

Abstract
Reactive oxygen and nitrogen species (e.g. H2O2, nitric oxide) confer redox regulation of essential cellular signaling pathways such as cell differentiation, proliferation, migration and apoptosis. In addition, classical regulation of gene expression or activity, including gene transcription to RNA followed by translation to the protein level, by transcription factors (e.g. NF-κB, HIF-1α) and mRNA binding proteins (e.g. GAPDH, HuR) is subject to redox regulation. This review will give an update of recent discoveries in this field, and specifically highlight the impact of reactive oxygen and nitrogen species on DNA repair systems that contribute to genomic stability. Emphasis will be placed on the emerging role of redox mechanisms regulating epigenetic pathways (e.g. miRNA, DNA methylation and histone modifications). By providing clinical correlations we discuss how oxidative stress can impact on gene regulation/activity and vise versa, how epigenetic processes, other gene regulatory mechanisms and DNA repair can influence the cellular redox state and contribute or prevent development or progression of disease.
Keywords: Oxidative stress, Redox signaling, Gene regulation, Epigenetics, DNA repair
Abbreviations: 5-hmC, 5-hydroxymethylcytosine; 5-mC, 5-methylcytosine; AP-1, activator protein 1; Ape-1, apurinic/apyrimidinic endonuclease 1; AREs, AU-rich elements; BER, base excision repair; COPD, chronic obstructive pulmonary disorder; DNMT, DNA methyltransferase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GPx-1, glutathione peroxidase-1; Grx, glutaredoxin; HAT, histone acetyltransferase; HDAC, histone deacetylase; HIF-1α, hypoxia inducible factor-1α; HO-1, heme oxygenase-1; HuR, mRNA-binding protein in the 3′-untranslated region; JmjC, Jumonji C domain-containing histone demethylases; Keap1, kelch-like ECH-associated protein 1; MMP, matrix metalloproteinase; NF-κB, nuclear factor-κB; NER, nucleotide excision repair; Nox, member of the NADPH oxidase family; Nrf2, nuclear factor erythroid related factor 2; OxyR, transcription factor (hydrogen peroxide-inducible genes activator); PETN, pentaerithrityl tetranitrate; PHD, prolylhydroxylase; RNS, reactive nitrogen species; ROS, reactive oxygen species; SOD, superoxide dismutase; Trx, thioredoxin
Graphical abstract
Highlights
-
•
Classical and epigenetic pathways of gene regulation are summarized.
-
•
Pathways of DNA repair are summarized.
-
•
The impact of redox signaling and oxidative stress on these pathways is discussed.
-
•
Redox regulation of mRNA stability is discussed.
-
•
New therapeutic strategies related to genome stability are presented.
Introduction
The ability of every living organism to leave a progeny is encoded by its genome, which is a long DNA chain that contains all information about the particular organism. Genomes of living organisms can contain as few as 500 genes (e.g. mycoplasma) and as many as 20,000–25,000 for humans. Considering how valuable the integrity of a DNA molecule is, cells developed several protection systems. First, DNA is contained inside the nucleus, which is surrounded by a selectively permeable membrane. Secondly, DNA is also localized to the mitochondria, which can either be considered as a preservation strategy for the cell or a compartmentalization approach enabling better functional division between DNA molecules.
DNA located in the nucleus predominantly encodes for RNA molecules that later on can be translated to proteins. This means that every piece of information concerning a particular organism is encoded by nuclear DNA, starting from cell division parameters and finishing with information regarding programmed cell death. Information from DNA is processed via transcription mechanisms, leading to formation of another nucleic acid chain, namely RNA. Depending on the type of RNA molecule, translation to amino acid sequences, the building blocks for proteins, or other functional roles may occur. DNA located in mitochondria encodes all information necessary for its robust activity. In particular, mtDNA is responsible for storing, maintaining and successful implementation of information regarding among others, main components of mitochondrial electron transport chain (ETC) including cytochrome b, NADH dehydrogenase subunits, cytochrome oxidase subunits. In addition to direct transcriptional effects mediated by transcription factor binding to DNA epigenetic marks due to chemical modification of cytosine residues of DNA (DNA methylation) and histone proteins associated with DNA (histone modifications) can modulate gene activity and expression as well as chromatin.
Even though cells developed strategies to preserve the integrity of DNA, multiple factors can alter the structure of DNA [1], among which are UV irradiation [2], reactive oxygen and nitrogen species [3], or extrinsic chemical compounds [4]. On average, the DNA of a mammalian cell receives the following assaults per day: 200 cytosine deaminations, 3000 guanine methylations, 10,000 spontaneous depurinations, 10,000–100,000 oxidative lesions, 10,000 single-strand breaks, and 10–50 double-strand breaks [3,5–7]. Every type of DNA damage is source specific. For example, UV light is mostly known for strand breaks and/or DNA–DNA cross-links, as well as DNA–protein cross-link formation [8]. Reactive oxygen and nitrogen species predominantly induce specific base modification, such as 8-oxo-dG, 8-nitro-dG [9], or GC to TA transversions due to their high reactivity with strong nucleophilic sites on nucleobases [10]. External chemical compounds can introduce particular chemical groups, for example alkylation of DNA by methylnitrosourea [11] or N-methyl-N′-nitro-N-nitrosoguanidine [12] can cause DNA cross-links (e.g. mitomycin C, cisplatin) [13,14], or enhance formation of single- and double-strand breaks by sealing DNA-topoisomerase complexes [15]. One feature of the mammalian genome is the fact that every type of DNA aberration has a unique damage response in form of detection and repair systems. In conclusion, not only regulation of gene expression by transcription factors and epigenetic pathways, but also DNA damage/repair largely contributes to genome stability.
This review will outline that redox signaling and oxidative stress will affect expression, transcription and translation of genomic information not only by classical and epigenetic regulation of gene expression, but also by inflicting direct DNA damage and regulation of the activity of DNA repair enzymes. In the first section, we provide an overview on the different pathways and enzymatic systems that contribute to genome stability and read-out of genomic information. In the second section, we focus on the impact of redox biology and oxidative stress on these different pathways. In the third section, we correlate these findings to the clinical situation. In the fourth section, we summarize the impact of redox biology and oxidative stress on genome stability as well as transcription and translation of genomic information.
DNA repair
Depending on the type of DNA modification different repair mechanisms will be activated in order to remove such damage. Whenever a toxic modification on a specific nucleobase appears or leads to formation of abasic sites, base excision repair (BER) is activated to resolve this problem [16]. Key players of the BER are DNA glycosylases (uracil-DNA glycosylase (UNG) [17], 8-oxoguanine DNA glycosylase (OGG1) [18], nth endonuclease III-like 1 (NTHL1) [19] and nei endonuclease VIII-like 1, 2 or 3 (NEIL1/NEIL2/NEIL3) [20]), all of which recognize different base modifications; DNA endonucleases such as apurinic/apyrimidinic endonuclease 1 (APE1) [21]; DNA polymerases (Polβ) and DNA ligases (Lig1) [22]. Specificity of this repair pathway is achieved by activity of the glycosylases that scan DNA molecules by slightly pulling the nucleotide strain. If there is a distortion of the helix, caused by a lack of hydrogen bonding between damaged Watson–Crick base pairs, these enzymes will flip this nucleobase out, insert it into the catalytic pocket, consequently cleave the N-glycosidic bond between the damaged base and the 2′-deoxyribose, and generate an apurinic- or apyrimidinic-(AP) site. All, AP sites are then processed by apurinic/apyrimidinic endonuclease 1 (APE-1), leaving clean 3′ and 5′ ends that allow DNA polymerase β (Polβ) and DNA ligase I (Lig1) to insert and ligate the appropriate base [23].
Nucleotide excision repair (NER) on the other hand, is able to remove larger and more complex types of damage found on DNA, like intra-strand and DNA–protein cross-links, and bulky formations [24]. Xeroderma pigmentosum, complementation group C (XPC), xeroderma pigmentosum, complementation group G (XPG), RAD23 homolog B (RAD23B), excision repair cross-complementation group 6 (ERCC6) and others function as damage identification molecules [25]. If the distortion was recognized during the replication process, the stalling of the replication fork will serve as an identification signal [26]. Upon receiving first NER up-regulation signals, the complex consisting of XPA, XPG [27], ERCC1, ERCC4, ERCC3 and replication protein A (RPA) acts as an excinuclease making two incisions in the DNA strand on either sides of the lesion. In a next step polymerases are activated to insert the correct DNA segment in the missing section and ligases will finish the repair process by sealing the strand.
If distinct DNA modifications are not repaired on time by repair machineries, they might lead to the formation of mismatches after incorrectly performed transcription. Unfortunately, such unfavorable transcriptional outcome can happen even without DNA damage. Mismatch signals as well as small insertion and deletion loops are identified by the mismatch repair machinery (MMR) [28]. Major regulators of the MMR are two enzymatic families, mutator S (MutS) and mutator L (MutL). MutS recognizes base–base mismatches and small insertion/deletion loops, while MutL is able to detect longer loops [29]. After the recognition step, these enzymes initiate the downstream repair cascade, consisting of accessory proteins including proliferating cell nuclear antigen (PCNA), replication factor C (RFC), exonucleases (EXO-1) and DNA polymerases, thereby introducing correct nucleobases into the DNA gap. Finalization of the repair process is orchestrated by Lig1, which joins up flanking ends of the DNA molecule [30].
