

Abstract
Dendritic cells (DC) are professional antigen presenting cells (APC) that are traditionally divided into two distinct subsets: myeloid DC (mDCs) and plasmacytoid DC (pDCs). pDCs are known for their ability to secrete large amount of IFN-α. Apart from IFN-α production, pDCs can also process antigen and induce T-cell immunity or tolerance. In several solid tumors, pDCs have been shown to play a critical role in promoting tumor immunosuppression. We investigated the role of pDCs in the process of glioma progression in the syngeneic murine model of glioma. We show that glioma-infiltrating pDCs are the major APC in glioma and are deficient in IFN-α secretion (p < 0.05). pDC depletion leads to increased survival of the mice bearing intracranial tumor by decreasing the number of regulatory T-cells (Treg) and by decreasing the suppressive capabilities of Tregs. We subsequently compared the ability of mDCs and pDCs to generate effective anti-glioma immunity in a GL261-OVA mouse model of glioma. Our data suggest that mature pDCs and mDCs isolated from naïve mice can be effectively activated and loaded with SIINFEKL antigen in vitro. Upon intra-dermal injection in the hind leg, a fraction of both types of DCs migrate to the brain and lymph nodes.. Compared to mice vaccinated with pDC or control mice, mice vaccinated with mDCs generated a robust Th1 type immune response, characterized by high frequency of CD4+Tbet+ T-cells and CD8+Siinfekel+ T-cells. This robust anti-tumor T-cell response resulted in tumor eradication and long-term survival in 60% of the animals (p<0.001).
Keywords: Glioma, brain tumor, regulatory T cells, plasmacytoid dendritic cells, myeloid dendritic cells, vaccine, immunization
Introduction
Plasmacytoid dendritic cells (pDCs) are a unique subset of DCs, that have been traditionally defined as professional interferon-α (IFN-α) producing cells, which upon activation by infectious agents produce large amounts of powerful pro-inflammatory cytokine IFN-α. Due to its ability to effectively stimulate host immune response in the setting of various solid tumors (1–3), IFN-α was the first cytokine to be approved for cancer treatment (4). IFN-α production by pDCs is regulated by the downstream signaling via the universal adapter protein MyD88 (myeloid differentiation primary response 88), upon stimulation of toll-like receptor (TLR)-7 and 9, which in turn acts via the constitutively expressed transcription factor IRF7 and the inflammatory transcription factor NF-κB, thereby initiating transcription of type I IFN (5). Recent studies indicate that pDCs also express MHCII molecules, undergo a maturation process similar to that of mDCs, can function as APCs and promote T cell–mediated immunity or the maintenance of self-tolerance (6–8). Whether the maturation of pDCs leads to immunity or tolerance depends on the context of antigen presentation. On one hand, pDCs can promote tolerance by presenting antigen (Ags) to CD4+ T cells and inhibiting their activation (7) or inducing Treg, which confer tolerance to cardiac allografts (9), prevent asthmatic reactions to inhaled Ags (10), and promote tumor progression in several solid tumors (11–13). On the other hand, activated pDCs can produce large amounts of IFN-α (1), induce strong allogeneic T-cell responses (14) and prime CD4+ and CD8+ T cells against viruses (15) or tumor antigens (16) and induce effective antitumor response (17, 18).
In several solid tumors, pDC infiltration has been associated with disease progression, poor prognosis and has been shown to promote tumor immunosuppression (11, 19, 20). Suggested mechanisms for tumor induced pDC dysfunction include: recruitment of immature pDCs, lack of expression of co-stimulatory molecules such as CD80, CD83 and CD86 (21, 22) and altered functioning of existing pDCs, such as diminished IFN-α secretion (21), mediated by tumor associated immunosuppressive cytokines. pDCs also contribute to tumor-associated immunosuppression by mediating mature Treg accumulation (23) and by releasing indoleamine 2,3-dioxygenase (IDO) which is a powerful promoter of Treg activation (24). Impaired production of IFN-α and accumulation of Tregs significantly impair local immune surveillance allowing tumors to escape IFN- α associated immune responses.
Although pDC’s role as an impaired professional IFN-α producing cell is well characterized in the context of tumors, their role as a professional APCs is not. Both in humans and mice, entire DC pool can be divided into two-subset, a larger subset consisting of mDCs and a smaller subset consisting of pDCs. Murine pDCs are characterized by expression of CD11c and BST-2 (CD317) and mDCs by expression of CD11c and are very distinct in their function and characteristics. Classically, mDCs have been shown to play the critical role in the induction of immunity or tolerance. Because of this decisive role played by mDCs in the induction of immunity, DCs have been widely used as a vehicle for DC-mediated immunotherapy, in which DCs loaded with tumor antigens are injected into patients with cancer to stimulate the anti-tumor T-cell response (25). Since the number of circulating natural DCs is low, virtually all vaccination studies use DCs differentiated ex vivo from monocytes or CD34+ progenitors (25). However, recent reports suggest that DCs matured ex vivo are less effective than their natural counterparts in activating T-cells and inducing effective anti-tumor immunity (26–28). There has been no study so far that compares the capacity of mDCs and pDCs to activate and prime naïve T cells.
Malignant gliomas (MGs), consisting of anaplastic astrocytoma (WHO grade III) and glioblastoma multiforme (GBM) (WHO grade IV), are the most common primary brain tumors in adults and are associated with dismal prognosis (29). MGs are associated with a potently immunosuppressive tumor microenvironment and efficiently evade the host antitumor response. We have previously shown that one of the hallmark features of glioma immunosuppression is the presence of Tregs (30–34). Besides presence of Tregs, several immune modulating mechanisms have been implicated in potentiating the immunosuppressive glioma microenvironment including the suppression of APC function(s) via expression of immunosuppressive cytokines, such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-b), which contributes to the inhibition of the effector T cells (30). Even though the paradigm of immune privilege suggests that classical DCs are absent from the brain (35), recent reports have revealed that both pDCs and mDCs are present in human brain which may contribute to orchestration of the local immune response (36–39).
In this study, we show that human grade III MGs have the highest infiltration of pDCs. In the murine model of glioma, intracranial (ic) tumor implantation leads to selective maturation of pDCs, characterized by up-regulation of MHC-II and B7-H1 (CD 274) on pDCs. Glioma infiltrating pDCs are deficient in producing IFN-α and the selective depletion of pDCs during the course of disease in BDCA2-DTR transgenic (Tg) mice (in which injection of diphtheria toxin (DT) in the mice results in selective depletion of pDCs) (40) results in increased median survival of the mice bearing ic tumor. pDC depletion also leads to decrease in the number of ICOS+ Tregs in the brain of glioma bearing mice and the Tregs from BDCA-2 DTR Tg mice are less suppressive compared to the Tregs from WT mice. These finding suggest, that in the initial stages of glioma progression, pDCs skew the immune response towards tolerance, rather than the efficient induction of anti-glioma immunity. Since our results show that selectively pDCs undergo maturation in the context of glioma and contribute to glioma-mediated immunosuppression, we compared the immune response generated by pDCs vs. mDCs in a DC-based vaccine strategy. We found that mDCs are much better at inducing the anti-glioma Th-1 immune response, when compared to pDCs and 60% of the mice vaccinated with mDCs survived long term. In conclusion, our study indicates that host pDCs promote glioma progression in the murine model of glioma and in the context of DC-based vaccination pDCs are less effective than mDCs in generating anti-glioma response.
Materials and Method
Mice
C57BL/6 (WT) and BDCA-2-DTR (B6 background) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained in the University of Chicago Carlson Barrier Facility. BDCA-2-DTR is a transgenic mouse model where pDC specific promoter, BDCA-2 is under the control of diphtheria toxin receptor (DTR) promoter. Administration of intra-peritoneal (ip) diphtheria toxin (DT) in these mice results in selective depletion of pDCs (40). All mice were intracranially (ic) injected with syngeneic GL261 or GL261-OVA cells (4 × 105 or 2 × 105 cells) between the ages of 6 and 8 weeks, as described previously (30). All animal work was reviewed and approved by the University of Chicago Institutional Animal Care and Use Committee. All surgical procedures were completed in accordance with NIH guidelines on the care and use of laboratory animals for research purposes. Mice were euthanized by CO2 and then by cervical dislocation. Following ic injection pDC depletion was carried out by ip injection of DT (Sigma-Aldrich; St. Louis MO) at 100–120 ng/mouse as previously described (40). For vaccination, mice were intra-dermally (id) injected in the hind leg (41) with pDCs or mDCs in PBS.
Tumor Cells
GL261 cells were obtained from the NCI Frederick National Tumor Repository Lab and cultured in Dulbecco’s Modified Eagle’s Media supplemented with 10% fetal calf serum, as well as streptomycin (100 mg/mL) and penicillin (100 U/mL) at 37°C in a humidified atmosphere of 95% air/5% CO2. GL261-OVA cells, GL261 line stably expressing pCDNA 3.1 containing artificial model antigen, chicken Ovalbumin Ag (OVA), were generated in house by selecting the stably transfected cells with media containing 200 µg/ml G418 (geneticin). Chicken OVA cDNA was amplified by PCR using pAc-neo-OVA as a template. The 5′ primer sequence was 5′-AACGCGGATCCACCATGGGCTCCATCGGCGC-3′, and the 3′ primer sequence was 5′-GAGCACCGCTCGAGTTTTTAAGGGGAAACACATC-3′. OVA expression plasmid was created by ligation of OVA cDNA into pcDNA3.1/Hygro(+) (Invitrogen) vector between BamHI and XhoI sites. To create GL261-OVA cell lines, OVA plasmid was used to transfect GL-261 cell lines and selected using hygromycin B (Invitrogen). OVA expression (45 kDa) in GL261-OVA cell lines was confirmed by Western blotting (data not shown) using anti-OVA mAbs obtained from Sigma-Aldrich. After transfection, stable clones were isolated by a combination of drug selection, flow cytometry sorting with the H-2Kb-SIINFEKL–specific Ab 25.D1.16, and cell cloning at limiting dilution. The OVA expression was validated by immunoblot analysis. All cell culture products were purchased from Gibco Invitrogen (Grand Island; NY).
