

Abstract
Purpose
Amphotericin B (AmB) is an effective anti-fungal and anti-leishmanial agent. However, AmB has low oral bioavailability (0.3%) and adverse effects (e.g., nephrotoxicity). The objectives of this study were to improve the oral bioavailability by entrapping AmB in pegylated (PEG) poly lactide co glycolide copolymer (PLGA–PEG) nanoparticles (NPs). The feasibility of different surfactants and stabilizers on the mean particle size (MPS) and entrapment efficiency were also investigated.
Materials and methods
NPs of AmB were prepared by a modified emulsification diffusion method employing a vitamin E derivative as a stabilizer. Physicochemical properties and particle size characterization were evaluated using Fourier Transform Infra-Red spectroscopy (FTIR), differential scanning calorimetry, scanning electron microscopy and transmission electron microscopy. Moreover, in vitro dissolution profiles were performed for all formulated AmB NPs.
Results
MPS of the prepared spherical particles of AmB ranged from 26.4 ± 2.9 to 1068 ± 489.8 nm. An increased stirring rate favored AmB NPs with a smaller MPS. There was a significant reduction in MPS, drug content and drug release, when AmB NPs were prepared using the diblock polymer PLGA–PEG with 15% PEG. Addition of three emulsifying agents poly vinyl pyrrolidone (PVP), Vitamin E (TPGS) and pluronic F-68 to AmB formulations led to a significant reduction in particle size and increase in drug entrapment efficiency (DEE) compared to addition of PVP alone. FTIR spectroscopy demonstrated a successful loading of AmB to pegylated PLGA–PEG copolymers. PLGA–PEG copolymer entrapment efficiency of AmB was increased up to 56.7%, with 92.7% drug yield. After a slow initial release, between 20% and 54% of AmB was released in vitro within 24 h phosphate buffer containing 2% sodium deoxycholate and were best fit Korsmeyer–Peppas model. In conclusion, PLGA–PEG diblock copolymer with 15% PEG produced a significant reduction (>70%) in MPS with highest drug content. The percentage of PEG in the copolymer and the surfactant/stabilizer used had a direct effect on AmB release in vitro, entrapment efficiency and MPS. These developed formulations are feasible, effective and improved alternatives to other carriers for oral delivery of AmB.
Keywords: Amphotericin B, Oral delivery, Nanoparticles, Emulsification–diffusion, PLGA–PEG copolymer
1. Introduction
Amphotericin B (AmB) is a macrocyclic polyee antibiotic (Fig. 1). A deoxycholate-soluble salt of AmB (Fungizone®) is marketed for use as an intravenous infusion (IVI) formulation and has been the gold standard drug treatment for systemic fungal infections since 1953 (Dutcher, 1968). This parenteral formulation is usually associated with several adverse effects (AE) including fever, chilling, vomiting, headache and nausea during administration (80% of the patients) and nephrotoxicity (30% of the patients) after dosing (Torrado et al., 2008, Van de Ven et al., 2012).
Figure 1.
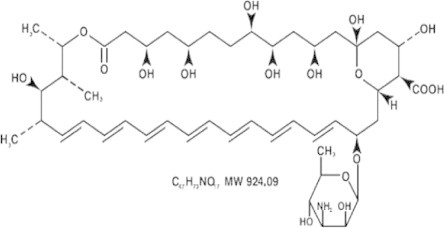
Structure of AmB (a), 3-D model of AmB (Lemke et al., 2005).
AmB possesses both hydrophobic (polyene hydrocarbon chain) and hydrophilic (polyhydroxyl chain) domains as shown in Fig. 1 (Lemke et al., 2005). This amphoteric nature is responsible for its poor solubility in both aqueous and organic solvents. It is classified (Biopharmaceutical Classification System (BCS)) as a class IV compound (Ménez et al., 2007) with limited solubility and permeability properties due to its high molecular weight of 924 Da, leading to a low bioavailability if given orally 0.3% (Ouellette et al., 2004).
Different tactics have been investigated to reduce AmB associated nephrotoxicity and other AE during Fungizone® administration. These include saline loading, alternative day dosing and dose reduction, all with limited success. In the early 1990s, new generations of AmB formulations were developed by replacing sodium deoxycholate with either phospholipids (Abelcet®) or cholesterol (Amphocil®) or both (AmBisome®) (Adler-Moore and Proffitt, 2008, Torrado et al., 2008). These AmB formulations have shown improved therapeutic indexes in comparison with Fungizone®. However, the high cost of these formulations (12–40 times more expensive than Fungizone®), the need of hospitalization for parenteral administration and the acute AE have limited the widespread use of these new formulations (Adler-Moore and Proffitt, 2008, Ibrahim et al., 2012).
Since oral preparations would reduce the requirement for hospitalization during administration, and therefore the costs associated with its use, the development of a safe oral formulation of AmB has been investigated.
For oral administration, the drug must be able to cross the gastrointestinal (GI) epithelium. Several formulations have been described for AmB oral delivery. These include lipid-based formulations, either a self-emulsifying drug delivery system (SEDDS) or cochleates (multilayer structure consisting of a continuous solid lipid bilayer sheet), nanosuspensions solid lipid nanoparticles (SLNs) polymeric NPs and polymeric lipid hybrid NPs (Patel et al., 2013).
In most cases, these formulations failed to increase the absorption of orally administered AmB. However, the cochleate formulation of AmB (Biodelivery Sciences, Inc., USA) was reported to successfully facilitate the oral absorption, and bioavailability of AmB (Ibrahim et al., 2012) and provided protection from both gastric acid degradation and efflux via P-glycoprotein (P-gp). After showing a favorable outcome of AmB formulation during phase I trial (February 2009) (Ibrahim et al., 2012), the manufacturer has ascertained that further AmB clinical trials would be underway. Another oral lipid-based AmB formulation is currently in development (iCo Therapeutics Inc., Canada), showing a 50-fold increase in AmB solubility over its non-lipid equivalent formulation (Ibrahim et al., 2012). This formulation and the cochleate formulation were reported to result in reduced nephrotoxicity compared to AmB intravenous infusion (Thornton and Wasan, 2009, Wasan et al., 2009). However, neither of these two formulations have undergone full clinical trials and to date, it is unclear whether these formulations of AmB will lead to commercial pharmaceuticals (Yang et al., 2012).
During the past few decades there has been an increasing interest in the development of polymeric biodegradable NPs, either natural or synthetic, for effective drug delivery (Barratt, 2003, Ma et al., 2008). The polyesters are the most widely used polymers in this class because of their biocompatibility, non-immunogenicity and low toxicity. Typical polymeric biodegradable NPs include poly (lactic acid) (PLA), poly (glycolic acid) (PGA), polycaprolactone (PCL) and their copolymers such as poly (lactide)-co-(glycolide) (PLGA). They are approved for use in vivo by the Food and Drug Administration (FDA) (Parveen and Sahoo, 2008).
One of the major problems with the polymeric drug carrier NPs is that they are rapidly eliminated from the blood stream through phagocytosis by the reticuloendothelial system (RES) (Stolnik et al., 1995, Owens and Peppas, 2006). Avoidance of phagocytosis can be achieved through pegylation using Poly(ethylene glycol) (PEG), i.e., formation of the so-called “stealth” NPs, which abrogate the rapid uptake by phagocytic cells. PEG has been the most common hydrophilic blocking agent used since it is readily hydrated, it has a high degree of conformational flexibility and is biocompatible. The inner hydrophobic block has shown greater variability depending on the system being studied (Jee et al., 2012).
AmB has been conjugated to either PEG (Conover et al., 2003) or PLGA alone (Fig. 2) (Nahar and Jain, 2009, Van de Ven et al., 2012), which resulted in an increased water solubility, increased efficacy and less toxicity when compared to the commercial AmB products.
