

Abstract
Improved treatments for heart failure patients will require the development of novel therapeutic strategies that target basal disease mechanisms. Disrupted cardiomyocyte Ca2+ homeostasis is recognized as a major contributor to the heart failure phenotype, as it plays a key role in systolic and diastolic dysfunction, arrhythmogenesis, and hypertrophy and apoptosis signaling. In this review, we outline existing knowledge of the involvement of Ca2+ homeostasis in these deficits, and identify four promising targets for therapeutic intervention: the sarcoplasmic reticulum Ca2+ ATPase, the Na+-Ca2+ exchanger, the ryanodine receptor, and t-tubule structure. We discuss experimental data indicating the applicability of these targets that has led to recent and ongoing clinical trials, and suggest future therapeutic approaches.
Keywords: Cardiac myocytes, calcium homeostasis, heart failure, SR Ca2+ ATPase, Na+/Ca2+ exchanger, ryanodine receptor, t-tubules.
I. INTRODUCTION
Heart failure treatment has historically undergone several paradigm shifts. Early pharmacological approaches included cardiac glycosides, which may be effective in relieving patients' symptoms, but carry a notoriously high risk of toxicity. In the 1980s, adrenergic agonists and phosphodiesterase inhibitors were introduced with great expectations, as it seemed intuitive to treat impaired contractility with positive inotropes. However, these agents were observed to increase mortality as they induced maladaptive cardiac responses and exhausted the failing heart. Consequently, the mainstay of today’s treatment is based on inhibitors of the chronic neurohumoral activation in heart failure, such as β-adrenergic blockers and angiotensin converting enzyme (ACE) inhibitors. Unfortunately, these treatments often only provide heart failure patients with symptomatic relief and temporarily impede disease progression. Therefore, new treatment strategies are needed that target the basal pathogenic mechanisms of this disease. The two main causes of death in heart failure patients, declining cardiac pump function and arrhythmias, have both been linked to disrupted Ca2+ homeostasis in cardiac muscle cells (cardiomyocytes) [1]. While the details of these deficiencies continue to be unraveled, existing data suggest that targeting deficient Ca2+ handling in failing cardiomyocytes has enormous therapeutic potential. In this review, we will examine the potential benefits of modulating several key players in Ca2+ homeostasis, with discussion of recent and ongoing clinical trials. However, to place these proposed therapies in their proper context, we will first briefly outline the involvement of Ca2+ in disrupted contraction and relaxation, arrhythmogenesis, and signaling in failing cardiomyocytes.
Impaired Cardiomyocyte Contraction and Relaxation
There is general agreement that smaller and slower contraction of individual cardiomyocytes contributes to reduced left ventricular contraction (systole) in heart failure [1, 2]. Contractility is regulated by a process known as excitation-contraction (EC) coupling [3]. This process is initiated as the action potential propagates over the surface membrane and into invaginations called t-tubules, triggering the opening of L-type Ca2+ channels (LTCCs). The resulting Ca2+ influx in turn triggers additional Ca2+ release from the sarcoplasmic reticulum (SR) via Ca2+ release channels called ryanodine receptors (RyRs) (Fig. 1A). This process, known as Ca2+ induced Ca2+ release (CICR), is made possible by close proximity between LTCCs and RyRs in functional units known as dyads. Ca2+ release from a dyad is called a Ca2+ spark, and the spatiotemporal summation of Ca2+ sparks constitutes the Ca2+ transient. Contraction is triggered as Ca2+ binds to the myofilaments.
Fig. (1).
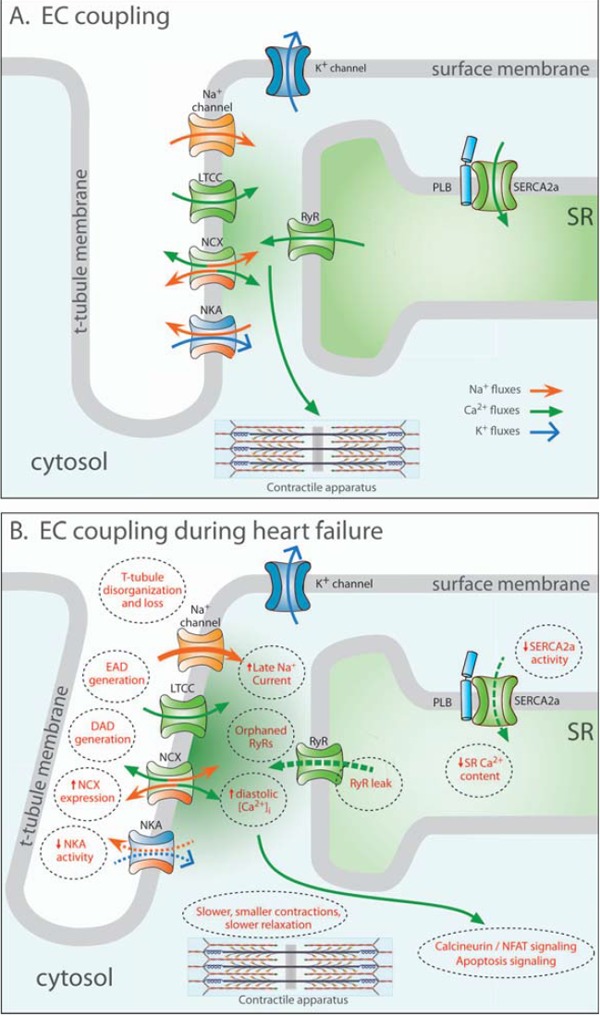
Excitation-contraction (EC) contraction coupling in the normal and failing heart. In healthy cardiomyocytes (A), the action potential propagates into the t-tubules, opening voltage-gated L-type Ca2+ channels (LTCCs). The resulting Ca2+ influx triggers the opening of Ca2+ release channels (ryanodine receptors, RyRs) in the membrane of the sarcoplasmic reticulum (SR). Released Ca2+ initiates contraction as it binds to the contractile apparatus, and relaxation occurs as Ca2+ is recycled into the SR by the SR Ca2+ ATPase 2a (SERCA2a), and removed from the cell via the Na+-Ca2+ exchanger (NCX). During heart failure (B), Ca2+ release is impaired, leading to slower and smaller contractions. Reduced SR Ca2+ content results from decreased SERCA2a activity, increased RyR leak and, in some cases, increased NCX activity. Systolic dysfunction also results from disruption of T-tubule structure, which functionally “orphans” some RyRs from LTCCs. Slowed and incomplete Ca2+ removal from the cytosol impairs cardiomyocyte relaxation, and promotes hypertrophy and apoptosis signaling. Triggered cardiac arryhythmia has been linked to RyR leak, and removal of released Ca2+ by NCX, causing DADs. EADs may result from inappropriate re-opening of Ca2+ channels. Deficient Ca2+ cycling is also linked to altered Na+ homeostasis, following downregulation of the Na+-K+ ATPase (NKA) and increased late Na+ current.
Fundamental to the reduction in contractility in heart failure is reduced EC-coupling gain, meaning that less Ca2+ is released from the SR upon LTCC activation, resulting in decreased magnitude of Ca2+ transients and contractions [4, 5]. This reduction in Ca2+ release in failing cells largely results from decreased SR Ca2+ content which, in turn, results from a) reduced SERCA2a function, b) increased RyR leak and/or c) increased NCX function, which competes with SERCA2a for Ca2+ [1] (Fig. 1B). Reduced EC-coupling gain additionally results from impaired communication between LTCCs and RyRs. In heart failure, t-tubule organization is disrupted, leading to “orphaned” RyRs which no longer have opposed LTCCs (Fig. 1B). Thus, Ca2+ release is de-synchronized across the cell, which slows the rising phase of the Ca2+ transient (Fig. 2A), and may contribute to reduced contractile power in this condition [2]. Ca2+ release is also de-synchronized in failing cells due to a sub-population of Ca2+ sparks with slowed kinetics, which we have hypothesized result from dispersion of RyRs in the dyad [6]. Finally, alterations in action potential configuration may de-synchronize Ca2+ release [7-9], as a prolonged action potential reduces the magnitude and kinetics of the Ca2+ current, making triggering of SR Ca2+ release less efficient. Thus, the nature of reduced EC coupling gain in heart failure is complex.
Relaxation of cardiomyocytes is reported to be slowed and/or less extensive in failing cells, which impairs the ability of the ventricle to fill with blood before the next heartbeat. This phase of the cardiac cycle, known as diastole, has gained appreciable attention in recent years with the realization that a significant proportion of heart failure patients exhibit impaired ventricular filling but normal systole, a condition known as heart failure with preserved ejection fraction (HFpEF) or diastolic heart failure [10]. At the cardiomyocyte level, for efficient relaxation to occur, Ca2+ needs to be efficiently removed from the cytosol following release. The main pathways for removal are Ca2+ recycling into SR by the SR Ca2+ ATPase 2a (SERCA2a), and Ca2+ extrusion mediated by the plasma membrane Na+/Ca2+-exchanger (NCX). Depending on the specific heart failure phenotype, both routes for Ca2+ removal may be impaired (Fig. 1B) [11]. Thus, targeting cardiomyocyte Ca2+ homeostasis has the potential to improve both systolic and diastolic function in heart failure patients.
Ca2+-Dependent Arrhythmogenesis
30-50% of heart failure patients are reported to die from sudden cardiac death, and the majority of these deaths are linked to ventricular tachycardia [12]. Ventricular tachycardia can result from spontaneous electrical activity of cardiomyocytes. In some cases, a spontaneous action potential may be triggered by a phasic depolarization during the downstroke of the action potential, called an early afterdepolarization (EAD). While the precise mechanisms underlying EAD generation remain debated and may vary in different settings, there is general agreement that many EADs result from inappropriate re-opening of LTCCs or other depolarizing currents [13] (Fig. 1B). Abnormal Ca2+ homeostasis can also promote arrhythmogenesis via delayed afterdepolarizations (DADs). These events are triggered by spontaneous Ca2+ release from the SR, resulting in depolarization as the released Ca2+ is removed from the cell by NCX [13]. Thus, targeting cardiomyocyte Ca2+ homeostasis has great potential for the prevention of triggered arrhythmias in heart failure patients as it is involved in the generation of both EADs and DADs.
Ca2+-Dependent Signaling
Beyond its role in triggering contraction/relaxation and contributing to the electrical stability of the cardiomyocyte, intracellular Ca2+ ([Ca2+]i) is also critically involved in signaling. For example, the Ca2+-dependent calcineurin-NFAT pathway is centrally involved in mediating pathologic hypertrophy [14] and inflammation [15]. While the precise nature of the Ca2+ signal which triggers this pathway remains unclear, accumulating evidence suggests that Ca2+ levels in the dyadic cleft may be critically involved [16, 17] (Fig. 1B). Ca2+-dependent signaling is also involved in cardiomyocyte stress-responses and apoptosis, which additionally contribute to impaired myocardial function. Indeed, reduced SR Ca2+ content, as is known to occur in failing myocytes, has recently been linked to activation of the unfolded protein response, apoptosis, and altered SR structure [18]. Finally, disrupted Ca2+ homeostasis is known to offset the finely tuned balance between energy demand and availability in the heart. Energy wasting during heart failure results from futile Ca2+ cycling, and ATP production is reduced following Ca2+-mediated mitochondrial dysfunction [19]. As will be described in the following sections, several proposed therapies aimed at improving contraction/relaxation in cardiomyocytes or arrhythmia suppression may simultaneously inhibit detrimental Ca2+ signaling.
Based on the above discussion, several key regulators of Ca2+ homeostasis emerge as potential therapeutic targets in failing cardiomyocytes, namely i) SERCA2a, ii) NCX, iii) RyR and iv) t-tubule structure. In the remainder of this review we will discuss the functional control of these targets, their dysregulation in heart failure, and ongoing efforts to reverse these impairments. This discussion will highlight opportunities to specifically target different heart failure phenotypes on a patient-to-patient basis.
II. SERCA
SERCA2a Function
SERCA2a contributes to Ca2+ removal from the cytosol by recycling Ca2+ into the SR. SERCA2a is thus an important regulator of diastolic function since, together with NCX, it sets the rate of Ca2+ transient decline and resting Ca2+ levels, and thereby the rate and extent of cardiomyocyte relaxation [11]. SERCA2a function also contributes to control of systolic function, by regulating the SR Ca2+ load available for release. SERCA2a activity is primarily determined by cytosolic Ca2+ levels and its endogenous inhibitor phospholamban (PLB). Since Ca2+ is usually the rate limiting factor for SERCA2a activity, increased [Ca2+]i directly enhances SR Ca2+ pumping [20]. PLB is a reversible inhibitor of SERCA2a, which acts by decreasing the Ca2+ affinity, but not the maximum pumping capacity (Vmax) of SERCA2a. Phosphorylation of PLB by protein kinase A (PKA), a cAMP- dependent kinase, relieves this inhibition (Fig. 2B). PKA is activated by intracellular cAMP, which is amplified via the β-adrenergic pathway. As PLB is one of the major substrates for PKA, PLB phosphorylation is accordingly one of the primary mechanisms for increasing SR Ca2+ content and relaxation rate following β-adrenergic stimulation [11, 21]. The main mechanism that terminates the β-adrenergic cascade is cAMP degradation by a class of enzymes called phosphodiesterases (PDEs), which importantly modulate SERCA2a function. The inhibitory effects of PLB are also relieved via phosphorylation by Ca2+/calmodulin-dependent kinase type II (CaMKII). As CaMKII is activated by increased [Ca2+]i, this mechanism enables SERCA2a stimulation at high pacing rates [11]. It is currently unclear whether the Ca2+ signal for activation of CaMKII is Ca2+ levels near the dyad, and/or integrated [Ca2+]i during the Ca2+ transient. Dephosphorylation of PLB is mediated mainly by protein phosphatase 1 [22] (Fig. 2B).
Fig. (2).
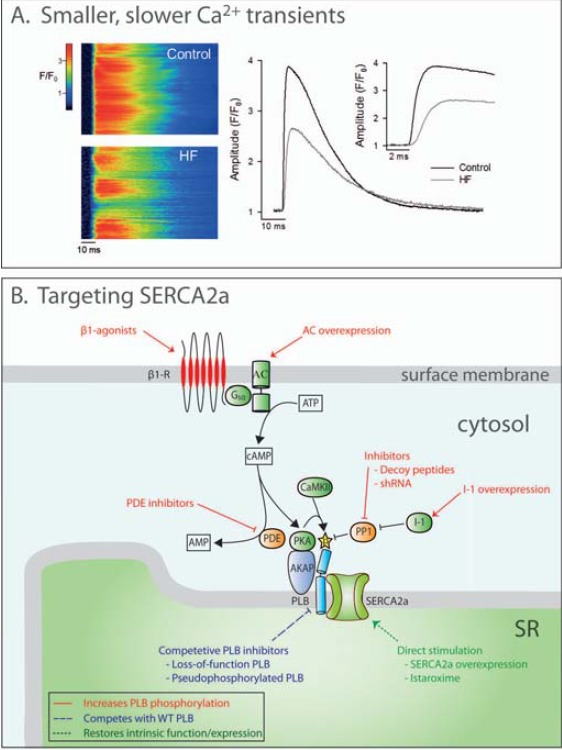
Disrupted Ca2+ homeostasis in failing cardiomyocytes and therapeutic targeting of SERCA2a. A: Representative confocal line-scan images (left) show that Ca2+ release is de-synchronized across failing cells, resulting in smaller and slower Ca2+ transients (centre panel, magnified in inset at right). Unpublished data are from a rat model of heart failure following myocardial infarction and sham-operated controls (fluo-4 AM loading). Smaller transients additionally result from declining SR content due to reduced SERCA2a activity. B: SERCA2a activity may be therapeutically increased in heart failure by gene therapy-mediated overexpression or pharmacological stimulation. Phospholamban (PLB)-dependent inhibition of SERCA could be relieved by increasing PLB phosphorylation by: 1) elevating cAMP levels (stimulating β-adrenergic signaling or preventing cAMP breakdown by phosphodiesterases (PDEs)), or 2) preventing PLB dephosphorylation by inhibiting protein phosphatase 1 (PP1), or increasing activity of inhibitor 1 (I-1). Alternatively, competitive inhibition of PLB could be employed.