Double strand breaks are considered to be the most severe type of DNA damage and if not promptly repaired, might contribute to programmed cell death. Evolutionary, two repair mechanisms emerged in order to resolve such pathological state, homologous recombination (HR) and non-homologous end joining (NHEJ) [31]. Recombination mechanisms have to be initiated by endonucleases that are able to cleave one of the partner DNA duplexes. This is a complex process which can be accompanied by the activity of the DNA helicases. After 3′ ends of the DNA molecule have been cleared from the residual damaged nucleobase they anneal with the homologous regions in another DNA duplex. In order to allow this process the homologous duplex is extended to provide extra space for the approximation of the strands. The loop, which is formed as a result of this reaction, is extended by adjuvant repair enzymes, filling the gap that was left by the damaged nucleobase. Finally, this loop gets resolved from this repair stage either with or without the cross-over of the genetic information, leaving behind a footprint of the genetic code from the homologous region [32]. When homologous strands are not present for the recombination pathway, a process called non-homologous end joining (NHEJ) is activated. It is initiated by Ku70 and Ku80 complexes [33] that bring the DNA strands together and attract the Mre11–Rad50–Nbs1 (MRN) complex that finalizes the approximation process. Joining of the broken ends is performed by a specific DNA ligase IV (LigIV). X-ray repair cross-complementing protein 4 (XRCC4) supports LigIV in the ligation process [34]. Homologous recombination and non-homologous end joining pathways are highly mutation-prone due to the already missing section of the DNA molecule and potential loss of certain open reading frames, which will later cause functional abnormalities. The major DNA repair pathways are summarized in Fig. 1.
Fig. 1.
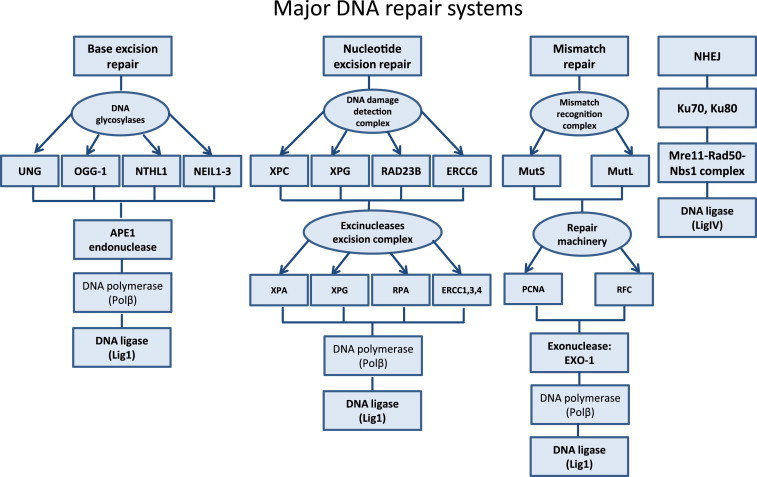
Summary of major DNA repair mechanisms in the mammalian cells. The genome of the mammalian cell is well protected from the plethora of extracellular and intracellular damaging factors by different DNA repair pathways. Depending on the type of DNA modification that has been introduced to the nucleic acid, appropriate repair mechanism will be activated for removal (BER, NER, mismatch repair, non-homologous end joining (NHEJ)). Major DNA glycosylases in BER pathway are: uracil-DNA glycosylase (UNG), 8-oxoguanine DNA glycosylase (OGG1), nth endonuclease III-like 1 (NTHL1), nei endonuclease VIII-like 1–3 (NEIL1-3). Specificity of NER is achieved by its DNA damage detection complex: xeroderma pigmentosum, complementation group C, G (XPC, XPG), RAD23 homolog B (RAD23B) and excision repair cross-complementation group 6 (ERCC6). Excision of the effected DNA section is conducted by members of the excinuclease excision complex: xeroderma pigmentosum, complementation group A, G (XPA, XPG), replication protein a (RPA) and excision repair cross complementation group 1, 3, 4 (ERCC1,3,4). MMR is directed by the recognition complex: mutator S (MutS) and mutator L (MutL) and by specific repair machinery:proliferation cell nuclear antigen (PCNA) and replication factor c (RFC). Concept according to Luo et al. [106].
Classical gene regulation
A DNA molecule is a carrier of information, covering most aspects of the organism's development and sustainability (role of miRNA and other noncoding RNAs will be discussed below). Although DNA contains all these data, it has tight regulation over the information release through the activation or repression of genes. Such regulation happens through many distinct pathways, which generally can be summarized into two sub-classes: classical gene regulation, which takes place through the activity of transcription factors and novel gene regulation through epigenetic regulators.
Transcription factors constitute a diverse class of proteins [35]. Among some of the most well-studied, redox-sensitive transcription factors are NF-κB [36], Nrf2 [37], estrogen receptor [38], hypoxia inducible factor-1 (HIF-1) [39], and transcription cofactors RNA helicases [40]. The estrogen receptor (ER) is a ligand-dependent transcription factor. It exhibits its effect on DNA through binding to discriminatory DNA motifs known as the estrogen response elements (EREs), half-EREs, or through protein–protein interactions with other transcription factors including specificity protein 1 (SP1), activator protein 1 (AP1), paired-like homeodomain 1 (Pitx1), and Runt-related transcription factor 1 (Runx1). ER association with DNA requires recruitment of pioneering factors, in particular forkhead box protein A1 (FOXA1), which binds to ER responsive sites before cell activation takes place in order to create favorable epigenetic conditions for ER binding and transcriptional activation [41].
NF-κB signaling is involved in a plethora of cellular pathways including inflammatory responses, cell cycle regulation, and immunity development [42]. The NF-κB transcription factor family consists of five proteins, p65, RelB, cRel, p105/p50 and p100/p52. These proteins can form different aggregates and are able to induce or repress gene expression of specific target genes. As NF-κB is involved in such complex and multi-dimensional signaling networks, its activation is triggered by different pathways, the canonical (classical) branch and the non-canonical (alternative) branch. They differ in the initial receptor activation and the subsequent signaling cascades, ultimately leading to NF-κB activation. Besides the canonical and the alternative pathway, additional pathways of NF-κB activation exist, sometimes termed atypical activation pathways [43,44]. NF-κB activation is also highly redox-regulated and thiol oxidation via activator protein 1 (AP-1), thioredoxin and IκB degradation activates NF-κB [45–47].
The nuclear factor erythroid related factor 2 (Nrf2), is a member of the cap ‘n’ collar family of transcription factors [48]. Under physiologic stationary conditions, Nrf2 is constantly degraded by the proteasome. This pathway is enabled by direct activity of kelch-like ECH associated protein 1 (Keap1). Keap1 couples with Cullin3 ubiquitin E3 ligase and afterwards poly-ubiquitinates Nrf2, tagging it for proteasomal degradation. The major function of Nrf2 is the maintenance of redox homeostasis. Upon stimulation with various exogenous and endogenous factors, and in particular by oxidative stress, cytosolic Nrf2 will not be ubiquitinated and degraded, but translocates into the nucleus, where it binds to the antioxidant responsive elements (ARE) located in promoter regions of genes, and activates the transcription of several antioxidant genes including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase quinone oxidoreductase 1 (NQO1), heme oxygenase-1 (HO-1), glutathione S-transferase, superoxide dismutase (SOD2), catalase, and γ-glutamate cysteine ligase (GCL). Up-regulation of all these protective enzymes enables a strong response to oxidative stress [49].
Hypoxia-inducible transcription factors of the HIF family are major regulators of the cellular adaptation to hypoxia, but are also sensitive to reactive oxygen species (ROS) and nitric oxide (•NO) [39]. The α-subunit of this heterodimer is hydroxylated at specific prolines by prolyl hydroxylases (PHD) under normoxic conditions. This allows binding of the ubiquitin E3 ligase von Hippel Lindau protein (pVHL) and subsequent degradation by the proteasome. PHDs are non heme Fe(II)- and 2-oxoglutarate-dependent dioxygenases, which require ascorbate as cofactor [50]. Under hypoxia PHD activity is decreased, thus preventing degradation of the HIF-1α subunits and subsequent transcriptional regulation of hundreds of target genes. While ROS sensitivity of HIF-1α is partially mediated by its transcriptional regulator NF-κB, ROS also can act via modulating PHD activity [51–53] either by interaction of ROS with iron by Fenton chemistry or by direct oxidation of the iron center itself. Recently, it has been shown that FIH, an asparagine hydroxylase, which regulates transcriptional activity of HIF-1α, is highly sensitive to peroxides [54]. Additionally, •NO was reported to induce HIF-1α under normoxic conditions. While S-nitros(yl)ation of HIF-1α was demonstrated in vitro, the biological significance and any role in HIF-1α stability regulation by nitrous(yl)ation awaits clarification [55]. Moreover, Chowdhury and colleagues proposed that the catalytic domain of PHD2 can react in vitro with •NO at the active site Fe(II) and at cysteine residues [56] (note by the authors: •NO is unlikely to react with thiols directly but requires either so-called “oxidative nitrosation” conditions, e.g. by the presence of ROS or high •NO concentration plus oxygen tension, or preformation of nitrosating species such as N2O3 or nitroso-thiols with trans-nitrosating reactivity [57]).
Finally, the successful “activation” of a specific gene and its translation to a protein is also determined by post-transcriptional regulation of mRNA stability depending on sequences found in the 3′-untranslated (3′-UTR) region of the mRNA [58]. For most species AU-rich elements (AREs) are highly conserved in the 3′-UTR region, representing binding sites for proteins like HuR or KSRP, which stabilize or destabilize mRNAs. According to observations by Kleinert and colleagues this mechanism contributes to the induction of HO-1 expression by the nitrovasodilator pentaerithrityl tetranitrate [58].
Epigenetic changes by direct DNA or histone modifications
Epigenetic regulators are able to pose a specific effect on the chromatin structure by reversible chemical modification of the DNA molecule itself or on co-localized histone proteins. One of the most studied epigenetic marks is DNA methylation. This modification is considered gene repressive, preventing the transcription process [59]. The most common sites at which DNA methylation is detected, are CpG islands, which are cytosine–guanine pairs that are present in particular high abundancy at certain sections of the DNA [60]. The 5th position on cytosine is the preferred methylation point [61]. A general overview of most common DNA modifications is shown in Fig. 2. Differential methylation states of DNA have been implicated in the maintenance of the normal physiology like genomic imprinting, inactivation of the X chromosome, as well as in pathological states like Angelman syndrome, Beckwith–Wiedemann syndrome and several types of cancer [62]. On the other hand, emerging data suggest that DNA methylation should not be plainly considered as a silencing mark: Depending on the location in the chromatin environment and the presence of transcription markers that are associated with this particular gene cluster, methylation can be either a transcription activating or repressing mark [63].