Figure 2.

The chemical structure poly[(d,l-lactide-co-glycolide)-co-PEG] diblock.
Block copolymers comprise two or more homopolymer subunits linked by covalent bonds. Block copolymers with two or three distinct blocks are called diblock copolymers and triblock copolymers, respectively. Amphiphilic block copolymers have the ability to form various types of NPs referred to as micelles, nanocapsules, nanospheres or core-corona NPs (Chen et al., 2011). Block copolymer micelles are water-soluble, biocompatible nano-containers in the size range of 10–100 nm characterized by a core-corona architecture in which the core serves as a reservoir for the incorporation of poorly water-soluble drugs, while the hydrophilic corona provides a protective interface between the core and the external medium (Gaucher et al., 2010).
In this study, recently introduced copolymers (PLGA–PEG copolymer “Resomer® PEG” copolymers), that shown to be biodegradable and biocompatible (Sinha et al., 2004) were used. The development of PLGA–PEG copolymer NPs as an AmB carrier for oral delivery formulations with improved bioavailability and lower toxicity was investigated. Phase I, which is presented here, has been focused on the in vitro optimization of the NPs formulations. Screening processes have utilized differential scanning calorimetry, particle size determination using Zetasizer, surface morphology using scanning electron microscopy and transmission electron microscopy as well as Fourier Transform Infra-Red (FTIR) characterization. NP yield, drug loading and in vitro AmB release have been studied.
2. Materials and methods
2.1. Polymer and chemicals
Amphotericin B (AmB) (99.8% purity) and Nicardipine Hydrochloride (98% purity), the internal standard (IS), were purchased from Sigma–Aldrich (St. Louis, MO, USA different types of Poly(d,l-lactide-co-glycolide) (Table 1) were supplied by Boehringer Ingelheim (Ingelheim, Germany). Miglyol®-812 neutral oil was obtained from Sasol (GmbH, Germany,). Vitamin E (TPGS-NF) grade was purchased from Peboc (Eastman, UK). Pluronic F68 Prilled was obtained from Ruger Chemical Co. Inc. (Irvington, New Jersey, USA). All other reagents and chemicals were HPLC analytical grade, and were used as received. Water was deionized and purified by a Milli-Q Reagent Grade water system from Millipore Corporation (Bedford, MX, USA).
Table 1.
Composition of the use pegylated PLGA–PEG copolymers types.
| Symbol | Name | Polymer type | Composition | Lactic to glycolic acid ratio | Content of PEG (%) | Molecular weight (Dalton) |
|---|---|---|---|---|---|---|
| A | RGPd 50105 | Diblock | PLGA–PEG 6000 | 1:1 | 10 | 5000 |
| B | RGPt 50106 | Triblock | PLGA–PEG 6000-PLGA | 1:1 | 10 | 6000 |
| C | RGPd 50155 | Diblock | PLGA–PEG 6000 | 1:1 | 15 | 6000 |
| D | RGPd 5055 | Diblock | PLGA–PEG 6000 | 1:1 | 5 | 5000 |
| E7 | R 203 H | Monoblock | Poly(d,l-lactide) | – | – | 18,000–28,000 |
2.2. Preparation of Amphotericin B loaded-nanoparticles
This study employed a modified emulsification–diffusion technique (Mora-Huertas et al., 2010). Briefly, the method requires the preparation of three phases: organic, aqueous and dilution phases. The organic phase was prepared by dissolving 20 mg of AmB in 5 ml of the organic phase (acetone/dichloromethane; (ACT/DCM: 6/1)) containing aliquot of PLGA–PEG copolymer and 200 μl of 5 N HCl. The composition of the studied AmB-NPs formulations is listed in Table 2. All the vehicles used in this study were protected from light, either by using amber containers or through covering containers with aluminum foil. Miglyol was added to the organic phase, then sonicated for 5 min and stirred until a clear solution was obtained using a magnetic stirrer. The aqueous phase was prepared by adding two types of stabilizer; polyvinyl pyrrolidone (PVP) and pluronic (F-68), TPGS was added also. The organic solution was slowly added (drop wise, for five minutes) to the aqueous phase using a high speed stirrer (Ultra-Turrax T25, IKA Labotecnik, Staufen, Germany). The organic emulsion was then added to 200 ml of distilled water to induce diffusion of the organic phase into the continuous phase, with mechanical stirring at 60 °C for 3 h. Each batch was prepared in triplicate. All formulations were freeze-dried under vacuum (1.25 mBar) at −52 °C (Labconco-Free zone 4.5, Kansas, US) until complete dryness. Formulated dry NPs were stored in the freezer (−20 °C) prior to further investigation.
Table 2.
Composition of AmB nanoparticles produced by emulsification–diffusion method using 24,000 rpm stirring rate (n = 3).
| Batch No. | Copolymer type | Phases |
|||
|---|---|---|---|---|---|
| Organic phasea |
Aqueous phaseb |
||||
| Drug (mg) | Miglyol (%) | PVA (%) | TPGS (%) | ||
| A3 | Diblock, 10% PEG RGPd 50105 | 20 | – | 1 | – |
| A4 | 20 | 2.5 | 1 | – | |
| A5 | 20 | 2.5 | 1 | 5 | |
| A6 | 20 | 2.5 | 4 | 5 | |
| B3 | Triblock, 10% PEG RGPt 50106 | 20 | – | 1 | – |
| B4 | 20 | 2.5 | 1 | – | |
| B5 | 20 | 2.5 | 1 | 5 | |
| B6 | 20 | 2.5 | 4 | 5 | |
| C3 | Diblock, 15% PEG RGPd 50155 | 20 | – | 1 | – |
| C4 | 20 | 2.5 | 1 | – | |
| C5 | 20 | 2.5 | 1 | 5 | |
| C6 | 20 | 2.5 | 4 | 5 | |
| C7 | 40 | 2.5 | 4 | 5 | |
| E7 | R 203 H | 40 | 2.5 | 4 | 5 |
| D3 | Diblock, 5% PEG RGPd 5055 | 20 | – | 1 | – |
| D4 | 20 | 2.5 | 1 | – | |
| D5 | 20 | 2.5 | 1 | 5 | |
| D6 | 20 | 2.5 | 4 | 5 | |
Each formula contains 200 mg polymer and 200 μl of 2 NHCL.
Contains 0.5% pluronic-68.
For optimizing the formulation process, the effect of various factors on the AmB-NPs formulations were investigated. The effect of the stirrer speed (8000, 13,500 and 24,000 rpm) on four different PLGA–PEG copolymer NPs were studied. The effect of PVP concentration, miglyol®-812 and TPGS were also studied.
Furthermore, in this study polymer R 203 H (Poly d,l-lactide) was included to assess the effect of pegylated copolymer.
2.3. Physicochemical characterization of the nanoparticles
2.3.1. Particle size analysis and polydispersity index (PDI)
The cumulative mean of particle size and polydispersity index (PDI) were determined by dynamic light scattering using Zetasizer (Malvern Instruments, Worcestershire, UK). Typically, after freeze drying of formulated batches of AmB-NPs, the dried powder samples were suspended in deionized water and sonicated before measurement. The obtained homogeneous suspension was examined to determine the mean diameter, size distribution and polydispersity index. Viscosity and the refractive index of the continuous phase were set to those specific to water.
2.3.2. Fourier Transform Infra-Red Spectroscopy
The Fourier Transform Infra-Red (FTIR) spectra of each pure component as well as the prepared freeze-dried AmB-NPs formulations were analyzed using a FTIR spectrophotometer (Perkin–Elmer-Spectrum 1600, Norwalk, USA). The FTIR measurements were performed at wave numbers ranging from 450 to 4000 cm−1 at constant rate of 10 °C/min under an argon purge. AmB samples were diluted with KBr powder. The IR spectra were obtained in a KBr disk at ambient temperature.