SERCA2a in Heart Failure
Beginning in the late 1980s, SERCA2a levels were reported to be reduced in animal models of hypertrophy [23, 24], and in 1990 Mercadier et al were first to report reduced mRNA levels of SERCA2a in human heart failure [25]. The relation between SERCA2a levels and loss of contractile force in heart failure was later demonstrated by Hasenfuss et al [26]. Numerous subsequent studies have confirmed the significance of reduced SERCA2a levels in heart failure pathogenesis. While some studies have reported reduced mRNA levels but unaltered protein levels [27, 28], different disease etiologies, stages and animal models employed may contribute to this discrepancy. Regional differences in SERCA expression have also recently been suggested to underlie these conflicting results [29].
SERCA2a activity may also be reduced in heart failure due to altered protein regulation, as a number of studies have reported augmented PLB inhibition. Although most studies have failed to demonstrate alterations in PLB levels in heart failure [27, 28, 30], SERCA2a downregulation gives a relative increase in PLB compared to SERCA2a, and this may increase inhibition of the remaining pumps. Most important, however, seems to be a reduced phosphorylation state of PLB observed in human heart failure [27, 31] and experimental models of heart failure [28]. In the failing human heart, phosphorylation at thr17 has been suggested to be reduced despite increased CaMKII activity, due to increased activity of the phosphatase calcineurin [32]. Furthermore, reduced ser16 phosphorylation has been attributed to increased activity of protein phosphatase 1 in both human heart failure [33, 34] and animal models [28].
As expected based on above discussions, downregulation of SERCA2a levels, and thus activity, have been correlated with both systolic and diastolic dysfunction in human heart failure [26, 35]. Similar findings have been made in mouse models with SERCA2a ablation. Mice with heterozygous SERCA2 knockout (+/-) showed reduced levels of SERCA2a, slowed SR Ca2+ uptake, and impaired in vivo cardiac contractility and relaxation [36]. Furthermore, conditional cardiomyocyte SERCA2a gene knockout caused decreased rates of cytosolic Ca2+ removal, reduced SR Ca2+ content and Ca2+ transient magnitude (Fig. 3A), and development of end-stage heart failure at several weeks following gene deletion [37-39]. These experiments illustrate the direct relationship between SERCA2a levels, Ca2+ handling, and cardiac pathology.
Fig. (3).
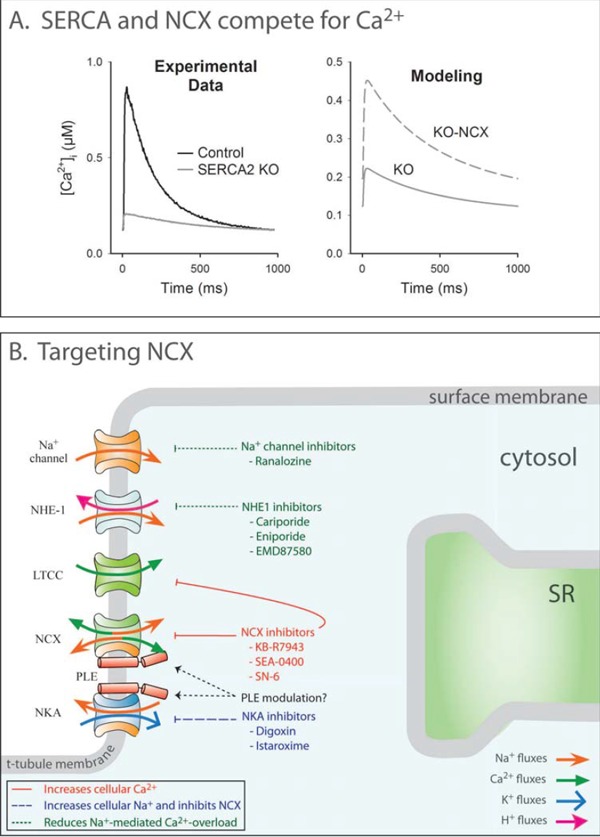
Altered NCX activity as a therapeutic target in heart failure. A: Experimental Ca2+ transients from SERCA2 knockout mice are dramatically reduced (left panel). Modeling data predict that simultaneous NCX ablation increases Ca2+ transient magnitude (right). This indicates that NCX competes with SERCA2a for the same pool of Ca2+ and reduces SR Ca2+ content and release. Data are adapted from [227], with permission. B: NCX activity could be therapeutically modulated by direct targeting or altering electrochemical gradients. NCX inhibitors attenuate cellular Ca2+ extrusion and thereby increase cellular Ca2+ load and ultimately contractility. Inhibition of NKA similarly inhibits NCX-mediated Ca2+ extrusion by increasing cellular Na+ levels. However, prevention of Ca2+ overload is desirable in patients at risk for arrhythmia, and this may be attained by inhibition of Na+ influx pathways, which augments Ca2+ extrusion.
Reduced SERCA2a function also has important implications for other aspects of the failing phenotype, including arrhythmogenesis, mechanoenergetics and hypertrophic and apoptotic signaling [40]. SERCA2a activity has been implicated in both early and late afterdepolarizations (EADs and DADs). Reduced SERCA2a activity decreases the magnitude of Ca2+ release, resulting in decreased inactivation of L-type Ca2+ channels and predisposition for EAD generation. Effects on DADs however, may depend on the interaction between SERCA2a activity and that of other Ca2+ handling proteins. Data from the SERCA2 knockout mouse have shown that reduced SERCA2a function is associated with a decreased threshold for RyR opening [41], which appears to be due to CaMKII-dependent phosphorylation of the channel. At baseline, these cells exhibited fewer Ca2+ waves and DADs, however β1-adrenergic stimulation increases SR Ca2+ content above the threshold for RyR opening, and may explain the increased incidence of arrhythmias in this setting [42].
Targeting Reduced SERCA2a Activity in Heart Failure
As the detrimental effects of reduced SERCA2a function have become evident, its potential as a target in heart failure has emerged. Transgenic mice overexpressing SERCA2a exhibit enhanced cardiac function at baseline in parallel to improved cardiomyocyte Ca2+ handling (increased Ca2+ transient magnitude, more rapid Ca2+ decline) and faster cardiomyocyte shortening and re-lengthening [43]. Work done in transgenic rats has consistently reported similar benefits of SERCA2a overexpression, and these rats are less prone to heart failure development after myocardial insults [44, 45].
The SERCA2a/PLB ratio has also been increased experimentally in heart failure models by overexpressing SERCA2a via adenoviral gene transfer. Isolated cardiomyocytes from failing human hearts transduced with the SERCA2a gene exhibited restored Ca2+ homeostasis and contractile function [46]. Similarly, in vivo gene delivery of SERCA2a in a rat model of heart failure restored SERCA2a expression and improved systolic and diastolic function [47]. SERCA2a restoration also positively influences myocardial arrhythmogenesis, mechanoenergetics, hypertrophic remodeling and apoptosis. A concern about SERCA2a restoration was that increased SR Ca2+ content would trigger spontaneous Ca2+ release in the setting of leaky RyRs. However, this has proven otherwise, as Ca2+ leak and ventricular arrhythmias were reduced by increasing SERCA2a levels in a rat model of heart failure [48]. A proposed explanation is that reversal of elevated diastolic [Ca2+]i attenuates arrhythmogenic NCX currents and/or reduces RyR Ca2+ leak.
The group of Hajjar has demonstrated that mechanoenergetic wasting in aortic banded rats was reduced by SERCA2a gene transfer, and survival improved, probably due to reduction in RyR Ca2+ leak (futile Ca2+ cycling) and improved mitochondrial function [49, 50]. SERCA2a overexpression also decreases diastolic [Ca2+]i, and a resulting attenuation of calcineurin/NFAT activation may be responsible for attenuated hypertrophic and apoptotic signaling [40]. SERCA2a overexpression in rat heart failure also reduced pathologic expression of miR-1, a micro RNA involved in regulation of transcription factors, receptor ligands, apoptosis regulators and ion channels [51].
Following these positive experimental results, human trials with SERCA2a gene therapy have been initiated. A phase 1 trial with an adeno-associated viral-1 vector (AAV1) was used to deliver SERCA2a gene by intracoronary infusion. The results were published in 2009, establishing safety and feasibility [52]. A phase 2 trial, the Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) study, was then conducted to further evaluate clinical benefits [53]. The CUPID study was a randomized, double blind, placebo controlled trial that included 39 patients with advanced heart failure. The effects of either low, mid or high dose AAV1-SERCA2a gene infusion were compared with placebo. The study confirmed safety, and the high dose group demonstrated therapeutic response with decreased symptoms of heart failure, improved functional status, decreased levels of natriuretic peptides and attenuated adverse remodeling of the left ventricle. A three-year follow-up study of these patients showed reduced mortality in those that received high dose gene therapy [54]. High titers of adenoviral neutralizing antibodies against the AAV1 are, however, a problem in some patients that needs to be addressed.
As an alternative to gene therapy, pharmacologic agents may directly act to restore SERCA2a function. Istaroxime produces both inotropic and lusitropic effects due to its dual mechanism of action [55]. First, it directly stimulates SERCA2a, increasing both SR Ca2+ content and cytosolic Ca2+ removal (Fig. 2B). Second, it is a non-glycoside inhibitor of the Na+/K+ ATPase, and thereby increases intracellular Na+ levels and indirectly inhibits NCX function. The resulting rise in [Ca2+]i makes more Ca2+ available for SERCA2a and increases the rate of Ca2+ removal and SR Ca2+ content. In vivo animal experiments have shown that both acute and chronic istaroxime treatment restore cardiac function with no increase in arrhythmic events or cardiac energetics [56-58]. This is contrary to the harmful profile of established inotropic drugs. The HORIZON-HF trial was a phase-2, randomized, double-blind, placebo-controlled clinical trial that evaluated hemodynamic, echocardiographic and neurohormonal effects of acute administration of istaroxime on heart failure deterioration [59, 60]. The results were promising, and included improved systolic and diastolic function, lower pulmonary capillary wedge pressure, increased blood pressure and decreased heart rate.
Another potential method of restoring SERCA2a activity is to reduce the inhibitory effects of PLB. Several studies of PLB-deficient mice [61-64] have demonstrated enhanced Ca2+ cycling, improved lusitropy and inotropy, no increase in mortality and abrogated progression to heart failure. Furthermore, inhibition of wild-type PLB by gene transfer of pseudophosphorylated PLB prevented heart failure development in several animal models [65-67] (Fig. 2B). Inhibition of wild-type PLB has also been demonstrated with loss-of-function PLB mutants [68]. In human heart failure, isolated cardiomyocytes were transduced with adenoviral vectors encoding PLB antisense RNA, with a resulting improvement of Ca2+ handling and contraction and relaxation velocities [69]. Somewhat worrying, however, is the link between human PLB mutations and cardiac disease. In fact, PLB null mutations cause lethal forms of hereditary dilated cardiomyopathy [70]. This suggests that knockdown or competitive inhibition of PLB inhibition should be approached with caution in humans.
Alternatively, relieved SERCA2a inhibition can also be accomplished by therapeutically increasing PLB phosphorylation. Protein phosphatase 1 (PP1), which dephosphorylates PLB, exhibits increased activity in heart failure due to i) increased expression [28, 33] and ii) reduced activity of inhibitor-1 [71] (Fig. 2B). Inhibiting the activity of PP1 is therefore a putative strategy to increase PLB phosphorylation. Indeed, gene delivery of inhibitor-1 or inhibitor-2 has been associated with reduced incidence of arrhythmia, hypertrophy, heart failure and death in animal models of heart failure [72, 73]. Delivery of short hairpin RNA targeting PP1 by adenoassociated viral vectors was also observed to prevent ventricular remodeling [74]. Recently, a decoy peptide mimicking phosphorylated PLB was shown to competitively inhibit PP1 from interacting with wild-type PLB, and improved cardiac contractility resulted [75]. However, since protein phosphatase 1, together with protein phosphatase 2a, exert the majority of total phosphatase activity in the heart, inhibition of its function will affect many phosphorylated cardiac proteins and may produce undesirable side-effects. The ryanodine receptor is such a target that poses a specific concern, as hyperphosphorylation of this receptor has been hypothesized by some to be arrhythmogenic [76] (see discussion in the RyR section).
PLB phosphorylation may also be increased by enhancing kinase activity. PKA activity is dependent on cAMP levels which could be raised by either increased synthesis (via adenylyl cyclase), or reduced degradation (via phosphodiesterases) (Fig. 2B). One can envision several possible approaches to increase adenylyl cyclase activity. Although the enzyme is activated by traditional inotropic β-agents such as dobutamine, the β1-receptor signaling pathway is associated with increased mortality. Gene therapy aiming at increasing adenylyl cyclase expression could bypass the detrimental β1-receptor cascade (Fig. 2B). Adenylyl cyclase isoform 6 is activated by the β1-receptor, and overexpression of this enzyme is reported to improve contractility and survival in animal models of heart failure [77, 78]. A human trial has been initiated to evaluate the potential of adenoviral delivery of adenylyl cyclase 6 by intracoronary infusion in human heart failure (ClinicalTrials.gov Identifier: NCT00787059). Another method to increase cAMP bioavailability, but avoid the β1-receptor cascade, is to inhibit cAMP degradation by phosphodiesterases (PDEs). Most cardiac PDE activity has been attributed to PDE3, PDE4 and, more recently, PDE1 [79] (Fig. 2B). PDE3 inhibition is known to have inotropic effects in heart [80, 81]. However, the benefits of PDE3 inhibition have only been sustained in short-term therapy, and long-term mortality is increased predominantly due to ventricular arrhythmias [80, 81]. It is likely that the underlying mechanisms are the same as for β1-receptor stimulation, as increased Ca2+ cycling results in the generation of arrhythmic currents and unfavorable metabolic effects. Interestingly, PDE3 inhibiton is also reported to contribute to the inotropic effects of the myofilament sensitizer Levosimendan [82].
II. NCX
NCX Function
While SERCA2a recycles Ca2+ into the SR, NCX is the main route for Ca2+ extrusion from the cardiomyocyte. NCX plays an important role in setting resting Ca2+ levels, and thus the extent of cardiomyocyte relaxation. However, since resting [Ca2+]i regulates SERCA2a activity, SR content, and Ca2+ transient magnitude, the net effect of altered NCX activity on the rate of Ca2+ decline can be difficult to predict, and may vary between species [11]. This functional interaction between NCX and SERCA2a is illustrated in Fig. 3A.
Although NCX functions predominantly to extrude Ca2+, it may also work in reverse mode resulting in Ca2+ influx. It is generally agreed upon that the NCX exchanges one molecule of Ca2+ for three molecules of Na+ [83], making it electrogenic. Therefore, forward-mode NCX operation results in a depolarizing current, while reverse mode carries a repolarizing current. The driving force which determines NCX direction and function are the electrochemical gradients, namely membrane potential and transmembrane gradients of Ca2+ and Na+ [84]. Negative membrane potential and low intracellular Na+ concentration ([Na+]i) promote forward operation mode. Since low cytosolic Na+ levels are actively maintained by the Na+/K+-ATPase (NKA), NCX function and NKA function are closely coupled (Fig. 1A). Indeed, experimental data suggest that NCX and the α2-isoform of NKA are closely localized in the t-tubules, which allows their function to be linked by [Na+]i in a restricted microdomain [85-87]. For example, reducing NKAα2 activity can locally elevate [Na+]i which inhibits Ca2+ removal by NCX, while making reverse-mode NCX function more likely [88].
Another mechanism of NCX regulation is the small inhibitory protein phospholemman. This is a member of the FXYD family of ion transport regulators and is found to co-localize with both NKA [89] and NCX [90] in heart (Fig. 3B). PKA and PKC phosphorylation modulate its inhibitory effect, but in a different manner in NCX compared to NKA; phospholemman phosphorylation relieves NKA inhibition but increases NCX inhibition [91]. During phosphorylation, phospholemman therefore increases contractility, by inhibiting NCX and increasing [Ca2+]i [92], but also protects against Na+ overload by relieved inhibition of NKA [93].