Fig. 2.
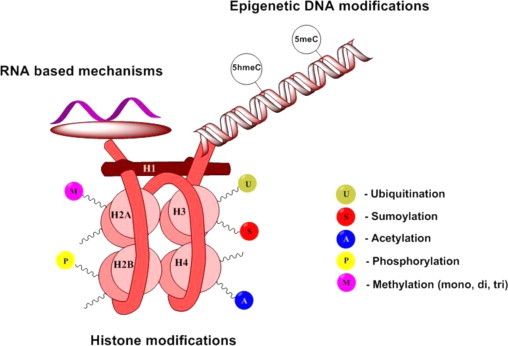
Overview of the epigenetic processes taking place in the cell. Histone structures are shown as H1, H2A, H2B, H3 and H4 according to the accepted terminology. Major modifications that are found on their lysine and arginine tails are methylation, acetylation, ubiquitination, phosphorylation and sumoylation. The DNA helix is subjected majorly to two epigenetic changes – methylation and hydroxymethylation of the 5th position of cytosine. Another epigenetic mechanism is the RNA-based pathway. Concept according to Wang et al. [72].
Another class of epigenetically regulated molecules are histones. These protein conglomerates are tightly associated with DNA to form the chromatin structure [64]. Among the most typical epigenetic marks observed on histones are methylation, acetylation, ubiquitination, sumoylation and ADP-ribosylation [65]. Predominantly, these modifications are introduced on lysine and/or arginine residues of the N-terminal tails due to their steric accessibility. Every moiety found on histones has a specific effect on the chromatin state and consequently downstream on gene expression levels. Histone acetylation is attributed to the formation of euchromatin, a more relaxed chromosomal state, which leads to transcriptional activity and inhibition of DNA methylation [66]. Histone deacetylation on the other hand will result in a condensed chromatin state and is associated with transcriptional repression [67]. Histone methylation often serves as a read out mark for DNA binding proteins or transcription factors, which in turn can activate versatile signaling cascades [68,69]. A summary of currently known histone modifications is shown in Fig. 2. Many studies in different cancers observed dysregulation of histone׳s epigenetic marks, although there is increasing evidence that epigenetic alterations also contribute to other pathological conditions, including cardiovascular diseases [70].
Epigenetic changes by non-coding RNA
DNA methylation and histone modifications alone cannot orchestrate the whole plethora of epigenetic changes that have been detected. Such observations led to an extensive search for other players in the intricate epigenetic cascades. Current information shows that only 2% of the human genome is being transcribed into protein-encoding RNA, while the rest of the genetic material, approximately 70–90%, is transcribed into non-coding RNAs (ncRNAs) [71]. There is high abundancy and variability among these molecules including ribosomal RNAs (rRNAs), microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). miRNAs are short RNA molecules, commonly with 22 nucleotides [72]. The maturation process of miRNA is very similar to mRNA, being governed by Drosha–DGCR8 complex [73]. During extraction from the nucleus, dicer cleavage of the pre-miRNA into the mature miRNA in the cytoplasm and later assembly of the miRNA–RNA-induced silencing complex (RISC) duplex leads to the formation of the active miRNA molecule [73]. The details of the miRNA maturation process are shown in Fig. 3. miRNA have been described to play a role in repression of target mRNA translation, de-adenylation of mRNA tails, as well as in regulation through the recruitment of specific proteins to the nucleus. lncRNAs are a broad family of molecules, each having structural and functional distinctiveness. Among functions attributed to lncRNAs are involvement in embryonic pluripotency, cell differentiation and development, and chromosomal stability [74]. Additionally, lncRNA have been implicated in several pathological conditions such as Russel–Silver and Prader–Willi syndrome, cardiac diseases and neurodegenerative disorders [75].
Fig. 3.
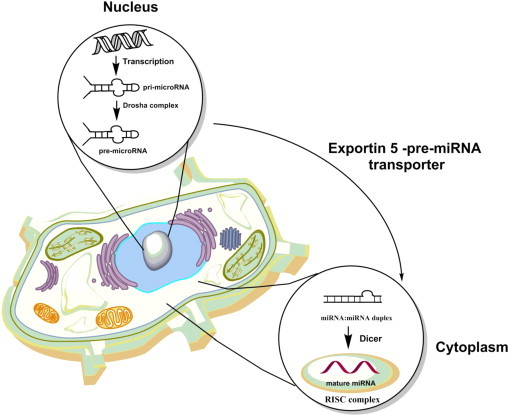
Schematic representation of the miRNA maturation process. As a result of the successful transcription and activity of the Drosha complex in the nucleus pre-microRNA is formed and is exported to the cytoplasm for further processing. Dicer and RNA-induced silencing complex (RISC) complexes are finalizing formation of the mature miRNA which will be catalytically active. Concept according to Wang et al. [72].
Impact of redox signaling and oxidative stress on gene regulation and DNA repair
Redox regulation versus oxidative stress
Many diseases and drug-induced complications are associated or even based on an imbalance between the formation of reactive oxygen and nitrogen species (ROS and RNS), and antioxidant enzymes catalyzing the break-down of harmful oxidants [76,77]. An imbalance between the oxidant formation and detoxification will ultimately lead to accumulation of oxidative damage in many biomolecules (e.g. lipid peroxidation with subsequent formation of reactive aldehydes and ketones, protein carbonylation, nitration and sulfoxidation, DNA lesions such as 8-oxo-dG) and impaired redox signaling (e.g. interference with H2O2 signaling that regulates cell differentiation, proliferation and migration). This condition is termed oxidative stress [78,79]. Many cardiovascular, neurodegenerative, and inflammatory diseases as well as cancer are associated or even triggered by oxidative stress [80–83]. Oxidative stress is a well-established hallmark of cardiovascular disease [84] and strong evidence for a causal role of ROS and RNS in these processes is based on improvement of cardiovascular complications by genetic deletion of enzymes involved in the synthesis of these reactive species as well as overexpression of antioxidant enzymes detoxifying these reactive species [76,85]. In accord, in most animal studies overexpression of reactive oxygen and nitrogen species producing enzymes as well as deletion of antioxidant enzymes resulted in aggravation of cardiovascular complications [76,85].
Based on these considerations, the oxidant/antioxidant equilibrium is an attractive target for therapeutic intervention and large scale clinical studies have been conducted to prove the efficacy of antioxidants (namely vitamins) by oral treatment of patients (e.g. HOPE, HOPE-TOO; for review see [76,86]). It was a great disappointment to realize that these antioxidant trials revealed in most cases no effects or in some patients even detrimental outcomes (e.g. for vitamin E) [86–89]. In contrast, a number of small scale studies using acute, high-dose intravenous infusion of antioxidants demonstrated remarkable benefits in various diseases (reviewed in [86]). Even more convincing are experimental data on the effects of antioxidants in several animal disease models [85]. Potential explanations for this obvious discrepancy could be that either the achievable dose of oral antioxidant therapy was not high enough or that systemic therapy with non-specific antioxidants interferes with redox signaling pathways controlled by ROS and RNS (reviewed in [76,86]). Antioxidant therapy might be more successful when using antioxidants with tissue- or cell organelle-specificity (e.g. mitochondria-targeted compounds such as mitoQ) [90], activators of endogenous antioxidant pathways (e.g. Nrf2/HO-1) [91] or source specific inhibitors (e.g. different Nox isoforms) [92], thus leaving important cellular redox signaling mechanisms intact. In the subsequent sections we will discuss the contribution of redox signaling and oxidative stress to gene regulation and DNA repair in order to highlight that systemic antioxidant therapy could have significant positive or negative impact on these essential cellular functions.
Redox impact on classical gene regulation
The notion that redox signaling and oxidative stress can change gene transcription and translation as well as the integrity of the DNA repair systems is well supported by the literature [93]. Many transcription factors are regulated in a redox-sensitive fashion involving zinc-finger motifs or transition metal centers [94,95]. For example, the bacterial oxidative stress regulator OxyR is activated by hydrogen peroxide-induced thiol oxidation or S-nitrosation by nitric oxide-derived nitrosating species and induces antioxidant genes [96]. The transcription factor Nrf2 is activated upon thiol oxidation of its regulator protein kelch-like ECH-associated protein 1 (Keap1) [97] and confers antioxidant protection (e.g. by up-regulation of HO-1). Thiol oxidation via activator protein 1 (AP-1), thioredoxin and IκB degradation activates NF-κB [45–47], while AP-1 is activated upon thiol oxidation via thioredoxin and regulates antioxidant genes [45,98] (for AP-1 molecular mechanism see Fig. 4). Further, hydroxylation of HIF-1α by prolyl hydroxylases, which might be inactivated by oxidation or nitrosylation of the ferrous iron-containing active site [99], or by the peroxide sensitive asparagine hydroxylase FIH [54], results in its degradation by the proteasome or in transcriptional inactivation, respectively. Redox regulation of transcription factors is summarized in Fig. 5.
Fig. 4.
