where p<0.05 was considered significant. *vs control (scrambled siRNA).
Discussion
In the present study, we demonstrated the involvement of cofilin in ischemia-induced neuronal death. This was achieved by using PC12 cells, mouse primary cultures of cortical neurons, and different in vitro stress models: chemical induced oxidative stress OGD and OGD/R. The expression profile studies demonstrated a decrease in phosphocofilin levels in all models of stress. Using two non-specific cofilin inhibitors, FK-506 and sodium vanadate, we showed that cofilin activation leads to neuronal death, and its inhibition rescued differentiated PC12 neuronal cells. Finally, using mouse primary cultures of cortical neurons, cofilin was found to be significantly activated after 1 h of OGD, and after knocking down cofilin by siRNA, neurons showed a significant reduction of cofilin levels in all groups (control, OGD and OGD/R), subsequently rescuing neuronal death. Additionally, cofilin siRNA reduced cofilin mitochondrial translocation and caspase 3 cleavage, with a concomitant increase in neuronal viability. These results suggest the active role of cofilin in ischemia induced-neuronal death and apoptosis.
Cofilin, an important cytoskeletal protein, is responsible for controlling actin dynamics in a cell. Cofilin functions beyond these regulatory activities are responsible for aberrations in cellular activity. Phosphatases were found to play a key role in the activation of cofilin during ATP depletion states in endothelial cells [17]. This activation, which is an energy conservation mechanism, results in the bundling of actin with cofilin, eventually forming rods but also potentially predisposing neurons to synaptic deficits [18]. Cofilin hyperactivation can also affect the mitochondrial functions by altering the mitochondrial membrane permeability and facilitating the release of cell death mediators like cytochrome C [15]. Furthermore, oxidative stress and ATP depletion states were found to oxidize cofilin and translocate it to mitochondria to induce cell death [8]. Cofilin is essential for neuronal development, which is why complete deletion of n-cofilin resulted in an embryonic-lethal phenotype [19]. Cofilin expression during the embryonic phase is slightly higher than that in the postnatal phase [20]. However, cofilin rods were detectable in the brains of normal aging rats, and this indicates that cofilin expression in the elderly is higher and results in cofilin rod formation [18].
In our studies using PC12 cells, cofilin was found to be hyper-activated by prominent dephosphorylation against the oxidative and OGD insults in a time-dependent manner. The undetectable phosphorylated cofilin levels after 4 h of OGD imply a complete activation of cofilin in response to OGD stress. Moreover, phosphatase inhibitor pretreatment inhibited cofilin dephosphorylation and activation in OGD conditions. This observation was complemented by our immunofluorescence studies, wherein morphological changes in the actin cytoskeleton, and thereby phosphocofilin expression in OGD, was demonstrated. Prominently, marked changes in actin filaments during OGD, such as aggregation of actin filaments in the cell periphery and the intense staining of FITC (green) in these regions, was also noted, confirming the enhanced actin-severing activity of cofilin. This inhibition was also found to ameliorate cell death during OGD. We have also successfully attributed the role of signaling mediators, SSH1L and calcineurin phosphatases, in the dephosphorylation and activation of cofilin in OGD. These results explain the cofilin predominance in ischemia conditions by dephosphorylation, which was mainly mediated by SSH1L phosphatase upon activation by calcineurin. Together, our results implicate that actin cytoskeletal degenerations in cerebral ischemia are mediated by cofilin in vitro and, if subsequently proven in animal studies, targeting this protein could be beneficial in ischemia related neuronal damage.
Cofilin differentially modulates actin dynamics depending on the ratio of cofilin to actin. At low cofilin to actin ratios, cofilin acts to sever actin filaments, whereas at high cofilin to actin ratios, cofilin nucleates actin assembly and stabilizes F-actin [3]. Accordingly, in OGD conditions with low ATP, cofilin stabilizes F-actin and leads to the formation of cofilin-actin rods which are neuroprotective, but only transiently because they cause neurite degeneration when the conditions persist for longer durations. In addition to controlling actin dynamics, cofilin also plays an important role in oxidative stress-induced cell death via the mitochondrial mechanism [15,8]. During oxidative stress, the oxidized cofilin loses its affinity for actin and then subsequently translocates to mitochondria, where it induces the opening of the permeability transition pore and cytochrome C release [8]. Because of the deleterious effects of cofilin during ischemia, targeting this protein could improve neuronal survival and reduce neurodegeneration. Cofilin1 siRNA-treated primary cultures of cortical neurons showed a highly significant increase in neuronal viability compared to scrambled siRNA-treated neurons. The number of viable neurons was approximately doubled in the cofilin1 siRNA-treated group. Along with the increase in the cell survival, significant reduction in cofilin mitochondrial translocation and caspase 3 cleavage was detected in cofilin1 siRNA-treated neurons, and these results are consistent with previously published work [15,8,16]. Cofilin dynamics in PC12 cells and primary cultures of cortical neurons showed a differential pattern. In primary cultures of cortical neurons, Western blot analysis showed a significant upregulation of cofilin after 1 h of OGD, an affect which was not seen in PC12 cells. In primary cultures of cortical neurons, nuclear cofilin was observed to be downregulated during OGD, which led us to conclude that nuclear phosphocofilin was activated by dephosphorylation and then translocated to cytosol and subsequently to mitochondria, which is the final destination involved in the induction of apoptosis (Figure 7). The translocation of activated nuclear cofilin to the cytosol contributes to downregulation in the nuclear fractions and upregulation in the cytosolic fractions, as observed in our study.