Antibodies and flow cytometry
Single cell suspensions were made from brain, cervical lymph node (cLN) or spleen as described in our previous publication (30). For cellular staining, cells were incubated with: anti-CD4 (RM4-5; eBioscience), anti-CD3 (145-2C11; eBioscience), anti-CD8 (53-6.7; eBioscience), PD1 (J43; eBioscience), CD45 (30-F11; eBioscience), BST-2 (eBio927; eBioscience), ICOS (C398-4A; eBioscience), TLR-9 (M9.D6; eBioscience), MHC-II (M5/114.15.2; eBioscience), Lag-3 (C9B7W; eBioscience), TIM (8B.2C12; eBioscience), CD11c (N418; eBioscience), B7-H1 (MIH5; eBioscience), CD45.2 (104; eBioscience), TGF-β (TW7-20B9; Biolegend; San Diego CA), and CD45.1 (A20; eBioscience) in PBS + 2 % bovine serum albumin (Sigma-Aldrich) for 30 min on ice. For intracellular cytokine analysis, T-cells were treated with cell stimulation cocktail with protein transport inhibitors (PMA/ionomycin with brefeldin A and monensin) (eBioscience; San Diego CA) for 5 hrs. and pDCs were stimulated with type A CpG with protein transport inhibitors for 7 hrs. at 37°C in a humidified atmosphere of 95% air/5% CO2. Cells were then fixed and permeabilized overnight at 4°C using fix-perm buffer (eBioscience) according to manufacturer’s instructions and stained with intracellular antibodies: anti-FoxP3-FITC/APC (FJK-16s; eBioscience), IFN-α (RMMA-1; Interferon Source; Piscataway NJ), Tbet (eBio4B10; eBioscience), IL-10 (JES5-16E3; BD-biosciences) and TGF-β (TW7-20B9; Biolegend; San Diego CA) for 30 min on ice. All the flow cytometric analysis was done using BD FACSCalibur (BD biosciences; San Jose CA) flow cytometer. For in vitro T-cell suppression assay, single cell suspensions were made from lymph nodes (LN) and spleens and then were sorted into CD4+CD25+ and CD4+ CD25− cells.
DC stimulation for vaccination
pDCs (CD3-CD45+CD11c+BST-2+) and mDCs (CD3-CD45+CD11c+BST-2-) were directly isolated from pooled spleen and LN of mice using the fully closed BD FACSAria Cell Sorter (BD biosciences; San Jose CA). This procedure resulted in clinically applicable purified pDCs, which had an average purity of 85%. Following isolation, pDCs and mDCs were cultured overnight at a concentration of 106 cells/mL in X-VIVO 15 (Lonza; Hopkinton MA) containing 2% FBS, supplemented with 10 ng/mL recombinant murine interleukin-3 (rIL-3; Peprotech; Rocky Hill NJ) or GM-CSF (Peprotech). For the vaccination, pDCs and mDCs were subsequently activated for 6 hours by addition of type A CpG (ODN 1585; Invivogen; San Diego CA) or LPS-EB (tlrl-eblps; Invivogen). During the last 3 hours of activation, pDCs and mDCs were loaded with the ova 257–264 peptide (Anaspec; Frement CA) (42). The peptide- loaded pDCs (5,000 cells) and mDCs (5, 000 cells) were administered intradermally (id) in the hind leg of the mouse. This procedure gave rise to mature pDCs and mDCs meeting the following release criteria; more than 50% viability, IFN-a secretion, high expression of MHC class I, MHC class II, CD83, CD80 and CD86 as previously described by Tel et al (42).
T cell suppression assay
In vitro suppression assays were carried out in RPMI/10% FCS in 96-well V-bottom plates (Costar; Corning, NY) with 1 × 106 CD4+ responder cells, titrated amounts of FACS-sorted CD4+CD25+ cells and 4 × 106 irradiated (2500rad) sorted antigen-presenting cells (CD4- spleenocytes). CD4+ cells were labeled with CellTrace Violet (C34557; Life Technologies; Carlsbad CA). Stimulation was carried out with plate-bound anti-CD3 (145-2C11; 1 µg/mL) and after 72 hours at 37°C, proliferation was determined using a FACS.
In-vitro DC stimulation and activation
mDC and pDC populations were sorted from mice 1 week after GL261 ic. tumor implantation. DCs were then pulsed with SIINFEKL (2µm) for 4 hours. MACS purified OT1 T-cells were then labeled with CTV proliferation dye and co-cultured with the pulsed DCs for 48 hours. Cells were then harvested and assessed via flow cytometry for number of cell divisions.
Human tissue microarray and histology
A human glioma microarray with normal brain control (25 tumor samples plus normal brain control) was obtained from US Biomax, Rockville MD, was deparaffinized in xylene and then rehydrated. After deparaffinization and rehydration, the microarray was treated with antigen retrieval buffer (S1699, DAKO; Carpenteria CA) in a steamer for 20 minutes. Human anti-BDCA-2 monoclonal antibody (Clone: 10E6.1) was obtained from Millipore, Billerica MA. The antibody was applied (1:100) on the tissue microarray for 1-hour at room temperature. Normal human tonsil tissue was used as positive control (as suggested by the manufacturer’s instruction). The antigen-antibody binding was detected by Bond Polymer Refine Detection system (DS9800, Leica Microsystems; Buffalo Grove IL).
Statistics
Survival was defined as the time from injection of GL261/GL261-OVA cells to day 150 of the time course. Kaplan-Meier curves were generated and the survival distributions were compared using log-rank test. All other data are presented as Mean±SEM. Comparisons between two groups were conducted using Student’s t test or Mann Whitney test as appropriate, and differences between more than two groups were assessed using ANOVA with Tukey’s post-hoc test. All analyses were conducted using GraphPad Prism version 4.0 (GraphPad Software, Inc.). All reported p values were two sided and were considered to be statistically significant at * p<0.05, **p<0.01, ***p<0.001.
Results
Infiltration of pDCs in human glioma
pDCs have been implicated to play a critical role in many solid tumors (11, 13, 43), however, the role of pDCs in glioma progression is unknown. Hussain et al. analyzed DC infiltration in human GBM sample and normal brain by flow cytometry and showed that there were less pDCs in GBM (grade IV) specimen than in normal brain (44). To establish the correlation between pDC infiltration and all glioma grades, human glioma tissue microarray, containing 25 samples of all representative glioma grades, was stained with anti-BDCA-2 antibody. The level of staining was initially graded by the author and then by an independent, blinded neuro-pathologist on a scale of 0 to 5, where 0 signifies no staining and 5 signifies the highest amount of staining (positive control). In human glioma, pDC infiltration steadily and significantly increased from grade I to III (Fig. 1A–D), however, in accordance with previous observation, no identifiable pDC staining was observed in grade IV (Fig. 1E). There was a statistically significant difference in pDC staining between low grade (grade I & II) and high-grade anaplastic (grade III) glioma (p < 0.05) (Fig. 1F).
Fig 1. Infiltration of pDCs in human glioma.
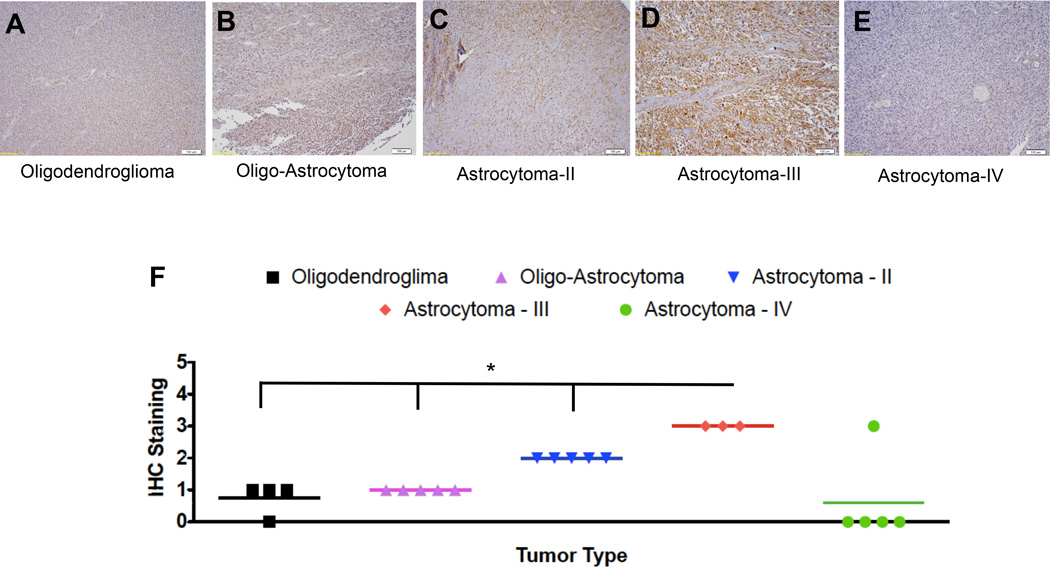
Human tissue microarray, containing 25 different glioma samples of all grades of glioma, was stained for the presence of pDCs by anti-BDCA-2 antibody. Level of staining was graded on a scale of 0 to 5 by an independent clinical pathologist, where 0 signifies no staining and 5 signifies highest amount of staining (positive control). Immunohistochemical (IHC) staining for pDCs in A) Oligodendroglioma, B) Oligo-Astrocytoma, C) Grade II Astrocytoma, D) Grade III Astrocytoma and E) Grade IV Astrocytoma. F) Comparison of the level of BDCA-2 staining amongst all grades. Each grade contains 3–5 different specimens. * p<0.05, **p<0.01, ***p<0.001.