NCX in Heart Failure
NCX upregulation is a common feature of both human heart failure [94-96] and animal models of heart failure [97, 98]. As such changes often occur simultaneously with SERCA2a downregulation, a marked increase in NCX/SERCA2a ratio is commonly reported, and has been implicated in both contractile dysfunction and arrhythmogenesis [99]. Reduced contractility is caused by increased transsarcolemmal Ca2+ extrusion relative to SR Ca2+ recycling, which depletes SR Ca2+ stores. Accordingly, experimental overexpression of NCX in cardiomyocytes is associated with reduced SR Ca2+ load and contractile dysfunction [100]. Thus, in many heart failure models, NCX upregulation potentiates the loss of SR Ca2+ caused by reduced SERCA2a activity (Fig. 3A). On the other hand, increased NCX function may actually be beneficial for diastolic function, by correcting for defective Ca2+ removal due to reduced SERCA2a function. In support of this concept, Hasenfuss et al showed that NCX levels predicted diastolic function in human heart failure, with preserved relaxation in subjects with upregulated NCX [101].
However, NCX function is dependent not only on its expression, but also by local Na+ and Ca2+ levels and action potential configuration – all of which are altered in heart failure [102]. [Na+]i is reported to be increased in both human heart failure and animal models [103], due to either decreased Na+ extrusion or increased Na+ influx. NKA is the major Na+ extrusion pathway in cardiomyocytes, and in several, but not all models of heart failure NKA expression is found to be decreased [103] (Fig. 1B). Downregulation of the α2 NKA isoform is of particular significance, due to its functional coupling with NCX [104, 105]. Important Na+ influx mechanisms believed to contribute to Na+ loading are NCX[102], Na+/H+ exchange (NHE) [106] and increased late Na+ current [107, 108]. With sufficient Na+ loading in end-stage heart failure, NCX-mediated Ca2+ extrusion may be impaired despite increased NCX expression [37, 39]. Prolongation of action potential duration in failing myocytes can exacerbate this deficit as positive membrane potentials reduce the driving force for Ca2+ extrusion by NCX [9, 39]. With respect to arrhythmogenesis, NCX activity may be implicated in several ways [13]. As described previously, spontaneous Ca2+ release during diastole is a feature of heart failure, and extrusion of released Ca2+ elicits DADs. When NCX expression is increased, the depolarizing current will be larger and DADs are more likely to trigger action potentials, ectopic beats and arrhythmias. In addition, inward NCX current associated with Ca2+ extrusion during the downstroke of the action potential may prolong action potential duration, leading to EADs and spontaneous beats [13].
Through its regulation of resting Ca2+ levels, NCX is also an important control point for hypertrophy signaling. In this regard, marked upregulation of Ca2+ extrusion may effectively compensate for SERCA2a loss and prevent hypertrophy, as we recently reported in SERCA2 KO mice [36, 109]. Without such compensation, increased diastolic [Ca2+]i and hypertrophic remodeling are expected [16] (Fig. 3A). Since Na+ and Ca2+ levels are coupled, increased [Na+]i in heart failure may indirectly promote hypertrophy, as reported following NHE-1 activation [110, 111].
Targeting NCX in Heart Failure
The above discussion illustrates that there is great potential for targeting NCX in heart failure, to prevent arrhythmia and improve pump function. Current pharmacological inhibitors of NCX are the drugs KB-R7943 [112], SEA-0400 [113] and SN-6 [114] (Fig. 3B). While KB-R7943 also blocks Na+, K+ and Ca2+ channels, SEA-0400 and SN-06 have more selective profiles. Still, regardless of pharmacological profile, NCX inhibitors indirectly inhibit LTCCs by increasing [Ca2+]i [115]. Even so, NCX inhibition is expected to have inotropic effects by shifting the balance from Ca2+ extrusion to SR Ca2+ recycling. Furthermore, NCX blockade is expected to block generation of the arrhythmogenic inward INCX.
NCX inhibition has been investigated in a range of different cardiac disease models, including heart faiilure. In a rabbit rapid-pacing model of heart failure, in vivo SEA-0400 administration was observed to reduce action potential duration, repolarization dispersion and the number of EADs and ventricular tachycardias [116]. In canine heart failure, DADs were also attenuated by SEA-0400 [117]. Inotropic effects of reduced NCX function have been experimentally demonstrated by intracellular application of the exchange inhibitory peptide (XIP) in a canine model of heart failure, with restoration of SR Ca2+ reuptake and release [118]. Also, pharmacological NCX blockade by SEA-0400 showed positive inotropic effects in mice with heart failure following transverse aortic constriction [115], although relaxation was slowed. This is an expected drawback of NCX inhibition for heart failure etiologies where increased NCX function compensates for impaired Ca2+ removal by SERCA2a. Thus, NCX inhibition may be best suited for patients which predominantly exhibit systolic dysfunction. However, based on presently available data, Antoons et al recently concluded that the effects of NCX inhibition have thus far been predominantly beneficial in reducing arrhythmias and restoring contractility [119]. An added benefit of NCX inhibition may be attenuation of myocardial fibrosis and stiffening, as reported by Kamimura et al in a rat model of heart failure with preserved ejection fraction. They hypothesized that the underlying mechanism was reduced Ca2+ entry into fibroblasts which attenuates collagen production [120].
Rather than directly modulating NCX function, an alternative approach is to indirectly modify NCX activity via changes in [Na+]i. Cardiac glycosides function, at least in part, by inhibiting NKA and increasing [Na+]i, which attenuates NCX-mediated Ca2+ removal and augments SR Ca2+ load (Fig. 3B). However, such action also increases the propensity for triggered arrhythmias, and Ca2+ overload is implicated in myocardial dysfunction, remodeling and failure. Istaroxime, which is a non-glycoside NKA inhibitor, bypasses these side effects of Ca2+ overload by also stimulating SERCA2a (as discussed in the SERCA section). Alternatively, the opposite strategy of targeting pathological increases in [Na+]i may reduce NCX mediated Ca2+ overload and improve survival. As discussed above, increased NHE-1 activity has been implicated in Na+ gain in failing cells, and NHE-1 inhibition has been shown to experimentally reverse cardiac hypertrophy [110, 121]. However, several clinical trials have not been able to reproduce the cardioprotective effects of the NHE-1 inhibitors cariporide or eniporide in patients with acute coronary syndromes [122-124]. In fact, the most recent of these trials demonstrated cariporide-induced toxicity [124]. Targeting the late Na+ current is another possible approach (Fig. 3B). Ranolazine, an inhibitor of this current, has been shown to reduce [Na+]i, and reverse diastolic dysfunction in tissue preparations from failing hearts [107]. Unfortunately, the recent RALI-DHF study failed to demonstrate improved left ventricular relaxation following Ranolazine treatment in patients with heart failure with preserved ejection fraction [125].
The final putative strategy for targeting NCX function that we will consider is modulation of phospholemman. Both up- and downregulation of phospholemman expression and phosphorylation are reported in different heart failure models [91]. Further complicating interpretation of the suitability of phospholemman as a drug target is its roving inhibition of NCX and NKA. Overexpression of phosphomimetic phospholemman in mice resulted in premature death due to heart failure and arrhythmias. Ca2+ removal was slower and diastolic Ca2+ increased [126], indicative of NCX inhibition. One might expect that the opposite intervention, that is reducing phospholemman phosphorylation to activate NCX, might be a suitable therapeutic approach in heart failure. However, expected reductions in diastolic [Ca2+]i and Ca2+-dependent signaling would likely come at the price of reduced contractility, as has been reported in studies examining NCX overexpression [100].
III. RyR
As described above, increased NCX activity and reduced SERCA2a activity are considered to be important contributors to reduced SR Ca2+ content in heart failure. The third pathway by which SR Ca2+ content may be reduced is via increased RyR Ca2+ leakage. As with SERCA2a and NCX, RyR is also recognized as a therapeutic target. In order to understand its potential in therapy, we will first outline its function in normal and diseased hearts.
RyR Function
RyR is a large tetrameric protein localized to the SR membrane. RyR2, which is the major cardiac RyR isoform, acts as a scaffolding protein that associates with a number of proteins to form a macromolecular complex [127, 128]. This complex, which is important for RyR regulation and integrity, includes regulatory proteins such as protein kinase A, protein phosphatase 1 and 2a, calmodulin, calmodulin kinase II and phosphodiesterase 4D3 (PDE4D3) (Fig. 4B). This structure allows tight control of RyR function via several phosphorylation sites, as well as Ca2+ activation and inactivation sites. The RyR2 binding protein FKBP12.6 (calstabin 2) stabilizes the tetrameric conformation.
Fig. (4).
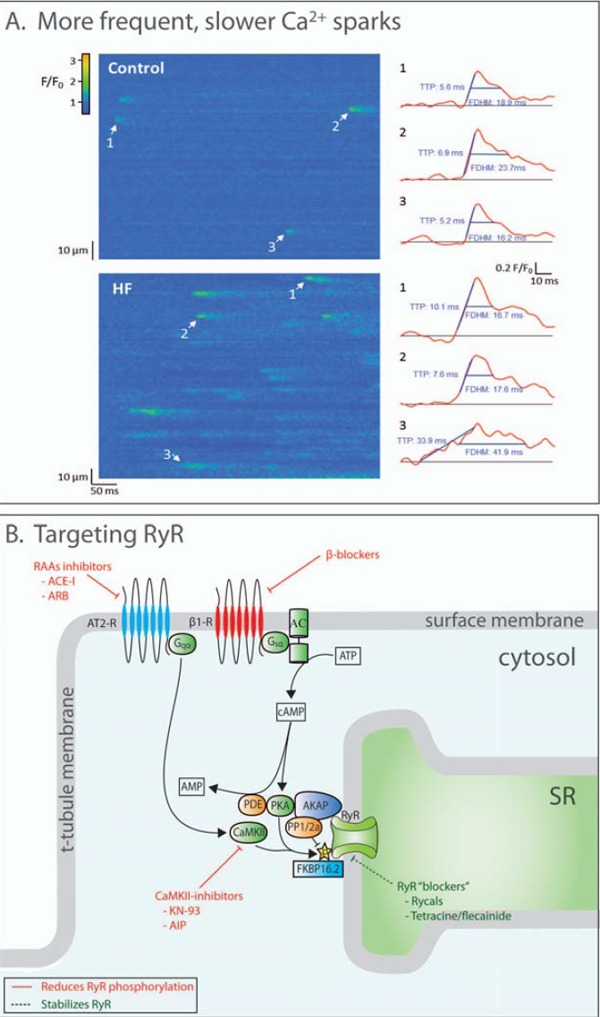
Altered Ca2+ sparks in failing cardiomyocytes and therapeutic targets of RyR activity. A: Line scan confocal imaging of failing cardiomyocytes (post-infarction mouse) reveals more frequent and slower Ca2+ sparks (temporal profiles of indicated sparks shown at right; reproduced from [6], with permission). Thus, disrupted RyR function in heart failure promotes SR Ca2+ leak and dyssynchronous Ca2+ release. B: RyR function is regulated by a large protein complex. Phosphorylation (by PKA and CaMKII) and dephosphorylation (by protein phosphatase 1 or 2a) are important regulatory pathways. Strategies to inhibit RyR phosphorylation, such as CaMKII inhibitors, are demonstrated to reduce SR Ca2+ leak. RyRs “blockers” such as rycals are an alternative approach to reducing leak.
RyR in the Failing Heart
There is general agreement that RyRs become leaky during heart failure, resulting in increased frequency of Ca2+ sparks (Fig. 4A) and waves, which promote arrhythmogenesis as released Ca2+ is removed by NCX, causing DADs [13]. Leak-induced elevation of [Ca2+] in or near the dyad is also believed to activate pathologic signaling leading to hypertrophy and myocardial dysfunction [17, 129]. However, the effect of RyR leakage on mechanical function remains a point of discussion. Many have reported that leak-induced reduction of SR Ca2+ content may decrease Ca2+ transient amplitude and contractility. However, the concept of autoregulation put forward by the Eisner group predicts that the decrease in SR Ca2+ content will be counterbalanced by a larger fractional SR Ca2+ release due to increased RyR sensitivity, leaving Ca2+ transients unaltered [130]. This concept is in accordance with the phenotype of human catecholaminergic polymorph ventricular tachycardia (CPVT), where RyR leak is increased, but contractile force is preserved. However, in heart failure RyR leak is also accompanied by disturbed SERCA2a and NCX function, which is likely critical for mediating depressed contractile function. The Eisner group has also shown that increased leak does not alter global diastolic [Ca2+]i [131], although even local increases in resting Ca2+ levels might inhibit relaxation.
Increased RyR open probability in heart failure has been attributed to PKA or CaMKII phosphorylation. Marx et al reported that increased β-adrenergic signaling caused PKA-mediated “hyperphosphorylation” at Ser2808 of RyR, resulting in FKBP12.6 dissociation from the channel, destabilization of the closed conformation of RyR, and increased Ca2+ leak [132]. Follow-up work by this group proposed that stress-induced phosphorylation, oxidation, and nitrosylation of RyR [133] also result in depletion of protein phosphatases and PDE4D3 [134], which further potentiates RyR hyperphosphorylation and FKBP12.6 dissociation. Recently, Lundby et al mapped downstream phosphorylation sites of β1-receptor signaling, and RyR Ser2808 was indeed identified as one phosphorylation target [135]. However, PKA phosphorylation of RyR and its relevance to heart failure pathogenesis are highly controversial [136]. Of particular note, the groups of Houser and Valdivia have been unable to reproduce PKA phosphorylation at Ser2808 as a relevant mechanism in the cardiac fight-flight response and heart failure development [137-140].
Work done in the Bers group showed that CaMKII phosphorylation of RyR in mice altered Ca2+ sparks [141], whereas PKA phosphorylation did not [142]. In agreement with this finding, increased RyR leak during heart failure has been linked to CaMKII phosphorylation at Ser2814 [143]. Recent evidence indicates that CaMKII-mediated S2814 phosphorylation actually promotes heart failure development [129, 144, 145]. Importantly, CaMKII activity is enhanced during chronic β1-adrenergic and angiotensin receptor II stimulation, as seen in heart failure, and expression of CaMKII is also increased in this disease [146].
While there has clearly been much study of RyR phosphorylation in heart failure, there is also some evidence to suggest that RyR expression may be altered, at least in some models [6, 147]. In mice with heart failure following myocardial infarction, we recently observed reduced RyR expression and Ca2+ sparks with slow kinetics [6] (Fig. 4A). Slow spark regions also exhibited slowed Ca2+ release during the action potential, which contributed to de-synchronized SR Ca2+ release in failing cells. We have hypothesized that reduced RyR density or sporadic RyR distribution may underlie these abnormal sparks.
Targeting RyR Leak to Restore Cardiac Function
Despite the controversies described above, there is clearly great potential for drugs that might act to reduce RyR-mediated Ca2+ leak, and thus prevent arrhythmias, hypertrophic signaling and possibly contractile dysfunction. Therapeutic agents may either directly interact with RyR, or indirectly reduce RyR phosphorylation by targeting upstream signaling pathways.
Several compounds, collectively called rycals, are known to bind and modulate RyR directly (Fig. 4B). One of the first drugs shown to restore abnormal RyR function was JTV519 (K201). This agent stabilizes the closed RyR conformation, but whether this is mediated by FKBP12.6 binding is still controversial. An early study in a canine model of heart failure showed that JTV519-treated hearts exhibited attenuated PKA-mediated phosphorylation of RyR, restored FKBP12.6 binding, inhibited SR Ca2+ leak, and ultimately rescued left ventricular function [148]. Further evidence that KFBP12.6 binding to RyR underlies these drug actions came from the observation that JTV519 effects were observed in transgenic FKBP12.6+/- but not FKBP12.6-/- mice [149, 150]. However, later studies [151, 152] have questioned such a requirement for FKBP12.6, as Yamamoto et al proposed that JTV519 acts to stabilize RyR by restoring normal RyR interdomain interactions [152]. There are early indications that JTV519 may have therapeutic promise, as experiments in human myocardium indicated that it improved both diastolic and systolic function under Ca2+ overload conditions [153]. However, it is not clear that these effects stem solely from RyR stabilization, as the drug also inhibits ICa, INa and IK1 [154]. A new derivate of JTV519, S107, has a more selective profile [155], and has been reported to increase the binding of FKBP12.6 to RyR, and to reduce abnormal diastolic Ca2+ release, arrhythmias [155], and heart failure progression in mice [133]. In a current phase 2, placebo-controlled randomized trial, the anti-arrhythmic properties of another RyR modulating drug, S44121, are being evaluated in patients with congestive heart failure (ISCRTN reg nr 14227980).
