

Abstract
Medulloblastoma (Med) is the most common malignant brain tumor in children. The role of ESR2 [estrogen receptor (ER)-β] in promoting Med growth was comprehensively examined in three in vivo models and human cell lines. In a novel Med ERβ-null knockout model developed by crossing Esr2−/− mice with cerebellar granule cell precursor specific Ptch1 conditional knockout mice, the tumor growth rate was significantly decreased in males and females. The absence of Esr2 resulted in increased apoptosis, decreased B-cell lymphoma 2 (BCL2), and IGF-1 receptor (IGF1R) expression, and decreased levels of active MAPKs (ERK1/2) and protein kinase B (AKT). Treatment of Med in Ptch1+/− Trp53−/− mice with the antiestrogen chemotherapeutic drug Faslodex significantly increased symptom-free survival, which was associated with increased apoptosis and decreased BCL2 and IGF1R expression and signaling. Similar effects were also observed in nude mice bearing D283Med xenografts. In vitro studies in human D283Med cells metabolically stressed by glutamine withdrawal found that 17β-estradiol and the ERβ selective agonist 2,3-bis(4-hydroxyphenyl)-propionitrile dose dependently protected Med cells from caspase-3-dependent cell death. Those effects were associated with increased phosphorylation of IGF1R, long-term increases in ERK1/2 and AKT signaling, and increased expression of IGF-1, IGF1R, and BCL2. Results of pharmacological experiments revealed that the cytoprotective actions of estradiol were dependent on ERβ and IGF1R receptor tyrosine kinase activity and independent of ERα and G protein-coupled estrogen receptor 1 (G protein coupled receptor 30). The presented results demonstrate that estrogen promotes Med growth through ERβ-mediated increases in IGF1R expression and activity, which induce cytoprotective mechanisms that decrease apoptosis.
Neoplasms of the central nervous system (CNS) are the second most common childhood malignancy, accounting for greater than 16% of all childhood cancers (1). Among these, the primitive neuroectodermal tumor medulloblastoma (Med) is the most common (1). Five-year survival rates for Med patients are approximately 70% due to advances in the multimodal treatment using surgery, radiotherapy, and chemotherapy (2). However, survivors often develop a debilitating spectrum of neurological, endocrine, and cognitive side effects that are primarily associated with the dose of radiation (3–5). Along with increasing the cure rates for Med, there is a need for adjuvant therapies that further improve outcomes and decrease the adverse side-effects of treatment (6).
Medulloblastoma most commonly arise from cerebellar granule cell precursors (GCPs) that have escaped terminal differentiation (7). We previously demonstrated that like GCPs and mature granule cells, malignant Med cells express estrogen receptor (ER)-β, and estrogen can regulate growth, viability, and migration in normal GCPs and Med cells (8–10). In the nude mouse xenograft model of Med used here, it was previously found that estrogen can increase growth rate of these human-derived tumors. The growth-promoting effects of estrogen in these human D283Med tumors were dependent on changes in estrogen-responsive gene expression, effects that were blocked by the antiestrogen chemotherapeutic drug Faslodex (8). Those studies revealed for the first time that estrogens and the nuclear receptor activities of ERβ played an important role in Med and established antiestrogen therapy as a potentially efficacious clinical treatment. Highlighting the need to understand in more detail the role of estrogen and ERβ in Med, subsequent studies in a Ptch1+/− mouse model of Med found that ERβ activity was associated with decreased Med tumor incidence induced by ionizing radiation (11–13). Results from in vitro studies also suggested that inhibition of ERβ activity in some Med cell lines may decrease sensitivity to cisplatin by enhancing Rad51-mediated DNA repair mechanisms (14).
The binding of 17β-estradiol (E2) at intracellular and membrane-associated ERs regulates estrogen-responsive gene expression in estrogen-responsive cells. During critical developmental periods of cerebellar granule cell maturation, physiological concentrations of E2 regulate GCP mitogenesis and cell death through classical ERβ-dependent mechanisms and concomitant increases in rapid ERK1/2 signaling and protein phosphatase 2 activity (10, 15–17). Classical ER-dependent mechanisms active in the nervous system also regulate the developmental and cytoprotective actions of IGF. Normal IGF signaling is critical during cerebellar development and treatment with IGF-1 can protect GCP from pathological cell death (18–20). The importance of IGF signaling in tumorigenesis and progression of Med and other CNS malignancies is also well established (21–23). In transgenic models of Med, the disruption of the IGF and sonic hedgehog signaling pathways act synergistically to increase Med incidence (24). In human Med, increased expression of IGF-1 receptor (IGF1R) and increased IGF signaling are associated with increased tumor growth and decreased apoptosis (21, 25). Inhibition of IGF1R has also been shown to decrease Med growth and to increase sensitivity of Med cells to cytotoxic chemotherapeutics (26, 27).
Although the role of ERβ in estrogen-responsive cancers is controversial, an important role of ERα in carcinogenesis and progression of estrogen responsive tumors of the breast, prostate, and female reproductive tract is well established (28, 29). For ER-positive breast cancer, the selective estrogen receptor modulator (SERM) tamoxifen is used clinically as the first-line treatment to block ER activity and inhibit breast tumor growth. The ER antagonist fulvestrant (Faslodex) is also used as an adjuvant therapy for treating advanced or metastatic breast cancer (30). Although it is known that ERα promotes the growth of estrogen-dependent cancers, results from studies investigating the role of ERβ in estrogen-responsive tumors from other tissues are less clear-cut, and the mechanisms underlying the reported differential effects of ERβ remain unclear. As in normal estrogen-sensitive tissues, a variety of factors including whether ERα and ERβ are coexpressed, the specific ERβ isoforms expressed, and the subcellular localization of expressed receptors can influence the effects of estrogen in different responsive cancers (29, 31, 32).
Interactions between classical ER-mediated mechanisms and the IGF-signaling pathway, a phenomena referred to as cross talk, have been established by studies demonstrating an interdependence between ERα transactivation and increased IGF/IGF1R activity in mediating the neuroprotective actions of estrogen in the brain (33, 34). Estrogen induced increases in IGF signaling activity resulting from increased IGF1R expression in Med and cerebellar precursors are due to the autocrine and paracrine actions of the locally high levels of expressed ligands (eg, IGF1/2), which drive increases in downstream signaling activity (23). The ability of ERβ to mediate cross talk between estrogen and IGF1 signaling pathways in Med is unknown. Similarly, interactions between ER activity and IGF signaling in cerebellum have not been established. However, ERβ and IGF1R expression is coordinately regulated in adult rat cerebellum, suggesting a link between ERβ-responsive gene expression and modulation of the IGF signaling pathway (35).
The aim of the current study was to establish directly whether ERβ promotes Med growth by increasing tumor cell proliferation and/or decreasing apoptosis and to examine whether the effects of E2 were mediated by cross talk between ERβ and IGF signaling. To address these aims, an ERβ-null Med knockout model was developed by selectively crossing Esr2−/− mice with a GCP-specific Ptch1 conditional knockout mouse model of Med (36, 37). The effect of Esr2 ablation on Med growth was evaluated by comparison of tumor growth rates in wild-type (WT) and Esr2−/− knockout Med mice. Quantitative histological and immunohistochemical approaches were also used to assess the impact of Esr2 loss of function on tumor cell proliferation and apoptosis and assess the effects on IGF1R and B-cell lymphoma 2 (BCL2) expression. Impacts on cytoprotective MAPK and AKT signaling pathways were similarly assessed. Additional studies using a different knockout mouse model of Med (Ptch1+/− Trp53−/−) and human Med xenografts were performed to investigate whether the effects of estrogen were generalizable across models and to establish the therapeutic efficacy of the antiestrogen drug Faslodex on tumor progression (38). Cell culture-based studies using human D283Med cells were performed to further understand the mechanisms mediating the actions of estrogen in Med.
Materials and Methods
Steroids and pharmacological agents
Dimethylsulfoxide (DMSO) and E2 were from Sigma-Aldrich. Faslodex (Fulvestrant 500 mg injection) was from AstraZeneca. 7α,17β-[9-[(4,4,5,5,5-Pentafluoropentyl) sulfinyl]nonyl]estra-1,3,5(10)-triene-3, 17-diol (ICI 182, 780), 4,4′,4′-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT), 2,3-bis(4-hydroxyphenyl)-propionitrile (DPN), G-1, 4-[2-phenyl-5,7-bis (trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP), 1,3-bis (4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole dihydrochloride (MPP), benzyloxycarbonyl-Asp (OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethylketone (Z-DEVD-fmk), and LY294002 were from Tocris Bioscience. U0126 was from Promega, wortmannin was from Calbiochem, and NVP-AEW541 was from Cayman Chemical.