2.3.3. Morphology of the nanoparticles
The dry samples of each prepared AmB-NPs formulation were mounted onto metal stubs using double-sided adhesive tape. The stubs were then vacuum coated with gold using fine coat ion sputter under reduced pressure to render them electrically conductive prior to examination by SEM (SEM, JEOL JSM-6060 LV, Jeol Ltd., Tokyo, Japan). The accelerating voltage was kept constant at 15 kV under an argon atmosphere.
Transmission electron microscopy (TEM, JEM-1011, Jeol Ltd., Tokyo, Japan) was used to examine the morphology of each prepared AmB-NPs formulation. The aqueous (water) dispersion of the freeze-dried NPs were placed over a 400 mesh copper grid covered with carbon film.
2.3.4. Differential scanning calorimetry (DSC)
Samples of AmB loaded nanoparticles were scanned to determine the thermal properties of the AmB in its NP formulations. Approximately 2 mg samples were accurately weighed and placed into standard aluminum pans, which were hermetically sealed. An empty pan was used as a reference. The transition temperature (Tc) and enthalpy of transition (ΔH) were determined from the thermogram, generated by a differential scanning calorimeter (Perkin Elmer DSC7, Perkin Elmer Ltd., Norwalk, USA). The heating rate was 10 °C/min from 30 to 200 °C, with a closed-pan system under a stream of argon gas flow, after which the system was cooled down at the same rate from 200 to 30 °C. The apparatus was calibrated with indium 99.99%. In addition, DSC scans were performed for each pure component of the formulations. All the samples were freeze-dried before the measurements.
2.3.5. Determination of drug entrapment efficiency and nanoparticles yield
Drug entrapment efficiency (DEE) of AmB in the NPs was determined directly by measuring the amount of AmB entrapped in the NPs. Briefly, a 10 mg sample of the freeze-dried NPs was dissolved in 5 ml of DCM: ACN (1:6 v/v), 200 μl of 5 N HCl and the volume was made up to10 ml with methanol:water (75:25 v/v). An aliquot of 1.25 ml of the stock solution was mixed with 1 ml of nicardipine (IS, 10 μg/ml) and the volume was 5 ml with methanol: water (75:25 v/v). Absorbance at 382 nm of diluted stock solution was determined and the AmB concentration was calculated using a validated high performance liquid chromatography (HPLC) method as described below.
DEE (%w/w) = 100 * (Mass of recovered AmB-NPs/Initial mass of drug used in formulation)
Yield (% w/w) = (mass of AmB-NPs/total mass of polymer and drug added) × 100
2.3.6. Drug release study
The quantitative in vitro release test was performed using US Pharmacopeia XXXII dissolution apparatus 2 (paddle). The dissolution was carried out in 500 ml of phosphate buffer containing 2% sodium deoxycholate (pH 6.8 ± 0.1) at 50 rpm and the temperature was maintained at 37 ± 0.5 °C. A sample of AmB NPs equivalent to 5.0 mg of AmB was placed on the surface of the dissolution medium. At appropriate time intervals (0.25, 0.5, 1, 2, 3, 4, 5, 6 and 24 h), 2.0 ml samples were withdrawn from each vessel, mixed with 1.0 ml of IS (10 μg/ml) and 5.0 ml with methanol:water (75:25). The solution was filtered through a 0.22 μm Millipore membrane filter and analyzed using a validated HPLC assay as described below. The volume was replaced each time with 2 ml of fresh medium kept at 37 ± 0.5 °C to maintain a sink condition.
2.3.7. HPLC assay for in vitro study
The concentration of AmB was measured using a Waters HPLC system equipped with a Waters 484 variable UV absorbance detector and a Waters 717 plus autosampler. Waters 515 solvent delivery system was used to operate the gradient flow through a Novapak C18 column (3.9 × 150 mm) packed with 5 μm spherical particles. The mobile phase consisted of acetonitrile (40.5%): methanol (4.5%) acetic acid (0.2%) and 0.1% triethylamine. Degassing was achieved by filtration through a 0.22 μm Millipore membrane filter and sonication for 10 min. The injection volume was 75–100 μl and detection was at 382 nm. The mobile phase flow rate was 1.2 ml/min and the run time was 5.0 min. Data were collected with an Empower Pro Chromatography Manager Data Collection System. The HPLC system was operated at ambient temperature. A daily standard calibration curve (6 standards ranging from 0.5 to 10 μg/ml) was performed to determine the unknown AmB concentration for DEE and release. The assay was fully validated for accuracy and precision according to USP guidelines (US Food and Drug Administration (2006)). The percent intra-day relative standard deviation (R.S.D.%) was <5%, and the inter-day deviation (R.S.D.%) was <6% at three different days (P > 0.05). The mean retention times of AmB and IS were about 2.8 and 4.2 min, respectively. The detection limit was 200 ng/ml.
2.3.8. Drug release kinetics
Drug release kinetics of AmB formulations were analyzed using various dissolution models including; zero, first, Higuchi and Korsmeyer–Peppas order. Release profile data were processed and plotted according to the equations of different models followed by regression analyses. The criterion for selecting the most appropriate model was based on best goodness-of-fit (R2 values). The slope of each plot and its release rate constant for each particular model were used to describe the release rate mechanism.
2.4. Data and statistical analysis
All in vitro results were expressed as mean ± SD of at least three replicates. The HPLC results of AmB were calculated using linear regression without weighting, according to the equation: Y = 0.041 + 0.379X, where, Y is the area under the peak (AUP) ratio of the drug to the internal standard, a is the intercept, b is the slope, and X is the concentration of AmB. The R.S.D.% was calculated for all values. The Student’s t-test was used to examine the concentration difference at each day and one-way analysis of variance (ANOVA) was employed to evaluate the reproducibility of the assay and the dissolution from batch to batch using IBMSPSS Statistics 20. The level of confidence was 95%.
3. Results
3.1. Optimization of the AmB-NPs formulation process
A particulate method of NPs preparation is usually employed considering the physicochemical properties of the drug to be entrapped and route of administration to be given. For hydrophobic drugs, nano precipitation (Bilati et al., 2005) or an emulsion–diffusion method is best suited (Dillen et al., 2006, Italia et al., 2009, Mora-Huertas et al., 2010).
Selection of the proper organic solvent was the most challenging issue for ing AmB acetone/dichloromethane (6:1) was found to be the most suitable agent for the drug and PLGA–PEG copolymer, with the addition of 200 μl of 5 N HCl, to ensure complete solubilization of AmB.
3.2. Factors affecting the particle size distribution
3.2.1. Effect of shearing rate
The relationship between stirring speed and mean particle size (MPS) is shown in Table 3. Different speeds of mechanical stirring – 8000, 13,500 and 24,000 rpm – were tested. The NPs size was inversely proportional to the stirring speed. A significant reduction in the NPs size (from 1068.1 ± 489.8 to 400.2 ± 62.1 nm) was observed on increasing the shearing rate from 8000 to 24,000 rpm for formulations A1 to A3 using RGPd 50105 polymer. The same trend was observed using different polymers, therefore throughout the study, the 24,000 rpm was used as the shearing rate for the nanoparticles preparations.
Table 3.