Flecainide and tetracaine have also been identified as possible RyR inhibitors. Flecainide is a class Ic antiarrhythmic clinically used to treat atrial fibrillation. The drug has additionally been shown to inhibit the open-state of the RyR, and thus experimentally prevent Ca2+ waves and arrhythmias in both mouse and human CPVT [156, 157]. However, this finding is controversial as Liu et al found that flecainide prevented triggered activity in CPVT mice primarily by Na+ channel block, while Ca2+ homeostasis was minimally affected [158]. Tetracaine, on the other hand, is a closed-state blocker of RyR [159], and could thereby reduce diastolic Ca2+ leak without interfering with systolic Ca2+ release.
Finally, there is great therapeutic potential in targeting signaling pathways that control RyR phosphorylation via PKA or CaMKII. Indeed, several existing heart failure therapies may ultimately target RyR phosphorylation via actions on these kinases. For instance, one important mechanism behind the beneficial effects of β1-antagonism may be reduced PKA- and/or CaMKII-dependent RyR phosphorylation [133] [160] (Fig. 4B). In addition, ACE inhibitors have been shown to reverse both elevated CaMKII expression and the hypertrophic phenotype in spontaneously hypertensive rats [161]. Angiotensin receptor blockers may similarly reduce CaMKII-derived leak. Specific inhibition of CaMKII by agents such as KN-93 or AIP also reduce RyR leak [143] (Fig. 4B), and have been shown to improve the force frequency-relationship in trabeculae isolated from failing human hearts [162]. KN-93 was additionally shown to prevent arrhythmia in CaMKII∂ overexpressing mice [163]. Finally, interventions such as SERCA2a overexpression which reduce diastolic Ca2+ levels can inhibit RyR opening directly and/or via reduced CaMKII activation [48]. The benefits of CaMKII inhibition may extend beyond effects on RyRs, as CaMKII targets a range of dysfunctional processes in heart failure, including gene transcription, inflammatory signaling and fibroblast activation (reviewed in [164]).
IV. T-TUBULE STRUCTURE
T-Tubule Structure and Function
As described in the introduction, the transverse tubules (t-tubules) are invaginations of the cellular membrane which form an organized, primarily transverse, network that enables the formation of dyadic junctions between the cellular membrane and the SR [165]. T-tubules have been identified in ventricular cardiomyocytes in a wide variety of mammalian species (for a thorough review see [166]) and recent studies on larger mammals such as pig, sheep, cow, horse, and human [167-170] also found t-tubules in atrial myocytes (for review see [171]). The majority of t-tubules lie in close proximity to the Z-lines of healthy ventricular cells [172-174]. However, in addition to the transverse elements of the t-tubule system, a smaller proportion of longitudinal elements extend between Z-lines [6, 175, 176]. T-tubule geometry and distribution vary between species, and it has been suggested that ventricular cells from smaller species (with higher heart rates) have thinner t-tubules but higher overall t-tubule densities [5, 174, 177, 178].
T-tubule density and organization play an important role in determining the homogeneity of SR Ca2+ release. The high degree of t-tubule organization in rodent ventricle cells facilitates very synchronous Ca2+ release [5, 6, 179]. In cells with lower t-tubule density, such as pig ventricular myocytes, Ca2+ release is less synchronous [180], and in de-tubulated cells a wave-like inward propagation from the periphery to the center of the myocyte is observed [181]. In addition, cultured ventricular cells lose t-tubules progressively over time leading to dyssynchronous release of Ca2+ [182]. Thus, in healthy cardiomyocytes, a well-organized t-tubule network ensures close localization between LTCCs and RyRs, tight control of CICR, and uniform Ca2+ release across the cell.
T-Tubules in the Failing Heart
We and others have shown that T-tubule structure is altered in pathological states such as heart failure (for review see [2]). Depending on species, the specific heart failure etiology, and the timepoint during disease progression, these alterations may include: i) disorganization of transversely-oriented t-tubules (Fig. 5A) [6, 105, 179, 183-193], ii) loss of t-tubules [5, 105, 185, 187, 188, 191, 194-200], iii) increase in the longitudinal fraction of tubules [6, 109, 179, 183, 189, 190, 193, 201, 202], and iv) dilation of t-tubules [173, 192, 199-201]. T-tubule loss or drift of transverse elements causes spatial dissociation between LTCCs in the t-tubules and RyRs in the SR, leading to the formation of orphaned RyRs (Fig. 1B). Such changes impair the ability of LTCCs to trigger SR Ca2+ release [6, 183], but also de-synchronize the Ca2+ transient, as Ca2+ release from orphaned RyRs can be triggered only after diffusion of Ca2+ from intact dyads [6, 159, 178, 188, 196, 203-205]. In the majority of studies, the relationship between T-tubule disruption and Ca2+ release dyssynchrony has been merely correlative. However, we recently examined this issue quantitatively in a mathematical model describing spatiotemporal dynamics of Ca2+ in the cytosol and SR. With progressive disorganization of T-tubule structure during heart failure development, the model confirmed greater Ca2+ release dyssynchrony (Fig. 5A). However, the model and experiments additionally showed that the magnitude of Ca2+ release and RyR Ca2+ sensitivity also affect release synchrony [193]. Of note, reduced SR content in failing cells exacerbates dyssynchrony of Ca2+ release resulting from T-tubule disruption. The less homogeneous Ca2+ transient is slower and of lower amplitude in failing cells, which has been linked to slower and smaller contraction [2].
In addition to enabling dyad formation, the integrity of t-tubules plays a crucial role for action potential propagation through the myocyte. T-tubule detachment from the surface membrane prevents those tubules from receiving action potentials, and effectively orphans RyRs in their dyadic junctions. However, interesting new data indicate that detached t-tubules also impede action potential propagation into t-tubules which remain in contact with the cell surface, worsening dyssynchrony of Ca2+ release [206]. Furthermore, important ion channels and transporters present in the t-tubules are expected to exhibit dramatically altered distribution following t-tubule re-organization. Such changes likely contribute to altered action potential configuration, which exacerbates impairments in CICR in failing cells [9, 207, 208]. Since the LTCC and NCX are more prominently expressed in t-tubules compared to the sarcolemmal membrane [175, 209], t-tubule disruption has been linked to deficient trans-sarcolemmal Ca2+ fluxes [179, 182, 183]. We have additionally linked impaired NCX-mediated Ca2+ removal in failing cells to loss of the alpha-2 NKA isoform during t-tubule disorganization, and disruption of a Na+ microdomain shared by these proteins [105].
While there are clearly a number of detrimental consequences of altered t-tubule organization in failing cells, our recent data indicate that the increased longitudinal fraction of t-tubules frequently observed during heart failure may be compensatory. We observed that in SERCA2 knockout mice, newly grown longitudinal elements form dyadic junctions with the SR [109]. Although LTCCs are not present in these tubules, they do contain NCX in close apposition to RyRs. Our mathematical modeling data suggest that such changes facilitate a greater reliance on trans-sarcolemmal Ca2+ cycling, by facilitating NCX-mediated Ca2+ entry and removal, and that Ca2+ entry via NCX can elicit CICR. This hypothesis has not yet been confirmed by functional data, and it also remains to be determined whether a similar adaptive role of newly grown tubules occurs in other heart failure models.
It is currently unclear whether t-tubule disarray during heart failure is pro-arrhythmic. Data from the Wehrens group have indicated that orphaned RyRs exhibit increased activity [186], which might theoretically contribute to greater SR Ca2+ leak and/or Ca2+ waves. However, our own data have indicated that Ca2+ sparks almost exclusively occur at intact dyads in failing cells [6]. This finding is supported by previous reports from failing [210, 211] and de-tubulated [181] cardiomyocytes.
Possible Therapeutic Targets to Recover the t-Tubule Network
To date, t-tubule structure has not been deliberately targeted in clinical practice. However, Sachse et al. [202] recently showed that resynchronization therapy of failing canine hearts improved the organization of t-tubules, suggesting that similar therapies in patients may also promote reverse remodeling of t-tubules. A suggested explanation for this action is that re-synchronization relieves the mechanical stress present in the late-activated lateral wall, and attenuates stress- or strain-dependent signaling which may lead to t-tubule disruption. Support for this hypothesis comes from the observation that Sildenafil treatment during pulmonary artery hypertension was observed to improve t-tubule structure by reducing afterload on the right ventricle [188]. Similar effects have been reported following pressure unloading by administration of β1-receptor blockers [212] and mechanical unloading by heterotopic transplantation of post-infarction failing hearts [195]. Thus, it appears that the load and/or stretch placed on the myocardium are key factors in determining the organization/disorganization of the t-tubule network. The t-tubules themselves may transduce these signals as they contain stretch-sensitive transmembrane proteins and channels, and exhibit altered geometry during stretch and contraction of the myocyte [213, 214].
Several molecules have been described to influence t-tubule structure and function [2]. One of the more extensively studied molecules is Junctophilin-2 (JP2) which plays an important role in anchoring t-tubules to the SR (Fig. 5B). Knockout (KO) models have shown that JP2 is critical for normal cardiomyocyte function [215]. This led to several investigations of JP2 in heart failure, and it is now well documented that JP2 is markedly down-regulated in failing mice [212], rats [185, 188, 216], and humans [217]. JP2 down-regulation was observed to correlate well with t-tubule loss [185], and recent studies have shown that JP2 knockdown or transgenic expression of JP2 shRNA causes t-tubule remodeling [194]. Thus, although the molecular mechanisms underlying JP2 down-regulation in heart failure remain unclear, existing data indicate that stabilization of JP2 levels may have therapeutic potential. Interestingly, caveolin-3, a muscle-specific caveolae-related protein which associates with JP2, may be important for anchoring JP2 in the t-tubule, and is down-regulated along with JP2 in pathological states including heart failure [218] (Fig. 5B). Furthermore, caveolin-3 KO mice exhibit structural remodeling of the t-tubule network in skeletal muscles [219]. Caveolin-3 over-expression may therefore serve as a therapeutic approach to combat JP2-dependent t-tubule remodeling.
Fig. (5).
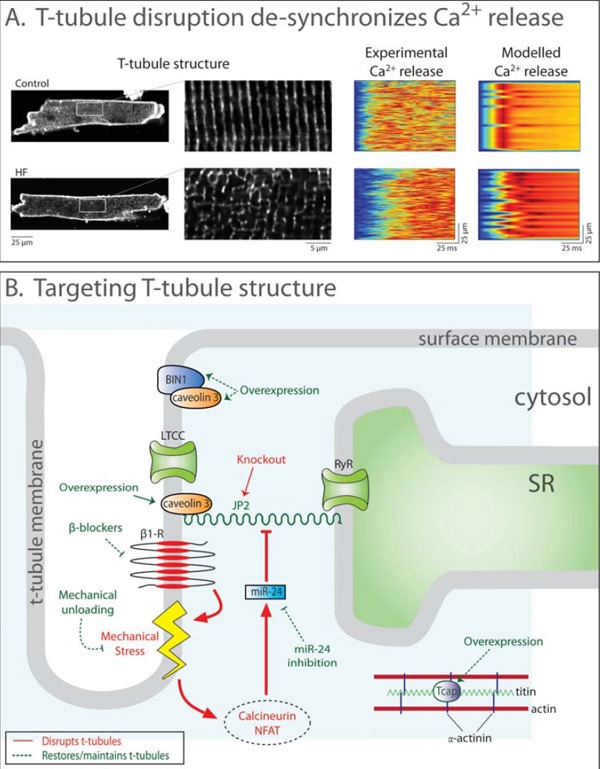
T-tubule disruption in heart failure and pathways to restore t-tubule structure. A: Confocal images post-infarction murine cardiomyocytes stained with di-8-ANEPPS show disrupted t-tubule structure in heart failure (left panel, magnified in insets). Accordingly, SR Ca2+ release was desynchronized in comparison with sham-operated controls. Mathematical modeling quantitatively reproduced the dyssynchronous pattern of Ca2+ release when changes in t-tubule organization, RyR threshold, and SR Ca2+ content were accounted for (right panels). Data are adapted from [193], with permission. B: Ttubule integrity and function is dependent on several proteins. JP2, along with caveolin 3, anchors t-tubules to the SR membrane. Increased calcineurin-NFAT signaling resulting from mechanical stress downregulates JP2 and disrupts t-tubules. JP2 expression may be restored by mechanical unloading, blockade of calcineurin-NFAT signaling, or by overexpressing the stretch-sensitive protein Tcap. BIN1 is involved in t-tubule growth and is downregulated in heart failure. Overexpression of BIN1 may therefore attenuate t-tubule loss.
Based on the growing consensus that mechanical stress regulates t-tubule structure and function, it is perhaps not surprising that recent work has implicated a role for calcineurin-NFAT signaling. It is well-established that this pathway transduces mechanical stimuli during hypertrophy [220] through a number of effectors, including microRNAs. MicroRNA-24 (miR-24), a member of the miR-23a-27a-24-2 cluster which is up-regulated in heart failure [221], has been identified as a prominent regulator of JP2 expression in cardiac tissue [221] (Fig. 5B). Overexpression of miR-24 in cultured cardiomyocytes led to JP2 down-regulation, disrupted dyadic structure, and dysfunctional CICR. The same group has since shown that miR-24 suppression protects against heart failure progression, and associated disruption of t-tubules and Ca2+ homeostasis [222]. Thus, therapeutic inhibition of miR-24 or other components of the NFAT signaling pathway holds great potential for maintaining dyadic integrity in heart failure.
Another protein of interest is telethonin (Tcap), a stretch-sensitive protein located in the Z-disc of cardiomyocytes. Tcap KO mice were observed to exhibit progressive disruption of the t-tubule network during development [191]. These defects were especially apparent in hearts working against increased mechanical load (thoracic aortic constriction). In addition, increased Tcap expression was associated with recovery of t-tubules during reverse remodeling induced by SERCA2a gene therapy [192]. Further studies are, however, needed to elucidate the precise interplay between myocardial load, expression of Tcap, and t-tubule organization.
Amphyphisin-2 (BIN1) is a protein involved in the formation of t-tubule invaginations and the trafficking of LTCCs to the t-tubules [223, 224]. Hong et al. [225] found that BIN1 was downregulated in failing human cardiomyocytes and that this was linked to decreased expression of LTCCs in the cell membrane. However, it is important to point out that in many heart failure models t-tubule density is unaltered or even increased, including a larger fraction of longitudinally-oriented tubules [109, 179, 183, 189, 190, 201, 202]. Whether BIN1 is involved in the growth of these new tubules remains unknown, but raises the possibility that BIN1 overexpression might have therapeutic potential.
FUTURE PERSPECTVES
As is evident from the above discussion, a growing body of preclinical data suggest that targeting abnormal Ca2+ handling in heart failure may be beneficial. However, much work remains to be done. Our current understanding of Ca2+ homeostasis during heart failure is largely focused on the end-stage of the disease, with surprisingly little known about changes in Ca2+ homeostasis during disease development. Existing data indicate that Ca2+ cycling is initially increased at early stages [226], but it is unclear if such alterations may be beneficial if maintained over longer periods, or if these apparent “compensations” in fact drive disease progression. Investigating the time course of changes in Ca2+ handling will generate complex data sets, and it will be a considerable challenge to integrate and interpret these data effectively. Therefore, we believe that the emerging fields of systems biology and mathematical modeling will play an increasingly important role in accurately identifying drug targets and predicting the consequences of their modulation. Since our ultimate goal is to reduce morbidity and mortality in heart failure patients, such mathematical models should be aimed at predicting function from the cardiomyocyte level up to the intact heart. Greater clinical translation can also be attained by increased experimentation on large animal heart failure models and human heart failure tissue. Close collaboration between experimentalists and clinicians will be essential for testing novel, cellular-level therapies in clinical trials.