Animal procedures and tissue preparation
All animal procedures were performed in accordance with approved Institutional Animal Care and Use Committee protocols. Mice were maintained ad libitum on a reduced phytoestrogen diet (2019S Teklad Global 19% protein extruded rodent diet; Teklad Diets). Mouse strains B6.129S2-Trp53tm1Tyj/J; STOCK Ptch1tm1Mps/J; B6.Cg-Tg(Atoh1-cre)1Bfri/J, B6.129P2-Esr2tm1Unc/J; B6N.129-Ptch1tm1Hahn/J were obtained from Jackson Laboratory. Selective breeding was performed in-house to obtain the desired Ptch1+/− Trp53−/−, Ptch1C/C Atoh1-cre, and Esr2−/− Ptch1C/C Atoh1-cre mice (Figure 1A). Genomic DNA isolated from tail biopsies was used to genotype pups as previously described (36, 37, 39, 40). Ptch1+/− Trp53−/− mice were randomly assigned to either the control or Faslodex treatment groups and received weekly 20-μL im injections of castor oil vehicle or Faslodex (1 mg Fulvestrant injection; AstraZeneca) beginning on postnatal day (PND) 21. Mice were observed daily for abnormal gait, ataxia, or signs of intracranial pressure and cranial doming. At symptom onset, study animals were euthanized by CO2 asphyxiation and perfused with 4% paraformaldehyde, and the whole brain was dissected and placed in 4% paraformaldehyde, which was renewed 24 hours later. WT and Esr2−/− Ptch1C/C Atoh1-cre mice were euthanized by CO2 asphyxiation on PND45. After the euthanasia, all tumors were isolated, weighed, and fixed in 4% paraformaldehyde. Methods describing the generation, isolation, and initial characterization of the D283Med xenografts used for these studies was described previously (8). Fixed tissues were washed several times in 70% ethanol prior to tissue processing and embedding in paraffin (Histocenter3; Thermo-Shandon). Microtome sections were cut at 5 μm from blocks at 4°C.
Figure 1.
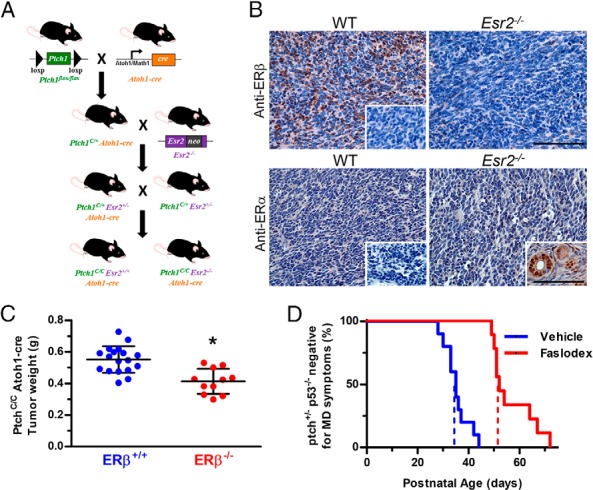
Effects ERβ knockout and ER Inhibition in mouse models of medulloblastoma. A, Details of the selective breeding schema used to generate ERβ-knock Ptch1C/C Atoh1-cre Med mice. B, Shown are representative micrographs of Med tumor sections from Esr2+/+ Ptch1C/C Atoh1-cre (WT) or Esr−/− Ptch1C/C Atoh1-cre (Esr2−/−). Sections in the top panels were immunostained with antiserum recognizing ERβ1 and ERβ2 (PA1–310B; 1 μg/mL), and sections in bottom panel were stained for ERα (SC-542; 1 μg/mL). Insets show control staining of Esr2+/+ Ptch1C/C Atoh1-cre tumor sections identically stained following incubation with normal serum and positive control ERα staining of mouse uterus. Scale bar, 50 μm. C, The effect of ERβ expression on the growth of Med in Ptch1C/C Atoh1-cre Med males and female mice was assessed by determination of mean weights for tumors isolated from WT (Esr2+/+) and ERβ-knockout (Esr2−/−) on PND45. A 25% reduction in the mean tumor weight of the ERβ-null Med mice (413.9 ± 79.4 mg; n = 11) was observed. The weight of tumors from ERβ-knockout mice was significantly less than the weight of tumors in WT Med mice (551.9 ± 84.3 mg; n = 18). Significant differences between the mean weight of tumors from WT and ERβ-knockout mice was determined using a Student's t test (P = .0002). D, The median symptom-free survival of untreated Ptch1+/− Trp53−/− male Med mice that received weekly mock injections of castor oil vehicle beginning at PND21 was 35 days (n = 10; blue hatched vertical line). Treatment with Faslodex significantly increased median symptom-free survival to 52 days (n = 9; red hatched vertical line). Median symptom-free survival was statistically increased by Faslodex treatment (Mantel-Cox log rank test; P < .0001).
Immunohistochemical analysis
Mounted sections were deparaffinized in xylene and rehydrated through graded ethanol into PBS (pH 7.4). Endogenous peroxidase activity was blocked with 3% H2O2 in PBS and washed in H2O. Heat-mediated epitope retrieval was performed at 97°C in Tris-EDTA buffer (10 mM Tris-HCl; 1 mM EDTA, pH 9.0) with 0.05% Tween 20. Sections were incubated at room temperature for 1 hour with 3% normal goat or horse serum in PBS and incubated overnight at 4°C with primary antibodies. Primary antibodies and concentrations used are listed in Supplemental Table 1. Controls establishing specificity primary antisera were described previously (8–10, 14, 16, 17), and additional controls included replacement of primary antibodies with nonspecific serum and preincubation of the cleaved caspase-3 antibody with blocking peptide (1050; Cell Signaling). Mouse spleen or uterus was used as positive controls for cleaved caspase-3 and ERα. Immunoreactivity was visualized with 0.05% 3′3-diaminobenzidine by the avidin-biotin peroxidase complex method (Vector Laboratories). Stained sections were counterstained in hematoxylin, dehydrated, and mounted in Permount (Fisher Scientific). Digital photomicrographs were collected using a Nikon Eclipse 55i microscope with a DS-Fi1 charge-coupled device camera and Digital Sight software (Nikon). Cell counts were scored by an investigator blinded to the treatment from 10 random ×40 fields from a single section of the tumor outside the tumors necrotic core. Apoptotic and mitotic index was expressed as number of positive cells divided by the total cell count multiplied by 100. Final photomicrograph graphics were generated and labeled using Adobe Photoshop.
Cell culture methods
All cell lines used were acquired directly from the American Type Culture Collection. Human D283Med cells (HTB-185, initially isolated from a 5 y old male) were maintained in suspension at a density of 0.5–1 × 106 cells/mL in MEM with Earle's balanced salt solution. Human PFSK1 cells (CRL-2060), established from a cerebral CNS primitive neuroectodermal tumor (PNET) from a 22-month-old male were maintained at a confluence density of 20%–80% in RPMI 1640 with media renewed every 2–3 days. Culture media were supplemented with 10% fetal bovine serum or 10% charcoal-stripped fetal bovine serum (CSS), 100 U/mL penicillin, and 100 μg/mL streptomycin, plus or minus L-glutamine as indicated (Fisher Scientific).
Viability and caspase-3 activity analysis
For viability analysis, D283Med cells were seeded at an initial density of 1.25 × 105 cells/mL and PFSK1 cells at 300 cells/mm2. At the 96-hour time point, cells were collected, stained with trypan blue, and viable (trypan blue excluding) cells counted with a hemocytometer. For caspase-3 activity analysis, D283Med cells were seeded at a density of 1 × 106 cells/mL and PFSK1 cells were seeded at an initial density of 300 cells/mm2. At the 48-hour time point, cells were lysed in 20 mM Tris-HCl (pH 7.5) with 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, and 1% Triton X-100. Cell lysates were assayed for protein concentration using the BioRad Dc protein assay (Bio-Rad Laboratories). Caspase-3 activity [picomoles of p-nitroaniline (pNA) per hour−1 per milligram of protein−1] was determined from a standard curve derived from known concentrations of pNA using 10 μg of lysate and liberation of pNA from Ac-DEVD-pNA (Enzo Life Sciences). At 24, 48, 72, and 96 hours after the start of the L-glutamine withdrawal and treatment, the amount of 5-bromo-2′-deoxyuridine (BrdU) incorporated into the DNA was monitored using an ELISA-based approach (8, 10).
Western blot analysis
After the indicated treatments, D283Med cells were pelleted and resuspended in lysis buffer [20 mM Tris-HCl (pH 7.5) with 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, and 1% Triton X-100] containing protease (Roche) and phosphatase (Sigma-Aldrich) inhibitors as previously described (41). Proteins were fractionated in 12% Tris-HCl sodium dodecyl sulfate-polyacrylamide gels and electrotransferred to nitrocellulose membranes (LI-COR). Membranes were blocked with 5% nonfat dry milk and then incubated overnight at 4°C with primary antiserum (Supplemental Table 1). Washed blots were incubated at room temperature with 0.1 μg/mL of goat antirabbit 800CW or 0.1 μg/mL of goat antimouse 680RD secondary antibodies, and specific immunoreactivity was quantified using an Odyssey Clx scanner and Image Studio software (Li-Cor). Results were normalized to β-actin or total phosphor-independent antigens from the same membrane. The results from multiple experiments for each treatment condition and time point were reported as mean fold change relative to control.