Figure 7. Cofilin dynamics during ischemia.
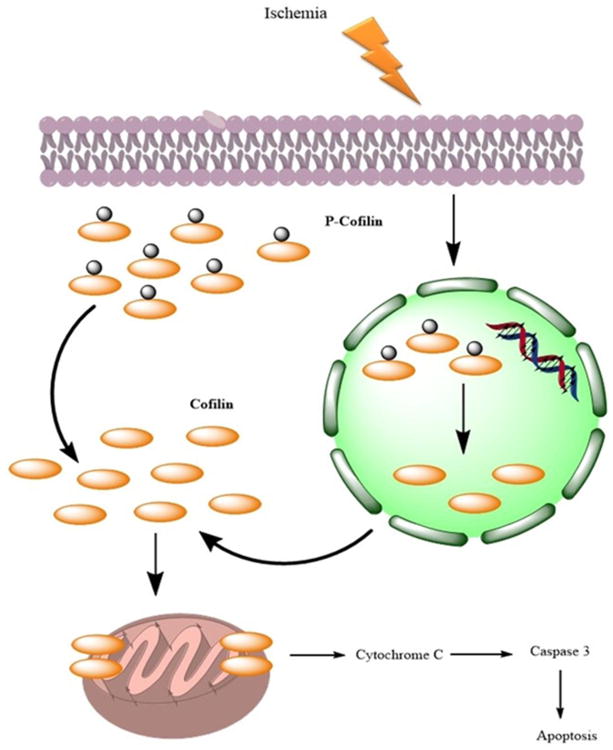
In normal conditions, a large proportion of cofilin exists in the nucleus and cytosol in phosphorylated form (phosphocofilin). However, during ischemia, nuclear phosphocofilin is dephosphorylated and translocated to the cytosol and then subsequently to the mitochondria, where it induces cytochrome C release and subsequent apoptosis.
The cytoskeleton of a neuron is the dynamic framework that has a potential to salvage the penumbra (partly viable neurons adjacent to core of the stroke injury) by regeneration, but the presence of certain mediators involved in the regulation outrage this dynamic nature, leading to neurodegeneration. Studies have demonstrated the active involvement of MAP (microtubule associated protein), tau and cofilin aggregations in the progression of disease states such as Alzheimer's and dementia [21]. These neurodegenerative pathologies deteriorate the disease state, and thereby also the regenerative strategies, due to their effect on the cytoskeleton. In addition to this, the cytoskeleton, which forms the major neuron structure, is essential for protein localizations and the transport of molecules to-and-fro between dendrites and axons [22]. Any gross changes associated with the cytoskeleton can immediately trigger necrosis or apoptosis. Supporting this, a study demonstrated that activated AC proteins were found to disrupt the actin cytoskeleton in response to ischemia in renal tubular epithelial cells [23]. Additionally, a recent study revealed the active involvement of cofilin in amyloid β-induced apoptosis and neurodegeneration [24]. These are consistent with our investigations, in which we elucidated cofilin's potential involvement in mediating neurocytoskeletal degenerations in a disease state such as ischemia. In astrocytes, calcineurin, a cofilin-activating protein, was found to be highly up-regulated by amyloid β [25]. Furthermore, up-regulation of AC and pyridoxal-5′-phosphate phosphatase/chronophin mediate astroglial apoptosis after status epilepticus [26]. In activated microglia, cofilin is involved in the regulation of NADPH oxidase activity [27] and phagocytosis [28,29]. Accordingly, future studies are warranted to elucidate the role of cofilin in microglial activation and astrocytes during ischemia.