pDCs are present in naïve mouse brain and predominantly up-regulate MHC-II expression in a mouse model of glioma
Presence of pDCs and their phenotype has been well characterized in murine BM and various other organs and their role has been implicated in the pathophysiology of several solid tumors (19, 45, 46). To delineate the DC population in the brain of naïve mice and compare that to other organs, DCs from brain, cLN and spleen were compared. The majority (>80%) of DCs in the cLN and spleen were CD11c+BST-2− mDCs. Interestingly in the brain, DCs were split almost equally between CD11c+BST-2+ pDCs and CD11c+BST-2− mDCs (Fig. 2A). The phenotype of the DCs from each tissue was analyzed for the presence of activating (MHC-II, CD80) or inhibitory markers (B7-H1). There was no statistically significant difference noted in the expression of the three molecules between pDCs and mDCs from the cLN and spleen. In the brain, there was no statistically significant difference between MHC-II and CD80 expression, however, there was a significantly higher expression of the inhibitory molecule B7-H1 (MFI 2636 +/− 500.5 pDC vs. 1300 +/− 185.1 mDC; p < 0.01) on pDCs, when compared to the mDCs (Fig. 2B). Thus, our data show that there are resident pDCs and mDCs in the naïve mice brain and at baseline, brain-resident pDCs express higher level of inhibitory marker B7-H1 when compared to mDCs.
Fig 2. pDCs are present in naïve mice brain.
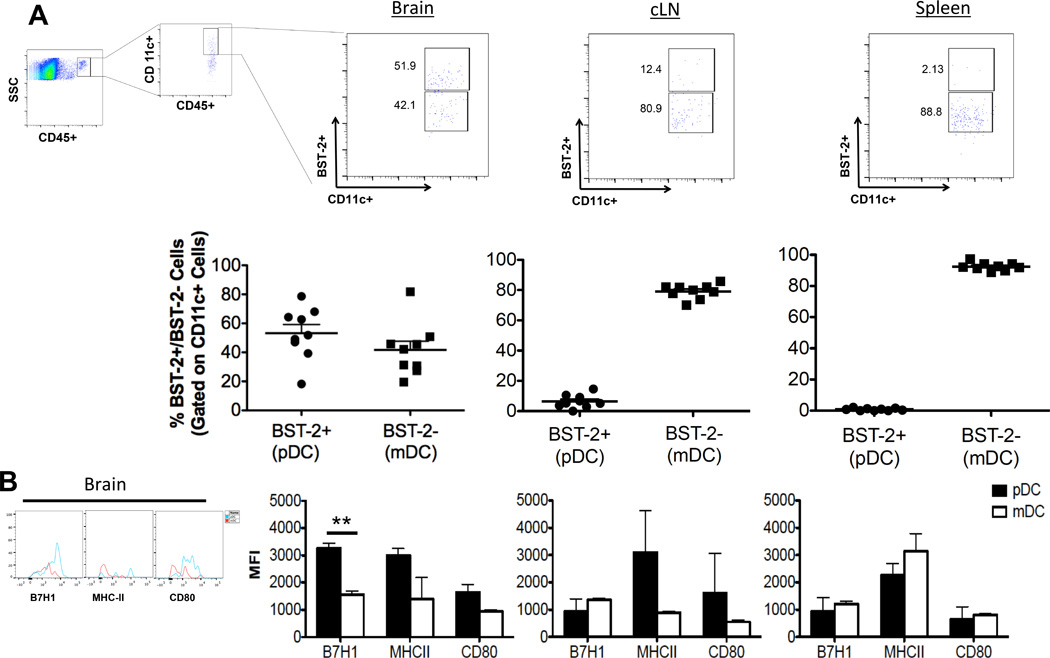
Dendritic cells were isolated from brain, cLN and spleen of naïve mice, stained for DC markers and analyzed using flow cytometry. A) DC distribution in the brain, cLN and spleen of naive mice. DCs were identified as CD45+CD11c+ cells, pDCs were defined as CD11c+BST-2+ and mDCs were identified as CD11c+BST-2−. This gating strategy was applied to identify pDCs and mDCs throughout the paper. For every pDC and mDC analysis 500,000 events were acquired. B) Phenotypical analysis of pDCs vs. mDCs from brain, cLN and spleen by comparing mean fluorescence intensity (MFI) of MHC-II, B7-H1 and CD80. pDCs and mDCs were defined by previously described gating strategy and MFI was calculated for both pDC and mDC population simultaneously (representative plots are shown). Same gating strategy was applied to calculate MFI’s throughout the paper. Error bars are derived from two to three separate experiments, each done in triplicate, representing the standard error between experiments throughout the manuscript. (n = 3–5 mice/group/experiment). * p<0.05, **p<0.01, ***p<0.001.
Next, to further outline which DC population acts as major APC in the orthotropic GL261 cell-based murine model of glioma, both DC cell populations were analyzed from the brain and cLN of tumor bearing mice at 1 week and 3 weeks post-tumor implantation (wpo) (Figure 3). There was no significant difference in the pDC or mDC frequency in the brain at 1week wpo. However, the frequency of pDCs in the tumor draining cLN increased at 1wpo (6.42% +/− 1.5% naïve vs. 13.45% +/− 1.67% 1 wpo; p < 0.01). By week 3 of tumor progression, the frequency pDCs decreased drastically in the brain compared to 1wpo (62.83% +/− 3.283% 1wpo vs. 3.09% +/− 2.09% 3wpo; p < 0.01) and frequency of mDCs increased (34.36% +/− 3.087 1 wpo vs. 95.10% +/− 2.34% 3 wpo; p < 0.001). The same trend in pDC (13.46% +/− 1.7% 1wpo vs. 6.6% +/− 1.1% 3wpo; p < 0.01) and mDC (83.97% +/− 2.0% 1wpo vs. 90.43% +/− 1.25% 3wpo; p < 0.05) frequency was observed in the cLN (Fig. 3A). There was a marked increase in MHC-II expression by pDCs in the brain (3000 +/− 249 Naive vs. 17509 +/− 3181 1wpo; p < 0.01) and cLN (3094 +/− 1539 Naive vs. 17293 +/−1767 1wpo; p < 0.01) at 1wpo compared to naïve control, however there was no difference in MHC-II expression by mDCs in the brain and a slight increase in MHC-II expression by mDCs in the cLN (884.7 +/− 67.7 Naive vs. 5794 +/− 744.6 1wpo; p < 0.01). There was also statistically significant increase in B7-H1 expression on tumor infiltrating pDCs (3261 +/− 187 Naive vs. 4712 +/− 387.7 1wpo; p < 0.05) in the brain and cLN (929.7 +/− 473.7 Naive vs. 2507 +/− 214.2 1wpo; p < 0.05). Both in naïve mice (3261 +/− 187 pDCs vs. 1557 +/− 132.1 mDCs ; p < 0.01) and 1wpo (4712 +/− 387.7 pDCs vs. 2062 +/− 143.1 mDCs ; p < 0.001) pDCs in the brain had higher expression of B7-H1 compared to mDCs (Fig. 3B). Collectively these in vivo data show that in the context of orthotopic murine model of glioma, pDCs undergo maturation by up-regulating MHC-II along with up-regulation of inhibitory molecule B7-H1. To analyze which group of DCs is better at antigen presentation and stimulate immune response vs. which group is more inhibitory, we compared the ability of pDCs and mDCs to present antigen and stimulate proliferation of OT-1 T cells in-vitro. Our results show that in-vitro pulsed mDCs from tumor bearing mice are significantly better at presenting antigen to cause T-cell proliferation than pDCs (Fig. 3C). Thus we concluded that pDCs are more inhibitory when compared to mDCs and most likely contribute to the profound immunosuppression associated with malignant glioma.
Fig 3. pDCs upregulate MHC-II in a mouse model of glioma.