Quantitative RT-PCR analysis
After 48-hour treatments, RNA was isolated from D283Med cells using the RNeasy kit (QIAGEN). RNA concentrations were measured (NanoDrop 2000; Thermo Scientific), and cDNA was generated using the high-capacity cDNA reverse transcription kit (Life Technologies). Quantitative PCR was performed using 10 ng of cDNA and gene-specific Taqman assays (Life Technologies). Relative expression of IGF1, IGF1R, IGF2, and BCL2 was analyzed using the ΔΔ cycle threshold method with normalization to 18S rRNA expression. Lactoferrin was used as a positive control estrogen-responsive gene.
Data and statistical analysis
Differences between control and treatment groups were analyzed with a Student's t test, Dunnett's multiple comparison test, or one-way ANOVA and Tukey-Kramer posttest as appropriate. Percentage data were arcsine transformed (arcsine of the square root of the value) prior to statistical analysis. A two-way ANOVA was used to determine whether sex and genotype factors significantly affected results for each end point analyzed in animals. Differences in symptom-free survival were analyzed using the Kaplan-Meier survival test and statistical significance determined using a Mantel-Cox log-rank test. Differences in IGF1R immunostaining was assessed using the immunohistochemical score (HSCORE) approach (42). The HSCORE was estimated according to the equation, HSCORE = ΣPi(I + 1), where I represents the intensity of staining (0, no staining; 1, mild; 2, moderate; 3, intense), and Pi represents the percentage of cells within each staining intensity category. A minimal level of statistical significance for differences was defined as P < .05 and is indicated in figures with an asterisk. Analysis was performed using Prism version 5 software (GraphPad Software).
Results
Loss of ERβ function decreases Med tumor growth
To determine whether ERβ regulates Med tumor growth Ptch1C/C Atoh1-cre ERβ+/+ and ERβ−/− mice were derived (Figure 1A). The absence of ERβ protein in Med tumors from ERβ−/− Ptch1C/C Atoh1-cre mice was confirmed (Figure 1B). Evident ERβ-like immunoreactivity was observed in tumors from the ERβ-WT Ptch1C/C Atoh1-cre mice with no staining observed in tumors from ERβ−/− Ptch1C/C Atoh1-cre mice. As observed in most human Med tumors (8), no specific ERα-like immunoreactivity was detected in tumors isolated from WT or ERβ−/− Ptch1C/C Atoh1-cre mice (Figure 1B). The analysis of Med tumor weight data by a two-way ANOVA revealed a significant main effect of genotype on tumor weight [F (1, 25) = 21.33, P = .0001] that was not influenced by sex [F (1, 25) = 2.76, P = .1089]. Tumor weight of ERβ-null mice (M = 413.9 mg, SD = 79.4 mg) was reduced significantly [t (27) = 4.37, P = .0002] by 25% compared with WT (M = 551.9 mg, SD = 84.3 mg) at PND45 (Figure 1C).
Using a different Med mouse model characterized by later postnatal onset of symptomatic Med, which allowed testing of antiestrogen efficacy beginning at weaning while limiting potential confounds related to maternal effects of treatments during gestational or lactation periods of development, the therapeutic efficacy of ER inhibition by Faslodex was determined by assessing time to symptom onset in Ptch1+/− Trp53−/− Med mice. Median symptom-free survival for vehicle treated controls was 35 days (M = 35.3 days; SD = 4.9; n = 10). Faslodex treatment significantly (log rank Mantel-Cox P < .0001) delayed Med progression, with median symptom-free survival increased to 52 days (M = 56.67 days, SD = 8.6; n = 9) (Figure 1D).
ERβ loss-of-function is associated with increased apoptosis and decreased Bcl2 expression
Faslodex treatment resulted in a significant increase in the number of active caspase-3 immunopositive profiles in treated human D283Med xenografts [t (9) = 4.625, P = .0021] and Ptch1+/− Trp53−/− tumors [t (9) = 4.246, P = .0022] (Figure 2A). Compared with WT, a similar increase in caspase-3 staining was detected in tumors from ERβ−/− Ptch1C/C Atoh1-cre mice [t (25) = 11.25, P < .0001] (Figure 2B). The observed increases in apoptotic profiles were associated with a decreased expression of the antiapoptotic protein BCL2. Faslodex significantly decreased BCL2 expression in D283Med xenografts [t (7) = 4.701, P = .0022] and in Ptch1+/− Trp53−/− Med tumors [t (4) = 3.223, P = .0322] (Figure 2C). A similar decrease in BCL2-expressing cells was found in ERβ−/− Ptch1C/C Atoh1-cre tumors [t (10) = 4.415 P = .0013] (Figure 2D). There were no detectable differences in proliferating cell numbers between control and Faslodex treated D283Med xenografts [t (7) = 0.7932, P = .4537], Ptch1+/− Trp53−/− tumors [t (11) = 1.099, P = .2952] or WT and ERβ−/− Ptch1C/C Atoh1-cre tumors [t (25) = 1.646, P = .1123] (Figure 3, A–C). Confirming the lack of major effects of ERβ loss of function on the tumor cell mitotic index, there were no significant differences [t (10) = 1.598, P = .1410] in mean numbers of Ki67-positive cells found in WT and ERβ−/− Ptch1C/C Atoh1-cre tumors (Figure 3, D and E). Together these results suggest that the inhibition of ER activity and loss of ERβ function increases apoptosis in both human and mouse Med tumors and that estrogen can regulate Med growth, at least in part, by cytoprotective mechanisms that act to decrease caspase-3-mediated cell death.
Figure 2.
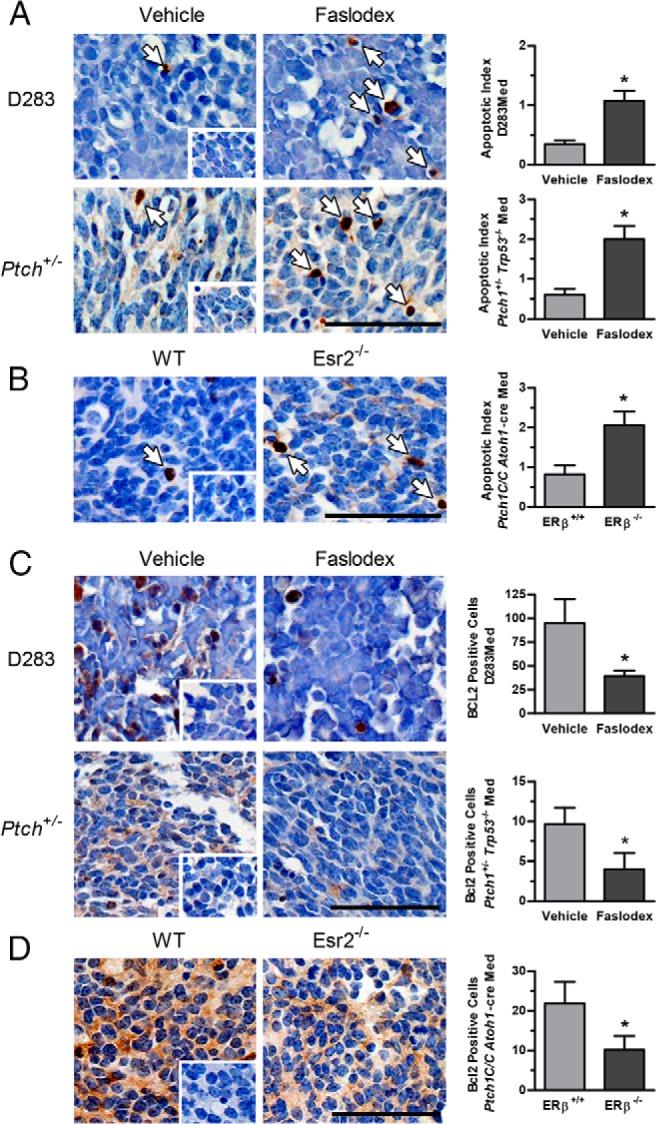
Immunohistochemical analysis of apoptosis and BCL2 expression in medulloblastoma tumors. Shown are representative micrographs of active cleaved caspase-3 immunostaining and the results from quantitative analysis of caspase-3 immunopositive apoptotic cell numbers from D283Med xenografts and Ptch1+/− Trp53−/− tumors from mice treated with vehicle or Faslodex (A) and Esr2+/+ Ptch1C/C Atoh1-cre and Esr−/− Ptch1C/C Atoh1-cre tumors (B). For D283Med xenograft groups, n = 6 for vehicle and n = 5 for Faslodex; Ptch1+/− Trp53−/− n = 5 for vehicle and n = 6 for Faslodex. For Esr2+/+ Ptch1C/C Atoh1-cre, n = 17 and Esr−/− Ptch1C/C Atoh1-cre, n = 10. Caspase-3-positive apoptotic cells are indicated with arrows. Representative micrographs of BCL-2 immunostained D283Med xenografts and Ptch1+/− Trp53−/− tumors from mice treated with vehicle or Faslodex (C) and Esr2+/+ Ptch1C/C Atoh1-cre and Esr−/− Ptch1C/C Atoh1-cre tumors (D) are shown. Quantitative analysis of BCL2-immunopositive cell numbers per 1000 cells counted is shown; for D283Med xenograft groups, vehicle, n = 5, Faslodex, n = 4; Ptch1+/− Trp53−/− vehicle, n = 3, Faslodex, n = 3; Esr2+/+ Ptch1C/C Atoh1-cre n = 6, Esr−/− Ptch1C/C Atoh1-cre, n = 6. Quantitative results are expressed as mean ± SD. Significant differences between mean values for control and experimental groups were determined using a Student's t test. Shown as insets are identically treated control sections from D283Med xenografts, Ptch1+/− Trp53−/− and Esr−/− Ptch1C/C Atoh1-cre tumors stained following incubation with normal serum. Scale bar, 50 μm.