Influence of shearing rate on the mean particle size and poly dispersity index of AmB-NPs prepared by emulsification–diffusion method.
| Batch number | Particle size (nm) ± SD | Polydispersity index ± SD | Stirring speed rpm | DEE% | Yield% |
|---|---|---|---|---|---|
| A1 | 1,068.1 ± 489.8 | 0.46 ± 0.1 | 8000 | 18.5 ± 3.3 | 67.3 ± 2.1 |
| A2 | 451.2 ± 84.2 | 0.46 ± 0.1 | 13,500 | 16.4 ± 3.4 | 73.7 ± 1.2 |
| A3 | 400.2 ± 62.1 | 0.64 ± 0.3 | 24,000 | 23.3 ± 7.3 | 75.0 ± 1.4 |
3.2.2. Effect of surfactant and emulsifier used
In optimization trials, the main basic constituents of AmB-NPs formulations were AmB (20 mg), PVP (1%) and F-68 (0.5%). The different composition of NPs investigated during the study and their effect on the DEE%, yield% and the size of the NPs are summarized in Table 3, Table 4. It was noticed that all the produced AmB NPs batches have homogenous distribution with PDI of <0.5, were in the nanometer size range with narrow size distribution and without polymer flakes or visible oil droplets.
Table 4.
Influence of different composition parameters on the mean particle size and poly dispersity index of AmB-NPs prepared by emulsification–diffusion method.
| Batch number | Particle size (nm) ± SD | Poly dispersity index ± SD | DEE% | Yield% |
|---|---|---|---|---|
| A3 | 400.2 ± 62.1 | 0.64 ± 0.3 | 23.3 ± 7.3 | 75.0 ± 1.4 |
| A4 | 126.8 ± 27.3 | 0.53 ± 0.2 | 23.9 ± 1.1 | 81.3 ± 2.2 |
| A5 | 105.2 ± 9.3 | 0.26 ± 0.1 | 24.8 ± 4.1 | 82.5 ± 1.3 |
| A6 | 93.3 ± 5.7 | 0.51 ± 0.3 | 37.8 ± 7.5 | 83.7 ± 1.4 |
| B3 | 107.4 ± 61.9 | 0.53 ± 0.3 | 20.4 ± 1.4 | 74.8 ± 1.5 |
| B4 | 97.4 ± 10.2 | 0.39 ± 0.1 | 21.2 ± 7.1 | 77.1 ± 2.3 |
| B5 | 69.4 ± 14.7 | 0.24 ± 0.1 | 36.9 ± 2.4 | 78.9 ± 2.4 |
| B6 | 55.4 ± 5.9 | 0.26 ± 0.1 | 37.5 ± 1.7 | 80.9 ± 1.7 |
| C3 | 57.2 ± 7.5 | 0.25 ± 0.1 | 25.9 ± 0.5 | 82.5 ± 1.1 |
| C4 | 36.8 ± 7.6 | 0.28 ± 0.1 | 29.9 ± 6.2 | 83.8 ± 1.2 |
| C5 | 27.0 ± 5.6 | 0.34 ± 0.1 | 40.6 ± 10.9 | 87.5 ± 1.1 |
| C6 | 23.8 ± 4.8 | 0.31 ± 0.0 | 48.3 ± 4.2 | 90.6 ± 0.5 |
| C7 | 25.3 ± 2.7 | 0.31 ± 0.0 | 56.5 ± 3.9 | 93.2 ± 0.5 |
| E7 | 539.9 ± 51.1 | 0.35 ± 0.1 | 27.2 ± 3.2 | 67.4 ± 0.8 |
| D3 | 515.6 ± 30.7 | 0.37 ± 0.0 | 24.3 ± 2.3 | 85.5 ± 1.4 |
| D4 | 418.8 ± 28.2 | 0.32 ± 0.1 | 24.4 ± 2.7 | 86.9 ± 2.2 |
| D5 | 344.4 ± 38.4 | 0.26 ± 0.10 | 27.4 ± 2.5 | 88.1 ± 1.7 |
| D6 | 318.4 ± 36.8 | 0.36 ± 0.10 | 39.4 ± 9.2 | 89.6 ± 1.0 |
A: RGPd 50105, B: RGPt 50106, C: RGPd 50155, D: RGPd 5055.
Four PLGA–PEG copolymer were investigated throughout the study RGPd 50105 (A), RGPt 50106 (B), RGPd 50155 (C) and RGPd 5055 (D). Note, the letter indicates the copolymer used, while numbers were used to indicate the composition of the formulation; the same composition will have same number.
For basic component formulations, batches containing copolymer C had the lowest MPS (Table 4) among the other tested formulations. Under the same condition copolymer D had the highest MPS within the tested batches.
The addition of miglyol-812 (2.5%) to the basic component of each copolymer (Batches A4, B4, C4, D4) during the preparation process (Table 4), led to a significant reduction in MPS (P < 0.05) in all batches. The size reduction was in the direction of A(68%) > C (35%) > D (18.9%) > B (8.5%). Therefore, miglyol-812 (2.5%) was added to the basic composition of the AmB-NPs starting from batches with number 5.
The same trend was observed on adding TPGS (5% w/v) causing further reduction in MPS of A and D (16.6% and 17.9%, respectively) and B and C (27.8% and 27%, respectively), TPGS was also added to the basic component of the AmB-NPs formulation starting from batches with number 6.
Moreover, increasing the concentration of PVP from 1% to 4%, the MPS of AmB-NPs formulation was also decreased the MPS in this order B, A, C, D (21.5%, 11.5%, 11.1% and 7.5% respectively).
Interestingly, changing the PLGA–PEG copolymer to 203H polymer (non-pegylated) formulation E7, caused a significant increase in the MPS > 95%.It should be mentioned that no more than 40 mg of AmB could be incorporated in these formulations (C7), with no significant effect on MPS. Therefore, formulations A6, B6, C6, C7, E7 and D6 with the lowest MPS were selected for the next in vitro characterizations steps.
3.3. Drug encapsulation efficiency and nanoparticles yield
The DEE of the AmB NPs was found to be inversely proportional to the MPS (Table 4). The minimum DEE for all formulated batches was 20% (B3) while the maximum encapsulation was 56.5% (C7) which is considered suitable for delivering a therapeutically active dose. Meanwhile, for the best selected formulations, the DEE was in the following order 56.5% > 48% > 39% > 37% > 32%in corresponding to C7, C6, D6, A6 and B6, respectively. The non-pegylated polymer (E7) shows a DEE of 27%. However the highest yield of AmB-NPs was 93% (C7) while the lowest one was 74% (B6).
3.4. Poly disparity index
PDI values, range from 0 to 1; a higher value indicates a less homogenous NP size distribution (Galindo-Rodriguez et al., 2004). Table 3, Table 4 indicate that PDI in all batches is almost <0.5; which means that all batches had an homogenous distribution of nanoparticles in water.
3.5. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM)
Representative SEM images of AmB-NPs selected formulations (Fig. 3, A6, B6, C6, C7, E7 and D6), demonstrates aspherical and discrete NPs of <1000 nm which consistent with the Zeta sizer measurement of MPS (Table 3, Table 4) and the TEM analyses of the freeze-dried AmB-NPs (Fig. 4, A6, B6, C6, C7, E7 and D6).
Figure 3.

SEM images for AmB-NPs containing 20 mg AmB and prepared with RGPd 50105 copolymer (A6); RGPt 50106 copolymer (B6); RGPd 50155 copolymer (C6); GPd 5055 copolymer (D6) and AmB-NPs containing 40 mg AmB and prepared with RGPd 50155 (C7) and R-203-H polymer (E7) batches.
Figure 4.

TEM images for AmB-NPs containing 20 mg AmB and prepared with RGPd 50105 copolymer (A6); RGPt 50106 copolymer (B6); RGPd 50155 copolymer (C6); GPd 5055 copolymer (D6) and AmB-NPs containing 40 mg AmB and prepared with RGPd 50155 (C7) and R-203-H polymer (E7) batches.
