

Abstract
Adoptive transfer of freshly isolated natural occurring CD4+CD25+FoxP3+ regulatory T cells (Treg) prevents graft versus host disease (GvHD) in several animal models and following hematopoietic cell transplantation (HCT) in clinical trials. Donor derived Treg have been mainly used as they share the same MHC with conventional CD4+ and CD8+ T cells (Tcon) that are primarily responsible for GvHD. Third-party derived Treg are a promising alternative for cellular therapy as they can be prepared in advance, screened for pathogens and activity and banked. We explored MHC disparities between Treg and Tcon in HCT to evaluate the impact of different Treg populations in GvHD prevention and survival. Third-party Treg and donor Treg are equally suppressive in ex vivo assays, while both donor and third-party but not host Treg protect from GvHD in allogeneic HCT with donor Treg being the most effective. In a MHC minor mismatched transplantation model (C57BL/6 → BALB/b) donor and third-party Treg were equally effective in controlling GVHD. Furthermore, using an in vivo Treg depletion mouse model, we found that Treg exert their main suppressive activity in the first two days after transplantation. Third-party Treg survive for a shorter period of time after adoptive transfer, but despite the shorter survival, they control Tcon proliferation in the early phases of HCT. These studies provide relevant insights on the mechanisms of Treg mediated protection from GvHD and support for the use of third-party Treg in clinical trials.
Introduction
Allogeneic hematopoietic cell transplantation (HCT) is a curative treatment for patients with hematological malignancies and many congenital and genetic disorders. One of the major complications of HCT is graft versus host disease (GvHD), a potentially lethal immune reaction caused by donor cells recognizing and destroying host tissues(1). Several studies have demonstrated that CD4+CD25+FoxP3+ regulatory T cells (Treg) control conventional CD4+ and CD8+ T cell (Tcon) proliferation limiting GvHD lethality yet retaining anti-viral and graft versus tumor activity thus promoting animal survival(2–6). Recently, these promising results have been translated into the clinic confirming that Treg based cellular therapy is a powerful approach for GvHD prevention(7–10).
Despite the promising results, there are several factors that are limiting the broader application of this treatment(11, 12). Treg from different sources such as the donor, recipient or third-party have been tested in preclinical and clinical transplantation studies, but no comparison between these different donor sources has been systematically reported, therefore, it is unclear which donor source has a greater impact on Tcon proliferation and prevention of GvHD. Several studies demonstrated that Treg exert their suppressive function through different mechanisms that can be contact or cytokine-mediated(13). In these animal models it has been demonstrated that Treg undergo expansion and control Tcon proliferation. Treg have been shown to directly home in primary lymphoid tissues after their adoptive transfer where they prevent Tcon proliferation and further homing in GvHD target tissues. Moreover timing of Treg adoptive transfer is extremely important as they require to be injected prior to Tcon for conferring the best GvHD protection(14–16).
It is yet unclear whether MHC disparities between Treg and Tcon impact Treg function. Furthermore several groups are investigating the clinical utility of ex vivo expanded Treg in order to increase their number since Treg are a rare cell population and others are improving culturing strategies to enhance Treg function(6, 17–20). Third-party Treg are particularly suitable for such studies as they can be prepared in advance and then banked for further use. Accordingly, their application may be particularly relevant in cases where the donor is not immediately available such as transplantation from an unrelated or umbilical cord blood donor. In this study we investigated the impact of MHC disparities between Treg and Tcon on alloreactive T cell proliferation and GvHD prevention aiming to establish the role of MHC disparate Treg sources, which is of central importance for their clinical application. Using a model of in vivo Treg depletion we further studied the timing of Treg function in vivo after adoptive transfer. We found that selective in vivo depletion of injected Treg at different time points has a different impact on GvHD onset and lethality, therefore our study introduces relevant insights to the complex mechanisms through which Treg protect from GvHD.
Material and Methods
Mice
Experiments used gender-matched mice between 7 and 12 weeks old. FVB/N (H-2q), BALB/c (H-2d, CD45.2) and C57BL/6 (H-2b, CD45.2) mice were purchased from Jackson Laboratories (Sacramento, CA). Luciferase-expressing (luc+) C57BL/6 (CD45.1, Thy1.1) and BALB/c (CD45.1, Thy1.1) mice were generated after backcrossing for more than 10 generations to the C57BL/6 or BALB/c background with the luc+ transgenic FVB/N L2G85 mice that were described previously(21). C57BL/6 albino FoxP3 mutant mice expressing diphtheria toxin (DT) receptor, GFP and luc (FoxP3DTR/GFP/luc) were a kind gift from Dr. Günter J. Hämmerling (Heidelberg, Germany) and Dr. Andreas Beilhack (Würzburg, Germany) and were bred in our animal facility. Animal protocols were approved by the Institutional Animal Care and Use Committee of Stanford University.
Cell isolation
CD4/CD8 conventional T cells (Tcon) were prepared from splenocytes and peripheral lymph node cells and enriched with anti-CD4 and anti-CD8 magnetic-activated cell sorting (MACS; Miltenyi Biotec, Auburn, CA). Purity was assessed by fluorescence-activated cell sorting (FACS) using a LSR II (BD Biosciences, San Jose, CA). T cell–depleted bone marrow (TCD-BM) was prepared by flushing bones and depleting T cells with anti-CD4 and anti-CD8 MACS beads. Treg were prepared from pooled spleens and lymph nodes, by staining for CD25-allophycocyanin and CD4, enriching with anti-allophycocyanin MACS beads and sorting for CD4+CD25hi cells or for CD4+CD25+GFP+ cells from C57BL/6 albino FoxP3DTR/GFP/luc mice in the experiments with in vivo Treg depletion on a FACS Aria or FACS Aida (BD Biosciences, San Jose, CA). Approximately 200,000 purified (>98% FoxP3+) Treg were routinely obtained per wild-type mouse and 400,000 purified Treg were obtained per C57BL/6 albino FoxP3DTR/GFP/luc mouse.
Flow cytometric analysis
The following anti-mouse antibodies were purchased from eBioscience (San Diego, CA) or Biolegend (San Diego, CA): PerCP/Cy5.5 anti-CD4 (GK1.5), APC/Cy7 anti-CD8a (53–6.7), Pacific Blue anti-CD44 (IM7), FITC anti-CD62L (MEL-14), APC anti-CD25 (3C7), Pacific Blue anti-FoxP3 (MF-14), FITC anti-H-2Kq (KH114), PE anti-H-2Dd (34-2-12), FITC anti-H-2Kb (AF6-88.5), PE/Cy7 anti-CD45.1 (A20), PE anti-CD45.2 (104), APC anti-CD90.1 (OX-7), APC anti-IL10 (JES5-16E3), PE anti-IFNγ (XMG1.2), Alexa Fluor 700 anti-IA/IE (M5/114.15.2), PE anti-CTLA4 (UC10-4B9). Isotype controls were purchased from the respective vendors. FoxP3 staining was performed with an anti-mouse/rat FoxP3 Staining Set (eBioscience, San Diego, CA). Fixable Viability Dye eFluor® 506 (eBioscence, San Diego, CA) was used for identification and removal of dead cells. Analysis was performed on a LSR II (Becton-Dickinson, San Jose, CA). Data were analyzed with FlowJo 10.0.7 (Tree Star, Ashland, OR).