Figure 3.

Analysis of mitotic index and Ki67 staining in medulloblastoma tumors. Results from quantitative cell count analysis of mitotic cells is shown as the mitotic index for D283Med xenografts (A) and Ptch1+/− Trp53−/− tumors (B) from mice treated with vehicle or Faslodex and Esr2+/+ Ptch1C/C Atoh1-cre and Esr−/− Ptch1C/C Atoh1-cre tumors (C). For D283Med xenograft groups, n = 4 for vehicle and n = 5 for Faslodex; Ptch1+/− Trp53−/−, n = 6 for vehicle and n = 7 for Faslodex; n = 17 for Esr2+/+ Ptch1C/C Atoh1-cre and n = 10 for Esr−/− Ptch1C/C Atoh1-cre. D, Shown are representative micrographs of sections from Esr2+/+ Ptch1C/C Atoh1-cre and Esr−/− Ptch1C/C Atoh1-cre tumors immunostained for Ki67. Immunopositive cells are indicated with arrows. E, The results from quantitative analysis of Ki67-immunopositive cell numbers per 1000 cells counted are shown for Esr2+/+ (WT) and Esr−/− tumors. Esr2+/+ Ptch1C/C Atoh1-cre, n = 6, Esr−/− Ptch1C/C Atoh1-cre, n = 6. All results are expressed as mean ± SD. No significant differences between mean values for control and experimental groups were identified using a Student's t test; calculated P values are indicated above each set of bars.
Effects of estradiol and ERβ loss of function on IGF1R and MAPK signaling
Faslodex treatment significantly decreased IGF1R immunoreactivity in D283Med xenografts [t (8) = 3.582, P = .0072; treated: M = 160, SD = 8.9; control: M = 232, SD = 18] and Ptch1+/− Trp53−/− tumors [t (6) = 3.677, P = .0104; treated M = 207.5, SD = 16.5; control M = 277.5, SD = 9.5] (Figure 4A). IGF1R immunoreactivity was also significantly decreased [t (10) = 5.301, P = .0003] in ERβ−/− Ptch1C/C Atoh1-cre tumors (M = 160, SD = 7.3) compared with WT control (M = 231.7, SD = 11.4) (Figure 4B). In tumors of Faslodex-treated Ptch1+/− Trp53−/− mice, a corresponding decrease in the number phospho-ERK [t (8) = 8.262, P < .0001] and phospho-AKT [t (6) = 4.321, P = .0050] immunopositive cells was also observed. Phosphorylation of p38 [t (8) = 0.090, P = .9303] and c-Jun N-terminal kinase (JNK) [t (4) = 0.553, P = .6095] was not affected by Faslodex treatment (Table 1). A significant decrease in numbers of phospho-ERK [t (10) = 7.210 P < .0001] and phospho-AKT [t (9) = 4.307 P = .002] immunopositive cells was also detected in ERβ−/− Ptch1C/C Atoh1-cre tumors. Likewise, no effect on phosphorylation of p38 [t (10) = 0.371 P = .7184] or JNK [t (10) = 0.461 P = .6547] was evident in the ERβ−/− Med tumors (Table 1).
Figure 4.

Immunohistochemical staining of IGF1R in medulloblastoma tumors. Shown are representative micrographs and the results from quantitative analysis of IGF1R immunostaining in D283Med xenografts and Ptch1+/− Trp53−/−tumors from mice treated with vehicle or Faslodex (A) and Esr2+/+ Ptch1C/C Atoh1-cre and Esr−/− Ptch1C/C Atoh1-cre tumors (B). Insets show staining of vehicle treated and Esr2+/+ tumors incubated with normal serum. Quantitative HSCORE results for IGF1R-immunostaing is shown as mean ± SD. Significant differences between mean values for control and experimental group were determined using a Student's t test. For D283Med xenograft groups, n = 5 for vehicle and n = 5 for Faslodex; Ptch1+/− Trp53−/−, n = 4 for vehicle, and n = 4 for Faslodex. For Esr2+/+ Ptch1C/C Atoh1-cre, n = 6 and Esr−/− Ptch1C/C Atoh1-cre, n = 6. Scale bar, 50 μm. Shown are representative results of time-course studies evaluating the temporal profile of ERK1/2, AKT, and IGF1R activation in D283Med cells exposed to 17β-estradiol for up to 60 minutes (C) and with 24- and 48-hour exposures to 17β-estradiol or DPN (D). Total and phospho-specific antigens were detected as indicated and equal loading of proteins were confirmed by simultaneous analysis for β-actin. Treatment times are indicated above each set of panels. Vehicle treated control groups (V) were treated with 0.01% DMSO for indicated times. E, Effects of ER agonists on IGF1, IGF1R, and BCL2 mRNA expression in D283Med cells. Analysis of quantitative RT-PCR analysis of IGF1R and BCL2 mRNA expression detected a 2- to 3-fold increases in the expression of IGF1, IGF1R, and BCL2 after 48 hours of exposure of D283Med cells to E2 or DPN. E2-induced increases in transcript levels were blocked by the ERβ selective antagonist PHTPP. E2 and DPN also induced expression of the positive control estrogen-responsive gene lactoferrin. Significant differences between mean values for expression of each gene target were determined using an ANOVA and Tucky-Kramer posttest from at least three independent experiments.
Table 1.
Immunohistochemical Analysis of Growth Factor Pathway Protein Expression
|
Ptch1+/−
Trp53−/− |
Ptch1C/C
Atoh1-cre |
|||
|---|---|---|---|---|
| Vehicle | Faslodex | ERβ+/+ | ERβ−/− | |
| Phospho-ERK | 23.8 ± 5.1 (n = 6) |
6.5 ± 1.0a (n = 4) |
16.3 ± 2.8 (n = 6) |
8.3 ± 1.0a (n = 6) |
| Phospho-p38 | 10.3 ± 3.5 (n = 3) |
10.0 ± 2.0 (n = 3) |
5.0 ± 2.8 (n = 6) |
5.7 ± 3.5 (n = 6) |
| Phospho-JNK | 3.0 ± 1.0 (n = 6) |
3.3 ± 0.6 (n = 4) |
2.2 ± 0.8 (n = 6) |
2.5 ± 1.2 (n = 6) |
| Phospho-AKT | 31.0 ± 12.6 (n = 4) |
7.3 ± 4.6a (n = 4) |
18.8 ± 5.6 (n = 5) |
7.0 ± 3.4a (n = 6) |
Values are mean number of immunopositive cells per 1000 cells ± SD.
Significant differences between mean values for control and treatment or genotype are indicated in bold.
ERβ-induced changes in IGF1R expression and MAPK signaling
Similar to previous results from primary cultured cerebellar granule cells and the CNS PNET cell line PFSK1 (10, 14), in serum-starved D283Med cells, 10 nM E2 induced a rapid 1.8- to 2-fold increase in phospho-ERK1/2 signaling that peaked 30 minutes after exposure (Figure 4C). Estradiol had no effect on AKT signaling, and IGF1R phosphorylation remained below detectable levels for up to 60 minutes after E2 exposure, suggesting that rapid E2 signaling in D283Med cells does not directly or indirectly involve the IGF1R (Figure 4C).
In metabolically stressed D283 Med cells cultured for 24 or 48 hours in CSS that lacked supplemental glutamine, E2 or the ERβ-selective agonist DPN increased expression of IGF1R by 2.04- ± 0.08-fold (Figure 4D). The increased expression of IGF1R was associated with a proportional 2.88- ± 0.32-fold increase in the actin-normalized levels of phospho-IGF1R. Although neither E2 nor DPN treatment changed total ERK1/2 expression at either time point, E2 increased the levels of activated phospho-ERK1 by 2.28- ± 0.08-fold and phospho-ERK2 by 2.66- ± 0.32-fold at 24 hours. The response induced by DPN was more robust with phospho-ERK1, and phospho-ERK2 levels increased 4.56- ± 0.19-fold and 5.90- ± 0.21-fold compared with vehicle (Figure 4D). At the 48-hour time point, phospho-ERK1 was increased by 2.39- ± 0.36-fold by E2 and 3.19- ± 0.11-fold by DPN, with similar 2.14- ± 0.06-fold and 3.74 ± -0.26 fold increases in phospho-ERK2 observed. At the 24-hour time point, phospho-AKT levels were comparable with control and became increased by 2.27- ± 0.32-fold relative to vehicle-treated controls at the 48-hour time point.