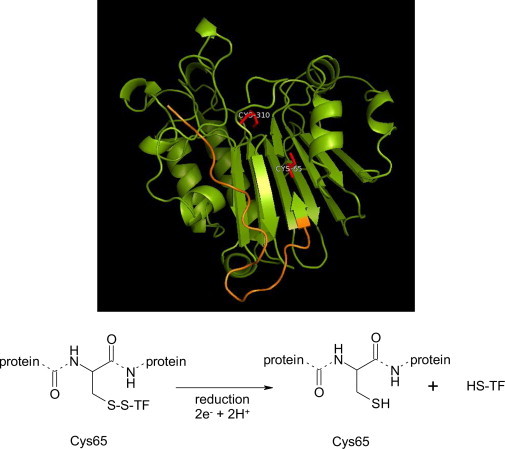
PyMOL representative image of the human isoform of the APE-1 endonuclease and its suggested mechanism of action. The redox N-terminal region of the enzyme is marked in orange. This section of the molecule is an evolutionary novelty for the human isoform. Redox components of the catalytic pocket, in particular Cys65 and Cys310 are highlighted in red. Based on the current knowledge of APE-1 enzymatic activity, we are suggesting a mechanism that emphasizes the importance of Cys65 for the process. TF – transcription factor. Concept according to Luo et al. [106]. Protein database file (1BIX) was used for rendering the structure with PyMOL (Schrödinger, USA).
Fig. 5.
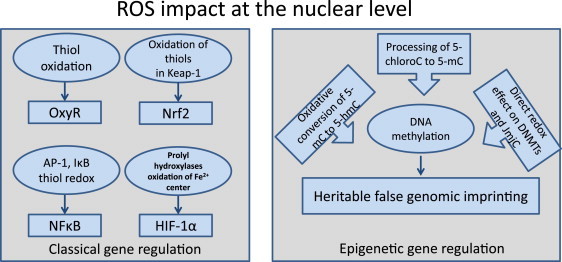
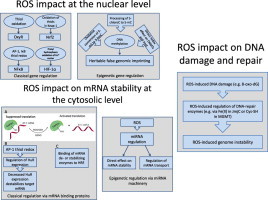
ROS effects at the nuclear level. Reactive oxygen species can display their regulatory effect on the classical gene regulatory machinery and on epigenetic processes. One of the prominent pathways attributed to oxidative stress is thiol oxidation, which is involved in OxyR, NF-κB and Keap1 signaling. Oxygen sensing prolyl hydroxylases represent another class of redox-dependent enzymes. For example, epigenetic involvement of ROS has been attributed to oxidative conversion of 5-mC to 5-hmC.
Likewise, ROS (in most cases hydrogen peroxide) and RNS (e.g. peroxynitrite via oxidation or nitric oxide via S-nitrosation) can affect mRNA stability by redox modification of proteins that bind to AU-rich elements (AREs) in their 3′-untranslated (3′-UTR) region [100]. Examples are (i) glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which binds to AREs of endothelin-1 mRNA and decreases its half-life, whereas under oxidative stress conditions binding of GAPDH is inhibited by S-glutathionylation, leading to increased endothelin-1 protein expression [101]; (ii) ROS and RNS activate AP-1 leading to decreased expression of the mRNA binding protein HuR and accordingly to a shorter half-life of the mRNA of soluble guanylyl cyclase [102]; (iii) nitric oxide and the nitrovasodilator pentaerithrityl tetranitrate improve HuR binding to HO-1 mRNA, increasing its half-life and HO-1 protein expression [58,103]; (iv) mRNA binding proteins such as KSRP and TTP promote the decay of oxidative stress-regulated genes such as c-fos, c-jun, inducible nitric oxide synthase (iNOS), GM-CSF, COX-2, IL-3, TNF-α and IL-2, probably by competing with mRNA stabilizing proteins such as HuR [100]. At least for TTP, increased expression levels were reported in oxidative stress conditions [104]. Redox regulation of mRNA binding proteins is summarized in Fig. 6.
Fig. 6.
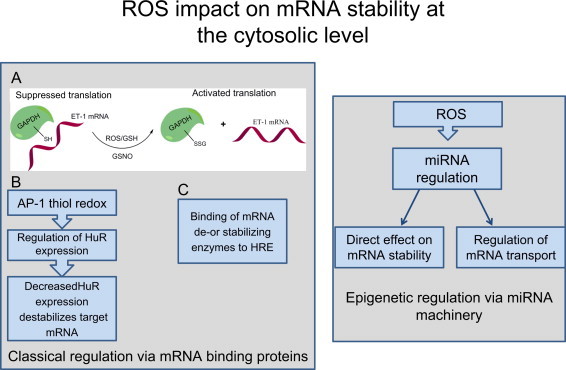
ROS impact on mRNA stability at the cytosolic level. Reactive oxygen species are involved in GAPDH signaling by directly altering its structure with the help of GSH or S-nitrosoglutathione (GSNO), and thus activate translation of endothelin-1 (ET-1) mRNA. AP-1 thiol redox regulation directly affects the gene regulating factor HuR by stability of its target mRNAs. ROS have been implicated in the regulation of miRNA pathways, altering mRNA stability and their transport inside the cytosol. HRE means hormone response element.
Redox regulation of DNA repair
A wide network of regulatory enzymes tasked with oxidation–reduction reactions exists in the nucleus. Major players maintaining the redox balance are glutathione (GSH) and thioredoxin-1 (Trx1), which often switch from cytosolic to nuclear localization [105]. Recent data indicate that these sulfhydryl biomolecules are not in a state of the chemical equilibrium, suggesting distinct targets [105]. Until lately, investigations of thiol/disulfide systems in DNA damage and repair have concentrated on determining their protective properties against ROS/RNS and only recently the role of redox signaling and the putative regulation of the DNA repair machinery through redox reactions has been explored [106]. Oxidative stress not only negatively impacts the DNA repair machinery by inhibiting a number of important DNA repair enzymes [9], but also increases the burden of oxidative DNA lesions such as single strand breaks, 8-nitro-dG and 8-oxo-dG [10].
In the past, nuclear GSH functions were studied in the context of DNA synthesis, management of the nuclear matrix organization and preservation of cysteine (Cys) residues on the DNA-binding motifs of nuclear proteins in a reduced state [107]. Recently though, there has been a shift in investigating involvement of GSH in the maintenance of the DNA repair machinery in the functional state. Major regulators of the DNA repair machinery are glutaredoxins (Grxs) [108]. These are thiol-disulfide oxidoreductases that perform GSH/GSSG oxidation–reduction reactions with Cys on selected proteins. Grxs are divided into two mechanistic groups, depending on the structure of their active motif: –Cys–Gly–Phe–Ser–, which are monothiol Grxs, and –Cys–Pro–Tyr–Cys– that are dithiol Grxs [109].
Thioredoxins are thiol-disulfide oxidoreductases of small molecular weight (12 kDa), which easily translocate into the nucleus. This class of enzymes is considered essential for overseeing and controlling redox signaling [110]. So far, two isoforms of mammalian thioredoxins have been detected, Trx1 and Trx2. Both of them tackle post-effects of oxidative stress in specific cellular compartments. Trx2 is predominantly detected in mitochondria, while Trx1 is found in the cytoplasm and nucleus. Upon detection of oxidative stress, Trx1 is transported from the cytoplasm to the nucleus to perform its antioxidant role [111].
GSH, Grx and Trx1 were reported to protect the nucleus from oxidative DNA damage by regulating induction of DNA repair enzymes [112], among other chemical reactions through S-glutathionylation. Trx1 was directly responsible for the reduction of the major DNA endonuclease APE1 [113]. APE1 has truly unique functions in the nucleus, as it has both repair and redox regulatory domains [114]. Redox function of this protein is a new evolutionary addition found only in mammals, while repair function is conserved through evolution. The endonuclease APE1 is an essential member of nucleotide excision repair, performing cleavage of the flanking DNA ends to prepare them for polymerase and ligase activities. Another redox active, ubiquitously expressed enzyme, GAPDH, has been reported to associate directly with APE1 via its active site cysteine 152, thereby reducing APE1 to its functional state and enabling restoration of its endonuclease activity [115]. A completely unexpected facet of APE1 is its redox activity that affects many major transcription factors such as p53 [116], HIF-1α [117], AP1 [114], NF-κB [118], cAMP response element-binding protein (CREB) [119] and many others. This redox activity of APE1 depends on Cys65, which was confirmed by multiple amino acid substitution studies [120]. The exact mode of interaction of this enzyme with such structurally distinct proteins is still enigmatic. Additional data suggest direct redox regulation of the DNA repair function of APE1 via oxidation/reduction of cysteine residues (e.g. Cys310 adjacent to a crucial His309 in the active site) [106,121].
One of the initiating enzymes of the non-homologs end joining (NHEJ) repair process is the Ku family of enzymes. Ku binding to the flanking ends of the broken DNA enables formation of the repair complex, consisting of several DNA dependent protein kinases and ligases [122]. Up to now, factors that were contributing to the enhanced or reduced affiliation of Ku proteins to DNA are still not fully determined, although it was shown that Ku enzymes bind to DNA in a sequence-independent manner and move along the DNA molecule in an ATP-independent manner [123]. It became apparent that the redox state of the enzyme is a fundamental driving force for its binding to DNA. Oxidative stress is able to inhibit Ku’s ability to colocalize with DNA and triggers specific conformational changes in the enzyme upon its oxidation that are increasing its Koff rate [124]. Currently, glutathione is thought to be the most likely regulator of the Ku enzyme redox state.
O-6-methylguanine-DNA methyltransferase (MGMT) is one of the most specific DNA repair enzymes and is recruited to remove alkyl modifications from guanine. In addition, this protein is redox regulated. In particular, MGMT is inhibited by S-nitrosation at the reactive, essential cysteine that performs the nucleophilic attack at alkylated nucleobases and confers dealkylation by alkyl transfer, thereby inactivating MGMT itself (suicide enzymatic reaction) [125]. It is very likely that MGMT is also inhibited in the setting of oxidative stress by direct oxidation or by S-glutathionylation.