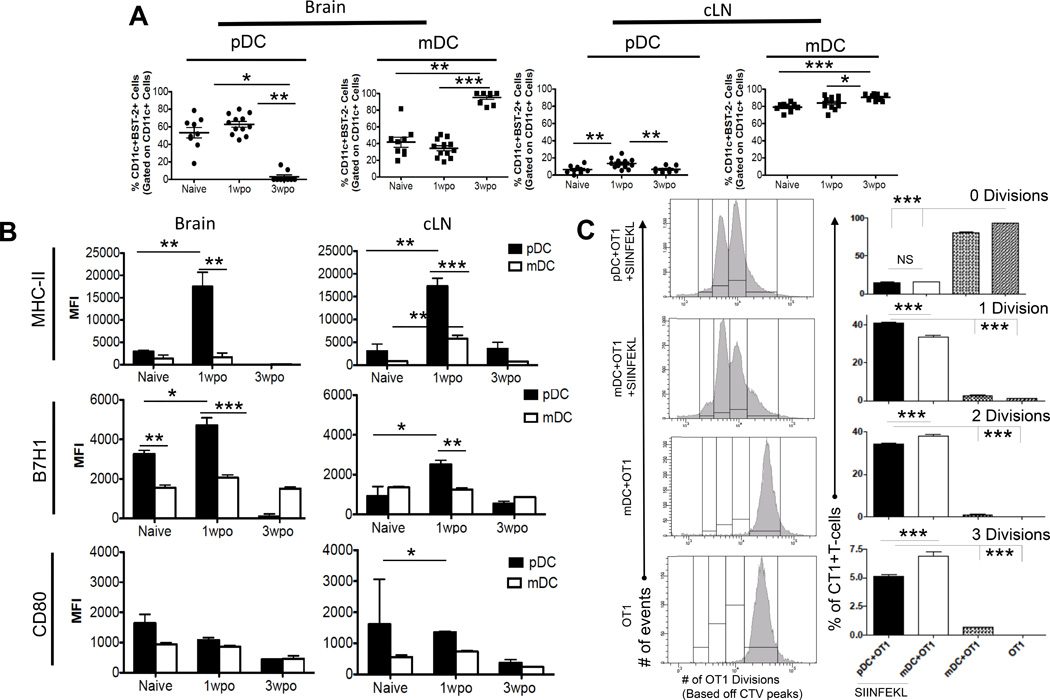
4 × 105 GL261 cells were implanted in the brain of WT mice and DC population from brain and cLN was analyzed at 1- and 3-wpo. A) Frequency of pDCs and mDCs in the brain and cLN. B) Phenotypic analysis of pDCs and mDCs with tumor progression. In the presence of tumor, pDCs upregulate expression of MHC-II and B7-H1. Graphs in A & B are shown as mean±SEM and are representative of 3–4 independent experiments (n = 3–5 mice/group). C) Panel on the left represents histograms demonstrating OT-1 T-cell proliferation. Panel on the right represents percentage of proliferating OT1 T-cells with each consecutive division. N=4–5 individually tested wells per group. Individual T-tests were performed to assess significance. *p≤0.05, **p≤0.01, ***p≤0.001.
The brain tumor decreases IFN-α secretion by pDCs
In response to variety of pathogenic stimulation, pDCs are known to produce large amount of IFN-α via TLR-9 and TLR-7 signaling (40) (47). Their ability to produce IFN- α varies depending on tissue location. In mice, BM resident pDCs are the major source of IFN-α, however the pDCs from LN or spleen are not (45). To analyze IFN-α production by brain pDCs in naïve mice and in tumor bearing mice, pDCs isolated from the brain of tumor bearing mice and naive mice were stimulated with type A CpG and stained for IFN-α. In accordance with the previously published literature (45), in naïve mice, high fraction of pDCs from BM (42.6% +/− 2.7%) produced IFN-α when stimulated with CpG, though pDCs from cLN (1.8% +/− 0.33%) did not. Interestingly, in naïve mice, majority of the pDCs from the brain (57.6% +/− 15.77%) also produced high level of IFN-α. One week post tumor implantation, there was a significant decrease in IFN-α production by pDCs from the brain (57.63% +/− 15.77 naïve vs. 10.9% +/− 6.4% 1 wpo; p < 0.05), BM (42.55% +/− 2.7% naïve vs. 1.4% +/− 0.16% 1 wpo; p < 0.001) and cLN (1.8% +/− 0.33% naïve vs. 0.5% +/− 0.2% 1 wpo; p < 0.05), compared to the naïve mice (Fig. 4A). To assess if the observed decrease in IFN-α secretion by tumor infiltrating pDCs is due to change in TLR-9/TLR-7 expression, pDCs from the brain, cLN and BM of naïve and 1wpo mice were analyzed for TLR-9/TLR-7 expression. There was no difference in TLR-7 expression by pDCs between naïve mice and tumor bearing mice, however compared to naïve pDCs, brain tumor infiltrating pDCs had significantly lower expression of TLR-9 (75.6% +/− 4.4% naïve vs. 3.7% +/− 1.5% 1 wpo; p < 0.001). The presence of brain tumor also decreased TLR-9 expression by pDCs from cLN (3.13% +/− 0.7% naïve vs. 2.61% +/− 0.61% 1 wpo) and BM (46.6% +/− 7.6% naïve vs. 30.5% +/− 8.8% 1 wpo) (Fig. 4B). These results indicate that presence of ic tumor leads to decreased expression of TLR-9 and decreased production of IFN-α by pDCs. In several solid tumors, tumor derived inhibitory cytokines, such as TGF-β and IL-10 have been shown to influence TLR-9 expression and decrease IFN-α production by pDCs (46). We have previously shown that, glioma infiltrating immune cells are major source of IL-10 and TGF-β (30). We thus conclude that in the setting of malignant glioma, glioma derived suppressive cytokines decrease TLR-9 expression and IFN-α production by pDCs.
Fig 4. Gliomas decrease IFN-α secretion by pDCs.
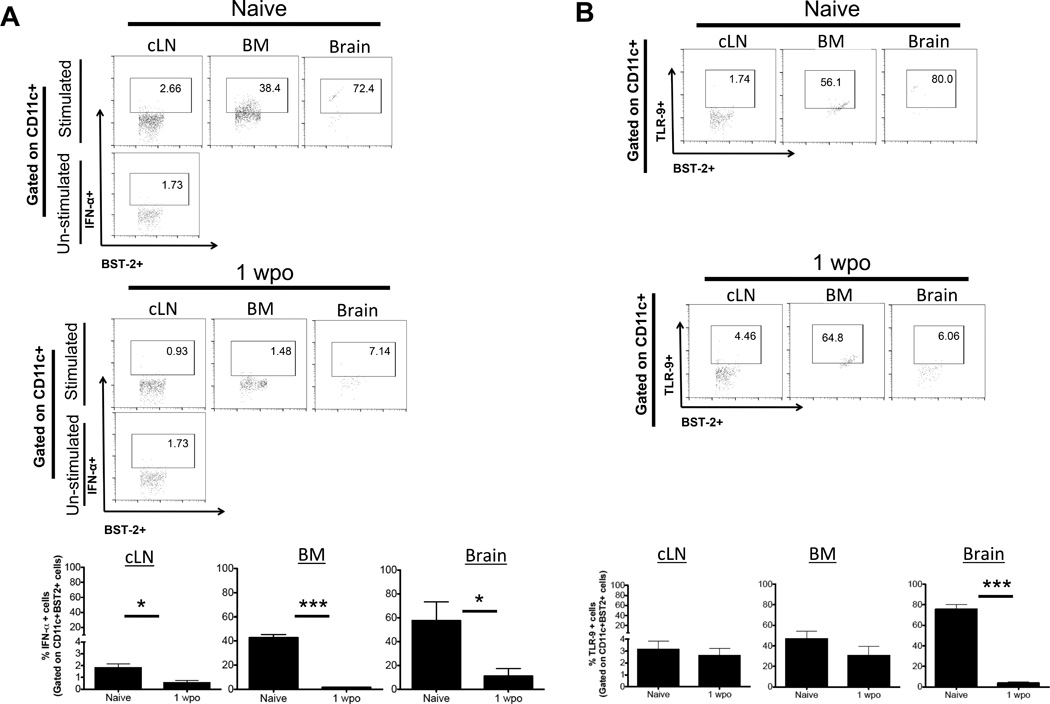
pDCs from the brain, BM and cLN of naïve mice and mice with ic tumor at 1wpo were analyzed for A) IFN-α secretion and B) TLR-9 expression. All pDC populations were identified by the same gating strategy as shown in Fig. 2 and for each analysis 500,000 events were collected. IFN-α gate is based on un-stimulated pDCs from cLN. Representative flow plot are shown. Bar graphs in A & B are shown as mean±SEM and are representative of 3–4 independent experiments (n = 3–5 mice/group). * p<0.05, **p<0.01, ***p<0.001.
pDC depletion results in increased survival by decreasing the number and suppressive function of Tregs
IFN-α secreted by pDCs has been known to be an effective anti-glioma therapeutic agent (1). In the study by Candolfi et al., the authors show that pDCs can be recruited in the glioma microenvironment by gene therapy using cytokine Flt3L, which effectively secreted IFN-α and led to increased survival in animal bearing intracranial glioma (1). Our results show that presence of brain tumor inhibits the capability of brain resident pDCs to produce IFN-α during the early priming phase of the glioma. Thus we hypothesized that presence of pDCs deficient in their ability to secrete IFN-α contributes to the tolerogenic glioma microenvironment. To test the hypothesis, pDCs were selectively depleted by injection of DT in the BDCA-2 DTR Tg mice bearing ic glioma and animals were followed for survival. Compared to the control group, pDC depletion in BDCA-2 DTR Tg mice with glioma, resulted in significant increase in median survival from 19 days to 30 days (p < 0.05) (Fig. 5A). Besides their ability to produce large amount of IFN-α, pDCs are also capable of internalizing, processing and presenting antigens to CD4+ T cells and cross- present antigens to CD8+ T cells (48–50).
Fig 5. pDC depletion results in increased survival by decreasing the number and suppressive function of Tregs.