Quantitative RT-PCR analysis of transcripts isolated from D283Med cells metabolically stressed by glutamine withdrawal for 48 hours confirmed that E2 or DPN significantly increased mRNA expression of IGF1R [F (3, 11) = 12.69, P < .0021] and its ligand IGF1 [F (3, 11) = 9.351, P < .0054] in D283Med cells (Figure 4E). Under these conditions the expression of IGF2 transcripts was not detected. Significant increases in BCL2 expression [F (3, 11) = 26.96, P < .0002] and increased expression of the known estrogen-responsive gene lactoferrin [F (3, 11) = 54.98, P < .0001] were also detected. Demonstrating a dependence on ERβ, these estrogen-induced increases in gene expression were blocked by the ERβ-selective antagonist PHTPP (Figure 4E).
ERβ-dependent mechanisms protect D283Med cells from metabolic stress-induced apoptosis
In D283Med cells metabolically stressed by glutamine withdrawal, significant increases in caspase-3 activity [F (2, 32) = 9.487, P = .0006] were first observed at 48 hours (Figure 5A) and was associated with decreased viable cell numbers (Figure 5B). The effects of glutamine withdrawal were reversed by 10 nM E2 or the caspase inhibitor Z-DEVD-fmk; Figure 5, B and C). The protective effects of E2 or Z-DEVD-fmk alone were not augmented by coexposure to both compounds.
Figure 5.

Analysis of the effects of glutamine withdrawal and E2 on D283Med viability and caspase-3 activity. A, Analysis of relative caspase-3 activity in D283Med cells cultured in MEM containing 10% CSS without supplemental L-glutamine for increasing times. NS, not significantly different from T0. Analysis of caspase-3 activity at 48 hours (B) and cell viability at 96 hours (C) for D283Med cells cultured in MEM 10% CSS supplemented with 5 mM L-glutamine [(+) L-Glut] or without L-glutamine and treated with to 0.01% DMSO vehicle [(−) L-Glut], 10 nM E2, 10 μM Z-DEVD-fmk (fmk) or 10 μM E2 and Z-DEVD-fmk. Significant differences between mean values of the supplemented and unsupplemented control groups were determined by an ANOVA and Tucky-Kramer post hoc analysis. The effects of E2 and Z-DEVD-fmk treatment were compared with the CSS control groups using Dunnett's multiple comparison test. The number of samples in each group was n = 3 for the analysis of viable cell numbers and n = 11 or 12 for caspase-3 activity analysis. D–H, Concentration-response analysis of the relative effects of ER agonists on caspase-3 activity in D283Med cells. For each vehicle control (0.01% DMSO) or indicated experimental treatment, caspase-3 activity was quantified in lysates from D283Med cells after a 48-hour incubation period in MEM 10% CSS lacking supplemental L-glutamine. Results are presented relative to the mean activity from vehicle-treated control cultures [(−) L-Glut]. All results are expressed as mean ± SEM. Significant differences between mean values of vehicle control and each experimental group were determined using Dunnett's multiple comparison test. The number of samples in each group was n = 10–12. I, Analysis of PFSK1 cell viability after 96 hours cultured in the presence [(+) L-Glut] or absence of 5 mM L-glutamine [(−) L-Glut] plus DMSO vehicle, 10 nM E2, DPN, or PPT. Significant differences between mean values of (−) L-Glut vehicle control and each experimental group were determined using a Dunnett's multiple comparison test. The number of samples in each group was n = 6. J, Analysis of relative caspase-3 activity in PFSK1 cells cultured after 48 hours in the absence of 5 mM L-glutamine [(−) L-Glut] plus DMSO vehicle, 10 nM E2, DPN, or PPT. Significant differences between mean values of (−) L-Glut vehicle control and each experimental group were determined using a Dunnett's multiple comparison test. The number of samples in the control group was n = 11 and n = 12 for each treatment group. K, Time-course analysis of BrdU incorporation in D283Med cultures in the presence [(+) L-Glut] or absence of 5 mM L-glutamine [(−) L-Glut] plus DMSO vehicle, 10 nM E2, DPN, or PPT. Significant differences between mean values of (−) L-Glut vehicle control and each experimental group were determined using a Dunnett's multiple comparison test. The number of samples in each group was n = 8.
Increasing concentrations of E2 significantly and dose dependently decreased caspase-3 activity in glutamine withdrawal stressed D283Med cells [F (4, 51) = 8.741, P < .0001], whereas 17α-estradiol did not affect activity [F (4, 50) = 0.5199, P < .7215] (Figure 5, D and E). The ERβ selective agonist DPN significantly reduced caspase-3 activity [F (4, 53) = 10.70, P < .0001] (Figure 5F). The calculate EC50 value for decreasing caspase-3 activity for E2 was 0.55 nM and 0.86 nM for DPN, which are consistent with the EC50 values reported for DPN transactivation in ERβ-luciferase reporter assays (0.85 nM for DPN vs 0.04 nM for E2) (41). Caspase-3 activity was unaffected by the ERα selective agonist PPT [F (4, 54) = 1.589, P = .1904] (Figure 5G); or the G protein-coupled estrogen receptor 1 (GPR30) agonist G-1 [F (3, 42) = 0.5507, P = .6505 (Figure 5H).
Migration, but not growth, of the PNET cell line PFSK1 was previously shown to be estrogen responsive (14). Like D283Med cells, PFSK1 cell growth was sensitive to glutamine withdrawal-induced metabolic stress (Figure 5I). In cultures lacking glutamine E2 or DPN significantly increased PFSK1 viability [F (3, 17) = 24.25, P = .0001] and decreased caspase-3 activity [F (3, 15) = 15.81, P = .0001] compared with vehicle-treated control cultures (Figure 5, I and J). The ERα selective agonist PPT had no effect on PFSK1 viability or caspase-3 activity.
Similar to previous findings in D283Med cells cultured in the presence of glutamine (8), 24 hours of treatment with E2 or DPN stimulated BrdU incorporation in cultures of D283Med cells lacking glutamine by 169% ± 8% and 177% ± 8% (Figure 5K). The ERα-selective agonist PPT was without effect at the 24-hour time point. Whereas D283 Med proliferation was maintained in cultures supplemented with 5 mM glutamine through the entire 96-hour time course, the ERβ-mediated mitogenic response to E2 or DPN was transient and not observed at later times (48–96 h) when increased caspase-3 activity and decreased cell viability were observed in cultures lacking E2 or DPN.
Confirming results observed with selective agonists, the inhibitory effects of E2 on caspase-3 activity were blocked by the nonselective ER antagonist ICI 182,780 [t (22) = 3.494, P = .0021] (Figure 6A) and the ERβ selective antagonist PHTPP [t (20) = 2.466, P = .0229] (Figure 6B), whereas the ERα selective antagonist MPP had no effect [t (22) = 1.408, P = .1732] (Figure 6C). Involvement of prosurvival IGF signaling in mediating the protective effects of E2 in D283Med cells was investigated using antagonists of the IGF1R, MAPK kinase (MEK1/2) and phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K). Inhibition of the IGF1R with NVP-AEW541 [t (19) = 3.136, P = .0054] (Figure 6D) or MEK1/2 with U0126 [t (19) = 2.598, P = .0177] blocked the effects of E2 (Figure 6E). Neither the irreversible PI3K inhibitor wortmannin nor the reversible inhibitor LY294002 blocked the ability of E2 to inhibit caspase-3 activity (Figure 6F). These data demonstrate that the ERβ-dependent protective effects of E2 are dependent on increased IGF1R activity and MEK1/2 activation of the ERK1/2 signaling cascade but are independent of PI3K activation.
Figure 6.

Analysis of selective antagonist effects on E2-induced inhibition of caspase-3 activity and IGF signaling. Caspase-3 activity was quantified after a 48-hour incubation period in MEM 10% CSS lacking supplemental L-glutamine for each 0.01% DMSO vehicle control [(−) L-Glut] or experimental treatment with 10 nM E2 plus or minus indicated concentrations of the nonselective ER antagonist ICI 182,780 (A), the ERβ selective antagonist PHTPP (B), the ERα selective antagonist MPP (C), the IGF1R inhibitor NVP-AEW541 (NVP) (D), the MEK 1/2 inhibitor U0126 (E), and the PI3K inhibitors wortmannin (Wort) or LY294002 (LY) (F). Final concentration of DMSO vehicle in each culture was 0.01%, and results were normalized to the relative caspase-3 activity of the vehicle control and expressed as mean percentage of control activity ± SEM. Significant differences between mean of Arc Sine-transformed values for vehicle control and each experimental group were determined using a Dunnett's multiple comparison test. The number of samples in each group was n = 10–12.