Redox regulation of epigenetic pathways
There is increasing support for the concept that redox signaling and oxidative stress can affect the epigenetic regulation of genes by changes in the function of histones and DNA modifying enzymes, thereby altering the phenotype of cells [126]. Likewise, epigenetic changes may alter the redox environment of a cell. Accordingly, epigenetic targeting of these different processes may provide new therapeutic strategies to prevent oxidative stress disorders including Alzheimer's disease, Parkinson's disease, cardiovascular disease and cancer [127–129]. Numerous examples of oxidative stress-associated disease show characteristic patterns of epigenetic changes. Neprilysin, a neutral endopeptidase (NEP), which degrades the amyloid beta peptide and thereby prevents its misfolding and aggregation in brain tissue, was found hypermethylated in Alzheimer’s disease. Neprilysin hypermethylation explained the decreased protein expression levels observed during the disease [130]. A vital interplay between oxidative stress and epigenetic changes (e.g. chromatin remodeling) was identified in various models of alveolar inflammation in general and chronic lung disease in particular [131,132]. Especially, histone deacetylases (e.g. class III HDAC, SIRT1) were identified as redox-sensitive key players in the development and progression of chronic obstructive pulmonary disorder (COPD) [133]. Increased ROS and RNS formation in an inflammatory setting will also favor the development of lung cancer, most likely involving similar epigenetic changes [134]. Finally, prominent miRNAs are regulated by oxidative stress [127] and vise versa can regulate antioxidant pathways (e.g. Nrf2/HO-1), and may even act in a redox sensitive manner (“redoximiRs”), allowing adjustment of their action to the cellular redox state or disease-associated oxidative stress condition [135]. For example, levels of miR-141, miR-429 and miR-200 family members are increased by ROS, leading to endothelial cell apoptosis and senescence [136]; miR-141 and miR-200a may participate in ovarian tumorigenesis via an oxidative stress response [137]; the miR-29 family plays a role in lung fibrosis via regulation of DNA methylation and all of these miRs are regulated by oxidative stress [138,139]; miR-433 targets gene activation of γ-glutamyl-cysteine ligase (GCL), the rate-limiting enzyme in the synthesis of GSH, thus decreases GSH levels, and thereby leads to changes in cellular redox state and alterations of endothelial cell function and fibrotic processes [140]. Impact of redox regulation on miRNAs is summarized in Fig. 6.
Epigenetic changes are also directly linked to ROS. ROS levels, most likely hydrogen peroxide from NADPH oxidases Duox1/2 and upon dismutation of Nox2 (CYBB)-derived superoxide can be directly regulated by epigenetic mechanisms, for example downregulated by DNA hypermethylation and histone hypoacetylation at the genomic level, while ROS generated by these sources can lead to increased DNA methyltransferase (DNMT) and HDAC activity, which in turn may “silence” the DUOX1/2 and CYBB genes in a feedback fashion [141,142]. Recently, the upregulation of Nox4 by epigenetic mechanisms was reported for cellular senescence in lung fibroblasts [143]. Likewise, ROS derived from NADPH oxidase or other sources such as xanthine oxidase, mitochondrial electron transport chain, uncoupled •NO synthase or P450 enzymes can alter the activity of other target genes [141], for example of the tumor suppressor E-cadherin. E-cadherin is silenced by DNA hypermethylation and histone hypoacetylation by interaction with the transcription factor SNAIL, DNMT1 and HDAC1 under oxidative stress conditions, which is a common carcinogenic event [144]. In another example Nox5-derived ROS were responsible for genetic silencing of the p16 tumor suppressor [145]. Matrix metalloproteinase-1 (MMP-1) expression contributes to the pathogenesis of numerous degenerative diseases. MMP-1 activity is increased under oxidative stress conditions and further augmented by SOD2 overexpression due to proteasomal degradation of HDAC2 and accumulation of histone H3 acetylation marks in the MMP-1 promoter [146]. All of these redox regulatory changes were reversed by catalase overexpression. SOD2 deficiency resulted in significant decrease in global histone epigenetic marks such as 2meH3K4, 3meH3K9, 2meH3K27, 3meH3K27 and AcH3K9 [147]. Interestingly, SOD1 and GPx1 deficiency differentially regulate epigenetic pathways. SOD1 deficiency increases histone H3 acetylation and H3K4 methylation in the promoter region of pdx1 and pdx1 binding to the transcription factor FOXA2 in islet cells, whereas GPx1 deficiency upregulates these parameters. Double knockout of SOD1 and GPx1 in mice displays the phenotype of SOD1 deficiency [148]. The data suggest opposite roles of superoxide and hydrogen peroxide in these epigenetic pathways.
Histone deacetylases (HDACs) represent a group of epigenetically active enzymes catalyzing the deacetylation of histone lysine residues [149,150]. An appreciable portion of HDACs are prone to redox regulation, among them sirtuins [93,133,151,152]. While most HDACs contain an Zn(II) active site and catalyze the simple hydrolysis of the amide group, sirtuins link the deacetylation to NAD+ hydrolysis yielding nicotinamide (an inhibitor of sirtuins), 2′-O-acetyl-ADP-ribose and the deacetylated histone as products. This allows specific regulation of sirtuins in a NAD+/NADH (or free nicotinamide)-dependent fashion, making them perfect sensors of the metabolic and energetic state of cells and regulators of a wide range of cellular processes like transcription, (oxidative) stress resistance, aging, inflammation, apoptosis, metabolic control, but also circadian clocks and mitochondrial biogenesis. Sirtuin-1, for example, can be regulated by S-glutathionylation, a redox regulatory mechanism that largely depends on the cellular redox state [153]. Glutaredoxin-2 mediated deglutathionylation increased sirtuin-1 activity under oxidative stress conditions [154] and a triple mutant of sirtuin-1 (with 3 cysteines exchanged by serines: C61S, C318S, C613S) was resistant to oxidative stress-induced inhibition of the enzyme [155]. In general, sirtuins can be regulated by toxic aldehydes such as acrolein or 4-hydroxynonenal (4-HNE) formed under oxidative stress conditions [156–158]. Of note, ROS increase the levels of 4-HNE and both species are strong activators of p38 MAPK, which regulates certain lysine acetyltransferases (e.g. KAT3A/KAT3B (CBP/p300)) [133,159].
Jumonji C (JmjC) domain-containing histone demethylases catalyze the demethylation of histone lysine residues in a two-step mechanism. First, a hydroxylation reaction occurs that converts the methyl group through Fe(II) and 2-oxoglutarate-dependent oxidation, followed by spontaneous elimination of formaldehyde and release of the demethylated lysine [160]. JmjC enzymes belong to the family of Fe(II)- and 2-oxoglutarate-dependent oxygenases similar to PHDs. Thus, ascorbate is an essential cofactor for this class of enzymes to maintain the ferrous (Fe(II)) state required for enzymatic activity [160,161] (Fig. 5). JmjC enzymes also require molecular oxygen for their function. However, in hypoxic conditions JmjC is strongly induced by HIF-1α and overall activity of JmjC is maintained in hypoxia despite the reduced oxygen tension [162]. The facilitated ferrous state of the JmjC enzyme under hypoxic conditions could also contribute to its conserved activity. Similar considerations with regard to vitamin C and oxygen tension apply for TET DNA hydroxylases, which catalyze the oxidation of methyl groups in the 5′-position of cytosine in DNA [160]. Although TET DNA hydroxylases might belong to the DNA repair machinery, they also directly change the pattern of DNA methylation.
In summary, both JmjC histone lysine demethylases and TET DNA hydroxylases confer direct epigenetic changes in histones and DNA. Both enzymes rely on a ferrous catalytic center that is easily oxidized to ferric state by superoxide, hydrogen peroxide and peroxynitrite as well as other ROS. Ascorbate was discussed for both enzymes as an essential cofactor to keep the ferrous catalytic center in the reduced state. Since ascorbate levels change with the cellular redox state this process could represent a redox switch for enzymatic activity of JmjC histone lysine demethylases and TET DNA hydroxylases. JmjC domain histone demethylases are not only regulated by cellular redox state, but can also confer antioxidant protection as shown for Ndy1 under oxidative stress conditions and cellular apoptosis [163]. In contrast, the mammalian flavin-dependent histone lysine-specific demethylase 1 (LSD1) generates hydrogen peroxide during its catalytic cycle, which can oxidize nearby deoxyguanidine (dG) nucleobases, leading to inefficient gene activation when not immediately coupled with the base excision repair (BER) enzyme OGG1 that removes the 8-oxo-dG lesions [164]. An example is LSD1/BER-dependent estrogen receptor (ER)-induced activation of the bcl-2 gene, which in the absence of OGG1 would be suppressed by LSD1-mediated oxidation of the nearby guanidine bases to 8-oxo-dG during H3K9me2 demethylation [165].
In addition, direct chemical effects of ROS and RNS on nucleotides are known. It was postulated that oxidative conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC) under oxidative stress conditions [166] changes the DNA methylation pattern by suppressing the activity of the maintenance methyltransferase DNMT1 and the methyl-CpG binding protein 2 (MeCP2) [167,168]. Peroxides can lead via peroxidase-catalyzed mechanisms in the presence of halogenide ions to chemical modification of nucleobases such as 5-chlorocytosine, which mimics 5-mC and can therefore induce inappropriate DNMT1 methylation within CpG sequences [169], thus providing a mechanistic link between oxidative stress, inflammation and epigenetic changes. Of note, these chemical DNA modifications and associated epigenetic changes are heritable [170]. Impact of ROS on false epigenetic imprinting is summarized in Fig. 5.