Mixed lymphocyte reaction
Tcon (1–2x105) with or without Treg (for Treg/Tcon ratio see text) were added per well to a 96-well U-bottom plate containing complete RPMI (cRPMI) and 1x106 BALB/c splenocytes, which were previously irradiated with 30-Gy. Tcon and Treg were isolated from different mouse strains as described in the text. Total volume per well was 300 μL and cells were cultured at 37°C and 5% CO2. For quantifying T cell proliferation, after 4 days of culture, cells were pulsed with 1 mCi/well [3H]thymidine (GE Healthcare Piscataway, NJ) for 16 hours. Cells were harvested onto filter membranes using a Wallac harvester (Perkin-Elmer, Shelton, CT), and the amount of incorporated [3H]thymidine was measured with a Wallac Betaplate counter (Perkin-Elmer, Shelton, CT).
Treg in vitro culture
For analyzing surface marker expression and for in vitro cytokine analysis, 5–10x104 Tregs were added per well of a 96-well U-bottom plate containing cRPMI and were stimulated with either interleukin-2 (IL-2, Chiron, Emeryville, CA) alone (5000 IU/ml) or IL-2 plus antiCD3/CD28 beads (Dynabeads® Mouse T-Activator CD3/CD28, Life-Technologies, Logan, UT, Treg/bead ratio 1:1) or were co-incubated with Tcon and irradiated splenocytes as described above. After 3–4 days of culture, supernatants were collected for cytokine analysis, while cells were harvested, stained and FACS analyzed as above described.
Transplantation models and in vivo bioluminescence imaging
For detecting T cell proliferation through in vivo bioluminescence imaging (BLI), BALB/c recipient mice were irradiated at day −2 with total body irradiation (TBI) 2 doses of 4 Gy, 4 hours apart with 200-Kv X-ray source. 5x105 host-type (BALB/c, H-2d), donor type (C57BL/6, H-2b) or third-party (FVB/N, H-2q) Treg were administered intravenously at day −2, 5x106 TCD-BM cells from CD45.2+ C57BL/6 mice and 1x106 Tcon from luc+ CD45.1+ C57BL/6 mice were injected intravenously at day 0. Transplanted animals were housed in autoclaved cages with antibiotic water or antibiotic food (sulfamethoxazole-trimethropim; Schein Pharmaceutical, Corona, CA). In vivo BLI was performed as described(22) with an IVIS 29 charge-coupled device imaging system (Xenogen, Alameda, CA). Images were analyzed with Living Image Software 3.4.1 (Xenogen, Alameda, CA).
For experiments with Treg in vivo depletion transplantation was performed as above described. Treg from C57BL/6 albino FoxP3DTR/GFP/luc mice were administered intravenously at day −2. Diphtheria Toxin (DT) was intaperitoneally injected at the dose of 50 mg/kg/mouse daily for two consecutive days with the timing described in the text. For Treg re-isolation after transplant, BALB/c recipient mice were transplanted as described above. Mice were euthanized at day 6 and 12 after transplantation and peripheral blood, spleen, lymph nodes and liver were harvested. Sera were collected from peripheral blood for cytokine analysis. Spleen and lymph nodes were disassociated for obtaining homogeneous cell suspensions. Liver was mashed and then was passed through a percoll gradient (GE Healthcare, Piscataway, NJ). The lymphocyte layer was collected. All cell suspensions were filtered, stained and FACS analyzed as described in the text. For survival experiments, BALB/c recipient mice were transplanted as described above. Mice were weighed weekly and GvHD score was calculated(23).
MHC minor mismatched model of transplantation was performed as above described with the only difference that BALB/b (H-2b) mice were used as recipients and 2.5x107 Tcon were injected for GvHD induction.
Cytokine analysis
For intracellular cytokine staining cells were stimulated with 20 ng/ml phorbol myristate acetate (PMA; Sigma-Aldrich, St. Louis, MO) and 1 μg/ml ionomycin (Sigma-Aldrich, St. Louis, MO) for 6 hours at 37°C and 5% CO2 in RPMI 1640 supplemented with 10% FCS, 2 mM L-glutamine (Mediatech, Manassas, VA), 100 U/ml penicillin (Thermo Fisher Scientific, Asheville, NC) and 100 μg/ml streptomycin (Thermo Fisher Scientific, Asheville, NC). Monensin (BD Biosciences, San Jose, CA) was used to block cellular protein transport. Cells were fixed and permeabilized (eBioscience, San Diego, CA) prior to staining of intracellular and intranuclear antigens. For quantitative measurement supernatants were collected from Treg in vitro cultures and sera were obtained from peripheral blood of transplanted mice at day 6 and 12 after transplantation. All the samples were analyzed for cytokine concentration through multiplex assay (Luminex, Life-Technologies, Logan, UT)
Confocal microscopy analysis
Spleens and livers from transplanted animals were harvested and fixed with 4% paraformaldehyde in PBS. Fixed organs were included in frozen blocks and cryosections were obtained. Slides were stained with PE anti-CD4 antibody. Data were analyzed though Leica SP2 confocal microscopy (Leica Microsystems, Mannheim, Germany)
Statistical analysis
Differences in animal survival (Kaplan-Meier survival curves) were analyzed with the log-rank test. Weight variation and GvHD score were analyzed with 2-way ANOVA test. All other comparisons were performed with the 2-tailed Student t test, and p < 0.05 was considered statistically significant.
Results
Donor derived and third-party Treg suppress conventional Tcon proliferation in vitro
In order to test the impact of MHC disparities on Treg suppressive function, we evaluated the ability of highly purified Treg derived from donors with different MHC backgrounds to suppress proliferation of C57BL/6 (H-2b) Tcon following exposure to irradiated stimulator splenocytes in the mixed lymphocyte reaction (MLR) in vitro. Both donor derived C57BL/6 (H-2b) or third-party FVB/N (H-2q) Treg suppressed Tcon proliferation induced by exposure to irradiated BALB/c (H-2d) splenocytes at different Treg/Tcon ratios (Figure 1A). Similar results were obtained by incubating different Tcon (BALB/c, H-2d or FVB/N, H-2q, third-party) with irradiated splenocytes from C57BL/6 (H-2b) mice and Treg from BALB/c (H-2d) mice. BALB/c Treg suppressed proliferation of both BALB/c and FVB/N Tcon equally (both p=0.04; Figure 1B) demonstrating that MHC disparities do not interfere with Treg suppressive function in vitro.