CONCLUSION
The past several decades of investigation have revealed that altered Ca2+ handling is as an important pathophysiological mechanism in heart failure, and that SERCA2a, NCX, RyR, and t-tubule structure are key players. Such insight has enabled the recent therapeutic targeting of these systems, resulting in attenuation or reversal of important facets of the heart failure phenotype, including mechanical dysfunction, arrhythmogenesis, and pathological signaling. Although the majority of these data are from experimental studies, recent and ongoing clinical trials have shown promise. Since heart failure is a diverse disease entity, with variations in etiology, phenotype, stage and molecular basis, it is anticipated that future therapies based on cardiomyocyte mechanisms will have the potential to individualize treatment, and improve patient outcomes.
SOURCES OF FUNDING
Generous funding was provided by the South-Eastern Norway Regional Health Authority, The Research Council of Norway, Anders Jahre's Fund for the Promotion of Science, The Norwegian Health Association, Oslo University Hospital Ullevål, University of Oslo, and European Union Project FP7-HEALTH-2010.2.4.2-4 (“MEDIA-Metabolic Road to Diastolic Heart Failure”).
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21:380–7. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- 2.Louch WE, Sejersted OM, Swift F. There goes the neighborhood pathological alterations in T-tubule morphology and consequences for cardiomyocyte Ca2+ handling. J Biomed Biotechnol. 2010;2010:503906–0. doi: 10.1155/2010/503906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 4.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–6. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 5.Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, Claus P, Dymarkowski S, Maes F, Bogaert J, Rademakers F, D'Hooge J, Sipido K. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res. 2008;102:338–46. doi: 10.1161/CIRCRESAHA.107.160085. [DOI] [PubMed] [Google Scholar]
- 6.Louch WE, Hake J, Mork HK, Hougen K, Skrbic B, Ursu D, Tonnessen T, Sjaastad I, Sejersted OM. Slow Ca2+ sparks de-synchronize Ca2+ release in failing cardiomyocytes Evidence for altered configuration of Ca2+ release units?. J Mol Cell Cardiol. 2013;58:41–52. doi: 10.1016/j.yjmcc.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 7.Sah R, Ramirez RJ, Backx PH. Modulation of Ca2+ release in cardiac myocytes by changes in repolarization rate role of phase-1 action potential repolarization in excitation-contraction coupling. Circ Res. 2002;90:165–73. doi: 10.1161/hh0202.103315. [DOI] [PubMed] [Google Scholar]
- 8.Harris DM, Mills GD, Chen X, Kubo H, Berretta RM, Votaw VS, Santana LF, Houser SR. Alterations in early action potential repolarization causes localized failure of sarcoplasmic reticulum Ca2+ release. Circ Res. 2005;96:543–50. doi: 10.1161/01.RES.0000158966.58380.37. [DOI] [PubMed] [Google Scholar]
- 9.Louch WE, Hake J, Jolle GF, Mork HK, Sjaastad I, Lines GT, Sejersted OM. Control of Ca2+ release by action potential configuration in normal and failing murine cardiomyocytes. Biophys J. 2010;99:1377–86. doi: 10.1016/j.bpj.2010.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanderson JE. Heart failure with a normal ejection fraction. Heart. 2007;93:155–8. doi: 10.1136/hrt.2005.074187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Louch WE, Stokke MK, Sjaastad I, Christensen G, Sejersted OM. No rest for the weary diastolic calcium homeostasis in the normal and failing myocardium. Physiology (Bethesda) 2012;27:308–23. doi: 10.1152/physiol.00021.2012. [DOI] [PubMed] [Google Scholar]
- 12.Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, Gregoratos G, Klein G, Moss AJ, Myerburg RJ, Priori SG, Quinones MA, Roden DM, Silka MJ, Tracy C, Priori SG, Blanc JJ, Budaj A, Camm AJ, Dean V, Deckers JW, Despres C, Dickstein K, Lekakis J, McGregor K, Metra M, Morais J, Osterspey A, Tamargo JL, Zamorano JL, Smith SC Jr, Jacobs AK, Adams CD, Antman EM, Anderson JL, Hunt SA, Halperin JL, Nishimura R, Ornato JP, Page RL, Riegel B. American College of C American Heart Association Task F European Society of Cardiology Committee for Practice G European Heart Rhythm A Heart Rhythm S ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Europace. 2006;8:746–837. doi: 10.1093/europace/eul108. [DOI] [PubMed] [Google Scholar]
- 13.Ter Keurs HE, Boyden PA. Calcium and arrhythmogenesis. Physiol Rev. 2007;87:457–506. doi: 10.1152/physrev.00011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molkentin JD, Lu Jr, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–28. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki J, Bayna E, Li HL, Molle ED, Lew WY. Lipopolysaccharide activates calcineurin in ventricular myocytes. J Am Coll Cardiol. 2007;49:491–9. doi: 10.1016/j.jacc.2006.10.043. [DOI] [PubMed] [Google Scholar]
- 16.Goonasekera SA, Molkentin JD. Unraveling the secrets of a double life contractile versus signaling Ca2+ in a cardiac myocyte. J Mol Cell Cardiol. 2012;52:317–22. doi: 10.1016/j.yjmcc.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Louch WE, Lyon AR. Mind the store modulating Ca2+ reuptake with a leaky sarcoplasmic reticulum. Cardiovasc Res. 2013;98:165–8. doi: 10.1093/cvr/cvt069. [DOI] [PubMed] [Google Scholar]
- 18.Liu XH, Zhang ZY, Andersson KB, Husberg C, Enger UH, Raeder MG, Christensen G, Louch WE. Cardiomyocyte-specific disruption of Serca2 in adult mice causes sarco(endo)plasmic reticulum stress and apoptosis. Cell Calcium. 2011;49:201–7. doi: 10.1016/j.ceca.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 19.Kohlhaas M, Maack C. Calcium release microdomains and mitochondria. Cardiovasc Res. 2013 doi: 10.1093/cvr/cvt032. [DOI] [PubMed] [Google Scholar]
- 20.Hove-Madsen L, Bers DM. Sarcoplasmic reticulum Ca2+ uptake and thapsigargin sensitivity in permeabilized rabbit and rat ventricular myocytes. Circ Res. 1993;73:820–8. doi: 10.1161/01.res.73.5.820. [DOI] [PubMed] [Google Scholar]
- 21.Chu G, Lester JW, Young KB, Luo W, Zhai J, Kranias EG. A single site (Ser16):phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to beta -agonists. J Biol Chem. 2000;275:38938–43. doi: 10.1074/jbc.M004079200. [DOI] [PubMed] [Google Scholar]
- 22.Luss H, Klein-Wiele O, Boknik P, Herzig S, Knapp J, Linck B, Muller FU, Scheld HH, Schmid C, Schmitz W, Neumann J. Regional expression of protein phosphatase type 1 and 2A catalytic subunit isoforms in the human heart. J Mol Cell Cardiol. 2000;32:2349–59. doi: 10.1006/jmcc.2000.1265. [DOI] [PubMed] [Google Scholar]
- 23.Komuro I, Kurabayashi M, Shibazaki Y, Takaku F, Yazaki Y. Molecular cloning and characterization of a Ca2+ + Mg2+-dependent adenosine triphosphatase from rat cardiac sarcoplasmic reticulum.Regulation of its expression by pressure overload and developmental stage. . J Clin Invest. 1989;83:1102–8. doi: 10.1172/JCI113989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagai R, Zarain-Herzberg A, Brandl CJ, Fujii J, Tada M, MacLennan DH, Alpert NR, Periasamy M. Regulation of myocardial Ca2+-ATPase and phospholamban mRNA expression in response to pressure overload and thyroid hormone. Proc Natl Acad Sc U S A. 1989;86:2966–70. doi: 10.1073/pnas.86.8.2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mercadier JJ, Lompre AM, Duc P, Boheler KR, Fraysse JB, Wisnewsky C, Allen PD, Komajda M, Schwartz K. Altered sarcoplasmic reticulum Ca2+-ATPase gene expression in the human ventricle during end-stage heart failure. J Clin Invest. 1990;85:305–9. doi: 10.1172/JCI114429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca2+-ATPase in failing and nonfailing human myocardium. Circ Res. 1994;75:434–42. doi: 10.1161/01.res.75.3.434. [DOI] [PubMed] [Google Scholar]
- 27.Schwinger RH, Bohm M, Schmidt U, Karczewski P, Bavendiek U, Flesch M, Krause EG, Erdmann E. Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca2+-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation. 1995;92:3220–8. doi: 10.1161/01.cir.92.11.3220. [DOI] [PubMed] [Google Scholar]
- 28.Sande JB, Sjaastad I, Hoen IB, Bokenes J, Tonnessen T, Holt E, Lunde PK, Christensen G. Reduced level of serine(16):phosphorylated phospholamban in the failing rat myocardium a major contributor to reduced SERCA2 activity. Cardiovasc Res. 2002;53:382–91. doi: 10.1016/s0008-6363(01)00489-8. [DOI] [PubMed] [Google Scholar]
- 29.Lou Q, Fedorov VV, Glukhov AV, Moazami N, Fast VG, Efimov IR. Transmural heterogeneity and remodeling of ventricular excitation-contraction coupling in human heart failure. Circulation. 2011;123:1881–90. doi: 10.1161/CIRCULATIONAHA.110.989707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacLennan DH, Kranias EG. Phospholamban a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–77. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt U, Hajjar RJ, Kim CS, Lebeche D, Doye AA, Gwathmey JK. Human heart failure cAMP stimulation of SR Ca2+-ATPase activity and phosphorylation level of phospholamban. Am J Physiol. 1999;277:H474–80. doi: 10.1152/ajpheart.1999.277.2.H474. [DOI] [PubMed] [Google Scholar]
- 32.Munch G, Bolck B, Karczewski P, Schwinger RH. Evidence for calcineurin-mediated regulation of SERCA 2a activity in human myocardium. J Mol Cell Cardiol. 2002;34:321–34. doi: 10.1006/jmcc.2001.1515. [DOI] [PubMed] [Google Scholar]
- 33.Neumann J, Eschenhagen T, Jones LR, Linck B, Schmitz W, Scholz H, Zimmermann N. Increased expression of cardiac phosphatases in patients with end-stage heart failure. J Mol Cell Cardiol. 1997;29:265–72. doi: 10.1006/jmcc.1996.0271. [DOI] [PubMed] [Google Scholar]
- 34.Mishra S, Gupta RC, Tiwari N, Sharov VG, Sabbah HN. Molecular mechanisms of reduced sarcoplasmic reticulum Ca2+ uptake in human failing left ventricular myocardium. J Heart Lung Transplant. 2002;21:366–73. doi: 10.1016/s1053-2498(01)00390-4. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt U, Hajjar RJ, Helm PA, Kim CS, Doye AA, Gwathmey JK. Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J Mol Cell Cardiol. 1998;30:1929–37. doi: 10.1006/jmcc.1998.0748. [DOI] [PubMed] [Google Scholar]
- 36.Periasamy M, Reed TD, Liu LH, Ji Y, Loukianov E, Paul RJ, Nieman ML, Riddle T, Duffy JJ, Doetschman T, Lorenz JN, Shull GE. Impaired cardiac performance in heterozygous mice with a null mutation in the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 (SERCA2):gene. J Biol Chem. 1999;274:2556–62. doi: 10.1074/jbc.274.4.2556. [DOI] [PubMed] [Google Scholar]
- 37.Li L, Louch WE, Niederer SA, Aronsen JM, Christensen G, Sejersted OM, Smith NP. Sodium accumulation in SERCA knockout-induced heart failure. Biphys J . 2012;102:2039–48. doi: 10.1016/j.bpj.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andersson KB, Birkeland JA, Finsen AV, Louch WE, Sjaastad I, Wang Y, Chen J, Molkentin JD, Chien KR, Sejersted OM, Christensen G. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J Mol Cell Cardiol. 2009;47:180–7. doi: 10.1016/j.yjmcc.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 39.Louch WE, Hougen K, Mork HK, Swift F, Aronsen JM, Sjaastad I, Reims HM, Roald B, Andersson KB, Christensen G, Sejersted OM. Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J Physiol. 2010;588:465–78. doi: 10.1113/jphysiol.2009.183517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lipskaia L, Chemaly ER, Hadri L, Lompre AM, Hajjar RJ. Sarcoplasmic reticulum Ca2+ ATPase as a therapeutic target for heart failure. Expert Opin Biol Ther. 2010;10:29–41. doi: 10.1517/14712590903321462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stokke MK, Hougen K, Sjaastad I, Louch WE, Briston SJ, Enger UH, Andersson KB, Christensen G, Eisner DA, Sejersted OM, Trafford AW. Reduced SERCA2 abundance decreases the propensity for Ca2+ wave development in ventricular myocyte. Cardiovasc Res. 2010;86:63–71. doi: 10.1093/cvr/cvp401. [DOI] [PubMed] [Google Scholar]
- 42.Stokke MK, Briston SJ, Jolle GF, Manzoor I, Louch WE, Oyehaug L, Christensen G, Eisner DA, Trafford AW, Sejersted OM, Sjaastad I. Ca2+ wave probability is determined by the balance between SERCA2-dependent Ca2+ reuptake and threshold SR Ca2+ content. Cardiovasc Res. 2011;90:503–12. doi: 10.1093/cvr/cvr013. [DOI] [PubMed] [Google Scholar]
- 43.He H, Giordano FJ, Hilal-Dandan R, Choi DJ, Rockman HA, McDonough PM, Bluhm WF, Meyer M, Sayen MR, Swanson E, Dillmann WH. Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest. 1997;100:380–9. doi: 10.1172/JCI119544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Y, Escoubet B, Prunier F, Amour J, Simonides WS, Vivien B, Lenoir C, Heimburger M, Choqueux C, Gellen B, Riou B, Michel JB, Franz WM, Mercadier JJ. Constitutive cardiac overexpression of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase delays myocardial failure after myocardial infarction in rats at a cost of increased acute arrhythmias. Circulation. 2004;109:1898–903. doi: 10.1161/01.CIR.0000124230.60028.42. [DOI] [PubMed] [Google Scholar]
- 45.Muller OJ, Lange M, Rattunde H, Lorenzen HP, Muller M, Frey N, Bittner C, Simonides W, Katus HA, Franz WM. Transgenic rat hearts overexpressing SERCA2a show improved contractility under baseline conditions and pressure overload. Cardiovasc Res. 2003;59:380–9. doi: 10.1016/s0008-6363(03)00429-2. [DOI] [PubMed] [Google Scholar]
- 46.del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, Gwathmey JK, Rosenzweig A, Hajjar RJ. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100:2308–11. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T, Guerrero JL, Gwathmey JK, Rosenzweig A, Hajjar RJ. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sc U S A. 20000 ;97:793–8. doi: 10.1073/pnas.97.2.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lyon AR, Bannister ML, Collins T, Pearce E, Sepehripour AH, Dubb SS, Garcia E, O'Gara P, Liang L, Kohlbrenner E, Hajjar RJ, Peters NS, Poole-Wilson PA, Macleod KT, Harding SE. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ Arrhythm Electrophysiol. 2011;4:362–72. doi: 10.1161/CIRCEP.110.961615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, Lewandowski ED, Hajjar RJ. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca2+-ATPase in a rat model of heart failure. Circulation. 2001;104:1424–9. doi: 10.1161/hc3601.095574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang LF, Tsuji T, Takewa Y, del Monte F, Peluso R, Zsebo K, Jeong D, Park WJ, Kawase Y, Hajjar RJ. Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J Mol Cell Cardiol. 2007;42:852–61. doi: 10.1016/j.yjmcc.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumarswamy R, Lyon AR, Volkmann I, Mills AM, Bretthauer J, Pahuja A, Geers-Knorr C, Kraft T, Hajjar RJ, Macleod KT, Harding SE, Thum T. SERCA2a gene therapy restores microRNA-1 expression in heart failure via an Akt/FoxO3A-dependent pathway. Eureart J. 2012;33:1067–75. doi: 10.1093/eurheartj/ehs043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15:171–81. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease I (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124:304–13. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, Hajjar RJ. Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure analysis of recurrent cardiovascular events and mortality. Circ Res. 2014;114:101–8. doi: 10.1161/CIRCRESAHA.113.302421. [DOI] [PubMed] [Google Scholar]