Clinical impact
Therapeutic targeting and biomarkers of the classical gene regulation and DNA repair machinery
Even though major concepts presented in the current review are describing knowledge gained from fundamental research, translation into clinic has been achieved in particular with regard to cancer as the best studied disease related to DNA damage and epigenetics [150,171]. For example with regard to the DNA repair machinery, many inhibitory approaches have been taken as an adjuvant or targeted anti-cancer therapy. A classical example of targeting the DNA repair machinery is inhibition of poly (ADP-ribose) polymerase 1 (PARP1) [172]. So far, two major concepts highlighted the clinical usefulness of PARP inhibition as an anti-neoplasia treatment. The first strategy uses a combination therapy approach, since PARP1 is an essential element of the DNA damage response (DDR), which cancer cells activate to resist DNA-damaging drugs. Also, as PARP1 is implicated in many DDR processes, inhibitors of this enzyme are expected to have multiple target points and will kill cancer cells more effectively [173]. Epigenetic modifications are becoming part of an emerging field of versatile biomarkers. For example, ten‐eleven translocation methylcytosine dioxygenase 2 (TET2) is mutated in more than 15% of all myeloid malignancies, potentially serving as a disease prognostic factor [174]. TET enzymes hydroxylate 5-methylcytosine to 5-hydroxymethylcytosine, thus being an import enzyme involved in DNA demethylation. In fact, low levels of 5-hydroxymethylcytosine have been found in the majority of patients with mutated Tet2. As of now, there are already 2 medical formulations (decitabine and azacitidine), approved by the FDA, that are able to remove 5-methylcytosine marks from DNA, leading to its hypomethylated state. These two drugs have been successfully used in the treatment of myelodysplastic syndrome and acute myeloid leukemia [175,176]. Considering transcription markers, the estrogen receptor (ER) α is the most common prognostic marker of breast cancer [36]. Tumors expressing ER are hormone-dependent and in the majority of cases will respond to endocrine therapies such as the selective estrogen receptor antagonist tamoxifen [177] or fulvestrant an estrogen receptor antagonist which works both by down-regulating and by degrading the estrogen receptor. Another anti-cancer therapeutic approach targets the nuclear glutaredoxins (Grx) and thioredoxins (Trx) reductases. Grx2b and thioredoxin reductase-1 mRNA levels were significantly elevated in patients with acute myeloid leukemia, suggesting that these enzymes may represent a promising target for anti-neoplasia treatments [178]. Further, nuclear forms of glutathione transferase (GST) were directly linked to anti-cancer drug resistance. Accumulation of this enzyme was observed in cell lines treated with cisplatin, 5-fluorouracil and several other compounds. Inhibition of GST might increase sensitization of the selective cancer lines to the treatment [179].
Therapeutic targeting of the epigenetic gene regulation pathways
Pharmacological targeting of epigenetic pathways is a two-edged sword. The DNA methyltransferase (DNMT) inhibitor zebularine as well as histone deacetylase (HDAC) inhibitors induce histone hypomethylation and hyperacetylation, which rescued SOD2 expression [180], but may also increase CYBB and DUOX1/2 expression [141]. Nevertheless, there are a number of successful therapeutic interventions at the epigenetic level [150,171] and most of them are also related to changes in the cellular redox state, as many of these drugs display indirect antioxidant properties. The DNMT inhibitor, 5-aza-2′-deoxycytidine, activates Keap1 and Nrf2 resulting in the up-regulation of highly beneficial HO-1 [181]. Further, 5-aza-2′-deoxycytidine prevented hypermethylation of CpG islands in an enhancer region of intron 2 and in the promoter region of SOD2 in the setting of pulmonary arterial hypertension (PAH), a disease with very high mortality [182]. The most relevant DNMTs are upregulated in PAH (Fig. 5). Normalized SOD2 levels prevented in PAH animals and patients the development of a pulmonary artery smooth muscle cell phenotype that was characterized by increased cell proliferation and decreased apoptosis. This phenotype was mimicked by deletion of SOD using siRNA and improvement was observed upon treatment of PAH cells with the SOD mimic MnTBAP, a manganese-porphyrin. Additionally, the 12-O-tetradecanoylphorbol-13-acetate-inducible expression of extracellular superoxide dismutase (SOD3) was increased by the DNMT inhibitor 5-azacytidine, whereas SOD3 was suppressed by pretreatment with the histone acetyltransferase (HAT) inhibitors, CPTH2 or garcinol, strongly supporting epigenetic regulation of this important antioxidant enzyme [183]. These findings were reinforced by data from pulmonary arteries, where HDAC inhibition by trichostatin A increased AcH3K27 and 3meH3K4 marks in the promoter region of SOD3 which correlated with 10-fold up-regulation at the protein level [184]. Interestingly, expression of the NADPH oxidase Nox4 was decreased by 95% by trichostatin A treatment, suggesting synergistic effects on ROS producing and detoxifying enzymes. Overall, the epigenetic regulation of SOD3 expression might have an important impact on cancer development and tumorigenesis [185] and similar considerations apply for epigenetic control of SOD2 expression in cancer [180]. The observation that the term “epigenetic therapy” is often related to cancer therapy is probably based on the fact that a major part of research in this area is directed to the epigenetic modulation of cancer. The natural alkaloid theophylline, a drug previously used to treat COPD, can induce HDAC2 and can have beneficial effects in COPD patients at low doses [186,187]. Likewise, the stilbenoid resveratrol causes changes in gene expression via miRNAs and epigenetic modifications [188–190], but also improves the function of the DNA repair machinery, thereby contributing to genome stability [191]. The broad effects of natural phytochemical antioxidants on epigenetic pathways are discussed in detail elsewhere and contribute to the novel disciplines nutrigenetics and nutrigenomics [192].
Besides these specific examples of epigenetically active drugs, pharmacological drugs with pleiotropic antioxidant effects that are often based on epigenetic mechanisms have been described (e.g. statins [193–195]). An interesting candidate is the nitrovasodilator pentaerithrityl tetranitrate (PETN), the only organic nitrate devoid of serious side effects such as endothelial dysfunction, nitrate tolerance and oxidative stress [196]. The molecular explanation for the beneficial effects of PETN is the induction of heme oxygenase-1 [197] in an Nrf2-dependent fashion [198]. Likewise, PETN therapy leads to the regulation of more than 1200 genes and up-regulates several cardio-protective genes [199], but also affects several miRNAs. More recently, PETN was shown to induce heritable epigenetic changes including enhanced AcH3K27 and 3meH3K4 with subsequent gene activation of endothelial nitric oxide synthase (eNOS), SOD2, glutathione peroxidase-1 (GPx-1) and HO-1 that cause blood pressure reduction in female offspring of PETN-treated hypertensive rats [200]. Of note, these beneficial effects were neither shared by other organic nitrates nor by classical •NO donors. Similarly, nitroglycerin-induced nitrate tolerance was improved by in vivo treatment with the HDAC pan-inhibitor trichostatin A (TSA), the specific HDAC1/3 inhibitor MS-27-275, as well as the histone acetylase (HAT) activator SPV106, whereas nitrate tolerance was mimicked by in vivo treatment with the HAT inhibitor anacardic acid (ANAC) [201]. In summary, these data support an essential role of histone acetylation in the vasodilatory properties of nitroglycerin and loss of these properties due to histone hypoacetylation in the setting of nitrate tolerance.
Conclusions
In conclusion, an ever increasing wealth of data indicates that classical DNA related pathways like gene regulation by transcription factors, DNA damage/repair mechanisms, as well as by more recently acknowledged pathways modulated by epigenetic mechanisms, are affected by the redox state of the organism and subsequently modulate genome stability. We are only starting to shed light on the intricate signaling cascades and regulatory domains of redox-dependent enzymes that have pronounced effects on aforementioned genetic and epigenetic systems as well as DNA damage/repair. An important consequence of current research into how redox biology is altering the overall genomic and epigenetic organization, is hopefully the emergence of more sophisticated and more specific clinical treatments for disease conditions, including chronic inflammation/fibrosis, cancer, neurological, metabolic and cardiovascular disorders [202].
Conflict of interest
None.
Acknowledgments
The present work was supported by continuous funding by Stufe1 and NMFZ programs of the Johannes Gutenberg-University Mainz as well as University Medical Center Mainz (A.D.), Science Foundation Ireland (U.G.K.) and German Research Foundation (DFG GO709/4-5 to A.G.). All authors of this review were supported by the European Cooperation in Science and Technology (COST Action BM1203/EU-ROS). Yuliya Mikhed holds a stipend from the International PhD Program on the “Dynamics of Gene Regulation, Epigenetics and DNA Damage Response” from the Institute of Molecular Biology gGmbH, (Mainz, Germany) funded by the Boehringer Ingelheim Foundation.