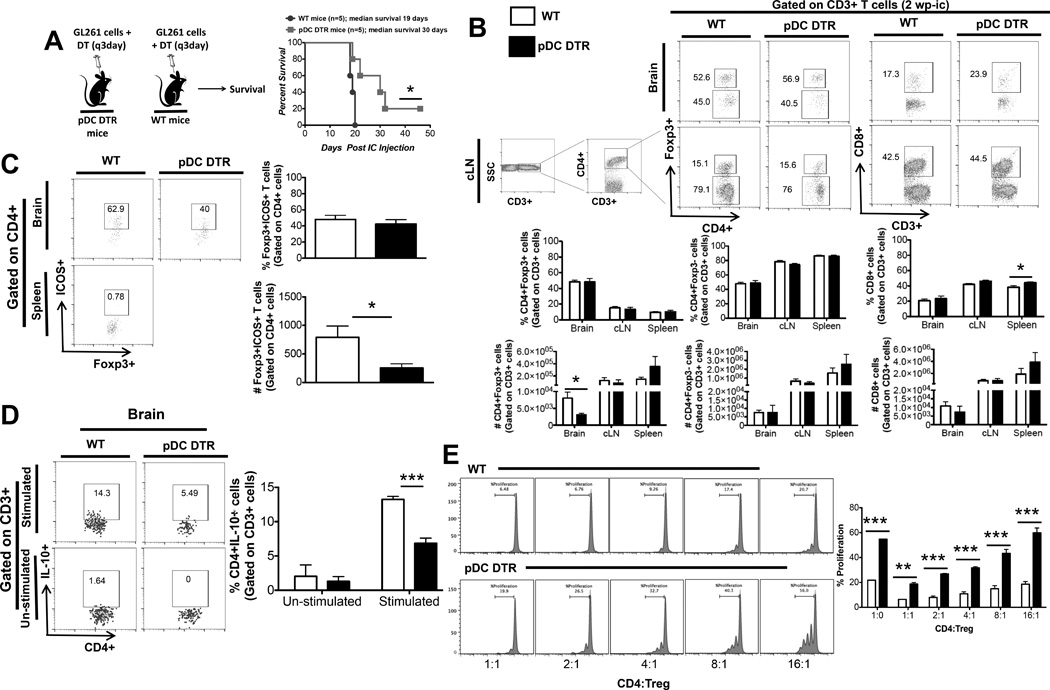
BDCA-2 DTR Tg mice and WT mice were ic. injected with 4×105 normal GL261 cells and were administered ip DT for 3 weeks. A) Animals were followed for survival B) The frequency and absolute numbers of total CD4+ T cells, CD8+ T cells, and CD4+FoxP3+ regulatory T cells isolated from the brain, cLN and spleen were analyzed at 2wpo. All T cell populations were initially gated on side scatter and CD3+ cells. CD4 and CD8 population was gated on CD3+ population. For all T-cell analysis 100,000 events were acquired. Representative flow cytometric plots shows the gating strategy used for the identification of total CD3+CD4+, CD3+CD8+, and CD3+CD4+FoxP3+ T cells. C) Frequency and absolute number of ICOS+ Tregs in the brain of WT and BDCA-2 DTR Tg at 2wpo. D) IL-10 expression by CD4+ T cells isolated from the brain of tumor-bearing WT and BDCA-2 DTR Tg mice was analyzed at 2wpo. All T cell populations were initially identified by the expression of CD3, then CD4 and IL-10 gate is based on un-stimulated CD4+ T-cells. E) In vitro suppression assay. Tregs and CD4+ cells were co-cultured for 72hrs and CD4+ cell proliferation was measures. Graphs are shown as mean±SEM and are representative of 2–3 independent experiments (n = 3–5 mice/group). Survival curve was repeated 5 times with n = 3–5 mice/experiment. * p<0.05, **p<0.01, ***p<0.001.
To understand the consequence of pDC depletion and the observed prolongation of survival, T-cells from brain, cLN and spleen were analyzed at 2wpo from WT mice and BDCA-2 DTR Tg mice bearing tumor. pDC depletion resulted in significant decrease in Treg numbers in the brain of the BDCA-2 DTR Tg mice compare to the WT mice (40,310 +/− 8,937 WT vs. 11,606 +/− 4178 BDCA-2 DTR; p < 0.05) (Fig. 5B). pDCs expressing inducible co-stimulatory (ICOS) molecule ligand have been reported to strongly favor ICOS+ Treg proliferation (51). Phenotypic analysis of the glioma infiltrating Tregs showed that there was a statistically significant decrease in ICOS+ Tregs numbers in BDCA-2 DTR mice compared to WT mice (3954.5 +/− 996.2 WT vs. 1265 +/− 376.7 BDCA-2 DTR; p < 0.05) (Fig. 5C). In terms of critical cytokine production, CD4+ cells from the brain of BDCA-2 DTR mice with glioma were found to produce significantly less of immunosuppressive cytokine IL-10 compared to WT mice (13.24% +/− 0.44% WT vs. 6.84% +/− 0.74% BDCA-2 DTR; p < 0.001) (Fig. 5D). Through an in vitro Treg suppression assay, where CD4+ cells and Tregs from the tumor bearing WT and BDCA-2 DTR mice were co-cultured and CD4+ cell proliferation was measured, we showed that Tregs from BDCA-2 DTR mice were significantly less suppressive and CD4+ cells are more proliferative compared to WT mice at every CD4:Treg ratio (Fig. 5E). Collectively, our data from the selective pDC depletion experiment in the setting of glioma shows that pDC depletion results in increased median survival of the mice bearing ic tumor. pDC depletion in the context of glioma results in decreased immunosuppressive cytokine, IL-10, production by CD4+ T cells, decreased Treg and ICOS+ Treg numbers in the brain and decreased suppressive capabilities of Tregs from the BDCA-2 DTR tumor bearing mice. Hence, in the context of glioma, pDCs deficient in IFN-α production. help to maintain large number of highly suppressive Tregs in the tumor.
Vaccinations with mDCs are superior to pDCs in prolonging survival
DCs have been harvested and used in anti-cancer therapeutic strategy for their unique capacity to process antigens and to present them to naïve T cells and activate targeted anti-tumor response. Our data collectively showed that in the context of glioma, pDCs set a tolerizing glioma microenvironment, which help promote glioma associated immunosuppression. Thus we hypothesized that pDCs and mDCs process antigen differentially and activate different arms of the immune system. Using a GL261-OVA antigen murine model of glioma, mice bearing GL261-OVA tumor (2 × 105 cells) were immunized with stimulated mature pDCs or mDCs. To analyze the migratory ability and distribution of DCs following immunization of tumor bearing mice, mature pDCs and mDCs were harvested from spleen and LN of WT (CD45.2) mice, DC’s were stimulated overnight and loaded with SIINFEKL peptide (42). Stimulated and loaded DCs were injected in the hind leg of CD45.1 tumor bearing mice at day -1, 2 and 6 post ic tumor implantation. Analysis of tissue distribution of injected CD45.2 DCs 1 week post immunization showed that both pDCs and mDCs migrated systemically to brain, cLN, spleen and inguinal LN (iLN) (Fig. 6A). Relative higher percentage of both pDCs and mDCs migrated to the brain compared to other lymphatic organs (Fig 6B). Vaccination with mDCs resulted in significant survival advantage in tumor bearing GL261-OVA tumor, where median survival of mice vaccinated with PBS was 26 days, pDCs was 36.5 days and 60% of the mice vaccinated with mDCs were alive at 60 days post tumor implantation (p < 0.001; mDC vs. pDC and PBS) (Fig 6C). These result show that both antigen loaded, pDCs and mDCs, when injected intra-dermally can migrate systemically and preferentially localize in the glioma microenvironment. Vaccination of tumor bearing mice with pDCs resulted in worse survival outcome compared to the mice vaccinated with mDCs. Thus in the setting of brain tumor, DC-based immunotherapy might benefit from selecting out pDCs and using only mDCs for antigen loading. However, further studies are necessary to solidify our findings.
Fig 6. Vaccination using mDCs is superior to pDCs in prolonging survival.
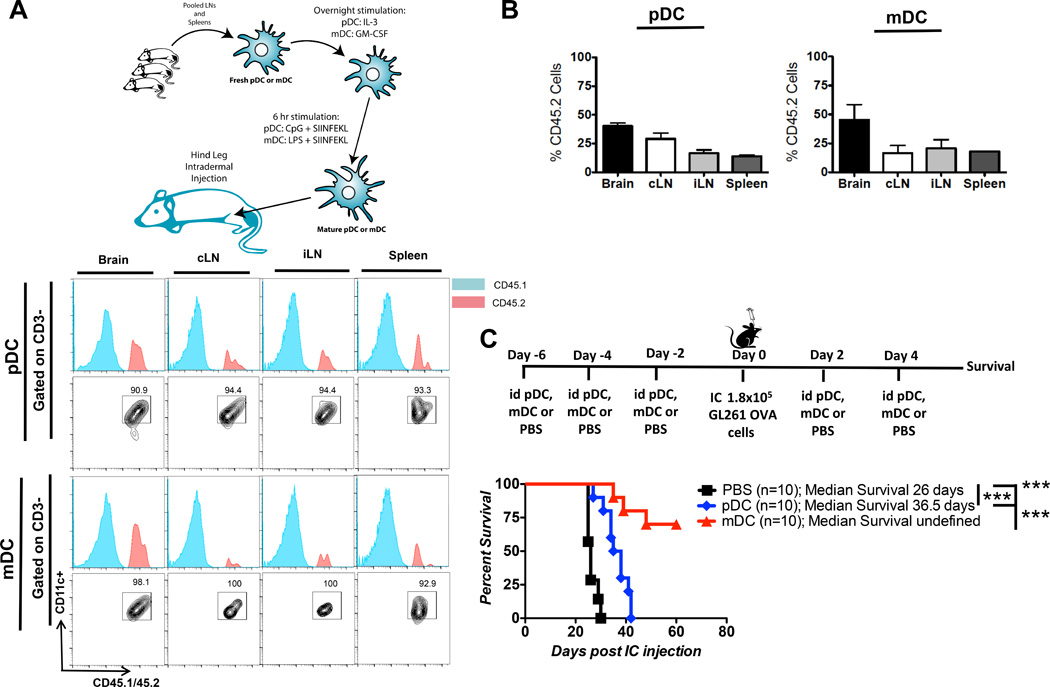
pDCs and mDCs isolated from spleen and cLN of WT CD45.2 mice, were stimulated overnight and loaded with SIINFEKL peptide. 5,000 antigen loaded DCs were used to vaccinate CD45.1 mice ic. injected with 2 × 105 GL261-OVA cells. A) DC distribution in various organs was analyzed at 1wpo. Host DCs and adoptively transferred DC were differentiated based on congenic marker CD45.1 and CD45.2. B) Percentage distribution of adoptively transferred pDCs and mDCs in brain and various lymphatic organs (n=6). C) Tumor bearing mice were vaccinated with pDCs or mDCs or PBS and followed for survival (n=10/group). * p<0.05, **p<0.01, ***p<0.001.