- 55.Rocchetti M, Besana A, Mostacciuolo G, Micheletti R, Ferrari P, Sarkozi S, Szegedi C, Jona I, Zaza A. Modulation of sarcoplasmic reticulum function by Na+/K+ pump inhibitors with different toxicity digoxin and PST2744 [(E,Z)-3-((2-aminoethoxy)imino)andro-stane-6,17-dione hydrochloride]. J Pharmacol Exp Ther. 2005;313:207–15. doi: 10.1124/jpet.104.077933. [DOI] [PubMed] [Google Scholar]
- 56.Mattera GG, Lo Giudice P, Loi FM, Vanoli E, Gagnol JP, Borsini F, Carminati P. Istaroxime a new luso-inotropic agent for heart failure. Am J Cardiol. 2007;99:33A–40A. doi: 10.1016/j.amjcard.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 57.Sabbah HN, Imai M, Cowart D, Amato A, Carminati P, Gheorghiade M. Hemodynamic properties of a new-generation positive luso-inotropic agent for the acute treatment of advanced heart failure. Am J Cardiol. 2007;99:41A–46A. doi: 10.1016/j.amjcard.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 58.Lo Giudice P, Mattera GG, Gagnol JP, Borsini F. Chronic istaroxime improves cardiac function and heart rate variability in cardiomyopathic hamsters. Cardiovasc Drugs Ther. 2011;25:133–8. doi: 10.1007/s10557-011-6283-y. [DOI] [PubMed] [Google Scholar]
- 59.Gheorghiade M, Blair JE, Filippatos GS, Macarie C, Ruzyllo W, Korewicki J, Bubenek-Turconi SI, Ceracchi M, Bianchetti M, Carminati P, Kremastinos D, Valentini G, Sabbah HN, Investigators H-H. Hemodynamic, echocardiographic, and neurohormonal effects of istaroxime, a novel intravenous inotropic and lusitropic agent a randomized controlled trial in patients hospitalized with heart failure. J Am Coll Cardiol. 2008;51:2276–85. doi: 10.1016/j.jacc.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 60.Shah SJ, Blair JE, Filippatos GS, Macarie C, Ruzyllo W, Korewicki J, Bubenek-Turconi SI, Ceracchi M, Bianchetti M, Carminati P, Kremastinos D, Grzybowski J, Valentini G, Sabbah HN, Gheorghiade M, Investigators H-H. Effects of istaroxime on diastolic stiffness in acute heart failure syndromes results from the Hemodynamic, Echocardiographic, and Neurohormonal Effects of Istaroxime, a Novel Intravenous Inotropic and Lusitropic Agent a Randomized Controlled Trial in Patients Hospitalized with Heart Failure (HORIZON-HF) trial. Ameart J. 2009;157:1035–41. doi: 10.1016/j.ahj.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 61.Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, Kranias EG. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res. 1994;75:401–9. doi: 10.1161/01.res.75.3.401. [DOI] [PubMed] [Google Scholar]
- 62.Hoit BD, Khoury SF, Kranias EG, Ball N, Walsh RA. In vivo echocardiographic detection of enhanced left ventricular function in gene-targeted mice with phospholamban deficiency. Circ Res. 1995;77:632–7. doi: 10.1161/01.res.77.3.632. [DOI] [PubMed] [Google Scholar]
- 63.Slack JP, Grupp IL, Dash R, Holder D, Schmidt A, Gerst MJ, Tamura T, Tilgmann C, James PF, Johnson R, Gerdes AM, Kranias EG. The enhanced contractility of the phospholamban-deficient mouse heart persists with aging. J Mol Cell Cardiol. 2001;33:1031–40. doi: 10.1006/jmcc.2001.1370. [DOI] [PubMed] [Google Scholar]
- 64.Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, Martone ME, Wang Y, Ross J Jr, Kranias EG, Giles WR, Chien KR. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999;99:313–22. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- 65.Iwanaga Y, Hoshijima M, Gu Y, Iwatate M, Dieterle T, Ikeda Y, Date MO, Chrast J, Matsuzaki M, Peterson KL, Chien KR, Ross J Jr. Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling after infarction in rats. J Clin Invest. 2004;113:727–36. doi: 10.1172/JCI18716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoshijima M, Ikeda Y, Iwanaga Y, Minamisawa S, Date MO, Gu Y, Iwatate M, Li M, Wang L, Wilson JM, Wang Y, Ross J Jr, Chien KR. Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat Med. 2002;8:864–71. doi: 10.1038/nm739. [DOI] [PubMed] [Google Scholar]
- 67.Kaye DM, Preovolos A, Marshall T, Byrne M, Hoshijima M, Hajjar R, Mariani JA, Pepe S, Chien KR, Power JM. Percutaneous cardiac recirculation-mediated gene transfer of an inhibitory phospholamban peptide reverses advanced heart failure in large animals. J Am Coll Cardiol. 2007;50:253–60. doi: 10.1016/j.jacc.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 68.Lockamy EL, Cornea RL, Karim CB, Thomas DD. Functional and physical competition between phospholamban and its mutants provides insight into the molecular mechanism of gene therapy for heart failure. Biochem Biophys Res Commun. 2011;408:388–92. doi: 10.1016/j.bbrc.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.del Monte F, Harding SE, Dec GW, Gwathmey JK, Hajjar RJ. Targeting phospholamban by gene transfer in human heart failure. Circulation. 2002;105:904–7. doi: 10.1161/hc0802.105564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW 2nd, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–76. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wittkopper K, Dobrev D, Eschenhagen T, El-Armouche A. Phosphatase-1 inhibitor-1 in physiological and pathological beta-adrenoceptor signalling. Cardiovasc Res. 2011;91:392–401. doi: 10.1093/cvr/cvr058. [DOI] [PubMed] [Google Scholar]
- 72.Yamada M, Ikeda Y, Yano M, Yoshimura K, Nishino S, Aoyama H, Wang L, Aoki H, Matsuzaki M. Inhibition of protein phosphatase 1 by inhibitor-2 gene delivery ameliorates heart failure progression in genetic cardiomyopathy. FASEB J. 2006;20:1197–9. doi: 10.1096/fj.05-5299fje. [DOI] [PubMed] [Google Scholar]
- 73.Pathak A, del Monte F, Zhao W, Schultz JE, Lorenz JN, Bodi I, Weiser D, Hahn H, Carr AN, Syed F, Mavila N, Jha L, Qian J, Marreez Y, Chen G, McGraw DW, Heist EK, Guerrero JL, DePaoli-Roach AA, Hajjar RJ, Kranias EG. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res. 2005;96:756–66. doi: 10.1161/01.RES.0000161256.85833.fa. [DOI] [PubMed] [Google Scholar]
- 74.Miyazaki Y, Ikeda Y, Shiraishi K, Fujimoto SN, Aoyama H, Yoshimura K, Inui M, Hoshijima M, Kasahara H, Aoki H, Matsuzaki M. Heart failure-inducible gene therapy targeting protein phosphatase 1 prevents progressive left ventricular remodeling. PLoS One. 2012;7:e35875. doi: 10.1371/journal.pone.0035875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oh JG, Kim J, Jang SP, Nguen M, Yang DK, Jeong D, Park ZY, Park SG, Hajjar RJ, Park WJ. Decoy peptides targeted to protein phosphatase 1 inhibit dephosphorylation of phospholamban in cardiomyocytes. J Mol Cell Cardiol. 2013;56:63–71. doi: 10.1016/j.yjmcc.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 76.Marks AR. Calcium cycling proteins and heart failure mechanisms and therapeutics. J Clin Invest. 2013;123:46–52. doi: 10.1172/JCI62834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou JY, Zhu J, Entrikin D, Hammond HK. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation. 1999;99:3099–102. doi: 10.1161/01.cir.99.24.3099. [DOI] [PubMed] [Google Scholar]
- 78.Lai NC, Tang T, Gao MH, Saito M, Takahashi T, Roth DM, Hammond HK. Activation of cardiac adenylyl cyclase expression increases function of the failing ischemic heart in mice. J Am Coll Cardiol. 2008;51:1490–7. doi: 10.1016/j.jacc.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Osadchii OE. Myocardial phosphodiesterases and regulation of cardiac contractility in health and cardiac disease. Cardiovasc Drugs Ther. 2007;21:171–94. doi: 10.1007/s10557-007-6014-6. [DOI] [PubMed] [Google Scholar]
- 80.Nony P, Boissel JP, Lievre M, Leizorovicz A, Haugh MC, Fareh S, de Breyne B. Evaluation of the effect of phosphodiesterase inhibitors on mortality in chronic heart failure patients A meta-analysis. Eur J Clin Pharmacol. 1994;46:191–6. doi: 10.1007/BF00192547. [DOI] [PubMed] [Google Scholar]
- 81.Packer M, Carver Jr, Rodeheffer RJ, Ivanhoe RJ, DiBianco R, Zeldis SM, Hendrix GH, Bommer WJ, Elkayam U, Kukin ML, et al. Effect of oral milrinone on mortality in severe chronic heart failure.The PROMISE Study Research Group. N Engl J Med. 1991;325:1468–75. doi: 10.1056/NEJM199111213252103. [DOI] [PubMed] [Google Scholar]
- 82.Hasenfuss G, Pieske B, Castell M, Kretschmann B, Maier LS, Just H. Influence of the novel inotropic agent levosimendan on isometric tension and calcium cycling in failing human myocardium. Circulation. 1998;98:2141–7. doi: 10.1161/01.cir.98.20.2141. [DOI] [PubMed] [Google Scholar]
- 83.Kang TM, Hilgemann DW. Multiple transport modes of the cardiac Na+/Ca2+ exchanger. Nature. 2004;427:544–8. doi: 10.1038/nature02271. [DOI] [PubMed] [Google Scholar]
- 84.Bers DM, Barry WH, Despa S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res. 2003;57:897–912. doi: 10.1016/s0008-6363(02)00656-9. [DOI] [PubMed] [Google Scholar]
- 85.Juhaszova M, Blaustein MP. Na+ pump low and high ouabain affinity alpha subunit isoforms are differently distributed in cells. Proc Natl Acad Sci U S A. 1997;94:1800–5. doi: 10.1073/pnas.94.5.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA, Lingrel JB. Identification of a specific role for the Na,K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol Cell. 1999;3:555–63. doi: 10.1016/s1097-2765(00)80349-4. [DOI] [PubMed] [Google Scholar]
- 87.Swift F, Tovsrud N, Enger UH, Sjaastad I, Sejersted OM. The Na+/K+-ATPase alpha2-isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc Res. 2007;75:109–17. doi: 10.1016/j.cardiores.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 88.Swift F, Tovsrud N, Sjaastad I, Sejersted OM, Niggli E, Egger M. Functional coupling of alpha(2)-isoform Na+/K+-ATPase and Ca2+ extrusion through the Na+/Ca2+-exchanger in cardiomyocytes. Cell Calcium. 2010;48:54–60. doi: 10.1016/j.ceca.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 89.Silverman B, Fuller W, Eaton P, Deng J, Moorman Jr, Cheung JY, James AF, Shattock MJ. Serine 68 phosphorylation of phospholemman acute isoform-specific activation of cardiac Na/K ATPase. Cardiovasc Res. 2005;65:93–103. doi: 10.1016/j.cardiores.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 90.Zhang XQ, Qureshi A, Song J, Carl LL, Tian Q, Stahl RC, Carey DJ, Rothblum LI, Cheung JY. Phospholemman modulates Na+/Ca2+ exchange in adult rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2003;284:H225–33. doi: 10.1152/ajpheart.00698.2002. [DOI] [PubMed] [Google Scholar]
- 91.Cheung JY, Zhang XQ, Song J, Gao E, Chan TO, Rabinowitz JE, Koch WJ, Feldman AM, Wang J. Coordinated regulation of cardiac Na+/Ca2+ exchanger and Na+-K+-ATPase by phospholemman (FXYD1). Adv Exp Med Biol. 2013;961:175–90. doi: 10.1007/978-1-4614-4756-6_15. [DOI] [PubMed] [Google Scholar]
- 92.Wang J, Gao E, Rabinowitz J, Song J, Zhang XQ, Koch WJ, Tucker AL, Chan TO, Feldman AM, Cheung JY. Regulation of in vivo cardiac contractility by phospholemman role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol. 2011;300:H859–68. doi: 10.1152/ajpheart.00894.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Despa S, Tucker AL, Bers DM. Phospholemman-mediated activation of Na/K-ATPase limits [Na]i and inotropic state during beta-adrenergic stimulation in mouse ventricular myocytes. Circulation. 2008;117:1849–55. doi: 10.1161/CIRCULATIONAHA.107.754051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, Just H, Holtz J, Drexler H. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–53. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- 95.Flesch M, Schwinger RH, Schiffer F, Frank K, Sudkamp M, Kuhn-Regnier F, Arnold G, Bohm M. Evidence for functional relevance of an enhanced expression of the Na+-Ca2+ exchanger in failing human myocardium. Circulation. 1996;94:992–1002. doi: 10.1161/01.cir.94.5.992. [DOI] [PubMed] [Google Scholar]
- 96.Reinecke H, Studer R, Vetter R, Holtz J, Drexler H. Cardiac Na+/Ca2+ exchange activity in patients with end-stage heart failure. Cardiovasc Res. 1996;31:48–54. [PubMed] [Google Scholar]
- 97.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–19. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 98.Sipido KR, Volders PG, de Groot SH, Verdonck F, Van de Werf F, Wellens HJ, Vos MA. Enhanced Ca2+ release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes potential link between contractile adaptation and arrhythmogenesis. Circulation. 2000;102:2137–44. doi: 10.1161/01.cir.102.17.2137. [DOI] [PubMed] [Google Scholar]
- 99.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–67. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 100.Schillinger W, Janssen PM, Emami S, Henderson SA, Ross RS, Teucher N, Zeitz O, Philipson KD, Prestle J, Hasenfuss G. Impaired contractile performance of cultured rabbit ventricular myocytes after adenoviral gene transfer of Na+-Ca2+ exchanger. Circ Res. 2000;87:581–7. doi: 10.1161/01.res.87.7.581. [DOI] [PubMed] [Google Scholar]
- 101.Hasenfuss G, Schillinger W, Lehnart SE, Preuss M, Pieske B, Maier LS, Prestle J, Minami K, Just H. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation. 1999;99:641–8. doi: 10.1161/01.cir.99.5.641. [DOI] [PubMed] [Google Scholar]
- 102.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–8. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 103.Bers DM, Despa S, Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006;1080:165–77. doi: 10.1196/annals.1380.015. [DOI] [PubMed] [Google Scholar]
- 104.Semb SO, Lunde PK, Holt E, Tonnessen T, Christensen G, Sejersted OM. Reduced myocardial Na+, K+-pump capacity in congestive heart failure following myocardial infarction in rats. J Mol Cell Cardiol. 1998;30:1311–28. doi: 10.1006/jmcc.1998.0696. [DOI] [PubMed] [Google Scholar]
- 105.Swift F, Birkeland JA, Tovsrud N, Enger UH, Aronsen JM, Louch WE, Sjaastad I, Sejersted OM. Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase alpha2-isoform in heart failure. Cardiovasc Res. 2008;78:71–8. doi: 10.1093/cvr/cvn013. [DOI] [PubMed] [Google Scholar]
- 106.Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R, Fiolet JW. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 2003;57:1015–24. doi: 10.1016/s0008-6363(02)00809-x. [DOI] [PubMed] [Google Scholar]
- 107.Sossalla S, Wagner S, Rasenack EC, Ruff H, Weber SL, Schondube FA, Tirilomis T, Tenderich G, Hasenfuss G, Belardinelli L, Maier LS. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts--role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol. 2008;45:32–43. doi: 10.1016/j.yjmcc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 108.Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes implications for repolarization variability. Eur J Heart Fail. 2007;9:219–27. doi: 10.1016/j.ejheart.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Swift F, Franzini-Armstrong C, Oyehaug L, Enger UH, Andersson KB, Christensen G, Sejersted OM, Louch WE. Extreme sarcoplasmic reticulum volume loss and compensatory T-tubule remodeling after Serca2 knockout. Proc Natl Acad Sci U S A. 2012;109:3997–4001. doi: 10.1073/pnas.1120172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nakamura TY, Iwata Y, Arai Y, Komamura K, Wakabayashi S. Activation of Na+/H+ exchanger 1 is sufficient to generate Ca2+ signals that induce cardiac hypertrophy and heart failure. Circ Res. 2008;103:891–9. doi: 10.1161/CIRCRESAHA.108.175141. [DOI] [PubMed] [Google Scholar]