References
- 1.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362(6422):709–715. doi: 10.1038/362709a0. 8469282 [DOI] [PubMed] [Google Scholar]
- 2.Alizadeh E., Orlando T.M., Sanche L. Biomolecular damage induced by ionizing radiation: the direct and indirect effects of low-energy electrons on DNA. Annu. Rev. Phys. Chem. 2015;66:379–398. doi: 10.1146/annurev-physchem-040513-103605. 25580626 [DOI] [PubMed] [Google Scholar]
- 3.Ames B.N., Shigenaga M.K., Hagen T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA. 1993;90(17):7915–7922. doi: 10.1073/pnas.90.17.7915. 8367443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehta A., Haber J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014;6(9):a016428. doi: 10.1101/cshperspect.a016428. 25104768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madabhushi R., Pan L., Tsai L.H. DNA damage and its links to neurodegeneration. Neuron. 2014;83(2):266–282. doi: 10.1016/j.neuron.2014.06.034. 25033177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haber J.E. DNA recombination: the replication connection. Trends Biochem. Sci. 1999;24(7):271–275. doi: 10.1016/s0968-0004(99)01413-9. 10390616 [DOI] [PubMed] [Google Scholar]
- 7.Vilenchik M.M., Knudson A.G. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA. 2003;100(22):12871–12876. doi: 10.1073/pnas.2135498100. 14566050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cadet J., Douki T., Ravanat J.L. Oxidatively generated damage to cellular DNA by UVB and UVA radiation. Photochem. Photobiol. 2015;91(1):140–155. doi: 10.1111/php.12368. 25327445 [DOI] [PubMed] [Google Scholar]
- 9.Liu R.H., Hotchkiss J.H. Potential genotoxicity of chronically elevated nitric oxide: a review. Mutat. Res. 1995;339(2):73–89. doi: 10.1016/0165-1110(95)90004-7. 7791803 [DOI] [PubMed] [Google Scholar]
- 10.Guetens G., De Boeck G., Highley M., van Oosterom A.T., de Bruijn E.A. Oxidative DNA damage: biological significance and methods of analysis. Crit. Rev. Clin. Lab. Sci. 2002;39(4–5):331–457. doi: 10.1080/10408360290795547. 12385502 [DOI] [PubMed] [Google Scholar]
- 11.Sharma V., Collins L.B., Clement J.M., Zhang Z., Nakamura J., Swenberg J.A. Molecular dosimetry of endogenous and exogenous O(6)-methyl-dG and N7-methyl-G adducts following low dose [D3]-methylnitrosourea exposures in cultured human cells. Chem. Res. Toxicol. 2014;27(4):480–482. doi: 10.1021/tx5000602. 24628573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomaszowski K.H., Aasland D., Margison G.P., Williams E., Pinder S.I., Modesti M., Fuchs R.P., Kaina B. The bacterial alkyltransferase-like (eATL) protein protects mammalian cells against methylating agent-induced toxicity. DNA Repair (Amst.) 2015;28:14–20. doi: 10.1016/j.dnarep.2015.01.009. 25703834 [DOI] [PubMed] [Google Scholar]
- 13.Yousef M.I., Hussien H.M. Cisplatin-induced renal toxicity via tumor necrosis factor-α, interleukin 6, tumor suppressor P53, DNA damage, xanthine oxidase, histological changes, oxidative stress and nitric oxide in rats: protective effect of ginseng. Food Chem. Toxicol. 2015;78:17–25. doi: 10.1016/j.fct.2015.01.014. 25640527 [DOI] [PubMed] [Google Scholar]
- 14.Gederaas O.A., Søgaard C.D., Viset T., Bachke S., Bruheim P., Arum C.J., Otterlei M. Increased anticancer efficacy of intravesical mitomycin C therapy when combined with a PCNA targeting peptide. Transl. Oncol. 2014;7(6):812–823. doi: 10.1016/j.tranon.2014.10.005. 25500092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montariello D., Troiano A., Di Girolamo D., Beneke S., Calabrò V., Quesada P. Effect of poly(ADP-ribose)polymerase and DNA topoisomerase I inhibitors on the p53/p63-dependent survival of carcinoma cells. Biochem. Pharmacol. 2015;94(3):212–219. doi: 10.1016/j.bcp.2015.01.012. 25667043 [DOI] [PubMed] [Google Scholar]
- 16.Kim Y.J., Wilson D.M., 3rd Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012;5(1):3–13. doi: 10.2174/1874467211205010003. 22122461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee D.H., Liu Y., Lee H.W., Xia B., Brice A.R., Park S.H., Balduf H., Dominy B.N., Cao W. A structural determinant in the uracil DNA glycosylase superfamily for the removal of uracil from adenine/uracil base pairs. Nucleic Acids Res. 2015;43(2):1081–1089. doi: 10.1093/nar/gku1332. 25550433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ba X., Aguilera-Aguirre L., Sur S., Boldogh I. 8-Oxoguanine DNA glycosylase-1-driven DNA base excision repair: role in asthma pathogenesis. Curr. Opin. Allergy Clin. Immunol. 2015;15(1):89–97. doi: 10.1097/ACI.0000000000000135. 25486379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarry-Adkins J.L., Martin-Gronert M.S., Fernandez-Twinn D.S., Hargreaves I., Alfaradhi M.Z., Land J.M., Aiken C.E., Ozanne S.E. Poor maternal nutrition followed by accelerated postnatal growth leads to alterations in DNA damage and repair, oxidative and nitrosative stress, and oxidative defense capacity in rat heart. FASEB J. 2013;27(1):379–390. doi: 10.1096/fj.12-218685. 23024373 [DOI] [PubMed] [Google Scholar]
- 20.Hegde M.L., Hegde P.M., Bellot L.J., Mandal S.M., Hazra T.K., Li G.M., Boldogh I., Tomkinson A.E., Mitra S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA. 2013;110(33):E3090–E3099. doi: 10.1073/pnas.1304231110. 23898192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park J.S., Kim H.L., Kim Y.J., Weon J.I., Sung M.K., Chung H.W., Seo Y.R. Human AP endonuclease 1: a potential marker for the prediction of environmental carcinogenesis risk. Oxid Med. Cell. Longev. 2014;2014:730301. doi: 10.1155/2014/730301. 25243052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dizdaroglu M. Oxidatively induced DNA damage: mechanisms, repair and disease. Cancer Lett. 2012;327(1–2):26–47. doi: 10.1016/j.canlet.2012.01.016. 22293091 [DOI] [PubMed] [Google Scholar]
- 23.Brenerman B.M., Illuzzi J.L., Wilson D.M., 3rd Base excision repair capacity in informing healthspan. Carcinogenesis. 2014;35(12):2643–2652. doi: 10.1093/carcin/bgu225. 25355293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dijk M., Typas D., Mullenders L., Pines A. Insight in the multilevel regulation of NER. Exp. Cell Res. 2014;329(1):116–123. doi: 10.1016/j.yexcr.2014.08.010. 25128816 [DOI] [PubMed] [Google Scholar]
- 25.Ziani S., Nagy Z., Alekseev S., Soutoglou E., Egly J.M., Coin F. Sequential and ordered assembly of a large DNA repair complex on undamaged chromatin. J. Cell Biol. 2014;206(5):589–598. doi: 10.1083/jcb.201403096. 25154395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamarthapu V., Nudler E. Rethinking transcription coupled DNA repair. Curr. Opin. Microbiol. 2015;24:15–20. doi: 10.1016/j.mib.2014.12.005. 25596348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scherly D., Nouspikel T., Corlet J., Ucla C., Bairoch A., Clarkson S.G. Complementation of the DNA repair defect in xeroderma pigmentosum group G cells by a human cDNA related to yeast RAD2. Nature. 1993;363(6425):182–185. doi: 10.1038/363182a0. 8483504 [DOI] [PubMed] [Google Scholar]
- 28.Muro Y., Sugiura K., Mimori T., Akiyama M. DNA mismatch repair enzymes: genetic defects and autoimmunity. Clin. Chim. Acta. 2015;442:102–109. doi: 10.1016/j.cca.2015.01.014. 25619773 [DOI] [PubMed] [Google Scholar]
- 29.Iyer R.R., Pluciennik A., Napierala M., Wells R.D. DNA triplet repeat expansion and mismatch repair. Annu. Rev. Biochem. 2015;84(1) doi: 10.1146/annurev-biochem-060614-034010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bak S.T., Sakellariou D., Pena-Diaz J. The dual nature of mismatch repair as antimutator and mutator: for better or for worse. Front. Genet. 2014;5:287. doi: 10.3389/fgene.2014.00287. 25191341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.House N.C., Koch M.R., Freudenreich C.H. Chromatin modifications and DNA repair: beyond double-strand breaks. Front. Genet. 2014;5:296. doi: 10.3389/fgene.2014.00296. 25250043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemaître C., Soutoglou E. DSB (Im)mobility and DNA repair compartmentalization in mammalian cells. J. Mol. Biol. 2015;427(3):652–658. doi: 10.1016/j.jmb.2014.11.014. 25463437 [DOI] [PubMed] [Google Scholar]
- 33.Grundy G.J., Moulding H.A., Caldecott K.W., Rulten S.L. One ring to bring them all − the role of Ku in mammalian non-homologous end joining. DNA Repair (Amst.) 2014;17:30–38. doi: 10.1016/j.dnarep.2014.02.019. 24680220 [DOI] [PubMed] [Google Scholar]
- 34.Jeggo P.A., Downs J.A. Roles of chromatin remodellers in DNA double strand break repair. Exp. Cell Res. 2014;329(1):69–77. doi: 10.1016/j.yexcr.2014.09.023. 25278484 [DOI] [PubMed] [Google Scholar]
- 35.Chandra V., Bhagyaraj E., Parkesh R., Gupta P. Transcription factors and cognate signalling cascades in the regulation of autophagy. Biol. Rev. 2015 doi: 10.1111/brv.12177. [DOI] [PubMed] [Google Scholar]
- 36.Frasor J., El-Shennawy L., Stender J.D., Kastrati I. NFκB affects estrogen receptor expression and activity in breast cancer through multiple mechanisms. Mol. Cell. Endocrinol. 2014 doi: 10.1016/j.mce.2014.09.013. 25450861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z., Zhou S., Jiang X., Wang Y.H., Li F., Wang Y.G., Zheng Y., Cai L. The role of the Nrf2/Keap1 pathway in obesity and metabolic syndrome. Rev. Endocr. Metab. Disord. 2015;16(1):35–45. doi: 10.1007/s11154-014-9305-9. 25540093 [DOI] [PubMed] [Google Scholar]