In summary, the question of cytoskeletal aberrations and cofilin involvement was addressed by our current investigations. We have successfully demonstrated cofilin's active role in ischemic conditions, and the cytoskeletal alterations studied by fluorescence methods confirmed cofilin's potential to alter the integrity of filaments in ischemia and thereby facilitate cell death. Furthermore, siRNA technique confirmed the active involvement of cofilin in apoptotic cell death during ischemia. We believe that targeting the actin-binding protein, cofilin, which is a subject of many adversaries and is involved in mediating cell death in ischemia, would be an effective strategy in treating human stroke.
Acknowledgments
The study was partly funded by a grant from NIH (R00AT004197) and start-up funds from The University of Toledo to ZAS. Qasim Alhadidi was supported by Higher Committee for Education Development in Iraq (www.hcediraq.org). The authors would like to thank Charisse N. Montgomery for her assistance in the manuscript editing.
References
- 1.Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. The New England journal of medicine. 1995;333(24):1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 2.Fuchs E, Cleveland DW. A Structural Scaffolding of Intermediate Filaments in Health and Disease. Science. 1998;279(5350):514–519. doi: 10.1126/science.279.5350.514. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein BW, Bamburg JR. ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 2010;20(4):187–195. doi: 10.1016/j.tcb.2010.01.001. doi:S0962-8924(10)00002-4 [pii]10.1016/j.tcb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, Nosworthy NJ. Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiological Reviews. 2003;83(2):433–473. doi: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- 5.Suurna MV, Ashworth SL, Hosford M, Sandoval RM, Wean SE, Shah BM, Bamburg JR, Molitoris BA. Cofilin mediates ATP depletion-induced endothelial cell actin alterations. Am J Physiol Renal Physiol. 2006;290(6):F1398–1407. doi: 10.1152/ajprenal.00194.2005. [DOI] [PubMed] [Google Scholar]
- 6.Bamburg JR, Wiggan OP. ADF/cofilin and actin dynamics in disease. Trends Cell Biol. 2002;12(12):598–605. doi: 10.1016/s0962-8924(02)02404-2. doi:S0962892402024042 [pii] [DOI] [PubMed] [Google Scholar]
- 7.Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nature Cell Biology. 2000;2(9):628–636. doi: 10.1038/35023579. [DOI] [PubMed] [Google Scholar]
- 8.Klamt F, Zdanov S, Levine RL, Pariser A, Zhang Y, Zhang B, Yu LR, Veenstra TD, Shacter E. Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat Cell Biol. 2009;11(10):1241–1246. doi: 10.1038/ncb1968. doi:ncb1968 [pii]10.1038/ncb1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nada SE, Shah ZA. Preconditioning with Ginkgo biloba (EGb 761(R)) provides neuroprotection through HO1 and CRMP2. Neurobiol Dis. 2012;46(1):180–189. doi: 10.1016/j.nbd.2012.01.006. doi:S0969-9961(12)00021-6 [pii]10.1016/j.nbd.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olechnowicz SW, Fedele AO, Peet DJ. Hypoxic induction of the regulator of G-protein signalling 4 gene is mediated by the hypoxia-inducible factor pathway. PLoS One. 2012;7(9):e44564. doi: 10.1371/journal.pone.0044564. doi:10.1371/journal.pone.0044564PONE-D-12-18040 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nada SE, Tulsulkar J, Raghavan A, Hensley K, Shah ZA. A derivative of the CRMP2 binding compound lanthionine ketimine provides neuroprotection in a mouse model of cerebral ischemia. Neurochem Int. 2012;61(8):1357–1363. doi: 10.1016/j.neuint.2012.09.013. doi:S0197-0186(12)00298-7 [pii]10.1016/j.neuint.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dimauro I, Pearson T, Caporossi D, Jackson MJ. A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res Notes. 2012;5:513. doi: 10.1186/1756-0500-5-513. doi:1756-0500-5-513 [pii]10.1186/1756-0500-5-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787(11):1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Shibasaki F, Mizuno K. Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J Biol Chem. 2005;280(13):12683–12689. doi: 10.1074/jbc.M411494200. [DOI] [PubMed] [Google Scholar]