3.6. Fourier Transform Infra-Red (FTIR) analysis
FTIR was used to identify any change in the drug in formulations compared to the pure drug. The important bands of AmB are listed in Table 5. Fig. 5, displays the FTIR spectra for AmB NPs prepared by RGPd 50155 copolymer (C). It can be observed that the FTIR spectrum for the pure AmB is different to that of the AmB-NPs, since pure AmB crystals show characteristic sharp bands at 1691, 1557 and 1009 cm−1, due to a C O stretch band; NH2 in-plane bend, polyene C C stretch band and C–H bend out of plane bend (trans polyene), respectively (Asher and Schwartzman, 1977, Nahar and Jain, 2009). In case of the freeze-dried batches of AmB-NPs, in particular, the carbonyl-stretching band was lost (Fig. 5) likely due to an interaction between the AmB and the polymer used. Similar interactions were observed with the other three copolymers (data not shown).
Table 5.
Important bands of IR spectrum of AmB.
| Frequencies (cm−1) | Functional group present |
|---|---|
| 3390 | C–H stretch (polyene) and O–H stretch (strongly H-bonded) |
| 2940 | C–H3 asymmetric shoulder stretch band |
| 1691 | Sharp C O stretch band, NH2 in-plane bend |
| 1557 | Polyene C C stretch band |
| 1402 | C–H bend in polyene ring |
| 1069 | C O asymmetric stretch |
| 1009 | C–H bend out of plane bend (trans polyene) |
| 851 | C–H bend in pyranose ring vibration |
Figure 5.
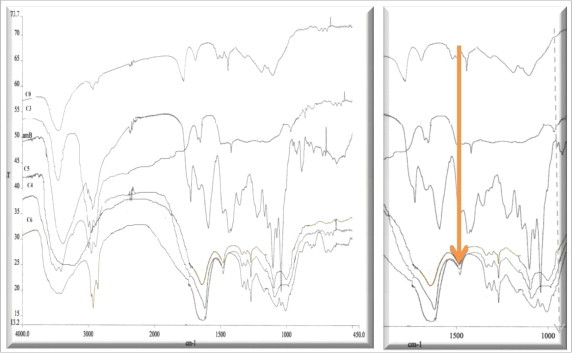
FTIR spectra for AmB-NPs formulations prepared by RGRGPd50155 copolymer (C).
3.7. Differential scanning calorimetry
Fig. 6 shows the thermogram of the drug and copolymer each separately. The DSC thermogram of drug showed a broad endothermic peak due to the loss of moisture starting from 30 to 100 °C attributed to the loss of adsorbed water (AmB is hygroscopic in nature). Two characteristic endothermic peaks were observed for AmB at 168.5 and 213.4 °C. Similar findings were observed others (Asher and Schwartzman, 1977, Janoff et al., 1988, Madden et al., 1990). The DSC traces for the pure copolymer did not show any endothermic peaks observed at in the melting region of the drug. Meanwhile, characteristic peaks for the copolymer at 66.3–68.7 °C were seen, corresponding to its phase transition temperature (tm). Similar findings have been obtained by (Vega et al., 2012). However, the complete disappearance of the characteristic peaks for AmB from the thermograms of the FD products (Fig. 6), indicating the loss of the crystalline lattice of the drug and formation of an amorphous state as a result of the inclusion of the drug inside the polymers (Al-Assady et al., 2013).
Figure 6.
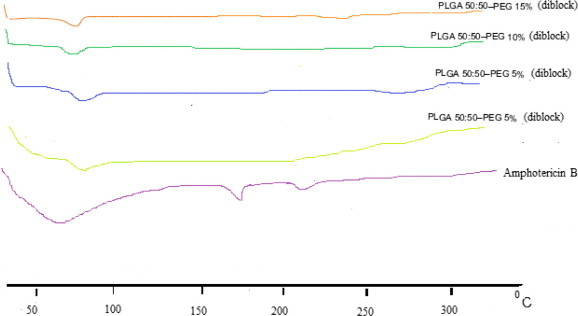
DSC thermograms of Amphotericin B and pure copolymer used.
3.8. In vitro release of Amphotericin
The in vitro release study of AmB was conducted in phosphate buffer with 2% sodium deoxycholate (Choi et al., 2008) was selected for release studies. This medium was able to maintain sink conditions, and thus was selected for release studies. The drug release pattern for 24 h is depicted in Fig. 7. Release profile indicated biphasic release of AmB from the NPs. In first phase (6 h, r > 0.9) there was an initial rapid release of about 21–51% according to the formulation followed a slow phase from 6 to 24 h where about 11% was released. According to the results obtained, it was found that there no lag time which indicates a burst in drug release during the first 15 min (about 4–10%) of the adsorbed drug on the NPs. The highest drug release within 24 h was obtained with C6 (61%), while the lowest was A6 (31%).
Figure 7.

In vitro drug release behavior for AmB-NPs. Data points with standard deviation error bars represent the mean values of three samples.
The results of modeling for the release profile are given in Table 6. The correlation coefficient (r) and release rate constant (K) were calculated for each release model. Higuchi, as well as Korsmeyer–Peppas (n = 0.5) models, show the highest correlation for all selected formulations. It should be mentioned that doubling the drug content in the formulation (C7) shows no significant difference (P > 0.5) on the drug release. Therefore, formula C6 was selected for the next in vivo (PO) study.
Table 6.
Modeling of Amphotericin release from different formulations.
| Formulation code | Zero-order (Qt = Q0 + kt) |
First-order LnQt = LnQ0 + kt |
Higuchi Qt = k√t |
Korsmeyer–Peppas Qt/Q∞ = Ktn (n = 0.5, 0.75, 1.25) |
||||
|---|---|---|---|---|---|---|---|---|
| R2 | K | R2 | K | R2 | K | R2 | K | |
| A1 | −0.1863 | 1.538 | 0.0395 | 0.020 | 0.7824 | 6.909 | 0.9229 | 1203.34 |
| A2 | −0.4214 | 1.203 | −0.2271 | 0.015 | 0.7861 | 5.433 | 0.9174 | 6283.477 |
| A3 | 0.1271 | 1.307 | 0.2817 | 0.016 | 0.9268 | 5.694 | 0.9162 | 1053.077 |
| A4 | −0.1558 | 1.510 | 0.0613 | 0.020 | 0.8232 | 6.784 | 0.8789 | 4329.172 |
| A5 | −0.1974 | 1.676 | 0.0651 | 0.023 | 0.8599 | 7.459 | 0.9730 | 15.335 |
| A6 | −0.7106 | 1.777 | −0.3564 | 0.025 | 0.7121 | 8.059 | 0.9671 | 6.660 |
| B3 | −0.3081 | 1.538 | −0.0615 | 0.020 | 0.7751 | 6.959 | 0.7479 | 5762.237 |
| B4 | −0.2749 | 1.941 | 0.0538 | 0.028 | 0.7967 | 8.750 | 0.8097 | 11841.066 |
| B5 | 0.0139 | 2.211 | 0.3448 | 0.034 | 0.8860 | 9.772 | 0.9095 | 109.169 |
| B6 | −0.4388 | 2.172 | −0.0314 | 0.033 | 0.8120 | 9.634 | 0.9719 | 8.122 |
| C3 | −0.4744 | 2.785 | 0.1308 | 0.054 | 0.7705 | 12.604 | 0.9568 | 12.331 |
| C4 | −0.5561 | 2.696 | 0.0627 | 0.053 | 0.7019 | 12.365 | 0.8887 | 1685.306 |
| C5 | −0.4713 | 2.671 | 0.1123 | 0.051 | 0.7341 | 12.200 | 0.9504 | 4592.819 |
| C6 | 0.0243 | 3.448 | 0.6637 | 0.089 | 0.8373 | 15.362 | 0.9036 | 38975.364 |
| C7 | −0.2879 | 3.229 | 0.4004 | 0.071 | 0.8329 | 14.378 | 0.9034 | 36540.313 |
| C9 | 0.2356 | 2.007 | 0.4714 | 0.029 | 0.8872 | 8.767 | 0.8762 | 1579.928 |
| D3 | −0.2474 | 1.622 | 0.0041 | 0.022 | 0.8471 | 7.235 | 0.9970 | 225.363 |
| D4 | −0.3310 | 1.491 | −0.0866 | 0.019 | 0.8234 | 6.680 | 0.8041 | 14294.643 |
| D5 | −0.0910 | 1.576 | 0.1378 | 0.021 | 0.8829 | 6.984 | 0.9648 | 9338.453 |
| D6 | −0.4197 | 2.094 | −0.0324 | 0.032 | 0.8111 | 9.410 | 0.9648 | 9338.453 |
R2, is the correlation coefficient; K, is the release rate constant for respective model. Where, Qt is the amount of drug released at time t, Q∞ is the initial amount of drug k is release rate constants of respective equation.