- 111.Cingolani HE, Ennis IL. Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation. 2007;115:1090–100. doi: 10.1161/CIRCULATIONAHA.106.626929. [DOI] [PubMed] [Google Scholar]
- 112.Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–7. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- 113.Matsuda T, Arakawa N, Takuma K, Kishida Y, Kawasaki Y, Sakaue M, Takahashi K, Takahashi T, Suzuki T, Ota T, Hamano-Takahashi A, Onishi M, Tanaka Y, Kameo K, Baba A. SEA0400, a novel and selective inhibitor of the Na+-Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J Pharmacol Exp Ther. 2001;298:249–56. [PubMed] [Google Scholar]
- 114.Iwamoto T, Inoue Y, Ito K, Sakaue T, Kita S, Katsuragi T. The exchanger inhibitory peptide region-dependent inhibition of Na+/Ca2+ exchange by SN-6 [2-[4-(4-nitrobenzyloxy)benzyl]thiazolidine-4-carboxylic acid ethyl ester], a novel benzyloxyphenyl derivative. Mol Pharmacol. 2004;66:45–55. doi: 10.1124/mol.66.1.45. [DOI] [PubMed] [Google Scholar]
- 115.Ozdemir S, Bito V, Holemans P, Vinet L, Mercadier JJ, Varro A, Sipido KR. Pharmacological inhibition of Na+/Ca2+ exchange results in increased cellular Ca2+ load attributable to the predominance of forward mode block. Circ Res. 2008;102:1398–405. doi: 10.1161/CIRCRESAHA.108.173922. [DOI] [PubMed] [Google Scholar]
- 116.Milberg P, Pott C, Frommeyer G, Fink M, Ruhe M, Matsuda T, Baba A, Klocke R, Quang TH, Nikol S, Stypmann J, Osada N, Muller FU, Breithardt G, Noble D, Eckardt L. Acute inhibition of the Na+/Ca2+ exchanger reduces proarrhythmia in an experimental model of chronic heart failure. Heart Rhythm. 2012;9:570–8. doi: 10.1016/j.hrthm.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 117.Nagy ZA, Virag L, Toth A, Biliczki P, Acsai K, Banyasz T, Nanasi P, Papp JG, Varro A. Selective inhibition of sodium-calcium exchanger by SEA-0400 decreases early and delayed after depolarization in canine heart. Br J Pharmacol. 2004;143:827–31. doi: 10.1038/sj.bjp.0706026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hobai IA, Maack C, O'Rourke B. Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res. 2004;95:292–9. doi: 10.1161/01.RES.0000136817.28691.2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Antoons G, Willems R, Sipido KR. Alternative strategies in arrhythmia therapy evaluation of Na/Ca exchange as an anti-arrhythmic target. Pharmacol Ther. 2012;134:26–42. doi: 10.1016/j.pharmthera.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 120.Kamimura D, Ohtani T, Sakata Y, Mano T, Takeda Y, Tamaki S, Omori Y, Tsukamoto Y, Furutani K, Komiyama Y, Yoshika M, Takahashi H, Matsuda T, Baba A, Umemura S, Miwa T, Komuro I, Yamamoto K. Ca2+ entry mode of Na+/Ca2+ exchanger as a new therapeutic target for heart failure with preserved ejection fraction. Eur Heart J. 2012;33:1408–16. doi: 10.1093/eurheartj/ehr106. [DOI] [PubMed] [Google Scholar]
- 121.Ennis IL, Garciarena CD, Escudero EM, Perez NG, Dulce RA, Camilion de Hurtado MC, Cingolani HE. Normalization of the calcineurin pathway underlies the regression of hypertensive hypertrophy induced by Na+/H+ exchanger-1 (NHE-1):inhibition. Can J Physiol Pharmacol. 2007;85:301–10. doi: 10.1139/y06-072. [DOI] [PubMed] [Google Scholar]
- 122.Zeymer U, Suryapranata H, Monassier JP, Opolski G, Davies J, Rasmanis G, Linssen G, Tebbe U, Schroder R, Tiemann R, Machnig T, Neuhaus KL, Investigators E. The Na+/H+ exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction.Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J Am Coll Cardiol. 2001;38:1644–50. doi: 10.1016/s0735-1097(01)01608-4. [DOI] [PubMed] [Google Scholar]
- 123.Theroux P, Chaitman BR, Danchin N, Erhardt L, Meinertz T, Schroeder JS, Tognoni G, White HD, Willerson JT, Jessel A. Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations.Main results of the GUARDIAN trial. Guard during ischemia against necrosis (GUARDIAN) Investigators. Circulation. 2000;102:3032–8. doi: 10.1161/01.cir.102.25.3032. [DOI] [PubMed] [Google Scholar]
- 124.Mentzer RM Jr, Bartels C, Bolli R, Boyce S, Buckberg GD, Chaitman B, Haverich A, Knight J, Menasche P, Myers ML, Nicolau J, Simoons M, Thulin L, Weisel RD, Investigators ES. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting results of the EXPEDITION study. Ann Thorac Surg. 2008;85:1261–70. doi: 10.1016/j.athoracsur.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 125.Maier LS, Layug B, Karwatowska-Prokopczuk E, Belardinelli L, Lee S, Sander J, Lang C, Wachter R, Edelmann F, Hasenfuss G, Jacobshagen C. RAnoLazIne for the treatment of diastolic heart failure in patients with preserved ejection fraction the RALI-DHF proof-of-concept study. JACC Heart Fail. 2013;1:115–22. doi: 10.1016/j.jchf.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 126.Song J, Gao E, Wang J, Zhang XQ, Chan TO, Koch WJ, Shang X, Joseph JI, Peterson BZ, Feldman AM, Cheung JY. Constitutive overexpression of phosphomimetic phospholemman S68E mutant results in arrhythmias, early mortality, and heart failure potential involvement of Na+/Ca2+ exchanger. Am J Physiol Heart Circ Physiol. 2012;302:H770–81. doi: 10.1152/ajpheart.00733.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–85. doi: 10.1146/annurev.biochem.76.053105.094237. [DOI] [PubMed] [Google Scholar]
- 128.Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol. 2004;37:417–29. doi: 10.1016/j.yjmcc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 129.van Oort RJ, Respress JL, Li N, Reynolds C, De Almeida AC, Skapura DG, De Windt LJ, Wehrens XH. Accelerated development of pressure overload-induced cardiac hypertrophy and dysfunction in an RyR2-R176Q knockin mouse model. Hypertension. 2010;55:932–8. doi: 10.1161/HYPERTENSIONAHA.109.146449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Eisner DA, Trafford AW, Diaz ME, Overend CL, O'Neill SC. The control of Ca release from the cardiac sarcoplasmic reticulum regulation versus autoregulation. Cardiovasc Res. 1998;38:589–604. doi: 10.1016/s0008-6363(98)00062-5. [DOI] [PubMed] [Google Scholar]
- 131.O'Neill SC, Eisner DA. A mechanism for the effects of caffeine on Ca2+ release during diastole and systole in isolated rat ventricular myocytes. J Physiol. 1990;430:519–36. doi: 10.1113/jphysiol.1990.sp018305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–76. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 133.Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375–87. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lundby A, Andersen MN, Steffensen AB, Horn H, Kelstrup CD, Francavilla C, Jensen LJ, Schmitt N, Thomsen MB, Olsen JV. In Vivo Phosphoproteomics Analysis Reveals the Cardiac Targets of beta-Adrenergic Receptor Signaling. Sci Signal. 2013;6:rs11. doi: 10.1126/scisignal.2003506. [DOI] [PubMed] [Google Scholar]
- 136.Bers DM. Ryanodine receptor S2808 phosphorylation in heart failure smoking gun or red herring. Circ Res. 2012;110:796–9. doi: 10.1161/CIRCRESAHA.112.265579. [DOI] [PubMed] [Google Scholar]
- 137.Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, Powers PA, Valdivia HH. Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–29. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- 138.MacDonnell SM, Garcia-Rivas G, Scherman JA, Kubo H, Chen X, Valdivia H, Houser SR. Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ Res. 2008;102:e65–72. doi: 10.1161/CIRCRESAHA.108.174722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang H, Makarewich CA, Kubo H, Wang W, Duran JM, Li Y, Berretta RM, Koch WJ, Chen X, Gao E, Valdivia HH, Houser SR. Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ Res. 2012;110:831–40. doi: 10.1161/CIRCRESAHA.111.255158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res. 2002;91:1015–22. doi: 10.1161/01.res.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- 141.Guo T, Zhang T, Mestril R, Bers DM. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 142.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–16. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 143.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–22. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 144.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Brown JH. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–40. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skardal K, Wisloff U, Wieland T, Ai X, Pogwizd SM, Dobrev D, Wehrens XH. Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ Res. 2012;110:1474–83. doi: 10.1161/CIRCRESAHA.112.268094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Anderson ME. CaMKII and a failing strategy for growth in heart. J Clin Invest. 2009;119:1082–5. doi: 10.1172/JCI39262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vatner DE, Sato N, Kiuchi K, Shannon RP, Vatner SF. Decrease in myocardial ryanodine receptors and altered excitation-contraction coupling early in the development of heart failure. Circulation. 1994;90:1423–30. doi: 10.1161/01.cir.90.3.1423. [DOI] [PubMed] [Google Scholar]
- 148.Yano M, Kobayashi S, Kohno M, Doi M, Tokuhisa T, Okuda S, Suetsugu M, Hisaoka T, Obayashi M, Ohkusa T, Kohno M, Matsuzaki M. FKBP12.-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation. 2003;107:477–84. doi: 10.1161/01.cir.0000044917.74408.be. [DOI] [PubMed] [Google Scholar]
- 149.Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304:292–6. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 150.Wehrens XH, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, Cheng Z, Deng SX, de Windt LJ, Landry DW, Marks AR. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sc U S A. 2005 ;102:9607–12. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hunt DJ, Jones PP, Wang R, Chen W, Bolstad J, Chen K, Shimoni Y, Chen SR. K201 (JTV519):suppresses spontaneous Ca2+ release and [3H]ryanodine binding to RyR2 irrespective of FKBP12. association. Bichem J. 2007;404:431–8. doi: 10.1042/BJ20070135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yamamoto T, Yano M, Xu X, Uchinoumi H, Tateishi H, Mochizuki M, Oda T, Kobayashi S, Ikemoto N, Matsuzaki M. Identification of target domains of the cardiac ryanodine receptor to correct channel disorder in failing hearts. Circulation. 2008;117:762–72. doi: 10.1161/CIRCULATIONAHA.107.718957. [DOI] [PubMed] [Google Scholar]
- 153.Toischer K, Lehnart SE, Tenderich G, Milting H, Korfer R, Schmitto JD, Schondube FA, Kaneko N, Loughrey CM, Smith GL, Hasenfuss G, Seidler T. K201 improves aspects of the contractile performance of human failing myocardium via reduction in Ca2+ leak from the sarcoplasmic reticulum. Basic Res Cardiol. 2010;105:279–87. doi: 10.1007/s00395-009-0057-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kimura J, Kawahara M, Sakai E, Yatabe J, Nakanishi H. Effects of a novel cardioprotective drug, JTV-519, on membrane currents of guinea pig ventricular myocytes. Jpn J Pharmacol. 1999;79:275–81. doi: 10.1254/jjp.79.275. [DOI] [PubMed] [Google Scholar]
- 155.Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, Lederer WJ, Kass RS, Morley G, Marks AR. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–45. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380–3. doi: 10.1038/nm.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hilliard FA, Steele DS, Laver D, Yang Z, Le Marchand SJ, Chopra N, Piston DW, Huke S, Knollmann BC. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol. 2010;48:293–301. doi: 10.1016/j.yjmcc.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Liu N, Denegri M, Ruan Y, Avelino-Cruz JE, Perissi A, Negri S, Napolitano C, Coetzee WA, Boyden PA, Priori SG. Short communication flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity. Circ Res. 2011;109:291–5. doi: 10.1161/CIRCRESAHA.111.247338. [DOI] [PubMed] [Google Scholar]
- 159.Laver DR, van Helden DF. Three independent mechanisms contribute to tetracaine inhibition of cardiac calcium release channels. J Mol Cell Cardiol. 2011;51:357–69. doi: 10.1016/j.yjmcc.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 160.Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart role of CaMKII. J Mol Cell Cardiol. 2010;48:322–30. doi: 10.1016/j.yjmcc.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hempel P, Hoch B, Bartel S, Karczewski P. Hypertrophic phenotype of cardiac calcium/calmodulin-dependent protein kinase II is reversed by angiotensin converting enzyme inhibition. Basic Res Cardiol. 2002;97(1):I96–101. doi: 10.1007/s003950200037. [DOI] [PubMed] [Google Scholar]
- 162.Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittkopper K, Renner A, Schmitto JD, Gummert J, El-Armouche A, Hasenfuss G, Maier LS. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. 2010;107:1150–61. doi: 10.1161/CIRCRESAHA.110.220418. [DOI] [PubMed] [Google Scholar]
- 163.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–11. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 164.Currie S, Elliott EB, Smith GL, Loughrey CM. Two candidates at the heart of dysfunction The ryanodine receptor and calcium/calmodulin protein kinase II as potential targets for therapeutic intervention-An in vivo perspective. Pharmacol Ther. 2011;131:204–20. doi: 10.1016/j.pharmthera.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 165.Fawcett DW, McNutt NS. The ultrastructure of the cat myocardium. I.; Ventricular papillary muscle. . J Cell Biol. 1969;42:1–45. doi: 10.1083/jcb.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Guo A, Zhang C, Wei S, Chen B, Song LS. Emerging mechanisms of T-tubule remodelling in heart failure. Cardiovasc Res. 2013 doi: 10.1093/cvr/cvt020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P, D'Hooge J, Heidbuchel H, Sipido KR, Willems R. Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ Res. 2009;105:876–85. doi: 10.1161/CIRCRESAHA.109.206276. [DOI] [PubMed] [Google Scholar]
- 168.Dibb KM, Clarke JD, Horn MA, Richards MA, Graham HK, Eisner DA, Trafford AW. Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ Heart Fail. 2009;2:482–9. doi: 10.1161/CIRCHEARTFAILURE.109.852228. [DOI] [PubMed] [Google Scholar]
- 169.Richards MA, Clarke JD, Saravanan P, Voigt N, Dobrev D, Eisner DA, Trafford AW, Dibb KM. Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am J Physiol Heart Circ Physiol. 2011;301 doi: 10.1152/ajpheart.00284.2011. H1996-2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Frisk M, Koivumaki JT, Norseng PA, Maleckar MM, Sejersted OM, Louch WE. Variable t-tubule organization and Ca2+ homeostasis across the atria. Am J Physiol Heart Circ Physiol. 2014;307:H609–20. doi: 10.1152/ajpheart.00295.2014. [DOI] [PubMed] [Google Scholar]
- 171.Trafford AW, Clarke JD, Richards MA, Eisner DA, Dibb KM. Calcium signalling microdomains and the t-tubular system in atrial mycoytes potential roles in cardiac disease and arrhythmias. Cardiovasc Res. 2013 doi: 10.1093/cvr/cvt018. [DOI] [PubMed] [Google Scholar]