Figure 1. Donor derived or third-party Treg equally suppress Tcon proliferation in vitro.
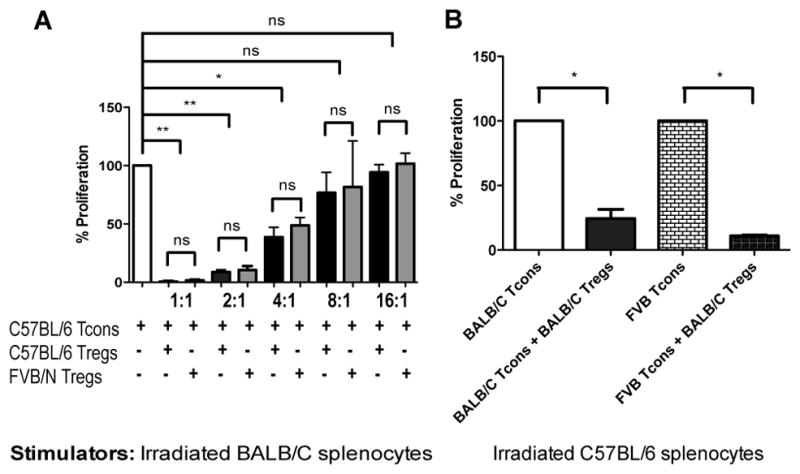
A: C57BL/6 Tcon were cultured with BALB/c irradiated splenocytes and C57BL/6 or FVB/N Treg at 1:1, 1:2, 1:4, 1:8 and 1:16 Treg/Tcon ratios. B: Either BALB/c Tcon or FVB/N Tcon were cultured with C57BL/6 irradiated splenocytes and BALB/c Treg at a ratio of 1:2. Normalized histograms are reported to allow comparison between different Tcon proliferations. For statistical analysis 2-tailed student t test was used, * p < 0.05, ** p < 0.01. One representative experiment is shown. Experiments were repeated three times.
LAG3 and CTLA4 are two key molecules that are up-regulated on Treg upon activation(13, 24–26). Moreover activated Treg release interleukin-10 (IL-10) and can reduce Tcon IFNγ production(5, 27, 28). We investigated the impact of MHC disparities on the activation of Treg in vitro. Treg were purified from either BALB/c (H-2d, matched with stimulators), C57BL/6 (H-2b, matched with responders) or FVB/N (H-2q, third-party) and incubated with IL-2 and anti-CD3/CD28 beads or were added to a MLR assay where C57BL/6 (H-2b) Tcon were the responders and BALB/c (H-2d) irradiated splenocytes were the stimulators for 3–4 days. In all of the conditions Treg maintained expression of CD25 and FoxP3 (Supplemental figure 1A–B). Third-party Treg had the same activation molecule expression patterns as MHC matched Tregs. LAG3 and CTLA4 surface expression was enhanced after stimulation with interleukin-2 (IL-2) and anti-CD3/CD28 beads or after 3–4 days of culture with Tcon and irradiated splenocytes, (Supplemental figure 1C–D). Furthermore third-party and MHC matched Treg produced the same levels of IL-10 and were similarly able to reduce Tcon IFNγ production (Supplemental figure 1E–F), thus in vitro Treg activation was not impaired by MHC disparities.
Third-party Treg suppress in vivo Tcon proliferation during early phases after transplantation
We translated these results to in vivo studies in animal models. In these studies TCD BM from C57BL/6 (H-2b) mice was injected into lethally irradiated (total body irradiation, TBI 8 Gy) BALB/c (H-2d) recipient mice to establish hematopoiesis. GvHD was induced by injecting luc+ donor derived Tcon (1x106/mouse). Using this model freshly isolated CD4+CD25+FoxP3+ Treg derived from BALB/c (H-2d, host type), C57BL/6 (H-2b, donor type) or FVB/N (H-2q, third-party) mice at the Treg/Tcon ratio of 1:2 were injected IV 2 days before Tcon injection immediately after irradiation (Figure 2A). Tcon proliferation was assessed with BLI. In vivo Tcon proliferation was similar in mice that received Tcon alone and in mice that received Tcon plus host type Treg (p>0.05, Figure 2B–D) at all time points analyzed demonstrating that host Treg lack in vivo suppressive activity. On the contrary Tcon proliferation was markedly reduced in mice that received either donor or third-party Treg 7 days after transplantation in comparison to mice that received Tcon alone (p=0.0002 and p=0.0001 respectively, Figure 2B). After 15 or 29 days Tcon proliferation was reduced in the mice that received donor Treg, but not in animals that received third-party Treg (at day 15 p=0.01 and p=ns and at day 29 p=0.04 and p=ns, Figure 2C–D) suggesting that third-party Treg can control in vivo Tcon proliferation in the early phases after transplantation but lose their activity over time in comparison to donor Treg. BLI of representative animals is shown in Figure 2E.
Figure 2. Donor type and third-party Treg reduce Tcon proliferation in vivo.

Transplantation scheme is shown as described in the text (A). In vivo BLI data are presented from representative animals at day +7 (B), day +15 (C) and day +29 (D) after transplantation. Each data point is the measurement of luminescence from one mouse (photons/sec/cm2). Mice that received only C57BL/6 luc+ Tcon (
 ), Tcon and donor type C57BL/6 Treg (◆), Tcon and third-party FVB/N Treg (◇) and Tcon and host type BALB/C Treg (△) are shown. E: Images of representative animals from each group (minimum 5 mice/group) are shown. For statistical analysis 2-tailed student t test was used, * p < 0.05, *** p < 0.001, ns = not significant. Data are representative of 3 independent experiments.
), Tcon and donor type C57BL/6 Treg (◆), Tcon and third-party FVB/N Treg (◇) and Tcon and host type BALB/C Treg (△) are shown. E: Images of representative animals from each group (minimum 5 mice/group) are shown. For statistical analysis 2-tailed student t test was used, * p < 0.05, *** p < 0.001, ns = not significant. Data are representative of 3 independent experiments.
Third-party Treg survive in vivo for a shorter period of time in comparison to donor type Treg
We previously demonstrated that during GvHD onset Treg home to secondary lymphoid tissues after injection where they are primed and induce suppression of Tcon proliferation(15, 29). Survival of Treg during this phase is essential for function. Using congenic markers that allows for discrimination of the infused donor Treg and the same transplantation model described above, we compared the in vivo survival of third-party and donor Treg by isolating spleen, lymph nodes, liver and peripheral blood of transplanted mice at different time points. Tissues from irradiated (TBI 8 Gy) BALB/c (H-2d) CD45.2 Thy1.2 mice injected with either donor C57BL/6 (H-2b) CD45.1, third party FVB/N (H-2q) CD45.1 Treg or host BALB/c (H-2d) Thy1.1 together with C57BL/6 CD45.2 Tcon and TCD BM were harvested at day 6 and day 12 after transplantation and analyzed in order to calculate the number of live injected Treg. Interestingly, in the mice that received third party Treg we found less viable injected Treg at day 6 and extremely few cells at day 12 in all the examined organs in comparison to the mice that received donor Treg (at day 6, lymph nodes p=0.03, spleen p=0.03, peripheral blood p=0.03, liver p=not significant; at day 12, lymph nodes p=0.04, spleen p=0.01, peripheral blood p=0.02, liver p=0.04) or host Treg (host versus donor Treg in all examined organs p=ns) where injected Treg were readily detected (Figure 3A–D). Injected donor Treg were found in secondary lymphoid organs such as spleen and lymph nodes and also in the liver, a GvHD target organ, in both the selected time points, while only an extremely limited number of third-party Treg survived and could be detected suggesting that these cells were rejected in vivo.