4. Discussions
The first challenge in developing any AmB drug delivery system was its poor solubility in any solvents, which has been overcome by co-solvation and through lowering of the pH (Venier-Julienne and Benoit, 1996, Nahar and Jain, 2009).
The second challenge was to improve the drug encapsulation efficiency (DEE) which determines the efficiency of the system to hold the drug as well as overall amount of polymer in carrier system required to deliver a particular amount of drug. The best developed formulation in this study has a DEE of 56.5% which is >5-fold better compared to the liposomal AmB (Ambisome) on the market, with a DEE of 11.72% (Jain and Kumar, 2010). Therefore, novel oral AmB-NPs have been developed using a modified emulsification diffusion method using pegylated (PLGA–PEG15% -diblock copolymer) which is characterized by a simple preparation procedure, high encapsulation efficiency and high reproducibility (Cauchetier et al., 2003, Mora-Huertas et al., 2011). The effect of main process variables on formulation parameters have been studied to obtain AmB-NPs with a narrow size distribution, low PDI and maximum DEE.
AmB formulation with PLGA–PEG diblock copolymer (RGPd 50155 with 15% PEG) has produced a significant reduction (>70%) in the MPS compared to the other developed AmB-NPs. This can be attributed to the higher percentages of the PEG loaded in the copolymer. Similar results were observed by (Buske et al., 2012). Additionally, the same polymer has shown that the highest drug content in comparison with other developed formulations. This is in agreement with (Mallardé et al., 2003, Buske et al., 2012) in that the percentage of PEG has a significant effect on the size reduction of formulated NPs. Meanwhile, in comparing diblock versus triblock with the same percentage of the PEG (10%) the study has shown that triblock copolymer is better by about 30% than the diblock copolymer (Buske et al., 2012, Zhang et al., 2014). Therefore, PLGA–PEG 15%diblock copolymer was the optimum formulation in these studies.
The addition of miglyol to the organic phase could lead to further reduction in particle size due to the fact that miglyol®-812 (Caprylic/Capric Triglyceride mixture) increases emulsification associated with the production of finely dispersed emulsions ((Pouton, 1985). Additionally, solvent capacity for less hydrophobic drugs can be improved by blending with triglycerides, so miglyol could dissolve higher amounts of AmB. This conclusion is in agreement with the finding of (Patil et al., 2004) with ketoprofen and (Garcia-Fuentes et al., 2005) with tripalmitin.
Proper selection of stabilizers or additives in the aqueous phase, with high affinity for hydrophobic NPs surface, is a key element in preventing such small particles with high surface area from aggregating. PVA is the most commonly used emulsifier because it generates particles that are relatively uniform, small and easy to re-disperse in aqueous medium (Sahoo et al., 2002, Saadati and Dadashzadeh, 2014). PVP is used in the present work as stabilizer to protect AmB-NPs from aggregation during the preparation.
Further reduction in MPS has been observed with the addition of a second emulsifier (TPGS) which is a water-soluble derivative of natural vitamin E, with amphiphilic bulk structure. Its large surface area makes it an excellent emulsifier, and solubilizer of hydrophobic drugs (Zhang et al., 2007) for fabricating nano/microparticles with high drug entrapment efficiency and high emulsification efficiency (Ke et al., 2005). This is in agreement with the study of (Zhang et al., 2007) on doxorubicin.
Therefore, the use of two emulsifying agents – PVP and Vitamin E (TPGS) – in an AmB formulation led to a significant reduction in particle size and increase in DEE than using PVP alone. This is quite possible since TPGS has tendency to migrate to the surface of nanoparticles (Si-Shen et al., 2004) and the amphiphilic nature of TPGS shows that its hydrophobic moiety was more influential than its hydrophilic moiety. Therefore, when TPGS was used as a second component of the matrix material with PVP, the interaction or affinity between the polymer matrix and the drug was enhanced and thus caused a further reduction in particle size (Si-Shen et al., 2004).
Pluronic F68 (also known as Poloxamers 188) is a hydrophilic non-ionic surfactant that has been widely used as wetting, solubilizing agent and surface adsorption excipient (Newa et al., 2007). They have been employed to enhance the solubility, dissolution and bioavailability of many poorly water soluble drugs – including ibuprofen – using various techniques (Yu et al., 2007). For some drugs, the improvement in solubility using Pluronic was higher compared to the other meltable polymers such as PEGs or complex forming agents such as cyclodextrins (Chutimaworapan et al., 2000). In this study, a similar finding (reduction in MPS) was observed by adding F68. Some reports demonstrated that Pluronic F68 performs better than other carriers (e.g., PVP and hydroxyl propyl methylcellulose (HPMC)) in terms of dissolution rate enhancement because it has dual roles in the solid dispersion formula, one as a polymeric carrier and the other as a surface active agent (He et al., 2011). Additionally, pluronic F68, offered an additional steric stabilization effect by preventing aggregation of the fine particles in the colloidal system (Mei et al., 2009). In this sense, Pluronic F68 may act as a co-emulsifier in the fabrication process, resulting in smaller particle size and narrow size distribution.
The morphology of AmB-NPs was spherical and the results of the TEM observations support this finding. Specific peaks associated with AmB were lost when present in the polymer as determined by FTIR. These results indicate that an interaction takes place between the drug and polymer. The FTIR spectral changes were attributed to electrostatic interaction between carboxylic acid group of the AmB and PLGA–PEG copolymer, turning the O–H bond of the carboxylic group into a new ether bond, which was represented by the peak at 1125 cm−1. The FTIR spectroscopy demonstrate the successful attachment of AmB to PLGA–PEG copolymer, similar finding with AmB attached to functionalized carbon nanotubes (f-CNTs) (Prajapati et al., 2011).
AmB-NPs formulations, which can be considered a matrix containing drug, is anticipated to release the drug by a diffusion mechanism. This finding is in agreement with (Italia et al., 2009).
Release study behavior of AmB-NPs shows biphasic release where the first rapid phase of release lasted for 6 h, followed by a slow diffusion order rate of release for the next 24 h. The first phase of AmB release may be due to dissolution and diffusion of the drug that was poorly entrapped in the Copolymer NPs matrix, while the slower and continuous release may be attributed to the diffusion of the drug localized in the core of the PLGA–PEG copolymer.