mDC vaccination generates a robust antigen specific Th1 response compared to pDCs
To further dissect the difference between the abilities of pDCs and mDCs in directing glioma specific immune response and define the type of immune response generated by each group of DCs, mice bearing ic GL261 tumor expressing OVA antigen, were vaccinated with stimulated and antigen loaded pDCs, mDCs or PBS as per the previous protocol and T-cell analysis was done at 2wpo. T cell analysis at 2wpo showed that mice vaccinated with mDCs had significantly higher frequency of CD8+ T cells in the brain compared to the pDC or PBS group (0.9% +/− 0.23% PBS vs. 0.96% +/− 0.13% pDC vs. 2.01% +/− 0.16% mDCs; p < 0.01) (Fig. 7A). Staining for SIINFEKL tetramer showed that there was a significantly higher frequency of antigen specific CD8+ T-cell in the brain of mice vaccinated with mDCs, compared to pDCs or PBS (6.75% +/− 0.56% PBS vs. 9.73% +/− 2.27% pDC vs. 14.82% +/− 1.24% mDCs; p < 0.001) (Fig. 7B). To investigate the ability of the DCs to induce effective anti-tumor Th1 immune response, T cells were analyzed for Th1 specific transcription factor (Tbet) (52). Compared to mice vaccinated with pDCs and PBS, mice vaccinated with mDCs had a significant increase in CD4+ Tbet+ (4.29 +/− 1.06% PBS vs. 4.87% +/− 2.28% pDC vs. 35.98% +/− 4.42% mDCs; p < 0.001) and CD8+ Tbet+ (2.15% +/− 0.73% PBS vs. 1.63% +/− 0.86% pDC vs. 30.48% +/− 8.13% mDCs; p < 0.001) T-cells in the brain (Fig. 7C). Previous reports have shown that tumor infiltrating CD8+ T-cells express high levels of inhibitory receptors and are incapable of tumor clearance (53). In our vaccination model, we show that CD8+ glioma infiltrating T cells have lower expression of the inhibitory molecules in the group vaccinated with mDCs compared to the group vaccinated with pDCs (Fig. 7D). Collectively the data suggest that mDCs are much more effective than pDCs in mounting effective anti-tumor antigen specific immune responses, which translates in significant survival benefit.
Fig 7. mDC vaccination generates a robust antigen specific Th1 response compared to pDCs.
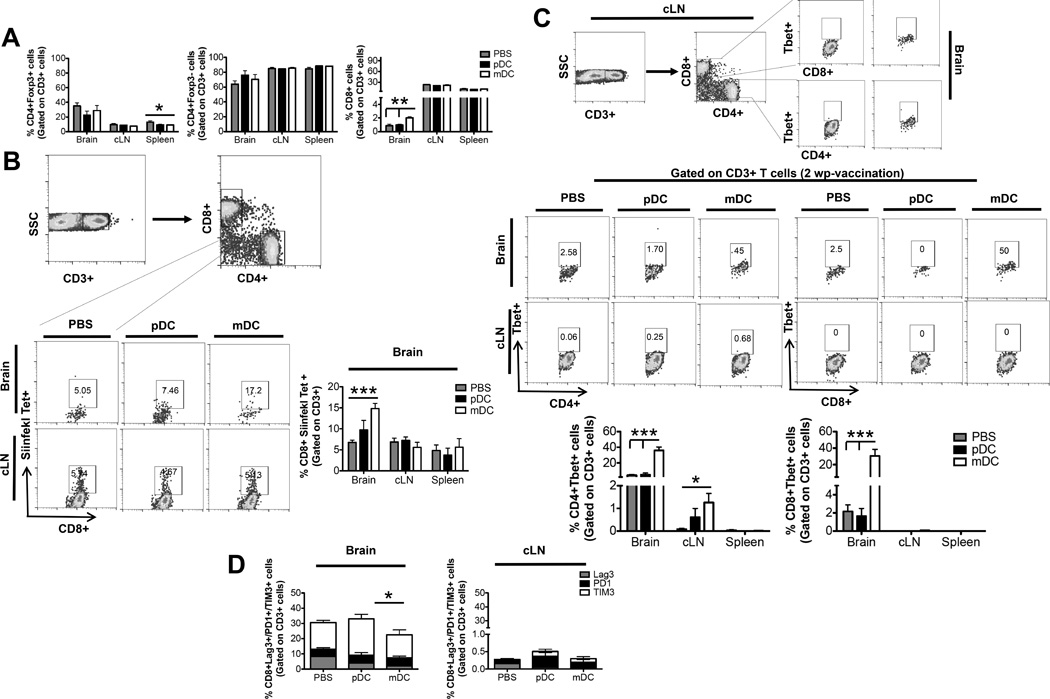
2 × 105 OVA cells were implanted in the brain of WT mice. The mice were vaccinated with SIINFEKL antigen loaded pDCs, mDCs or PBS and A) the frequency and absolute numbers of total CD4+ T cells, CD8+ T cells, and CD4+FoxP3+ regulatory T cells isolated from the brain, cLN and spleen were analyzed at 2wpo. B) Antigen specific anti-tumor response in the brain was analyzed by SIINFEKL tetramer staining. C) Systemic Th1 immune response was compared between the three groups by staining for Tbet+ cells. D) Comparison of CD8+ T-cell anergy marker on the CD8+ T-cells from brain and cLN between the three experimental groups. Graphs are shown as mean±SEM and are representative of 2–3 independent experiments (n = 3–5 mice/group). * p<0.05, **p<0.01, ***p<0.001.
Discussion
One of the major barriers to the development of effective anti-glioma immunotherapy is the profoundly immunosuppressive glioma microenvironment (54). Even though several processes have been implicated, the exact mechanism by which the presence of ic glioma leads to profound immunosuppression is not well understood. Self or foreign Ag processing and presentation by DCs is the critical step in determining if activated DC and naïve T cell interaction will result in T cell activation and induction of immunity or T cell anergy and induction of tolerance. For a long time it was believed that the CNS is an immune privileged site and this immune privilege was attributed to lack of presence of DCs in the CNS. However, recently this notion has been challenged and proven wrong (39). DCs have been shown to be present in human brain and thus may contribute to orchestration of the local immune response (38). Therefore, better understanding the immune milieu of glioma microenvironment is essential for the development of effective anti-glioma immunotherapy. In this study we investigated the role of pDCs in glioma progression and studied the T cell immune profile generated by the different subset of DCs in the context of DC-based vaccine. Our results show that pDCs are present in human glioma samples, with highest level of staining observed in grade III glioma and no staining was seen in grade IV glioma. Interestingly, in line with the observation in the human glioma, in the GL261 based murine model of glioma we noticed highest frequency of pDCs during the first week of glioma progression and almost no pDCs by week 3 of glioma progression. Even though both pDCs and mDCs are present in the brain of naïve mice, presence of ic tumor selectively induces maturation of pDCs, with up-regulation of MHC-II and B7-H1, but not mDCs. Thus they are potentially the major APCs in the brain tumor where they present antigen in the context of an inhibitory co-stimulatory molecule B7-H1 during the early priming phase of the glioma progression thus setting the stage for a tolerogenic glioma microenvironment that contributes to glioma progression. This observation that antigen presentation by pDCs in glioma leads to polarization of glioma microenvironment towards tolerization instead of immune activation needs to be further investigated to understand the exact mechanism of antigen presentation.
Phenotypic analysis of the tumor infiltrating pDCs showed that presence of ic tumor drastically impairs the ability of tumor-infiltrating pDCs to make IFN-α. This impairment in IFN-α secretion was noted systemically and not just in tumor infiltrating pDCs. Along with the decreased ability to make IFN-α, there was also a decrease in TLR-9 expression by pDCs mostly from the brain, but to some extent there was a systemic decrease in TLR-9 expression by pDCs. In the connection of several solid tumors, pDCs are known to defective in their ability to effectively produce IFN-α (13, 55). In accordance with these reports, our results also suggest that presence of glioma leads to decreased production of IFN-α by pDCs, which potentially could be due to downregulation of TLR-9 in the presence of glioma. Since the observed downregulation of TLR-9 and impairment of IFN-α production was not just localized to the glioma microenvironment but was systemic, suggesting that this is most likely mediated by soluble factors. This phenomenon of tumor-induced suppression of IFN-α has been previously described in several other cancers. In head and neck cancer, it has been demonstrated that tumor cells actively suppress IFN-α production by pDCs by decreasing TLR-9 expression and secreting inhibitory cytokine IL-10 (19). Breast cancer derived TGF-β and TNF-α has been shown to directly compromise IFN-α secretion by pDCs by influencing TLR-9 downstream signaling (46). Presence of inhibitory cytokines like TGF- β and IL-10 is a hallmark feature of malignant glioma (30). Thus, the defect in IFN-α production observed in glioma pDCs is most likely due to glioma generated inhibitory cytokine induced downregulation of TLR-9.