Figure 3. Third-party Treg survive for a shorter period of time compared to donor-derived Treg following in vivo transfer.

The absolute numbers of H2Kb+CD45.1+CD4+FoxP3+ donor type (●), H2Kq+CD45.1+CD4+FoxP3+ third-party (□) or H2Kd+Thy1.1+CD4+FoxP3+ host type (◆) reisolated Treg are shown in lymph nodes (A), spleen (B), peripheral blood (C) and liver (D) at day 6 and at day 12 after transplantation. No H2Kd+Thy1.1+CD4+FoxP3+ host type Treg are shown at day 12 as only few mice survived. For statistical analysis third-party Treg and host Treg have been individually compared with donor Treg (2-tailed student t test, * p < 0.05, ns = not significant). Pooled data from two consecutive experiments are shown.
Donor type and third-party, but not host type Treg reduce infiltration and cytokine production of donor T cells
CD62L is a key molecule that enables T cell homing into lymph nodes via endothelial venules. T cell receptor engagement induces T cells to lose CD62L expression, thus effector memory CD62L-CD44+ activated T cells leave lymphoid tissue and migrate to the targets to drive immune responses(30). After priming in lymphoid tissues such as spleen and lymph nodes, Tcon home to target organs such as liver, skin and gastrointestinal tract and induce GvHD in transplantation animal models. High numbers of CD44+ T cells have been found and proliferate in GvHD affected organs(31). Adoptive transfer of CD62L+CD4+CD25+FOXP3+ Treg has been shown to limit Tcon priming in lymphoid tissues consequently controlling their proliferation(16). Using the GvHD model described before, we detected the presence of CD4+CD62L+ and CD4+CD62L-CD44+ T cells in the lymph nodes and in the liver of BALB/c recipient mice that received either C57BL/6 donor Tcon alone or Tcon and host type BALB/c Treg or Tcon and C57BL/6 donor Treg or Tcon and FVB/N third-party Treg at day 6 after transplantation. Fewer effector memory CD4+CD62L-CD44+ activated T cells infiltrated the liver in mice that received donor or third-party Treg in comparison to mice that did not receive Treg (p=0.04 and p=0.03, respectively), while host type Treg did not reduce CD4+CD62L-CD44+ T cell liver infiltration (p > 0.05, Figure 4A). At the same time CD4+CD62L+CD44-/CD4+ T cell ratio was higher in the lymph nodes of mice that received donor and third-party Treg (both p=0.02), but not in the mice that received host type Treg (p > 0.05, Figure 4B). These results demonstrate that in the presence of both donor type and third-party Treg, a higher proportion of Tcon maintain CD62L and did not express CD44 in secondary lymphoid organs, therefore, donor type and third-party Treg reduce Tcon activation in lymph nodes thus limiting their proliferation and infiltration into peripheral tissues. We also analyzed LAG3 expression in Treg isolated from the spleen and lymph nodes at day 6 after transplantation and found that, while LAG3 was minimally expressed in residual host type Treg, expression was enhanced in donor and third party Treg (in lymph nodes, donor versus host type: p=0.006, donor versus third party: p > 0.05, third party versus host type: p=0.02; in spleen, donor versus host type: p=0.0002, donor versus third party: p > 0.05, third party versus host type: p=0.04. Figure 4C) demonstrating that both donor and third party Treg exhibit a suppressive phenotype after their adoptive transfer. Furthermore, as Treg treatment has been previously demonstrated to reduce IFNγ production by Tcon and increase IL-10 production in GvHD affected mice, we collected sera of transplanted mice where we found that injection of donor Treg induces lower IFNγ (p=0.006) and higher IL-10 (p=0.05) production in comparison to mice that were treated with third-party Treg (Figure 4D). Little or no difference was found in interleukin-4 (IL-4) and interleukin-5 (IL-5) production (data not shown). Therefore, donor Treg more effectively induce a suppressive cytokine profile.
Figure 4. Infiltration of effector memory CD4+CD62L-CD44+ donor T cells is reduced after donor type or third-party Treg adoptive transfer.

A: Numbers of live H2Kb+CD4+CD62L-CD44+ cells found in liver of BALB/c mice that received only donor C57BL/6 Tcon (white), Tcons and host type BALB/c Treg (black), Tcon and donor type C57BL/6 Treg (grey) and Tcon and third-party FVB/N Treg (white dotted) are shown after analysis of the samples at day 6 after transplantation. A representative gating strategy of CD4+CD62L-CD44+ cells is also reported. B: H2Kb+CD4+CD62L+ percentage over H2Kb+CD4+ cells is shown in lymph nodes. Histograms of CD62L expression on H2Kb+CD4+ cells in lymph nodes of mice that received only donor C57BL/6 Tcon (no dashing), Tcon and host type BALB/c Treg (dotted), Tcon and donor type C57BL/6 Treg (short dashed) and Tcon and third-party FVB/N Treg (long dashed) and a representative gating strategy of CD4+CD62L+ cells are also reported. C: LAG3+CD4+FoxP3+ cell percentage over CD4+FoxP3+ cells is reported in lymph nodes and spleen after analysis of the samples at day 6 after transplantation is reported. D: IFNγ and IL-10 concentrations (pg/ml) in the sera collected at day 6 after transplantation are shown. White histograms are representative of mice that received only donor type Tcon, black histograms are representative of mice that received Tcon and host type Treg, grey histograms are representative of mice that received Tcon and donor type Treg and white dotted histograms are representative of mice that received Tcon and third-party Treg. For statistical analysis 2-tailed student t test was used, *** p < 0.0005, ** p < 0.005, * p < 0.05, ns p=not significant. Pooled data from two consecutive experiments are shown.