The possible reason for the significantly higher release rate in C batches may be corresponds to a lowest MPS of the prepared AmB-NPs in comparison with other NPs formulations, providing maximum surface area for interaction with dissolution media. The mechanism of drug release may be assumed, in that AmB-NPs possess greater hydrophilicity, leading to a greater tendency for the release medium to penetrate the particle core to cause swelling. The release rate of the drug and its appearance in the dissolution medium was governed by diffusion of drug across NPs.
5. Conclusion
The selected AmB NPs formulation showed a narrow size distribution with mean particle size which could be systemically varied according to the preparation conditions. The variation of the NP mean particle size was also associated with the physicochemical properties of the content of the organic and aqueous phases used for NP formulation.
The feasibility of using three surfactants and stabilizers on the MPS and DEE was clear that the best significant. This was related to the percentage of PEG in the polymer as well as the solubilizing agent added to the formulations. Addition of higher concentrations of PVP1, Vitamin-E (TPGS) as well as miglyol led to significant increases in drug entrapment efficiency as well as to increases in the in vitro release in comparison to other formulations.
Based on these results, it can be concluded that the formulations developed could be a viable, effective and good alternative to other carriers as a novel oral delivery of AmB. Ongoing in vivo studies are underway which ascertain the oral absorption of the developed formulations and the possible future marketing as oral dosage form.
Disclosures
Authors have no conflict of interests to disclose other than what had been acknowledged above.
Acknowledgments
This research project was supported by a grant from the “Research Center of the Female Scientific and Medical Colleges”, Deanship of Scientific Research, King Saud University.
Footnotes
Peer review under responsibility of King Saud University.
References
- Adler-Moore J., Proffitt R. Amphotericin B lipid preparations: what are the differences? Clin. Microbiol. Infect. 2008;14(s4):25–36. doi: 10.1111/j.1469-0691.2008.01979.x. [DOI] [PubMed] [Google Scholar]
- Al-Assady N.A.H., Saad E., et al. Preparation, characterization and evaluation of controlled release microspheres containing Amphotericin B. J. Basrah Res. Sci. 2013;39(3) [Google Scholar]
- Asher I.M., Schwartzman G. Amphotericin B. Anal. Profiles Drug Subst. 1977;6:1–42. [Google Scholar]
- Barratt G. Colloidal drug carriers: achievements and perspectives. Cell. Mol. Life Sci. CMLS. 2003;60(1):21–37. doi: 10.1007/s000180300002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilati U., Allémann E., et al. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur. J. Pharm. Sci. 2005;24(1):67–75. doi: 10.1016/j.ejps.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Buske J., König C., et al. Influence of PEG in PEG–PLGA microspheres on particle properties and protein release. Eur. J. Pharma. Biopharm. 2012;81(1):57–63. doi: 10.1016/j.ejpb.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Cauchetier E., Deniau M., et al. Atovaquone-loaded nanocapsules: influence of the nature of the polymer on their in vitro characteristics. Int. J. Pharm. 2003;250(1):273–281. doi: 10.1016/s0378-5173(02)00556-2. [DOI] [PubMed] [Google Scholar]
- Chen S., Cheng S.X., et al. Self-assembly strategy for the preparation of polymer-based nanoparticles for drug and gene delivery. Macromol. Biosci. 2011;11(5):576–589. doi: 10.1002/mabi.201000427. [DOI] [PubMed] [Google Scholar]
- Choi K.-C., Bang J.-Y., et al. Amphotericin B-incorporated polymeric micelles composed of poly(d,l-lactide-co-glycolide)/dextran graft copolymer. Int. J. Pharm. 2008;355(1):224–230. doi: 10.1016/j.ijpharm.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Chutimaworapan S., Ritthidej G., et al. Effect of water-soluble carriers on dissolution characteristics of nifedipine solid dispersions. Drug Dev. Ind. Pharm. 2000;26(11):1141–1150. doi: 10.1081/ddc-100100985. [DOI] [PubMed] [Google Scholar]
- Conover C.D., Zhao H., et al. Utility of poly (ethylene glycol) conjugation to create prodrugs of amphotericin B. Bioconjug. Chem. 2003;14(3):661–666. doi: 10.1021/bc0256594. [DOI] [PubMed] [Google Scholar]
- Dillen K., Vandervoort J., et al. Evaluation of ciprofloxacin-loaded Eudragit® RS100 or RL100/PLGA nanoparticles. Int. J. Pharm. 2006;314(1):72–82. doi: 10.1016/j.ijpharm.2006.01.041. [DOI] [PubMed] [Google Scholar]
- Dutcher J.D. The discovery and development of amphotericin B. Chest. 1968;54(Suppl. 1):296–298. doi: 10.1378/chest.54.supplement_1.296. [DOI] [PubMed] [Google Scholar]
- Galindo-Rodriguez S., Allémann E., et al. Physicochemical parameters associated with nanoparticle formation in the salting-out, emulsification–diffusion, and nanoprecipitation methods. Pharm. Res. 2004;21(8):1428–1439. doi: 10.1023/b:pham.0000036917.75634.be. [DOI] [PubMed] [Google Scholar]
- Garcia-Fuentes M., Alonso M.J., et al. Design and characterization of a new drug nanocarrier made from solid–liquid lipid mixtures. J. Colloid Interface Sci. 2005;285(2):590–598. doi: 10.1016/j.jcis.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Gaucher G., Satturwar P., et al. Polymeric micelles for oral drug delivery. Eur. J. Pharma. Biopharm. 2010;76(2):147–158. doi: 10.1016/j.ejpb.2010.06.007. [DOI] [PubMed] [Google Scholar]
- He X., Pei L., et al. Comparison of spray freeze drying and the solvent evaporation method for preparing solid dispersions of baicalein with Pluronic F68 to improve dissolution and oral bioavailability. AAPS PharmSciTech. 2011;12(1):104–113. doi: 10.1208/s12249-010-9560-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim F., Gershkovich P., et al. Assessment of novel oral lipid-based formulations of amphotericin B using an in vitro lipolysis model. Eur. J. Pharm. Sci. 2012;46(5):323–328. doi: 10.1016/j.ejps.2012.02.008. [DOI] [PubMed] [Google Scholar]
- Italia J., Yahya M., et al. Biodegradable nanoparticles improve oral bioavailability of amphotericin B and show reduced nephrotoxicity compared to intravenous Fungizone®. Pharm. Res. 2009;26(6):1324–1331. doi: 10.1007/s11095-009-9841-2. [DOI] [PubMed] [Google Scholar]
- Jain J.P., Kumar N. Development of amphotericin B loaded polymersomes based on (PEG)3-PLA co-polymers: Factors affecting size and in vitro evaluation. Eur. J. Pharm. Sci. 2010;40(5):456–465. doi: 10.1016/j.ejps.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Janoff A., Boni L., et al. Unusual lipid structures selectively reduce the toxicity of amphotericin B. Proc. Natl. Acad. Sci. 1988;85(16):6122–6126. doi: 10.1073/pnas.85.16.6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jee J.-P., McCoy A., et al. Encapsulation and release of Amphotericin B from an ABC triblock fluorous copolymer. Pharm. Res. 2012;29(1):69–82. doi: 10.1007/s11095-011-0511-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke W.-T., Lin S.-Y., et al. Physical characterizations of microemulsion systems using tocopheryl polyethylene glycol 1000 succinate (TPGS) as a surfactant for the oral delivery of protein drugs. J. Control. Release. 2005;102(2):489–507. doi: 10.1016/j.jconrel.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Lemke A., Kiderlen A., et al. Amphotericin B. Appl. Microbiol. Biotechnol. 2005;68(2):151–162. doi: 10.1007/s00253-005-1955-9. [DOI] [PubMed] [Google Scholar]