Discussion
Estrogen-dependent breast cancers express functional ERs and respond clinically to SERMs that act by inhibiting ER transactivational activity (28, 30). Tumors from the prostate, colon, lung, and CNS, although not estrogen dependent, are also estrogen responsive; however, the role of ERβ in tumor progression is not clear (29). Conflicting results have demonstrated that ERβ can either stimulate or inhibit tumor proliferation and survival (43–48). The role of ERβ in cancer etiology and progression likely depends on a variety of factors that may include whether ERα is coexpressed, which isoforms of ERβ are expressed, and the subcellular localization of the expressed receptors (29, 31, 49). We previously defined Med as an ERβ-positive and estrogen-responsive tumor whose growth and migration could be stimulated by E2 both in vitro and in vivo (8). The results of the studies presented here have defined in more detail the nature of tumor growth-promoting activity of E2 and ERβ in Med. The long-term ability of E2 to increase tumor growth through ERβ-mediated changes in responsive gene expression in Med appears to result from mechanisms that decrease caspase-3 activity and protect Med cells from apoptosis. The tumor-promoting activity of ERβ was most clearly demonstrated by the results of studies in the Esr2−/− knockout Med mouse, in which the lack of ERβ limited tumor growth, resulting in a 25% decrease in Med tumor growth rate (see Figure 1). In vitro studies showing that E2 protected metabolically stressed Med cells through the activation of ERβ, effects that were independent of ERα or G protein-coupled estrogen receptor 1/GPR30, confirmed a mechanistic requirement for nuclear receptor activity of ERβ in the observed cytoprotective actions of E2. The in vivo relevance of those pharmacological studies were further confirmed by complementary findings in different genetic models of Med that demonstrated decreased tumor growth resulting from pharmacological inhibition of ER activity, effects that were similarly associated with an increase in tumor cell apoptosis. In both models, the effects of ERβ loss of function were associated with decreased IGF1R expression and decreased IGF-like signaling. Furthermore, the observed therapeutic efficacy of the antiestrogen drug Faslodex in the Ptch1+/− Trp53−/− mouse model of Med, which more than doubled the symptom-free survival time after initiation of therapy, suggests that inhibition of ERβ may be clinically beneficial for some Med patients. It is notable that the beneficial effects of Faslodex were observed even for treatments that were started relatively late at PND21. Without treatment Med symptoms were evident in untreated controls only 2 weeks later.
In apparent contrast to the results from our previous studies and those reported here, ERβ activation has also been reported to decrease the incidence of Med in an ionizing radiation-induced Ptch1+/− mouse model of Med (11–13). Those effects were attributed to estrogen activation of ERβ causing a decrease in proliferation of preneoplastic lesions. A lack of this apparent anticarcinogenic effect of estrogen in that Med model was proposed to explain the increased incidence of Med in human males (1, 11). As an alternative interpretation, the results of the studies presented here suggest instead that cytoprotective effects of estrogen mediated by ERβ may have decreased the incidence of Ptch1+/− Med through mechanisms that effectively limited the inductive effects of ionizing radiation in GCPs (10, 50). It is also notable that, unlike humans, Med incidence in the Ptch1+/− and Ptch1+/− Sufu+/− mouse models of Med is greater in females than males (1, 51). In the Ptch1+/− p53−/− Med model used for some of our studies, only males develop Med. The male bias in this model does not arise from sex-specific effects related to Med but rather because homozygous disruption of p53−/− causes female-specific embryonic lethality (39). Because of noted sex-specific differences between mouse models of Med and human, the appropriateness of their use for investigating the effect of sex on Med incidence is unclear.
The actions of estrogen and the IGF signaling pathways work in concert to elicit cytoprotection by mechanisms involving changes in estrogen-responsive gene expression. Direct ER binding at estrogen response elements or binding of transactivational complexes formed between specificity protein-1 transcription factors and ERs at half estrogen response element sites is responsible for the up-regulation of the IGF1, IRS1, and IGF1R genes by estrogen (52–54). The up-regulation of the IGF1, IRS1, and IGF1R genes by estrogen results in increased IGF pathway activity, and activation of AKT and ERK signaling pathways, which can each act as a primary mediator of increased cell survival (33, 34, 55). The cytoprotective actions of IGF1 and increased IGF-signaling are also observed in GCPs (19, 56–58). During cerebellar development elevated expression of IGF1 increases GCP survival, whereas loss of insulin receptor substrate-2 results in reduced GCP proliferation and decreased cerebellar volume (59–61). Similarly, ERβ is likely important in the developing cerebellar in which ERβ expression is tightly regulated in developing neuronal precursors, and estrogen can regulate viability and migration of GCPs (9, 10, 15).
The potential for involvement of IGF signaling in estrogen-mediated cytoprotective mechanisms prompted the investigation of involvement of IGF1R and downstream modulators in the potential mechanism responsible for the protective effects of E2 in Med cells. Our results demonstrated the inhibition of the IGF1R or MEK1/2, but not PI3K, blocked protective effects of E2 in Med cells and that E2 or the ERβ-selective agonist DPN increased expression of IGF1, IGF1R, and BCL2 mRNA. These findings suggest that the protective actions of estrogen in Med are dependent on ERβ, IGF1R, and ERK activity but not PI3K signaling. Those conclusions were supported in vivo by the observation of increased apoptosis in Med tumors treated with Faslodex and in ERβ-null tumors. Increased Med apoptosis coincided with decreased IGF1R expression, and importantly, activation of AKT and ERK, which are coordinately regulated by IGF1R, was also decreased in Med tumors treated with an ER inhibitor or those that had ERβ knocked out. Expression of the prosurvival protein BCL2, whose expression is regulated downstream of IGF1R, was also decreased by ER inhibition or in the absence of Esr2.
The ability of E2 to increase IGF1R expression and signaling though ERα is well characterized in ER-positive breast cancers (32, 62). In contrast to ERα+ breast cancer cells, estrogen appears to regulate IGF1R expression though ERβ in Med. It is now appreciated that the relative expression of specific ERβ splice variants and intracellular localization of the expressed receptors may influence breast cancer disease progression, which suggests that physiological responses of ERβ to E2 may vary, depending on cellular context (32, 62). Results of studies from ERα-negative/progesterone receptor-negative (ERα−/PR−) MDA-MB-453 and MDA-MB-468 breast cancer cell lines found that ERβ can increase proliferation and survival, suggesting that estrogen-dependent activation of ERβ in the absence of the overriding effects of ERα may promote tumor growth (45). Although there are relatively few studies examining the role of ERβ in tumors like Med that typically lack ERα, lung cancer stands as an example of another nonreproductive estrogen-responsive cancer that expresses ERβ and not ERα (63). Similar to Med, in human lung cancers, ERβ and IGF1R expression levels are positively correlated, and increased levels of their expression are associated with a poor prognosis (64). In ERβ-positive/ERα-negative non-small cell lung cancer cells, E2 acts via ERβ to increase IGF1R expression and signaling, and in a mouse model of lung adenocarcinoma, E2 promotes tumor development through this mechanism (64, 65). It is notable that ERα is typically not expressed, or expressed at very low levels, in both Med and lung cancer in which the tumor-promoting effects of E2 increases IGF1R expression and signaling via ERβ, thus raising the possibility that ERα status might be a central determinant in defining the tumor growth potential of ERβ.
Along with establishing a mechanism for estrogen's tumor-promoting actions in Med, results from this study also suggest that inhibition of ERβ activity may be a useful strategy for treating Med. Current treatments for Med have resulted in an overall 5-year survival rate of greater than 70%, although survivors frequently suffer an array of life-long side effects associated with the therapeutic use of radiation and chemotherapy (2–5). Therefore, focus needs to be placed on the development of novel adjuvant therapies that can limit side effects of radiation or chemotherapy while increasing the cure rate of Med. Inhibitors of the cytoprotective actions of ERβ, either alone or in combination IGF1R inhibitors, or downstream inhibitors that target protective IGF1R signaling, may be useful adjuvants for this purpose. Although not a specific inhibitor of ERβ-mediated cytoprotection, the SERM tamoxifen can sensitize Med cells to the cytotoxic effects of etoposide (66). Whereas further analysis is required, the effects of tamoxifen on the chemosensitivity of Med cells supports the potential utility for pharmacological inhibition of ERβ to sensitize Med tumors to cytotoxic chemotherapeutics, an effect that could increase cure rates and limit side effects of current therapy. However, studies with Doay Med cells have found that loss of ERβ function can decrease sensitivity to cisplatin by increasing Rad51-mediated DNA repair (14). Along with supporting the need to understand in more detail the actions of estrogens in Med, those findings further highlight the fact that the actions of estrogens in these estrogen-responsive tumors may involve the integration of multiple estrogen-induced mechanisms. Although ERβ selective agonists have been developed and evaluated in clinical trials for a variety of purposes, ERβ selective antagonists that may prove most useful as adjuvants for treating Med are not available clinically (67, 68). Whereas we demonstrated that the nonselective ER antagonist Faslodex is efficacious for limiting tumor progression in mouse models of Med and that benefits occur rapidly, long-term treatment with nonselective ER antagonists may not be therapeutically most useful because of their potential for causing nonspecific reproductive and developmental side effects through inhibition of other estrogen-responsive processes (69).