Donor and third-party but not host Treg provide GvHD protection
We tested the ability of Treg from different sources to protect mice from GvHD using the above described in vivo model. We evaluated GvHD onset and mouse survival following the adoptive transfer of freshly isolated CD4+CD25+FoxP3+ Treg derived from BALB/c (H-2d, host type), C57BL/6 (H-2b, donor type) or FVB (H-2q, third-party) at the Treg/Tcon ratio of 1:2 (Figure 5A). Donor Treg exerted the strongest dose dependent GvHD protection (p=0.028), while host Treg did not improve mouse survival (p = 0.58). Third-party Treg improved mouse survival (p=0.028), but these animals had worse GvHD score profiles (p < 0.001) and did not recover their weight as well as mice treated with donor Treg (p < 0.001, Figure 5A–C). To exclude a possible strain related effect we also transplanted BALB/c recipient with FVB/N derived TCD BM and Tcon and adoptively transferred either BALB/c (host type), FVB/N (donor type) or C57BL/6 (third-party) Treg. Even in this case donor and third-party, but not host Treg improved mouse survival (data not shown). These data confirm that third-party Treg can be a useful tool for GvHD prevention.
Figure 5. Donor and third-party but not host Treg prevent GvHD in allogeneic transplantation.

Survival (A), weight variation (B) and GvHD score (C) of allogeneic transplanted BALB/c recipient mice after injection of only C57BL/6 Tcon (
), Tcon and donor type C57BL/6 Treg (◆), Tcon and third-party FVB/N Treg (◇) and Tcon and host type BALB/c Treg (△) are shown. Mice that received TCD BM only (dashed line, ■) and mice that were lethally irradiated but not transplanted (dotted line, ●) were used as controls. For statistical analysis of mouse survival Kaplan-Meier test was used, for weight variation and GvHD score 2-way ANOVA test was used, * p < 0.05, ** p < 0.01, *** p < 0.001. Data is representative of one of three experiments (minimum 5 mice/group).
Since the major mismatch model produces the most aggressive GvHD we compared donor and third-party Treg for GvHD protection in a minor-mismatch mouse model of transplantation that more closely resembles clinical conditions in HLA matched human transplantation. In this model, TCD BM from C57BL/6 (H-2b) mice was injected into lethally irradiated (TBI 8 Gy) BALB/b (H-2b, minor mismatched) recipient mice. Because it was previously demonstrated that adoptive transfer of higher number of Tcon is needed to induce GvHD in minor mismatched compared to major mismatched mouse models(32), we injected 2.5x107 donor derived Tcon. To prevent GvHD onset and lethality we adoptively transferred 5x105 freshly isolated CD4+CD25+FoxP3+ Treg derived from C57BL/6 (H-2b, donor type) or FVB/N (H-2q, third-party) mice (Figure 6A). In these studies third-party and donor Treg equally prolonged survival (p=0.02 and p=0.04 vs Tcon alone group, respectively; p>0.05 when the donor Treg group was compared with third-party Treg group) of these mice (Figure 6B). No differences could be detected in weight recovery (Figure 6C) and GvHD score (Figure 6D) demonstrating that both donor and third-party Treg are able to protect mice from GvHD equally in this MHC minor mismatched model.
Figure 6. Treg equally protect from GvHD lethality in a MHC minor mismatched mouse model.

Transplantation scheme (A). Survival (B), weight (C) and GvHD score (D) of MHC minor mismatched transplanted BALB/b recipient mice after injection of C57BL/6 Tcon (
), Tcon and donor type C57BL/6 Treg (◆) and Tcon and third-party FVB/N Treg (◇) are shown. Mice that received TCD BM only (dashed line, ■) and mice that were lethally irradiated but not transplanted (dotted line, ●) were used as controls. For statistical analysis of mouse survival Kaplan-Meier test was used, for weight variation and GvHD score 2-way ANOVA test was used, * p < 0.05, ns = not significant. Pooled data from two experiments are shown.
Treg exert the main protection from GvHD in the very early phase after transplantation
While Treg dynamics in vivo after adoptive transfer have been widely studied, less is known regarding the timing of their in vivo function. We used a mouse model of in vivo Treg depletion to explore the timeframe during which injected Treg control Tcon proliferation and homing in GvHD target tissues after transplantation. We injected TCD BM from C57BL/6 mice into lethally irradiated (TBI 8 Gy) BALB/c (H-2d) recipient mice and GvHD was induced by injecting donor derived Tcon (1x106/mouse). Freshly isolated CD4+CD25+FoxP3+GFP+ Treg derived from C57BL/6 albino FoxP3DTR/GFP/luc mice at the Treg/Tcon ratio of 1:2 were injected IV 2 days before Tcon injection. In some animals we depleted the injected Treg pool through intraperitoneal administration of DT for two consecutive days at two different time points: immediately after their transfer (day −2 and day −1) or at the day of Tcon injection (day 0 and day +1, Figure 7A). GFP+ injected Treg were detectable only in the spleen and peripheral blood of the animals that did not receive DT treatment proving the efficacy of Treg depletion with DT in vivo (Figure 7B). Utilizing BLI we found high intensity signal from luc+ Treg only in mice that did not receive DT treatment further confirming that DT was effective in depleting injected Treg (Figure 7C). Mice that received DT treatment at day 0 had reduced infiltration of donor derived Tcon in the liver in comparison to mice that received DT treatment at day −2 and to mice that did not receive Treg adoptive transfer and similarly to mice that received Treg but no DT treatment (Supplemental Figure 2A–B). These results demonstrate that Treg exert their control on Tcon entry in the liver in the first two days after their injection.
Figure 7. Treg limit GvHD lethality in the very early phase of transplantation.

Transplantation scheme is shown as described in the text (A). Treg were efficiently depleted after DT treatment. Percentages of GFP+ Treg in spleen and peripheral blood of mice that did not receive Treg transfer (Tcon), that received Treg transfer and DT at day −2 and −1 (Tcon + Treg DT −2), that received Treg transfer and DT at day 0 and +1 (Tcon + Treg DT 0) and that received Treg transfer with no DT treatment (Tcon + Treg no DT) are shown. Representative samples are reported. GFP+ Treg were detectable only in the last group (red arrows, B). Images of representative mice that received Tcon + luc+ Treg + DT at day −2 and −1 (Tcon + Treg DT −2) or Tcon + luc+ Treg + DT at day 0 and +1 (Tcon + Treg DT 0) or only Tcon + luc+ Treg (Tcon + Treg no DT) are reported. Images were taken at day 0 and at day +6. Data demonstrate that Treg were effectively depleted first in mice that received DT at day −2 and −1 and successively in mice that received DT at day 0 and +1, while Treg were in vivo proliferating in mice that did not receive DT treatment (C). Survival (D), weight (E) and GvHD score (F) of transplanted BALB/c recipient mice after injection of C57BL/6 Tcon (
), Tcon + C57BL/6 albino FoxP3DTR/GFP/luc Treg + DT at day −2 and −1 (▽), Tcon + Treg + DT at day 0 and +1 (◇) and Tcon + Treg with no DT (◆) are shown. Mice that received TCD BM only (dashed line, ■) and mice that were lethally irradiated but not transplanted (dotted line, ●) were used as controls. For statistical analysis of mouse survival Kaplan-Meier test was used, for weight variation and GvHD score 2-way ANOVA test was used, * p < 0.05, ns = not significant. Pooled data from two experiments are shown.