- Ma L.L., Jie P., et al. Block copolymer ‘stealth’ nanoparticles for chemotherapy: interactions with blood cells in vitro. Adv. Funct. Mater. 2008;18(5):716–725. [Google Scholar]
- Madden T., Janoff A., et al. Incorporation of amphotericin B into large unilamellar vesicles composed of phosphatidylcholine and phosphatidylglycerol. Chem. Phys. Lipids. 1990;52(3):189–198. doi: 10.1016/0009-3084(90)90114-7. [DOI] [PubMed] [Google Scholar]
- Mallardé D., Boutignon F., et al. PLGA–PEG microspheres of teverelix: influence of polymer type on microsphere characteristics and on teverelix in vitro release. Int. J. Pharm. 2003;261(1):69–80. doi: 10.1016/s0378-5173(03)00272-2. [DOI] [PubMed] [Google Scholar]
- Mei L., Zhang Y., et al. A novel docetaxel-loaded poly (ε-caprolactone)/pluronic F68 nanoparticle overcoming multidrug resistance for breast cancer treatment. Nanoscale Res. Lett. 2009;4(12):1530–1539. doi: 10.1007/s11671-009-9431-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ménez C., Legrand P., et al. Physicochemical characterization of molecular assemblies of miltefosine and amphotericin B. Mol. Pharm. 2007;4(2):281–288. doi: 10.1021/mp0601143. [DOI] [PubMed] [Google Scholar]
- Mora-Huertas C., Fessi H., et al. Polymer-based nanocapsules for drug delivery. Int. J. Pharm. 2010;385(1):113–142. doi: 10.1016/j.ijpharm.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Mora-Huertas C., Fessi H., et al. Influence of process and formulation parameters on the formation of submicron particles by solvent displacement and emulsification–diffusion methods: critical comparison. Adv. Colloid Interface Sci. 2011;163(2):90–122. doi: 10.1016/j.cis.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Nahar M., Jain N.K. Preparation, characterization and evaluation of targeting potential of amphotericin B-loaded engineered PLGA nanoparticles. Pharm. Res. 2009;26(12):2588–2598. doi: 10.1007/s11095-009-9973-4. [DOI] [PubMed] [Google Scholar]
- Newa M., Bhandari K.H., et al. Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer 188. Int. J. Pharm. 2007;343(1):228–237. doi: 10.1016/j.ijpharm.2007.05.031. [DOI] [PubMed] [Google Scholar]
- Ouellette M., Drummelsmith J., et al. Leishmaniasis: drugs in the clinic, resistance and new developments. Drug Resist. Updat. 2004;7(4):257–266. doi: 10.1016/j.drup.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Owens D.E., III, Peppas N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006;307(1):93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Parveen S., Sahoo S.K. Polymeric nanoparticles for cancer therapy. J. Drug Target. 2008;16(2):108–123. doi: 10.1080/10611860701794353. [DOI] [PubMed] [Google Scholar]
- Patel P.A., Fernandes C.B., Pol A.S., Patravale V.B. Oral Amphotericin B: challenges and avenues. Int. J. Pharm. Biosci. Technol. 2013;1:1–9. [Google Scholar]
- Patil P., Joshi P., et al. Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system (SEDDS) of ketoprofen. AAPS PharmSciTech. 2004;5(3):43–50. doi: 10.1208/pt050342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouton C.W. Self-emulsifying drug delivery systems: assessment of the efficiency of emulsification. Int. J. Pharm. 1985;27(2):335–348. [Google Scholar]
- Prajapati V.K., Awasthi K., et al. Targeted killing of Leishmania donovani in vivo and in vitro with amphotericin B attached to functionalized carbon nanotubes. J. Antimicrob. Chemother. 2011;66(4):874–879. doi: 10.1093/jac/dkr002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadati R., Dadashzadeh S. Marked effects of combined TPGS and PVA emulsifiers in the fabrication of etoposide-loaded PLGA–PEG nanoparticles: in vitro and in vivo evaluation. Int. J. Pharm. 2014;464(1):135–144. doi: 10.1016/j.ijpharm.2014.01.014. [DOI] [PubMed] [Google Scholar]
- Sahoo S.K., Panyam J., et al. Residual polyvinyl alcohol associated with poly(d,(l-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J. Control. Release. 2002;82(1):105–114. doi: 10.1016/s0168-3659(02)00127-x. [DOI] [PubMed] [Google Scholar]
- Sinha V., Bansal K., et al. Polycaprolactone microspheres and nanospheres: an overview. Int. J. Pharm. 2004;278(1):1–23. doi: 10.1016/j.ijpharm.2004.01.044. [DOI] [PubMed] [Google Scholar]
- Si-Shen F., Li M., et al. Nanoparticles of biodegradable polymers for clinical administration of paclitaxel. Curr. Med. Chem. 2004;11(4):413–424. doi: 10.2174/0929867043455909. [DOI] [PubMed] [Google Scholar]
- Stolnik S., Illum L., et al. Long circulating microparticulate drug carriers. Adv. Drug Deliv. Rev. 1995;16(2):195–214. [Google Scholar]
- Thornton S.J., Wasan K.M. The reformulation of amphotericin B for oral administration to treat systemic fungal infections and visceral leishmaniasis. Expert Opin. Drug Deliv. 2009;6:271–284. doi: 10.1517/17425240902802861. [DOI] [PubMed] [Google Scholar]
- Torrado J., Espada R., et al. Amphotericin B formulations and drug targeting. J. Pharm. Sci. 2008;97(7):2405–2425. doi: 10.1002/jps.21179. [DOI] [PubMed] [Google Scholar]
- Van de Ven H., Paulussen C., et al. PLGA nanoparticles and nanosuspensions with amphotericin B: potent in vitro and in vivo alternatives to Fungizone and AmBisome. J. Control. Release. 2012;161(3):795–803. doi: 10.1016/j.jconrel.2012.05.037. [DOI] [PubMed] [Google Scholar]
- Vega E., Egea M.A., et al. Role of hydroxypropyl-β-cyclodextrin on freeze-dried and gamma-irradiated PLGA and PLGA–PEG diblock copolymer nanospheres for ophthalmic flurbiprofen delivery. Int. J. Nanomed. 2012;7:1357. doi: 10.2147/IJN.S28481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venier-Julienne M., Benoit J. Preparation, purification and morphology of polymeric nanoparticles as drug carriers. Pharm. Acta Helv. 1996;71(2):121–128. doi: 10.1016/0031-6865(95)00059-3. [DOI] [PubMed] [Google Scholar]
- Wasan E.K., Bartlett K., et al. Development and characterization of oral lipid-based Amphotericin B formulations with enhanced drug solubility, stability and antifungal activity in rats infected with Aspergillus fumigatus or Candida albicans. Int. J. Pharm. 2009;372(1):76–84. doi: 10.1016/j.ijpharm.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Yang Z., Tan Y., et al. Development of amphotericin B-loaded cubosomes through the solemuls technology for enhancing the oral bioavailability. AAPS PharmSciTech. 2012;13(4):1483–1491. doi: 10.1208/s12249-012-9876-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Chun M., et al. Preparation and characterization of piroxicam/poloxamer solid dispersion prepared by melting method and solvent method. J. Korean Pharm. Sci. 2007;37(1):1. [Google Scholar]
- Zhang Z., Huey Lee S., et al. Folate-decorated poly (lactide-co-glycolide)-vitamin E TPGS nanoparticles for targeted drug delivery. Biomaterials. 2007;28(10):1889–1899. doi: 10.1016/j.biomaterials.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Zhang K., Tang X., et al. PEG–PLGA copolymers: Their structure and structure-influenced drug delivery applications. J. Control. Release. 2014;183:77–86. doi: 10.1016/j.jconrel.2014.03.026. [DOI] [PubMed] [Google Scholar]