In conclusion, the results of this study demonstrate clearly that ERβ has cytoprotective actions that can increase Med tumor growth through a mechanism that likely involves cross talk with the IGF signaling pathway. Although an extensive literature supports an important role for IGF1R in mediating the reported effects of E2 in cerebellar granule cell precursors and medulloblastoma, study limitations do not fully exclude a potential involvement of insulin receptor-induced signaling. Further study is required to fully elucidate the nature of the mechanisms linking nuclear receptors and growth factor receptor tyrosine kinase receptor signaling and to determine how generalizable these cytoprotective mechanisms are across the different molecular class of Med, PNETs, and other ERβ+/ERα− cells and cancers.
Acknowledgments
We are indebted to Robin Gear and Jessica Kendziorski for their contribution to various aspects of this study, and we greatly appreciate the efforts of Xiaolan Ma, who generated the medulloblastoma xenograft tumors used in these analysis, and Michelle Kirby, who initially developed some of the medulloblastoma cell culture methods used here.
This work was supported by Research Grants R01ES015145, R03ES023098, and RC2ES018765 and Training Grant T32ES007250 from the National Institute of Environmental Health Sciences.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BCL2
- B-cell lymphoma 2
- BrdU
- 5-bromo-2′-deoxyuridine
- CNS
- central nervous system
- CSS
- charcoal-stripped fetal bovine serum
- DMSO
- dimethylsulfoxide
- DPN
- 2,3-bis(4-hydroxyphenyl)-propionitrile
- E2
- 17β-estradiol
- ER
- estrogen receptor
- GCP
- granule cell precursor
- ICI 182,780
- 7α,17β-[9-[(4,4,5,5,5-pentafluoropentyl) sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol
- IGF1R
- IGF-1 receptor
- JNK
- c-Jun N-terminal kinase
- Med
- medulloblastoma
- MEK1/2
- MAPK kinase
- MPP
- 1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole dihydrochloride
- PHTPP
- 4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol
- PI3K
- phosphatidylinositol-4,5-bisphosphate 3-kinase
- pNA
- p-nitroaniline
- PND
- postnatal day
- PNET
- primitive neuroectodermal tumor
- PPT
- 4,4′,4′-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol
- SERM
- selective estrogen receptor modulator
- WT
- wild type
- Z-DEVD-fmk
- Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethylketone.
References
- 1. Ries LAG, Smith MA, Gurney JG, et al. Cancer incidence and survival among children and adolescents: United States SEER program 1975–1995. NIH publication number 99-4649 Bethesda, MD: National Institutes of Health; 1999. [Google Scholar]
- 2. Smoll NR. Relative survival of childhood and adult medulloblastomas and primitive neuroectodermal tumors (PNETs). Cancer. 2012;118:1313–1322. [DOI] [PubMed] [Google Scholar]
- 3. Frange P, Alapetite C, Gaboriaud G, et al. From childhood to adulthood: long-term outcome of medulloblastoma patients. The Institut Curie experience (1980–2000). J Neurooncol. 2009;95:271–279. [DOI] [PubMed] [Google Scholar]
- 4. Mulhern RK, Merchant TE, Gajjar A, Reddick WE, Kun LE. Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol. 2004;5:399–408. [DOI] [PubMed] [Google Scholar]
- 5. Rose SR, Danish RK, Kearney NS, et al. ACTH deficiency in childhood cancer survivors. Pediatr Blood Cancer. 2005;45:808–813. [DOI] [PubMed] [Google Scholar]
- 6. Belcher SM. Blockade of estrogen receptor signaling to improve outlook for medulloblastoma sufferers. Future Oncol. 2009;5:751–754. [DOI] [PubMed] [Google Scholar]
- 7. Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annu Rev Neurosci. 2001;24:385–428. [DOI] [PubMed] [Google Scholar]
- 8. Belcher SM, Ma X, Le HH. Blockade of estrogen receptor signaling inhibits growth and migration of medulloblastoma. Endocrinology. 2009;150:1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jakab RL, Wong JK, Belcher SM. Estrogen receptor β immunoreactivity in differentiating cells of the developing rat cerebellum. J Comp Neurol. 2001;430:396–409. [DOI] [PubMed] [Google Scholar]
- 10. Wong JK, Le HH, Zsarnovszky A, Belcher SM. Estrogens and ICI182,780 (Faslodex) modulate mitosis and cell death in immature cerebellar neurons via rapid activation of p44/p42 mitogen-activated protein kinase. J Neurosci. 2003;23:4984–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mancuso M, Leonardi S, Ceccarelli M, et al. Protective role of 17β-estradiol on medulloblastoma development in Patched1 heterozygous mice. Int J Cancer. 2010;127:2749–2757. [DOI] [PubMed] [Google Scholar]
- 12. Mancuso M, Leonardi S, Giardullo P, et al. The estrogen receptor β agonist diarylpropionitrile (DPN) inhibits medulloblastoma development via anti-proliferative and pro-apoptotic pathways. Cancer Lett. 2011;308:197–202. [DOI] [PubMed] [Google Scholar]
- 13. Pazzaglia S, Mancuso M, Atkinson MJ, et al. High incidence of medulloblastoma following X-ray-irradiation of newborn Ptc1 heterozygous mice. Oncogene. 2002;21:7580–7584. [DOI] [PubMed] [Google Scholar]
- 14. Kirby M, Zsarnovszky A, Belcher SM. Estrogen receptor expression in a human primitive neuroectodermal tumor cell line from the cerebral cortex: estrogen stimulates rapid ERK1/2 activation and receptor-dependent cell migration. Biochem Biophys Res Commun. 2004;319(3):753–758. [DOI] [PubMed] [Google Scholar]
- 15. Belcher SM. Regulated expression of estrogen receptor α and β mRNA in granule cells during development of the rat cerebellum. Brain Res Dev Brain Res. 1999;115:57–69. [DOI] [PubMed] [Google Scholar]
- 16. Belcher SM, Le HH, Spurling L, Wong JK. Rapid estrogenic regulation of extracellular signal-regulated kinase 1/2 signaling in cerebellar granule cells involves a G protein- and protein kinase A-dependent mechanism and intracellular activation of protein phosphatase 2A. Endocrinology. 2005;146:5397–5406. [DOI] [PubMed] [Google Scholar]
- 17. Zsarnovszky A, Le HH, Wang HS, Belcher SM. Ontogeny of rapid estrogen-mediated extracellular signal-regulated kinase signaling in the rat cerebellar cortex: potent nongenomic agonist and endocrine disrupting activity of the xenoestrogen bisphenol A. Endocrinology. 2005;146:5388–5396. [DOI] [PubMed] [Google Scholar]
- 18. Harkins AB, Fox AP. Cell death in weaver mouse cerebellum. Cerebellum. 2002;1:201–206. [DOI] [PubMed] [Google Scholar]
- 19. Zhong J, Deng J, Ghetti B, Lee W-H. Inhibition of insulin-like growth factor I activity contributes to the premature apoptosis of cerebellar granule neuron in weaver mutant mice: in vitro analysis. J Neurosci Res. 2002;70:36–45. [DOI] [PubMed] [Google Scholar]
- 20. Zhong J, Deng J, Phan J, et al. Insulin-like growth factor-I protects granule neurons from apoptosis and improves ataxia in weaver mice. J Neurosci Res. 2005;80:481–490. [DOI] [PubMed] [Google Scholar]
- 21. Gualco E, Wang JY, Del Valle L, et al. IGF-IR in neuroprotection and brain tumors. Front Biosci. 2009;14:352–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zumkeller W, Westphal M. The IGF/IGFBP system in CNS malignancy. Mol Pathol. 2001;54:227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hartmann W, Koch A, Brune H, et al. Insulin-like growth factor II is involved in the proliferation control of medulloblastoma and its cerebellar precursor cells. Am J Pathol. 2005;166:1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rao G, Pedone CA, Del Valle L, Reiss K, Holland EC, Fults DW. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene. 2004;23(36):6156–6162. [DOI] [PubMed] [Google Scholar]
- 25. Valle LD, Enam S, Lassak A, et al. Insulin-like growth factor I receptor activity in human medulloblastomas. Clin Cancer Res. 2002;8:1822–1830. [PubMed] [Google Scholar]
- 26. Wang JY, Del Valle L, Gordon J, et al. Activation of the IGF-IR system contributes to malignant growth of human and mouse medulloblastomas. Oncogene. 2001;20:3857–3868. [DOI] [PubMed] [Google Scholar]
- 27. Zhou H, Rao J, Lin J, et al. The insulin-like growth factor-I receptor kinase inhibitor NVP-ADW742 sensitizes medulloblastoma to the effects of chemotherapy. Oncol Rep. 2011;25:1565–1571. [DOI] [PubMed] [Google Scholar]