To further understand the impact of Treg killing on their in vivo function and to explore the mechanism through which third party Treg are able to prevent GvHD even if rapidly rejected in vivo, we used the mouse model of Treg in vivo depletion as described above and followed animals for survival, weight and GvHD score. Mice that received Tcon and Treg adoptive transfer and DT treatment at day 0 had improved survival in comparison to mice that received Tcon and Treg adoptive transfer and DT treatment at day −2 and to mice that received Tcon alone; survival of these animals was comparable to animals that did not receive DT treatment (Figure 7D). Furthermore DT treatment at day 0 only mildly impacted mouse weight variation and GvHD score after transplantation (Figure 7E–F). We also treated mice that received Tcon adoptive transfer (1x106/mouse) without Treg with DT and observed similar survival indicating that DT toxicity was limited in vivo and had no impact on GvHD (Supplemental figure 3). These data demonstrate that Treg exert their in vivo function in the very early phase of transplantation and that their main activity is employed in the absence of Tcon.
Discussion
In this study we demonstrate that freshly isolated natural occurring third-party Treg are a valuable and useful alternative to Treg derived from the same donor as the Tcon, as both effectively suppress alloreactive T cell proliferation and GvHD. The major limitation to the adoptive transfer of third-party Treg is their shorter survival in vivo in comparison to donor derived Treg, which were present for longer periods of time in lymphoid tissues and GvHD target organs. Interestingly, donor derived Treg could be found in the peripheral blood as well as in lymphoid tissues. While donor cell rejection occurs because of host versus graft reactions that can be sustained by host T cells and recently described subsets of host natural killer cells(33), third party Treg may be also rejected by donor Tcon because they are MHC mismatched to both host and donor.
Despite their inferior survival compared to donor Treg, third party Treg demonstrated effective protection from GvHD. This observation prompted us to study the timing of Treg function in vivo to investigate the mechanism of third party Treg function. We found that adoptively transferred Treg exert their protection against GvHD mainly in the very early phase after transplantation, thus explaining why third party Treg are functional even if rejected early. Treg rapidly home to secondary lymphoid organs after injection and a few days later circulate to peripheral tissues(15). In our experiments, even if in vivo Treg depletion occurred only two days after Treg injection (day 0), Treg were able to reduce GvHD lethality proving that their function is mainly sustained by their early presence in lymphoid tissues. Donor and third-party Treg similarly reduce Tcon activation in lymphoid tissues limiting their further infiltration of GvHD target organs confirming that in vivo Treg control over Tcon proliferation mainly happens in lymph nodes and is an early event after Treg adoptive transfer. Early rejection of third party Treg, even if induces a worse GvHD score profile and a further reduction of body weight in the animals, is not enough to abrogate Treg function as third party Treg are anyway able to limit GvHD lethality in the first days after transplantation.
The fact that third-party Treg were efficacious in controlling GvHD also demonstrates that the main mechanisms through which Treg exert their in vivo function are independent of Treg-Tcon MHC identity. Others showed that post-transplant residual host type dendritic cells (DC) interact with donor Tcon triggering GvHD(34–36). Treg need to be present during the Tcon priming phase for effective suppressive(37) and recently they have been shown to disrupt DC-Tcon interactions inducing suppression of in vivo Tcon proliferation limiting GvHD (38). Our data demonstrate that these early events after transplant happen in the absence of MHC identity while Treg-Tcon MHC disparities are only responsible for in vivo Treg survival. Furthermore, in our model, Treg exert their function in the first two days after adoptive transfer preceding Tcon injection, therefore Treg function is mainly sustained by Treg interactions with lymphoid environment even in the absence of donor Tcon suggesting that Treg modify the ability of host antigen presenting cells to effectively prime Tcon and induce GvHD. Further studies are required to understand the cellular interactions involved and the mechanisms that underlie these early events after transplantation. As Treg exert their function before Tcon expansion, it can also explain why Treg adoptive transfer is more effective in preventing GvHD if executed before Tcon injection(15).
Our insights on timing of Treg function suggest that Treg adoptive transfer is mainly effective in the early phase of transplantation and demonstrates that timing of Treg injection is crucial for their function suggesting that delayed GvHD treatment with Treg may be minimally effective as Treg would not be able to interfere with the Tcon priming phase. Clinical trials are ongoing with the goal of expanding the Treg pool in vivo to reduce GvHD symptoms and possibly limiting lethality(10, 39).
Others demonstrated that Treg that are specific for a third party antigen are effective in controlling GvHD if reactivated in vivo by providing the specific antigen suggesting that specific Treg require previous activation to exploit a broader ability of suppression(40). We transferred fresh polyclonal Treg proving that antigen specificity may be not required or may be acquired in vivo for effective function that does not depend on MHC matching. Moreover Treg-Tcon MHC disparities strongly limit natural occurring Treg function when the bone marrow recipient is selected as the source of Treg. Previous studies showed that differently activated host type Treg are able to control donor Tcon proliferation thus reducing GvHD lethality(6, 41). We demonstrated that adoptive transfer of freshly isolated unmanipulated host type Treg do not prevent GvHD and do not have any impact on animal survival even if their in vivo detection is still possible several days after injection. Therefore, host Treg may require activation to exert their effect, while third party Treg are functional even in the absence of any ex vivo or in vivo manipulation but presumably are activated in vivo following GvHD induction.
Analyzing different mouse tissues after transplantation, we were also able to detect previously transferred Treg in the peripheral blood as well as spleen, lymph nodes and liver demonstrating that Treg presence in the peripheral blood is reflective of their infiltration of lymphoid and peripheral tissues. Such data allow for peripheral blood monitoring of Treg transfer providing a relevant tool for clinical translation.
When we induced GvHD in a MHC minor mismatch model of transplantation, there was no impact of Treg-Tcon MHC disparities on mouse survival and on GvHD onset. In this model higher numbers of Tcon are required for inducing lethal GvHD, but even Treg that have been injected at an extremely low Treg/Tcon ratio (1/50 in our study) resulted in protection and prolonged survival. Both donor type and third party Treg were effective in GVHD protection. As donor Tcon are activated in vivo by minor antigens and graft versus host reactions are weaker because of their MHC class I identity(42), we might expect that third-party Treg that are MHC major mismatched with the host environment would be more effective in this model. Somewhat surprisingly, we found that third party Treg protect mice from GvHD as well as donor Treg strongly suggesting that the role of MHC disparities in Treg activation and the survival of transferred Treg in vivo have limited impact on GvHD protection when minor antigens work as GvHD triggers. In this context third party Treg may be rejected less by the donor T cell pool, survive longer and possibly provide effective in vivo suppressive function even if limited in number, therefore, explaining the similar survival that we observed in mice that received donor or third party Treg. Because this model better resembles HLA conditions in human HLA-matched transplantation, these data promote third-party Treg cellular therapy for GvHD prevention in this clinical setting.