Our data cumulatively showed that, in the context of brain tumor, tumor infiltrating pDCs are: a) present during early priming phase of the tumor progression, b) selectively undergoing maturation and c) incapable of producing pro-inflammatory cytokine IFN-α. We investigated the effect of selective pDC depletion in the setting of glioma. Using BDCA-2 DTR Tg mice, we show that pDC depletion during the course of the disease progression provides a significant survival advantage. In the setting of several solid tumors, such as breast, ovarian, head and neck, pDCs have been shown to promote tolerance by inducing Tregs (11–13). In line with the current literature, our studies also show that pDC depletion results in decreased number of Tregs and ICOS+ Tregs in the brain tumor. In addition to that, in the absence of pDCs, Tregs are also less immunosuppressive as quantified by the in vitro suppression assay. Thus we show that in the murine model of glioma pDCs promote tumor tolerance by inducing Tregs number and function. The observed survival advantage was present when pDCs were depleted during the first 3 weeks after tumor implantation starting with day 0. However the survival advantage disappeared if the pDCs were depleted starting at week 2 or week 3 (data not shown). This suggests that pDCs are critical during early priming stage of glioma progression and needs to be depleted before the priming stage, however once they set the stage for immunosuppression their depletion is inconsequential for the course of disease progression. In the clinical setting it is impossible to manipulate the priming phase of the tumor, since the priming stage has passed by the time tumor is diagnosed. Hence clinically targeting or manipulating pDCs in MG is not an effective therapeutic strategy. However, following glioma resection use of DC-based immunotherapy might provide an attractive opportunity to re-set and re-prime the immune response. Thus we further went on to characterize the immune response generated by the two different subset of DCs.
DCs, used as vehicles in current immunotherapy regimens, dictate the effectiveness of the anti-tumor T cell response. Since pDCs and mDCs act differentially in the presence of glioma, we decided to study the antigen specific T cell response generated by the two subsets of DCs when used for DC-based vaccination. We show that both pDCs and mDCs effectively migrated to iLN, cLN, spleen and brain. Animals vaccinated with mDCs mounted a robust antigen specific Th1 type T cell response that translated into survival of 60% of the animals beyond 60 days. However pDCs were less effective in mounting effective anti-glioma response and pDC vaccination did not provide any survival advantage compared to vaccination with PBS. Natural mDCs isolated from WT mice can be effectively stimulated to induce an anti-glioma immune response and are more effective than pDCs when used in DC-based immunotherapy. This observation has tremendous clinical implications since virtually all current DC-based vaccines use ex vivo differentiated cells that are considered to be less effective than their natural counterparts (27). Thus using natural mDCs and selectively depleting pDCs from the DC pool can potentially enhance the efficacy of DC based immunotherapy. We for the first time show that mDCs and pDCs behave very differently in DC-based anti-glioma vaccine and the type of DCs used for the vaccination significantly influence the anti-tumor immune response. Although, this observation needs to further tested and optimized in terms of optimal antigen and timing of administration it provides the essential groundwork for further investigation and optimization of anti-glioma vaccine therapy.
Acknowledgments
Grant Support
This work was supported by NIH R01CA122930 (MSL), RR01CA138587 (MSL), R01NS077388 (MSL), U01NS077388 (MSL), R25NS065744 (MD) and ACS Resident Research Fellowship Grant (MD).
Footnotes
Disclosures
The authors have no financial conflicts of interest.
Authors' Contributions
Conception and design: M. Dey, D.A. Wainwright, M.S. Lesniak.
Development of methodology: M. Dey, A.L. Chang, D. A. Wainwright, A.U Ahmed, M.S. Lesniak.
Acquisition of data: M. Dey, A.L. Chang, J. Miska, I. V. Balyasnikova.
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): M. Dey, A. L. Chang, J. Miska, D. A. Wainwright, P. Pytel, L. Zhang, A. U. Ahmed, I. V. Balyasnikova, J. Qiao, M.S. Lesniak.
Writing, review, and/or revision of the manuscript: M. Dey, A.L. Chang, D. A. Wainwright, A. U. Ahmed, I. V. Balyasnikova, J. Qiao, M.S. Lesniak.
Administrative, technical, or material support: M. Dey, A.U. Ahmed, Y. Han, A. Tobias, M.S. Lesniak.
Study supervision: M.S. Lesniak.
References
- 1.Candolfi M, King GD, Yagiz K, Curtin JF, Mineharu Y, Muhammad AK, Foulad D, Kroeger KM, Barnett N, Josien R, Lowenstein PR, Castro MG. Plasmacytoid dendritic cells in the tumor microenvironment: immune targets for glioma therapeutics. Neoplasia. 2012;14:757–770. doi: 10.1593/neo.12794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nature reviews. Drug discovery. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sgorbissa A, Tomasella A, Potu H, Manini I, Brancolini C. Type I IFNs signaling and apoptosis resistance in glioblastoma cells. Apoptosis : an international journal on programmed cell death. 2011;16:1229–1244. doi: 10.1007/s10495-011-0639-4. [DOI] [PubMed] [Google Scholar]
- 4.Parmar S, Platanias LC. Interferons: mechanisms of action and clinical applications. Current opinion in oncology. 2003;15:431–439. doi: 10.1097/00001622-200311000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Lombardi VC, Khaiboullina SF, Rizvanov AA. Plasmacytoid dendritic cells, a role in neoplastic prevention and progression. European journal of clinical investigation. 2015;45(Suppl 1):1–8. doi: 10.1111/eci.12363. [DOI] [PubMed] [Google Scholar]
- 6.Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity. 2008;29:352–361. doi: 10.1016/j.immuni.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Irla M, Kupfer N, Suter T, Lissilaa R, Benkhoucha M, Skupsky J, Lalive PH, Fontana A, Reith W, Hugues S. MHC class II-restricted antigen presentation by plasmacytoid dendritic cells inhibits T cell-mediated autoimmunity. The Journal of experimental medicine. 2010;207:1891–1905. doi: 10.1084/jem.20092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lui G, Manches O, Angel J, Molens JP, Chaperot L, Plumas J. Plasmacytoid dendritic cells capture and cross-present viral antigens from influenza-virus exposed cells. PloS one. 2009;4:e7111. doi: 10.1371/journal.pone.0007111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ochando JC, Homma C, Yang Y, Hidalgo A, Garin A, Tacke F, Angeli V, Li Y, Boros P, Ding Y, Jessberger R, Trinchieri G, Lira SA, Randolph GJ, Bromberg JS. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nature immunology. 2006;7:652–662. doi: 10.1038/ni1333. [DOI] [PubMed] [Google Scholar]
- 10.de Heer HJ, Hammad H, Soullie T, Hijdra D, Vos N, Willart MA, Hoogsteden HC, Lambrecht BN. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. The Journal of experimental medicine. 2004;200:89–98. doi: 10.1084/jem.20040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conrad C, Gregorio J, Wang YH, Ito T, Meller S, Hanabuchi S, Anderson S, Atkinson N, Ramirez PT, Liu YJ, Freedman R, Gilliet M. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer research. 2012;72:5240–5249. doi: 10.1158/0008-5472.CAN-12-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu H, Huang X, Liu X, Jin H, Zhang G, Zhang Q, Yu J. Regulatory T cells and plasmacytoid dendritic cells contribute to the immune escape of papillary thyroid cancer coexisting with multinodular non-toxic goiter. Endocrine. 2013;44:172–181. doi: 10.1007/s12020-012-9853-2. [DOI] [PubMed] [Google Scholar]
- 13.Sisirak V, Faget J, Vey N, Blay JY, Menetrier-Caux C, Caux C, Bendriss-Vermare N. Plasmacytoid dendritic cells deficient in IFNalpha production promote the amplification of FOXP3 regulatory T cells and are associated with poor prognosis in breast cancer patients. Oncoimmunology. 2013;2:e22338. doi: 10.4161/onci.22338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nature immunology. 2000;1:305–310. doi: 10.1038/79747. [DOI] [PubMed] [Google Scholar]
- 15.Fonteneau JF, Gilliet M, Larsson M, Dasilva I, Munz C, Liu YJ, Bhardwaj N. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood. 2003;101:3520–3526. doi: 10.1182/blood-2002-10-3063. [DOI] [PubMed] [Google Scholar]
- 16.Salio M, Cella M, Vermi W, Facchetti F, Palmowski MJ, Smith CL, Shepherd D, Colonna M, Cerundolo V. Plasmacytoid dendritic cells prime IFN-gamma-secreting melanoma-specific CD8 lymphocytes and are found in primary melanoma lesions. European journal of immunology. 2003;33:1052–1062. doi: 10.1002/eji.200323676. [DOI] [PubMed] [Google Scholar]
- 17.Schlecht G, Garcia S, Escriou N, Freitas AA, Leclerc C, Dadaglio G. Murine plasmacytoid dendritic cells induce effector/memory CD8+ T-cell responses in vivo after viral stimulation. Blood. 2004;104:1808–1815. doi: 10.1182/blood-2004-02-0426. [DOI] [PubMed] [Google Scholar]
- 18.Salio M, Palmowski MJ, Atzberger A, Hermans IF, Cerundolo V. CpG-matured murine plasmacytoid dendritic cells are capable of in vivo priming of functional CD8 T cell responses to endogenous but not exogenous antigens. The Journal of experimental medicine. 2004;199:567–579. doi: 10.1084/jem.20031059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartmann E, Wollenberg B, Rothenfusser S, Wagner M, Wellisch D, Mack B, Giese T, Gires O, Endres S, Hartmann G. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer research. 2003;63:6478–6487. [PubMed] [Google Scholar]