- 172.McNutt NS. Ultrastructure of the myocardial sarcolemma. Circ Res. 1975;37:1–13. doi: 10.1161/01.res.37.1.1. [DOI] [PubMed] [Google Scholar]
- 173.Kostin S, Scholz D, Shimada T, Maeno Y, Mollnau H, Hein S, Schaper J. The internal and external protein scaffold of the T-tubular system in cardiomyocytes. Cell Tissue Res. 1998;294:449–60. doi: 10.1007/s004410051196. [DOI] [PubMed] [Google Scholar]
- 174.Soeller C, Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res. 1999;84:266–75. doi: 10.1161/01.res.84.3.266. [DOI] [PubMed] [Google Scholar]
- 175.Brette F, Orchard C. T-tubule function in mammalian cardiac myocytes. Circ Res. 2003;92:1182–92. doi: 10.1161/01.RES.0000074908.17214.FD. [DOI] [PubMed] [Google Scholar]
- 176.Song LS, Guatimosim S, Gomez-Viquez L, Sobie EA, Ziman A, Hartmann H, Lederer WJ. Calcium biology of the transverse tubules in heart. Ann N Y Acad Sci. 2005;1047:99–111. doi: 10.1196/annals.1341.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Savio-Galimberti E, Frank J, Inoue M, Goldhaber JI, Cannell MB, Bridge JH, Sachse FB. Novel features of the rabbit transverse tubular system revealed by quantitative analysis of three-dimensional reconstructions from confocal images. Biophys J. 2008;95:2053–62. doi: 10.1529/biophysj.108.130617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cannell MB, Crossman DJ, Soeller C. Effect of changes in action potential spike configuration, junctional sarcoplasmic reticulum micro-architecture and altered t-tubule structure in human heart failure. J Muscle Res Cell Motil. 2006;27:297–306. doi: 10.1007/s10974-006-9089-y. [DOI] [PubMed] [Google Scholar]
- 179.Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I, Sejersted OM. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol. 2006;574:519–33. doi: 10.1113/jphysiol.2006.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Heinzel FR, Bito V, Volders PG, Antoons G, Mubagwa K, Sipido KR. Spatial and temporal inhomogeneities during Ca2+ release from the sarcoplasmic reticulum in pig ventricular myocytes. Circ Res. 2002;91:1023–30. doi: 10.1161/01.res.0000045940.67060.dd. [DOI] [PubMed] [Google Scholar]
- 181.Brette F, Despa S, Bers DM, Orchard CH. Spatiotemporal characteristics of SR Ca2+ uptake and release in detubulated rat ventricular myocytes. J Mol Cell Cardiol. 2005;39:804–12. doi: 10.1016/j.yjmcc.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 182.Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K, Sipido KR. Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res. 2004;62:63–73. doi: 10.1016/j.cardiores.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 183.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sc USA. 2006;103:4305–10. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ibrahim M, Al Masri A, Navaratnarajah M, Siedlecka U, Soppa GK, Moshkov A, Al-Saud SA, Gorelik J, Yacoub MH, Terracciano CM. Prolonged mechanical unloading affects cardiomyocyte excitation-contraction coupling, transverse-tubule structure, and the cell surface. FASEB J. 2010;24:3321–9. doi: 10.1096/fj.10-156638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–31. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, De Almeida AC, Skapura DG, Rudy Y, Burns AR, Ackerman MJ, Wehrens XH. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–88. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wu CY, Jia Z, Wang W, Ballou LM, Jiang YP, Chen B, Mathias RT, Cohen IS, Song LS, Entcheva E, Lin RZ. PI3Ks maintain the structural integrity of T-tubules in cardiac myocytes. PLoS One. 2011;6:e24404. doi: 10.1371/journal.pone.0024404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Xie YP, Chen B, Sanders P, Guo A, Li Y, Zimmerman K, Wang LC, Weiss RM, Grumbach IM, Anderson ME, Song LS. Sildenafil prevents and reverses transverse-tubule remodeling and Ca2+ handling dysfunction in right ventricle failure induced by pulmonary artery hypertension. Hypertension. 2012;59:355–62. doi: 10.1161/HYPERTENSIONAHA.111.180968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wagner E, Lauterbach MA, Kohl T, Westphal V, Williams GS, Steinbrecher JH, Streich JH, Korff B, Tuan HT, Hagen B, Luther S, Hasenfuss G, Parlitz U, Jafri MS, Hell SW, Lederer WJ, Lehnart SE. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ Res. 2012;111:402–14. doi: 10.1161/CIRCRESAHA.112.274530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tao W, Shi J, Dorn GW 2nd, Wei L, Rubart M. Spatial variability in T-tubule and electrical remodeling of left ventricular epicardium in mouse hearts with transgenic Galphaq overexpression-induced pathological hypertrophy. J Mol Cell Cardiol. 2012;53:409–19. doi: 10.1016/j.yjmcc.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ibrahim M, Siedlecka U, Buyandelger B, Harada M, Rao C, Moshkov A, Bhargava A, Schneider M, Yacoub MH, Gorelik J, Knoll R, Terracciano CM. A critical role for Telethonin in regulating t-tubule structure and function in the mammalian heart. Hum Mol Genet. 2013;22:372–83. doi: 10.1093/hmg/dds434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lyon AR, Nikolaev VO, Miragoli M, Sikkel MB, Paur H, Benard L, Hulot JS, Kohlbrenner E, Hajjar RJ, Peters NS, Korchev YE, Macleod KT, Harding SE, Gorelik J. Plasticity of surface structures and beta(2)-adrenergic receptor localization in failing ventricular cardiomyocytes during recovery from heart failure. Circ Heart Fail. 2012;5:357–65. doi: 10.1161/CIRCHEARTFAILURE.111.964692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Oyehaug L, Loose KO, Jolle GF, Roe AT, Sjaastad I, Christensen G, Sejersted OM, Louch WE. Synchrony of Cardiomyocyte Ca2+ Release is Controlled by t-tubule Organization, SR Ca2+ Content, and Ryanodine Receptor Ca2+ Sensitivity. Biphys J . 2013;104:1685–97. doi: 10.1016/j.bpj.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wu HD, Xu M, Li RC, Guo L, Lai YS, Xu SM, Li SF, Lu QL, Li LL, Zhang HB, Zhang YY, Zhang CM, Wang SQ. Ultrastructural remodelling of Ca2+ signalling apparatus in failing heart cells. Cardiovasc Res. 2012;95:430–8. doi: 10.1093/cvr/cvs195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ibrahim M, Navaratnarajah M, Siedlecka U, Rao C, Dias P, Moshkov AV, Gorelik J, Yacoub MH, Terracciano CM. Mechanical unloading reverses transverse tubule remodelling and normalizes local Ca2+-induced Ca2+release in a rodent model of heart failure. Eur J Heart Fail. 2012;14:571–80. doi: 10.1093/eurjhf/hfs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kemi OJ, Hoydal MA, Macquaide N, Haram PM, Koch LG, Britton SL, Ellingsen O, Smith GL, Wisloff U. The effect of exercise training on transverse tubules in normal, remodeled, and reverse remodeled hearts. J Cell Physiol. 2011;226:2235–43. doi: 10.1002/jcp.22559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Stolen TO, Hoydal MA, Kemi OJ, Catalucci D, Ceci M, Aasum E, Larsen T, Rolim N, Condorelli G, Smith GL, Wisloff U. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ Res. 2009;105:527–36. doi: 10.1161/CIRCRESAHA.109.199810. [DOI] [PubMed] [Google Scholar]
- 198.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE, Gorelik J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A. 2009;106:6854–9. doi: 10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Schaper J, Froede R, Hein S, Buck A, Hashizume H, Speiser B, Friedl A, Bleese N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation. 1991;83:504–14. doi: 10.1161/01.cir.83.2.504. [DOI] [PubMed] [Google Scholar]
- 200.Maron BJ, Ferrans VJ, Roberts WC. Ultrastructural features of degenerated cardiac muscle cells in patients with cardiac hypertrophy. Am J Pathol. 1975;79:387–434. [PMC free article] [PubMed] [Google Scholar]
- 201.Kaprielian RR, Stevenson S, Rothery SM, Cullen MJ, Severs NJ. Distinct patterns of dystrophin organization in myocyte sarcolemma and transverse tubules of normal and diseased human myocardium. Circulation. 2000;101:2586–94. doi: 10.1161/01.cir.101.22.2586. [DOI] [PubMed] [Google Scholar]
- 202.Sachse FB, Torres NS, Savio-Galimberti E, Aiba T, Kass DA, Tomaselli GF, Bridge JH. Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ Res. 2012;110:588–97. doi: 10.1161/CIRCRESAHA.111.257428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Prestle J, Janssen PM, Janssen AP, Zeitz O, Lehnart SE, Bruce L, Smith GL, Hasenfuss G. Overexpression of FK506-binding protein FKBP12. in cardiomyocytes reduces ryanodine receptor-mediated Ca2+ leak from the sarcoplasmic reticulum and increases contractility. Circ Res. 2001;88:188–94. doi: 10.1161/01.res.88.2.188. [DOI] [PubMed] [Google Scholar]
- 204.He J, Conklin MW, Foell JD, Wolff MR, Haworth RA, Coronado R, Kamp TJ. Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc Res. 2001;49:298–307. doi: 10.1016/s0008-6363(00)00256-x. [DOI] [PubMed] [Google Scholar]
- 205.Balijepalli RC, Lokuta AJ, Maertz NA, Buck JM, Haworth RA, Valdivia HH, Kamp TJ. Depletion of T-tubules and specific subcellular changes in sarcolemmal proteins in tachycardia-induced heart failure. Cardiovasc Res. 2003;59:67–77. doi: 10.1016/s0008-6363(03)00325-0. [DOI] [PubMed] [Google Scholar]
- 206.Sacconi L, Ferrantini C, Lotti J, Coppini R, Yan P, Loew LM, Tesi C, Cerbai E, Poggesi C, Pavone FS. Action potential propagation in transverse-axial tubular system is impaired in heart failure. Proc Natl Acad Sci U S A. 2012;109:5815–9. doi: 10.1073/pnas.1120188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Orchard CH, Pasek M, Brette F. The role of mammalian cardiac t-tubules in excitation-contraction coupling experimental and computational approaches. Exp Physiol. 2009;94:509–19. doi: 10.1113/expphysiol.2008.043984. [DOI] [PubMed] [Google Scholar]
- 208.Pasek M, Simurda J, Christe G, Orchard CH. Modelling the cardiac transverse-axial tubular system. Prog Biophys Mol Biol. 2008;96:226–43. doi: 10.1016/j.pbiomolbio.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 209.Brette F, Salle L, Orchard CH. Quantification of calcium entry at the T-tubules and surface membrane in rat ventricular myocytes. Biophys J. 2006;90:381–9. doi: 10.1529/biophysj.105.069013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Biesmans L, Macquaide N, Heinzel FR, Bito V, Smith GL, Sipido KR. Subcellular heterogeneity of ryanodine receptor properties in ventricular myocytes with low T-tubule density. PLoS One. 2011;6:e25100. doi: 10.1371/journal.pone.0025100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Meethal SV, Potter KT, Redon D, Munoz-del-Rio A, Kamp TJ, Valdivia HH, Haworth RA. Structure-function relationships of Ca spark activity in normal and failing cardiac myocytes as revealed by flash photography. Cell Calcium. 2007;41:123–34. doi: 10.1016/j.ceca.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 212.Chen B, Li Y, Jiang S, Xie YP, Guo A, Kutschke W, Zimmerman K, Weiss RM, Miller FJ, Anderson ME, Song LS. beta-Adrenergic receptor antagonists ameliorate myocyte T-tubule remodeling following myocardial infarction. FASEB J. 2012;26:2531–7. doi: 10.1096/fj.11-199505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.McNary TG, Bridge JH, Sachse FB. Strain transfer in ventricular cardiomyocytes to their transverse tubular system revealed by scanning confocal microscopy. Biphys J. 2011;100:L53–5. doi: 10.1016/j.bpj.2011.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.McNary TG, Spitzer KW, Holloway H, Bridge JH, Kohl P, Sachse FB. Mechanical modulation of the transverse tubular system of ventricular cardiomyocytes. Prog Biophys Mol Biol. 2012;110:218–25. doi: 10.1016/j.pbiomolbio.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 216.Xu M, Zhou P, Xu SM, Liu Y, Feng X, Bai SH, Bai Y, Hao XM, Han Q, Zhang Y, Wang SQ. Intermolecular failure of L-type Ca2+ channel and ryanodine receptor signaling in hypertrophy. PLoS Biol. 2007;5:e21–0. doi: 10.1371/journal.pbio.0050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Landstrom AP, Kellen CA, Dixit SS, van Oort RJ, Garbino A, Weisleder N, Ma J, Wehrens XH, Ackerman MJ. Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ Heart Fail. 2011;4:214–23. doi: 10.1161/CIRCHEARTFAILURE.110.958694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Minamisawa S, Oshikawa J, Takeshima H, Hoshijima M, Wang Y, Chien KR, Ishikawa Y, Matsuoka R. Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem Biophys Res Commun. 2004;325:852–6. doi: 10.1016/j.bbrc.2004.10.107. [DOI] [PubMed] [Google Scholar]
- 219.Galbiati F, Engelman JA, Volonte D, Zhang XL, Minetti C, Li M, Hou H Jr, Kneitz B, Edelmann W, Lisanti MP. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem. 2001;276:21425–33. doi: 10.1074/jbc.M100828200. [DOI] [PubMed] [Google Scholar]
- 220.Zobel C, Rana OR, Saygili E, Bolck B, Saygili E, Diedrichs H, Reuter H, Frank K, Muller-Ehmsen J, Pfitzer G, Schwinger RH. Mechanisms of Ca2+-dependent calcineurin activation in mechanical stretch-induced hypertrophy. Cardiology. 2007;107:281–90. doi: 10.1159/000099063. [DOI] [PubMed] [Google Scholar]
- 221.Xu M, Wu HD, Li RC, Zhang HB, Wang M, Tao J, Feng XH, Guo YB, Li SF, Lai ST, Zhou P, Li LL, Yang HQ, Luo GZ, Bai Y, Xi JJ, Gao W, Han QD, Zhang YY, Wang XJ, Meng X, Wang SQ. Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circ Res. 2012;111:837–41. doi: 10.1161/CIRCRESAHA.112.277418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Li RC, Tao J, Guo YB, Wu HD, Liu RF, Bai Y, Lv ZZ, Luo GZ, Li LL, Wang M, Yang HQ, Gao W, Han QD, Zhang YY, Wang XJ, Xu M, Wang SQ. In vivo suppression of microRNA-24 prevents the transition toward decompensated hypertrophy in aortic-constricted mice. Circ Res. 2013;112:601–5. doi: 10.1161/CIRCRESAHA.112.300806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Hong TT, Smyth JW, Gao D, Chu KY, Vogan JM, Fong TS, Jensen BC, Colecraft HM, Shaw RM. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 2010;8:e1000312. doi: 10.1371/journal.pbio.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lee E, Marcucci M, Daniell L, Pypaert M, Weisz OA, Ochoa GC, Farsad K, Wenk MR, De Camilli P. Amphiphysin 2 (Bin1):and T-tubule biogenesis in muscle. Science. 2002;297:1193–6. doi: 10.1126/science.1071362. [DOI] [PubMed] [Google Scholar]
- 225.Hong TT, Smyth JW, Chu KY, Vogan JM, Fong TS, Jensen BC, Fang K, Halushka MK, Russell SD, Colecraft H, Hoopes CW, Ocorr K, Chi NC, Shaw RM. BIN1 is reduced and Cav1. trafficking is impaired in human failing cardiomyocytes. Heart Rhythm. 2012;9:812–20. doi: 10.1016/j.hrthm.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Mork HK, Sjaastad I, Sande JB, Periasamy M, Sejersted OM, Louch WE. Increased cardiomyocyte function and Ca2+ transients in mice during early congestive heart failure. J Mol Cell Cardiol. 2007;43:177–86. doi: 10.1016/j.yjmcc.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 227.Li L, Louch WE, Niederer SA, Andersson KB, Christensen G, Sejersted OM, Smith NP. Calcium dynamics in the ventricular myocytes of SERCA2 knockout mice A modeling study. Biophys J. 2011;100:322–31. doi: 10.1016/j.bpj.2010.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]