- 15.Chua BT, Volbracht C, Tan KO, Li R, Yu VC, Li P. Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat Cell Biol. 2003;5(12):1083–1089. doi: 10.1038/ncb1070. doi:10.1038/ncb1070ncb1070 [pii] [DOI] [PubMed] [Google Scholar]
- 16.Posadas I, Perez-Martinez FC, Guerra J, Sanchez-Verdu P, Cena V. Cofilin activation mediates Bax translocation to mitochondria during excitotoxic neuronal death. J Neurochem. 2012;120(4):515–527. doi: 10.1111/j.1471-4159.2011.07599.x. [DOI] [PubMed] [Google Scholar]
- 17.Huang TY, Minamide LS, Bamburg JR, Bokoch GM. Chronophin mediates an ATP-sensing mechanism for cofilin dephosphorylation and neuronal cofilin-actin rod formation. Developmental cell. 2008;15(5):691–703. doi: 10.1016/j.devcel.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cichon J, Sun C, Chen B, Jiang M, Chen XA, Sun Y, Wang Y, Chen G. Cofilin aggregation blocks intracellular trafficking and induces synaptic loss in hippocampal neurons. J Biol Chem. 2012;287(6):3919–3929. doi: 10.1074/jbc.M111.301911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurniak CB, Perlas E, Witke W. The actin depolymerizing factor n-cofilin is essential for neural tube morphogenesis and neural crest cell migration. Dev Biol. 2005;278(1):231–241. doi: 10.1016/j.ydbio.2004.11.010. doi:S0012-1606(04)00802-4 [pii]10.1016/j.ydbio.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 20.Bellenchi GC, Gurniak CB, Perlas E, Middei S, Ammassari-Teule M, Witke W. N-cofilin is associated with neuronal migration disorders and cell cycle control in the cerebral cortex. Genes Dev. 2007;21(18):2347–2357. doi: 10.1101/gad.434307. doi:21/18/2347 [pii]10.1101/gad.434307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi M, Tsujioka Y, Yamada T, Tsuboi Y, Okada H, Yamamoto T, Liposits Z. Glycosylation of microtubule-associated protein tau in Alzheimer's disease brain. Acta neuropathologica. 1999;97(6):635–641. doi: 10.1007/s004010051040. [DOI] [PubMed] [Google Scholar]
- 22.Lipton SA. Neuronal protection and destruction by NO. Cell death and differentiation. 1999;6(10):943–951. doi: 10.1038/sj.cdd.4400580. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz N, Hosford M, Sandoval RM, Wagner MC, Atkinson SJ, Bamburg J, Molitoris BA. Ischemia activates actin depolymerizing factor: role in proximal tubule microvillar actin alterations. The American journal of physiology. 1999;276(4 Pt 2):F544–551. doi: 10.1152/ajprenal.1999.276.4.F544. [DOI] [PubMed] [Google Scholar]
- 24.Woo JA, Jung AR, Lakshmana MK, Bedrossian A, Lim Y, Bu JH, Park SA, Koo EH, Mook-Jung I, Kang DE. Pivotal role of the RanBP9-cofilin pathway in Abeta-induced apoptosis and neurodegeneration. Cell death and differentiation. 2012;19(9):1413–1423. doi: 10.1038/cdd.2012.14. doi:cdd201214 [pii]10.1038/cdd.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim D, Iyer A, Ronco V, Grolla AA, Canonico PL, Aronica E, Genazzani AA. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia. 2013;61(7):1134–1145. doi: 10.1002/glia.22502. [DOI] [PubMed] [Google Scholar]
- 26.Kim JE, Ryu HJ, Kim MJ, Kim DW, Kwon OS, Choi SY, Kang TC. Pyridoxal-5′-phosphate phosphatase/chronophin induces astroglial apoptosis via actin-depolymerizing factor/cofilin system in the rat brain following status epilepticus. Glia. 2010;58(16):1937–1948. doi: 10.1002/glia.21063. [DOI] [PubMed] [Google Scholar]
- 27.Rasmussen I, Pedersen LH, Byg L, Suzuki K, Sumimoto H, Vilhardt F. Effects of F/G-actin ratio and actin turn-over rate on NADPH oxidase activity in microglia. BMC Immunol. 2010;11:44. doi: 10.1186/1471-2172-11-44. doi:1471-2172-11-44 [pii]10.1186/1471-2172-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hadas S, Spira M, Hanisch UK, Reichert F, Rotshenker S. Complement receptor-3 negatively regulates the phagocytosis of degenerated myelin through tyrosine kinase Syk and cofilin. J Neuroinflammation. 2012;9:166. doi: 10.1186/1742-2094-9-166. doi:1742-2094-9-166 [pii] 10.1186/1742-2094-9-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gitik M, Kleinhaus R, Hadas S, Reichert F, Rotshenker S. Phagocytic receptors activate and immune inhibitory receptor SIRPalpha inhibits phagocytosis through paxillin and cofilin. Front Cell Neurosci. 2014;8:104. doi: 10.3389/fncel.2014.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]