Steiner et al described third party Treg as an effective alternative tool when injected for overcoming experimental induced rejection(43). Our work confirms their observations highlighting third party Treg value in suppressing GvHD and introduces relevant insights on Treg mechanism of in vivo tolerance induction. In the Steiner et al. transplantation model donor and third party Treg are injected in mice that received host type T cells for rejection induction, therefore both may be rejected by host mediated alloreactions. In our model the host immune system is quickly overcome by donor T cells resulting in rapid engraftment and GvHD; even in these conditions that favor donor cell persistence, third party Treg are still effective in suppressing GvHD.
One of the major limitations to a broader clinical application of Treg adoptive transfer is the difficulty in obtaining enough cells from a donor due to Treg paucity in the periphery. The use of third party Treg may overcome this issue as they can be harvested from several donors, pooled if needed, prepared in advance and banked allowing for a readily available cell product for clinical use.
MHC matching is a key factor in transplantation and the new introduction of cellular therapies requires better understanding of MHC interactions and roles in this setting. We believe that our study elucidates critical impact of Treg/Tcon MHC disparities and provides important insights into the selection of the source of Treg for the clinical application of Treg cellular therapy in GvHD prevention. Although HLA matched sibling donors can often provide fresh natural occurring Treg, our findings demonstrate that the use of third party Treg is a valuable alternative to donor-derived Treg immunotherapy in other clinical settings, where HLA-matched donors are not readily available.
Our study also brings relevant insights about timing of in vivo Treg function and helps elucidate the mechanisms through which Treg suppress GvHD in preclinical and clinical settings. The very early phase of transplantation is extremely important and regulates transplantation tolerance and GvHD onset and lethality. Treg adoptive transfer plays a key role in this crucial phase of transplantation and treatment interventions that modify the balance between regulatory and effector interactions during this phase strongly impacts transplantation outcomes.
Supplementary Material
Acknowledgments
This work was supported by Fondazione Italiana per la Ricerca sul Cancro (AP) and by Program Project Grants CA49605 and HL075462 and an R01 HL114591 from the National Cancer Institute and National Heart, Lung and Blood Institute.
We thank Stanford Shared FACS Facility and Stanford Center for Innovation in In-Vivo Imaging (SCI3) for providing facilities for FACS analysis and in vivo imaging.
Footnotes
Authorship
AP designed and performed research, analyzed data and wrote the manuscript. LC, MA, DS, HN, JB, YP, MF and BSK performed research and reviewed the manuscript. RN helped write the manuscript and provided overall guidance.
Conflict of Interest
The authors declare no competing financial interest.
References
- 1.Ferrara JL, Deeg HJ. Graft-versus-host disease. The New England journal of medicine. 1991;324:667–674. doi: 10.1056/NEJM199103073241005. [DOI] [PubMed] [Google Scholar]
- 2.Jones SC, Murphy GF, Korngold R. Post-hematopoietic cell transplantation control of graft-versus-host disease by donor CD425 T cells to allow an effective graft-versus-leukemia response. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2003;9:243–256. doi: 10.1053/bbmt.2003.50027. [DOI] [PubMed] [Google Scholar]
- 3.Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL. CD4(+)CD25(+) immunoregulatory T Cells: new therapeutics for graft-versus-host disease. The Journal of experimental medicine. 2002;196:401–406. doi: 10.1084/jem.20020090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, Negrin RS. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nature medicine. 2003;9:1144–1150. doi: 10.1038/nm915. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. The Journal of experimental medicine. 2002;196:389–399. doi: 10.1084/jem.20020399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood. 2002;99:3493–3499. doi: 10.1182/blood.v99.10.3493. [DOI] [PubMed] [Google Scholar]
- 7.Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, Del Papa B, Zei T, Ostini RI, Cecchini D, Aloisi T, Perruccio K, Ruggeri L, Balucani C, Pierini A, Sportoletti P, Aristei C, Falini B, Reisner Y, Velardi A, Aversa F, Martelli MF. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117:3921–3928. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- 8.Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, Pierini A, Massei MS, Amico L, Urbani E, Del Papa B, Zei T, Iacucci Ostini R, Cecchini D, Tognellini R, Reisner Y, Aversa F, Falini B, Velardi A. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood. 2014;124:638–644. doi: 10.1182/blood-2014-03-564401. [DOI] [PubMed] [Google Scholar]
- 9.Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, Defor T, Levine BL, June CH, Rubinstein P, McGlave PB, Blazar BR, Wagner JE. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117:1061–1070. doi: 10.1182/blood-2010-07-293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, 3rd, Armand P, Cutler C, Ho VT, Treister NS, Bienfang DC, Prasad S, Tzachanis D, Joyce RM, Avigan DE, Antin JH, Ritz J, Soiffer RJ. Interleukin-2 and regulatory T cells in graft-versus-host disease. The New England journal of medicine. 2011;365:2055–2066. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martelli MF, Di Ianni M, Ruggeri L, Pierini A, Falzetti F, Carotti A, Terenzi A, Reisner Y, Aversa F, Falini B, Velardi A. “Designed” grafts for HLA-haploidentical stem cell transplantation. Blood. 2014;123:967–973. doi: 10.1182/blood-2013-10-531764. [DOI] [PubMed] [Google Scholar]
- 12.Schneidawind D, Pierini A, Negrin RS. Regulatory T cells and natural killer T cells for modulation of GVHD following allogeneic hematopoietic cell transplantation. Blood. 2013;122:3116–3121. doi: 10.1182/blood-2013-08-453126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nature reviews. Immunology. 2011;11:119–130. doi: 10.1038/nri2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colonna L, Sega EI, Negrin RS. Natural and expanded CD4(+)CD25(+) regulatory T cells in bone marrow transplantation. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2011;17:S58–62. doi: 10.1016/j.bbmt.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen VH, Zeiser R, Dasilva DL, Chang DS, Beilhack A, Contag CH, Negrin RS. In vivo dynamics of regulatory T-cell trafficking and survival predict effective strategies to control graft-versus-host disease following allogeneic transplantation. Blood. 2007;109:2649–2656. doi: 10.1182/blood-2006-08-044529. [DOI] [PubMed] [Google Scholar]