- 28. Flötotto T, Djahansouzi S, Gläser M, et al. Hormones and hormone antagonists: mechanisms of action in carcinogenesis of endometrial and breast cancer. Horm Metab Res. 2001;33:451–457. [DOI] [PubMed] [Google Scholar]
- 29. Thomas C, Gustafsson J-Å. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11:597–608. [DOI] [PubMed] [Google Scholar]
- 30. Ciruelos E, Pascual T, Arroyo Vozmediano ML, et al. The therapeutic role of fulvestrant in the management of patients with hormone receptor-positive breast cancer. Breast. 2014;23:201–208. [DOI] [PubMed] [Google Scholar]
- 31. Murphy LC, Leygue E. The role of estrogen receptor-β in breast cancer. Semin Reprod Med. 2012;30:5–13. [DOI] [PubMed] [Google Scholar]
- 32. Yee D, Lee AV. Cross talk between the insulin-like growth factors and estrogens in breast cancer. J Mammary Gland Biol Neoplasia. 2000;5(1):107–115. [DOI] [PubMed] [Google Scholar]
- 33. Garcia-Segura LM, Arevalo MA, Azcoitia I. Interactions of estradiol and insulin-like growth factor-I signalling in the nervous system: new advances. Prog Brain Res. 2010;181:251–272. [DOI] [PubMed] [Google Scholar]
- 34. Garcia-Segura LM, Sanz A, Mendez P. Cross-talk between IGF-I and estradiol in the brain: focus on neuroprotection. Neuroendocrinology. 2006;84:275–279. [DOI] [PubMed] [Google Scholar]
- 35. Cardona-Gómez GP, Mendez P, DonCarlos LL, Azcoitia I, Garcia-Segura LM. Interactions of estrogens and insulin-like growth factor-I in the brain: implications for neuroprotection. Brain Res Rev. 2001;37:320–334. [DOI] [PubMed] [Google Scholar]
- 36. Krege JH, Hodgin JB, Couse JF, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor β. Proc Natl Acad Sci USA. 1998;95:15677–15682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang ZJ, Ellis T, Markant SL, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched1. Cancer Res. 2001;61:513–516. [PubMed] [Google Scholar]
- 39. Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. [DOI] [PubMed] [Google Scholar]
- 40. Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. [DOI] [PubMed] [Google Scholar]
- 41. Le HH, Belcher SM. Rapid signaling actions of environmental estrogens in developing granule cell neurons are mediated by estrogen receptor β. Endocrinology. 2010;151(12):5689–5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McCarty KS, Jr, Miller LS, Cox EB, Konrath J, McCarty KS., Sr Estrogen receptor analyses. Correlation of biochemical and immunohistochemical methods using monoclonal antireceptor antibodies. Arch Pathol Lab Med. 1985;109(8):716–721. [PubMed] [Google Scholar]
- 43. Hershberger PA, Stabile LP, Kanterewicz B, et al. Estrogen receptor β (ERβ) subtype-specific ligands increase transcription, p44/p42 mitogen activated protein kinase (MAPK) activation and growth in human non-small cell lung cancer cells. J Steroid Biochem Mol Biol. 2009;116:102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hou Y-F, Yuan S-T, Li H-C, et al. ERβ exerts multiple stimulative effects on human breast carcinoma cells. Oncogene. 2004;23:5799–5806. [DOI] [PubMed] [Google Scholar]
- 45. Li W, Jia M, Qin X, Hu J, Zhang X, Zhou G. Harmful effect of ERβ on BCRP-mediated drug resistance and cell proliferation in ERα/PR-negative breast cancer. FEBS J. 2013;280:6128–6140. [DOI] [PubMed] [Google Scholar]
- 46. Nakajima Y, Akaogi K, Suzuki T, et al. Estrogen regulates tumor growth through a nonclassical pathway that includes the transcription factors ERβ and KLF5. Science Signaling. 2011;4:ra22. [DOI] [PubMed] [Google Scholar]
- 47. Ström A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson J-A. Estrogen receptor β inhibits 17β-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci USA. 2004;101:1566–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang G, Liu X, Farkas AM, et al. Estrogen receptor β functions through nongenomic mechanisms in lung cancer cells. Mol Endocrinol. 2009;23:146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miki Y, Abe K, Suzuki S, Suzuki T, Sasano H. Suppression of estrogen actions in human lung cancer. Mol Cell Endocrinol. 2011;340:168–174. [DOI] [PubMed] [Google Scholar]
- 50. Zorrilla Zubilete Ma, Guelman LR, Maur DG, et al. Partial neuroprotection by 17-β-estradiol in neonatal γ-irradiated rat cerebellum. Neurochem Int. 2011;58:273–280. [DOI] [PubMed] [Google Scholar]
- 51. Svärd J, Rozell B, Toftgård R, et al. Tumor suppressor gene co-operativity in compound Patched1 and suppressor of fused heterozygous mutant mice. Mol Carcinog. 2009;48:408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hewitt SC, Li Y, Li L, Korach KS. Estrogen-mediated regulation of Igf1 transcription and uterine growth involves direct binding of estrogen receptor α to estrogen-responsive elements. J Biol Chem. 2010;285:2676–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maor S, Mayer D, Yarden RI, et al. Estrogen receptor regulates insulin-like growth factor-I receptor gene expression in breast tumor cells: involvement of transcription factor Sp1. J Endocrinol. 2006;191:605–612. [DOI] [PubMed] [Google Scholar]
- 54. Panno ML, Mauro L, Marsico S, et al. Evidence that the mouse insulin receptor substrate-1 belongs to the gene family on which the promoter is activated by estrogen receptor α through its interaction with Sp1. J Mol Endocrinol. 2006;36:91–105. [DOI] [PubMed] [Google Scholar]
- 55. Alonso A, Gonzalez C. Neuroprotective role of estrogens: relationship with insulin/IGF-1 signaling. Front Biosci. 2012;4:607–619. [DOI] [PubMed] [Google Scholar]
- 56. D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Leski ML, Valentine SL, Baer JD, Coyle JT. Insulin-like growth factor I prevents the development of sensitivity to kainate neurotoxicity in cerebellar granule cells. J Neurochem. 2000;75:1548–1556. [DOI] [PubMed] [Google Scholar]
- 58. Villalba M, Bockaert J, Journot L. Concomitant induction of apoptosis and necrosis in cerebellar granule cells following serum and potassium withdrawal. Neuroreport. 1997;8:981–985. [DOI] [PubMed] [Google Scholar]
- 59. Chrysis D, Calikoglu AS, Ye P, D'Ercole AJ. Insulin-like growth factor-I overexpression attenuates cerebellar apoptosis by altering the expression of Bcl family proteins in a developmentally specific manner. J Neurosci. 2001;21:1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schubert M, Brazil DP, Burks DJ, et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci. 2003;23:7084–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ye P, Xing Y, Dai Z, D'Ercole AJ. In vivo actions of insulin-like growth factor-I (IGF-I) on cerebellum development in transgenic mice: evidence that IGF-I increases proliferation of granule cell progenitors. Dev Brain Res. 1996;95:44–54. [DOI] [PubMed] [Google Scholar]
- 62. Fagan D, Yee D. Crosstalk between IGF1R and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13(4):423–429. [DOI] [PubMed] [Google Scholar]
- 63. Siegfried JM, Stabile LP. Estrogenic steroid hormones in lung cancer. Semin Oncol.41(1):5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tang H, Liao Y, Chen G, et al. Estrogen upregulates the IGF-1 signaling pathway in lung cancer through estrogen receptor-β. Med Oncol. 2012;29:2640–2648. [DOI] [PubMed] [Google Scholar]
- 65. Tang H, Liao Y, Xu L, et al. Estrogen and insulin-like growth factor 1 synergistically promote the development of lung adenocarcinoma in mice. Int J Cancer. 2013;133(10):2473–2482. [DOI] [PubMed] [Google Scholar]
- 66. Ramachandran C, Khatib Z, Petkarou A, et al. Tamoxifen modulation of etoposide cytotoxicity involves inhibition of protein kinase C activity and insulin-like growth factor II expression in brain tumor cells. J Neurooncol. 2004;67:19–28. [DOI] [PubMed] [Google Scholar]
- 67. Calabrese C, Praticò C, Calafiore A, et al. Eviendep® reduces number and size of duodenal polyps in familial adenomatous polyposis patients with ileal pouch-anal anastomosis. World J Gastroenterol. 2013;19:5671–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. National Library of Medicine. The efficacy and safety of a selective estrogen receptor β agonist (LY500307) for negative symptoms and cognitive impairment associated with schizophrenia. 2013. NLM identifier NCT01874756. ClinicalTrials.gov Accessed January 21, 2015.
- 69. Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116:561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]