- 20.Sawant A, Hensel JA, Chanda D, Harris BA, Siegal GP, Maheshwari A, Ponnazhagan S. Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells. Journal of immunology. 2012;189:4258–4265. doi: 10.4049/jimmunol.1101855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vermi W, Bonecchi R, Facchetti F, Bianchi D, Sozzani S, Festa S, Berenzi A, Cella M, Colonna M. Recruitment of immature plasmacytoid dendritic cells (plasmacytoid monocytes) and myeloid dendritic cells in primary cutaneous melanomas. The Journal of pathology. 2003;200:255–268. doi: 10.1002/path.1344. [DOI] [PubMed] [Google Scholar]
- 22.Beckebaum S, Zhang X, Chen X, Yu Z, Frilling A, Dworacki G, Grosse-Wilde H, Broelsch CE, Gerken G, Cicinnati VR. Increased levels of interleukin-10 in serum from patients with hepatocellular carcinoma correlate with profound numerical deficiencies and immature phenotype of circulating dendritic cell subsets. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:7260–7269. doi: 10.1158/1078-0432.CCR-04-0872. [DOI] [PubMed] [Google Scholar]
- 23.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. The Journal of clinical investigation. 2007;117:2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, Messina JL, Chandler P, Koni PA, Mellor AL. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. The Journal of clinical investigation. 2004;114:280–290. doi: 10.1172/JCI21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nature reviews. Immunology. 2007;7:790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 26.Kalinski P, Schuitemaker JH, Hilkens CM, Wierenga EA, Kapsenberg ML. Final maturation of dendritic cells is associated with impaired responsiveness to IFN-gamma and to bacterial IL-12 inducers: decreased ability of mature dendritic cells to produce IL-12 during the interaction with Th cells. Journal of immunology. 1999;162:3231–3236. [PubMed] [Google Scholar]
- 27.Schreibelt G, Benitez-Ribas D, Schuurhuis D, Lambeck AJ, van Hout-Kuijer M, Schaft N, Punt CJ, Figdor CG, Adema GJ, de Vries IJ. Commonly used prophylactic vaccines as an alternative for synthetically produced TLR ligands to mature monocyte-derived dendritic cells. Blood. 2010;116:564–574. doi: 10.1182/blood-2009-11-251884. [DOI] [PubMed] [Google Scholar]
- 28.Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nature immunology. 2000;1:311–316. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- 29.Dey M, Ulasov IV, Lesniak MS. Virotherapy against malignant glioma stem cells. Cancer letters. 2010;289:1–10. doi: 10.1016/j.canlet.2009.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dey M, Chang AL, Wainwright DA, Ahmed AU, Han Y, Balyasnikova IV, Lesniak MS. Heme oxygenase-1 protects regulatory T cells from hypoxia-induced cellular stress in an experimental mouse brain tumor model. Journal of neuroimmunology. 2014;266:33–42. doi: 10.1016/j.jneuroim.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El Andaloussi A, Han Y, Lesniak MS. Prolongation of survival following depletion of CD4+CD25+ regulatory T cells in mice with experimental brain tumors. Journal of neurosurgery. 2006;105:430–437. doi: 10.3171/jns.2006.105.3.430. [DOI] [PubMed] [Google Scholar]
- 32.El Andaloussi A, Lesniak MS. CD4+ CD25+ FoxP3+ T-cell infiltration and heme oxygenase-1 expression correlate with tumor grade in human gliomas. Journal of neuro-oncology. 2007;83:145–152. doi: 10.1007/s11060-006-9314-y. [DOI] [PubMed] [Google Scholar]
- 33.Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim C, Tobias AL, Cheng Y, Kim J, Zhang L, Qiao J, Han Y, Lesniak MS. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4 and PD-L1 in mice with brain tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014 doi: 10.1158/1078-0432.CCR-14-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, Tobias AL, Han Y, Lesniak MS. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:6110–6121. doi: 10.1158/1078-0432.CCR-12-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marland G, Bakker AB, Adema GJ, Figdor CG. Dendritic cells in immune response induction. Stem cells. 1996;14:501–507. doi: 10.1002/stem.140501. [DOI] [PubMed] [Google Scholar]
- 36.Santambrogio L, Belyanskaya SL, Fischer FR, Cipriani B, Brosnan CF, Ricciardi-Castagnoli P, Stern LJ, Strominger JL, Riese R. Developmental plasticity of CNS microglia. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6295–6300. doi: 10.1073/pnas.111152498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer HG, Reichmann G. Brain dendritic cells and macrophages/microglia in central nervous system inflammation. Journal of immunology. 2001;166:2717–2726. doi: 10.4049/jimmunol.166.4.2717. [DOI] [PubMed] [Google Scholar]
- 38.Pashenkov M, Huang YM, Kostulas V, Haglund M, Soderstrom M, Link H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain : a journal of neurology. 2001;124:480–492. doi: 10.1093/brain/124.3.480. [DOI] [PubMed] [Google Scholar]
- 39.D'Agostino PM, Gottfried-Blackmore A, Anandasabapathy N, Bulloch K. Brain dendritic cells: biology and pathology. Acta neuropathologica. 2012;124:599–614. doi: 10.1007/s00401-012-1018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity. 2010;33:955–966. doi: 10.1016/j.immuni.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohlfest JR, Andersen BM, Litterman AJ, Xia J, Pennell CA, Swier LE, Salazar AM, Olin MR. Vaccine injection site matters: qualitative and quantitative defects in CD8 T cells primed as a function of proximity to the tumor in a murine glioma model. Journal of immunology. 2013;190:613–620. doi: 10.4049/jimmunol.1201557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tel J, Aarntzen EH, Baba T, Schreibelt G, Schulte BM, Benitez-Ribas D, Boerman OC, Croockewit S, Oyen WJ, van Rossum M, Winkels G, Coulie PG, Punt CJ, Figdor CG, de Vries IJ. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer research. 2013;73:1063–1075. doi: 10.1158/0008-5472.CAN-12-2583. [DOI] [PubMed] [Google Scholar]
- 43.Aspord C, Leccia MT, Charles J, Plumas J. Plasmacytoid dendritic cells support melanoma progression by promoting Th2 and regulatory immunity through OX40L and ICOSL. Cancer immunology research. 2013;1:402–415. doi: 10.1158/2326-6066.CIR-13-0114-T. [DOI] [PubMed] [Google Scholar]
- 44.Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro-oncology. 2006;8:261–279. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bjorck P, Leong HX, Engleman EG. Plasmacytoid dendritic cell dichotomy: identification of IFN-alpha producing cells as a phenotypically and functionally distinct subset. Journal of immunology. 2011;186:1477–1485. doi: 10.4049/jimmunol.1000454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sisirak V, Vey N, Goutagny N, Renaudineau S, Malfroy M, Thys S, Treilleux I, Labidi-Galy SI, Bachelot T, Dezutter-Dambuyant C, Menetrier-Caux C, Blay JY, Caux C, Bendriss-Vermare N. Breast cancer-derived transforming growth factor-beta and tumor necrosis factor-alpha compromise interferon-alpha production by tumor-associated plasmacytoid dendritic cells. International journal of cancer. Journal international du cancer. 2013;133:771–778. doi: 10.1002/ijc.28072. [DOI] [PubMed] [Google Scholar]
- 47.Ito T, Wang YH, Liu YJ. Plasmacytoid dendritic cell precursors/type I interferon-producing cells sense viral infection by Toll-like receptor (TLR) 7 and TLR9. Springer seminars in immunopathology. 2005;26:221–229. doi: 10.1007/s00281-004-0180-4. [DOI] [PubMed] [Google Scholar]
- 48.Young LJ, Wilson NS, Schnorrer P, Proietto A, ten Broeke T, Matsuki Y, Mount AM, Belz GT, O'Keeffe M, Ohmura-Hoshino M, Ishido S, Stoorvogel W, Heath WR, Shortman K, Villadangos JA. Differential MHC class II synthesis and ubiquitination confers distinct antigen-presenting properties on conventional and plasmacytoid dendritic cells. Nature immunology. 2008;9:1244–1252. doi: 10.1038/ni.1665. [DOI] [PubMed] [Google Scholar]
- 49.Sapoznikov A, Fischer JA, Zaft T, Krauthgamer R, Dzionek A, Jung S. Organ-dependent in vivo priming of naive CD4+, but not CD8+, T cells by plasmacytoid dendritic cells. The Journal of experimental medicine. 2007;204:1923–1933. doi: 10.1084/jem.20062373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Pucchio T, Chatterjee B, Smed-Sorensen A, Clayton S, Palazzo A, Montes M, Xue Y, Mellman I, Banchereau J, Connolly JE. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nature immunology. 2008;9:551–557. doi: 10.1038/ni.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Faget J, Bendriss-Vermare N, Gobert M, Durand I, Olive D, Biota C, Bachelot T, Treilleux I, Goddard-Leon S, Lavergne E, Chabaud S, Blay JY, Caux C, Menetrier-Caux C. ICOS-ligand expression on plasmacytoid dendritic cells supports breast cancer progression by promoting the accumulation of immunosuppressive CD4+ T cells. Cancer research. 2012;72:6130–6141. doi: 10.1158/0008-5472.CAN-12-2409. [DOI] [PubMed] [Google Scholar]
- 52.Rengarajan J, Szabo SJ, Glimcher LH. Transcriptional regulation of Th1/Th2 polarization. Immunology today. 2000;21:479–483. doi: 10.1016/s0167-5699(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 53.Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, Rufer N, Speiser DE. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. The Journal of clinical investigation. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lesniak MS. Immunotherapy for glioblastoma: the devil is in the details. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:3105. doi: 10.1200/JCO.2011.34.9019. author reply 3105-3106. [DOI] [PubMed] [Google Scholar]
- 55.Labidi-Galy SI, Sisirak V, Meeus P, Gobert M, Treilleux I, Bajard A, Combes JD, Faget J, Mithieux F, Cassignol A, Tredan O, Durand I, Menetrier-Caux C, Caux C, Blay JY, Ray-Coquard I, Bendriss-Vermare N. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer research. 2011;71:5423–5434. doi: 10.1158/0008-5472.CAN-11-0367. [DOI] [PubMed] [Google Scholar]