- 16.Ermann J, Hoffmann P, Edinger M, Dutt S, Blankenberg FG, Higgins JP, Negrin RS, Fathman CG, Strober S. Only the CD62L+ subpopulation of CD4+CD25+ regulatory T cells protects from lethal acute GVHD. Blood. 2005;105:2220–2226. doi: 10.1182/blood-2004-05-2044. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann P, Eder R, Kunz-Schughart LA, Andreesen R, Edinger M. Large-scale in vitro expansion of polyclonal human CD4(+)CD25high regulatory T cells. Blood. 2004;104:895–903. doi: 10.1182/blood-2004-01-0086. [DOI] [PubMed] [Google Scholar]
- 18.Levings MK, Sangregorio R, Roncarolo MG. Human cd25(+)cd4(+) t regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. The Journal of experimental medicine. 2001;193:1295–1302. doi: 10.1084/jem.193.11.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veerapathran A, Pidala J, Beato F, Betts B, Kim J, Turner JG, Hellerstein MK, Yu XZ, Janssen W, Anasetti C. Human regulatory T cells against minor histocompatibility antigens: ex vivo expansion for prevention of graft-versus-host disease. Blood. 2013;122:2251–2261. doi: 10.1182/blood-2013-03-492397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veerapathran A, Pidala J, Beato F, Yu XZ, Anasetti C. Ex vivo expansion of human Tregs specific for alloantigens presented directly or indirectly. Blood. 2011;118:5671–5680. doi: 10.1182/blood-2011-02-337097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao YA, Wagers AJ, Beilhack A, Dusich J, Bachmann MH, Negrin RS, Weissman IL, Contag CH. Shifting foci of hematopoiesis during reconstitution from single stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:221–226. doi: 10.1073/pnas.2637010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edinger M, Cao YA, Verneris MR, Bachmann MH, Contag CH, Negrin RS. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood. 2003;101:640–648. doi: 10.1182/blood-2002-06-1751. [DOI] [PubMed] [Google Scholar]
- 23.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J, Jr, Crawford JM, Ferrara JL. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–3239. [PubMed] [Google Scholar]
- 24.Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S, Kowalski J, Levitsky HI, Powell JD, Pardoll DM, Drake CG, Vignali DA. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–513. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 25.Sega EI, Leveson-Gower DB, Florek M, Schneidawind D, Luong RH, Negrin RS. Role of lymphocyte activation gene-3 (Lag-3) in conventional and regulatory T cell function in allogeneic transplantation. PloS one. 2014;9:e86551. doi: 10.1371/journal.pone.0086551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 27.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr, Muller W, Rudensky AY. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 28.Shin HJ, Baker J, Leveson-Gower DB, Smith AT, Sega EI, Negrin RS. Rapamycin and IL-2 reduce lethal acute graft-versus-host disease associated with increased expansion of donor type CD4+CD25+Foxp3+ regulatory T cells. Blood. 2011;118:2342–2350. doi: 10.1182/blood-2010-10-313684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen VH, Shashidhar S, Chang DS, Ho L, Kambham N, Bachmann M, Brown JM, Negrin RS. The impact of regulatory T cells on T-cell immunity following hematopoietic cell transplantation. Blood. 2008;111:945–953. doi: 10.1182/blood-2007-07-103895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 31.Beilhack A, Schulz S, Baker J, Beilhack GF, Wieland CB, Herman EI, Baker EM, Cao YA, Contag CH, Negrin RS. In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood. 2005;106:1113–1122. doi: 10.1182/blood-2005-02-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korngold R, Wettstein PJ. Immunodominance in the graft-vs-host disease T cell response to minor histocompatibility antigens. J Immunol. 1990;145:4079–4088. [PubMed] [Google Scholar]
- 33.Sun K, Alvarez M, Ames E, Barao I, Chen M, Longo DL, Redelman D, Murphy WJ. Mouse NK cell-mediated rejection of bone marrow allografts exhibits patterns consistent with Ly49 subset licensing. Blood. 2012;119:1590–1598. doi: 10.1182/blood-2011-08-374314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duffner UA, Maeda Y, Cooke KR, Reddy P, Ordemann R, Liu C, Ferrara JL, Teshima T. Host dendritic cells alone are sufficient to initiate acute graft-versus-host disease. J Immunol. 2004;172:7393–7398. doi: 10.4049/jimmunol.172.12.7393. [DOI] [PubMed] [Google Scholar]
- 35.Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, McNiff J, Shlomchik WD. Donor APCs are required for maximal GVHD but not for GVL. Nature medicine. 2004;10:987–992. doi: 10.1038/nm1089. [DOI] [PubMed] [Google Scholar]
- 36.Reddy P, Maeda Y, Liu C, Krijanovski OI, Korngold R, Ferrara JL. A crucial role for antigen-presenting cells and alloantigen expression in graft-versus-leukemia responses. Nature medicine. 2005;11:1244–1249. doi: 10.1038/nm1309. [DOI] [PubMed] [Google Scholar]
- 37.Wang XN, Haniffa MA, Holtick U, Collin MP, Jackson G, Hilkens CM, Holler E, Edinger M, Hoffmann P, Dickinson AM. Regulatory T-cell suppression of CD8+ T-cell-mediated graft-versus-host reaction requires their presence during priming. Transplantation. 2009;88:188–197. doi: 10.1097/TP.0b013e3181ac14ce. [DOI] [PubMed] [Google Scholar]
- 38.Lin KL, Fulton LM, Berginski M, West ML, Taylor NA, Moran TP, Coghill JM, Blazar BR, Bear JE, Serody JS. Intravital imaging of donor allogeneic effector and regulatory T cells with host dendritic cells during GVHD. Blood. 2014;123:1604–1614. doi: 10.1182/blood-2013-09-526020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, Murase K, Cutler C, Ho VT, Alyea EP, Armand P, Blazar BR, Antin JH, Soiffer RJ, Ritz J. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Science translational medicine. 2013;5:179ra143. doi: 10.1126/scitranslmed.3005265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin GH, Gregoire S, Landau DA, Pilon C, Grinberg-Bleyer Y, Charlotte F, Mege JP, Chatenoud L, Salomon BL, Cohen JL. In vivo activation of transferred regulatory T cells specific for third-party exogenous antigen controls GVH disease in mice. European journal of immunology. 2013;43:2263–2272. doi: 10.1002/eji.201343449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chopra M, Riedel SS, Biehl M, Krieger S, von Krosigk V, Bauerlein CA, Brede C, Jordan Garrote AL, Kraus S, Schafer V, Ritz M, Mattenheimer K, Degla A, Mottok A, Einsele H, Wajant H, Beilhack A. Tumor necrosis factor receptor 2-dependent homeostasis of regulatory T cells as a player in TNF-induced experimental metastasis. Carcinogenesis. 2013;34:1296–1303. doi: 10.1093/carcin/bgt038. [DOI] [PubMed] [Google Scholar]
- 42.Reddy P, Negrin R, Hill GR. Mouse models of bone marrow transplantation. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2008;14:129–135. doi: 10.1016/j.bbmt.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steiner D, Brunicki N, Blazar BR, Bachar-Lustig E, Reisner Y. Tolerance induction by third-party “off-the-shelf” CD4+CD25+ Treg cells. Experimental hematology. 2006;34:66–71. doi: 10.1016/j.exphem.2005.10.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.