- 38.Vrtačnik P., Ostanek B., Mencej-Bedrač S., Marc J. The many faces of estrogen signaling. Biochem. Med. 2014;24(3):329–342. doi: 10.11613/BM.2014.035. 25351351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Semenza G.L. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. doi: 10.1016/j.cell.2012.01.021. 22304911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ariumi Y. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front. Genet. 2014;5:423. doi: 10.3389/fgene.2014.00423. 25538732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manavathi B., Samanthapudi V.S., Gajulapalli V.N. Estrogen receptor coregulators and pioneer factors: the orchestrators of mammary gland cell fate and development. Front. Cell Dev. Biol. 2014;2:34. doi: 10.3389/fcell.2014.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mothes J., Busse D., Kofahl B., Wolf J. Sources of dynamic variability in NF-κB signal transduction: a mechanistic model. BioEssays. 2015;37(4):452–462. doi: 10.1002/bies.201400113. 25640005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohamed M.R., McFadden G. NFkB inhibitors: strategies from poxviruses. Cell Cycle. 2009;8(19):3125–3132. doi: 10.4161/cc.8.19.9683. 19738427 [DOI] [PubMed] [Google Scholar]
- 44.Hayden M.S., Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26(3):203–234. doi: 10.1101/gad.183434.111. 22302935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schenk H., Klein M., Erdbrügger W., Dröge W., Schulze-Osthoff K. Distinct effects of thioredoxin and antioxidants on the activation of transcription factors NF-kappa B and AP-1. Proc. Natl. Acad. Sci. USA. 1994;91(5):1672–1676. doi: 10.1073/pnas.91.5.1672. 8127864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schreck R., Albermann K., Baeuerle P.A. Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells (a review) Free Radic. Res. Commun. 1992;17(4):221–237. doi: 10.3109/10715769209079515. 1473734 [DOI] [PubMed] [Google Scholar]
- 47.Meyer M., Schreck R., Baeuerle P.A. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993;12(5):2005–2015. doi: 10.1002/j.1460-2075.1993.tb05850.x. 8491191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uruno A., Yagishita Y., Yamamoto M. The Keap1–Nrf2 system and diabetes mellitus. Arch. Biochem. Biophys. 2015;566:76–84. doi: 10.1016/j.abb.2014.12.012. 25528168 [DOI] [PubMed] [Google Scholar]
- 49.Bhakkiyalakshmi E., Sireesh D., Rajaguru P., Paulmurugan R., Ramkumar K.M. The emerging role of redox-sensitive Nrf2–Keap1 pathway in diabetes. Pharmacol. Res. 2015;91:104–114. doi: 10.1016/j.phrs.2014.10.004. 25447793 [DOI] [PubMed] [Google Scholar]
- 50.Kaelin W.G., Jr., Ratcliffe P.J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 2008;30(4):393–402. doi: 10.1016/j.molcel.2008.04.009. 18498744 [DOI] [PubMed] [Google Scholar]
- 51.Pan Y., Mansfield K.D., Bertozzi C.C., Rudenko V., Chan D.A., Giaccia A.J., Simon M.C. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell. Biol. 2007;27(3):912–925. doi: 10.1128/MCB.01223-06. 17101781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerald D., Berra E., Frapart Y.M., Chan D.A., Giaccia A.J., Mansuy D., Pouysségur J., Yaniv M., Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118(6):781–794. doi: 10.1016/j.cell.2004.08.025. 15369676 [DOI] [PubMed] [Google Scholar]
- 53.Diebold I., Flügel D., Becht S., Belaiba R.S., Bonello S., Hess J., Kietzmann T., Görlach A. The hypoxia-inducible factor-2alpha is stabilized by oxidative stress involving NOX4. Antioxid. Redox Signal. 2010;13(4):425–436. doi: 10.1089/ars.2009.3014. 20039838 [DOI] [PubMed] [Google Scholar]
- 54.Masson N., Singleton R.S., Sekirnik R., Trudgian D.C., Ambrose L.J., Miranda M.X., Tian Y.M., Kessler B.M., Schofield C.J., Ratcliffe P.J. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 2012;13(3):251–257. doi: 10.1038/embor.2012.9. 22310300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sumbayev V.V., Budde A., Zhou J., Brüne B. HIF-1alpha protein as a target for S-nitrosation. FEBS Lett. 2003;535(1–3):106–112. doi: 10.1016/s0014-5793(02)03887-5. 12560087 [DOI] [PubMed] [Google Scholar]
- 56.Chowdhury R., Flashman E., Mecinović J., Kramer H.B., Kessler B.M., Frapart Y.M., Boucher J.L., Clifton I.J., McDonough M.A., Schofield C.J. Studies on the reaction of nitric oxide with the hypoxia-inducible factor prolyl hydroxylase domain 2 (EGLN1) J. Mol. Biol. 2011;410(2):268–279. doi: 10.1016/j.jmb.2011.04.075. 21601578 [DOI] [PubMed] [Google Scholar]
- 57.Daiber A., Schildknecht S., Müller J., Kamuf J., Bachschmid M.M., Ullrich V. Chemical model systems for cellular nitrous(yl)ation reactions. Free Radic. Biol. Med. 2009;47(4):458–467. doi: 10.1016/j.freeradbiomed.2009.05.019. 19477267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daiber A., Oelze M., Wenzel P., Bollmann F., Pautz A., Kleinert H. Heme oxygenase-1 induction and organic nitrate therapy: beneficial effects on endothelial dysfunction, nitrate tolerance, and vascular oxidative stress. Int. J. Hypertens. 2012;2012:842632. doi: 10.1155/2012/842632. 22506100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Smet C., Lurquin C., Lethé B., Martelange V., Boon T. DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol. Cell. Biol. 1999;19(11):7327–7335. doi: 10.1128/mcb.19.11.7327. 10523621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. 11782440 [DOI] [PubMed] [Google Scholar]
- 61.Moen E.L., Mariani C.J., Zullow H., Jeff-Eke M., Litwin E., Nikitas J.N., Godley L.A. New themes in the biological functions of 5-methylcytosine and 5-hydroxymethylcytosine. Immunol. Rev. 2015;263(1):36–49. doi: 10.1111/imr.12242. 25510270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weissman J., Naidu S., Bjornsson H.T. Abnormalities of the DNA methylation mark and its machinery: an emerging cause of neurologic dysfunction. Semin. Neurol. 2014;34(3):249–257. doi: 10.1055/s-0034-1386763. 25192503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wan J., Oliver V.F., Wang G., Zhu H., Zack D.J., Merbs S.L., Qian J. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genomics. 2015;16:49. doi: 10.1186/s12864-015-1271-4. 25652663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Le J.M., Squarize C.H., Castilho R.M. Histone modifications: targeting head and neck cancer stem cells. World J. Stem Cells. 2014;6(5):511–525. doi: 10.4252/wjsc.v6.i5.511. 25426249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. 17320507 [DOI] [PubMed] [Google Scholar]
- 66.Dekker F.J., Haisma H.J. Histone acetyl transferases as emerging drug targets. Drug Discov. Today. 2009;14(19–20):942–948. doi: 10.1016/j.drudis.2009.06.008. 19577000 [DOI] [PubMed] [Google Scholar]
- 67.Gallinari P., Di Marco S., Jones P., Pallaoro M., Steinkühler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17(3):195–211. doi: 10.1038/sj.cr.7310149. 17325692 [DOI] [PubMed] [Google Scholar]
- 68.Di Lorenzo A., Bedford M.T. Histone arginine methylation. FEBS Lett. 2011;585(13):2024–2031. doi: 10.1016/j.febslet.2010.11.010. 21074527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Völkel P., Angrand P.O. The control of histone lysine methylation in epigenetic regulation. Biochimie. 2007;89(1):1–20. doi: 10.1016/j.biochi.2006.07.009. 16919862 [DOI] [PubMed] [Google Scholar]
- 70.Mancuso M., Matassa D.S., Conte M., Colella G., Rana G., Fucci L., Piscopo M. H3K4 histone methylation in oral squamous cell carcinoma. Acta Biochim. Pol. 2009;56(3):405–410. 19753335 [PubMed] [Google Scholar]
- 71.Rodriguez A., Griffiths-Jones S., Ashurst J.L., Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14(10A):1902–1910. doi: 10.1101/gr.2722704. 15364901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang J., Gong L., Tan Y., Hui R., Wang Y. Hypertensive epigenetics: from DNA methylation to microRNAs. J. Hum. Hypertens. 2015 doi: 10.1038/jhh.2014.132. [DOI] [PubMed] [Google Scholar]
- 73.Jinek M., Doudna J.A. A three-dimensional view of the molecular machinery of RNA interference. Nature. 2009;457(7228):405–412. doi: 10.1038/nature07755. 19158786 [DOI] [PubMed] [Google Scholar]
- 74.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. 14744438 [DOI] [PubMed] [Google Scholar]
- 75.Villegas V.E., Zaphiropoulos P.G. Neighboring Gene Regulation by antisense Long non-coding RNAs. Int. J. Mol. Sci. 2015;16(2):3251–3266. doi: 10.3390/ijms16023251. 25654223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen A., Chen D., Daiber A., Faraci F., Li H., Rembold C., Laher I. Free radical biology of the cardiovascular system. Clin. Sci. 2012;123(2):73–91. doi: 10.1042/CS20110562. [DOI] [PubMed] [Google Scholar]
- 77.Yorek M.A. The role of oxidative stress in diabetic vascular and neural disease. Free Radic. Res. 2003;37(5):471–480. doi: 10.1080/1071576031000083161. 12797466 [DOI] [PubMed] [Google Scholar]
- 78.Griendling K.K., FitzGerald G.A. Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo monitoring of ROS. Circulation. 2003;108(16):1912–1916. doi: 10.1161/01.CIR.0000093660.86242.BB. 14568884 [DOI] [PubMed] [Google Scholar]
- 79.Sies H. Oxidative Stress: Oxidants and Antioxidants. Academic Press; London, UK: 1991. [DOI] [PubMed] [Google Scholar]
- 80.Karbach S., Wenzel P., Waisman A., Munzel T., Daiber A. eNOS uncoupling in cardiovascular diseases – the role of oxidative stress and inflammation. Curr. Pharm. Des. 2014;20(22):3579–3594. doi: 10.2174/13816128113196660748. 24180381 [DOI] [PubMed] [Google Scholar]
- 81.Münzel T., Gori T., Bruno R.M., Taddei S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010;31(22):2741–2748. doi: 10.1093/eurheartj/ehq396. 20974801 [DOI] [PubMed] [Google Scholar]
- 82.Nezis I.P., Stenmark H. p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxid. Redox Signal. 2012;17(5):786–793. doi: 10.1089/ars.2011.4394. 22074114 [DOI] [PubMed] [Google Scholar]
- 83.Ischiropoulos H., Beckman J.S. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J. Clin. Investig. 2003;111(2):163–169. doi: 10.1172/JCI17638. 12531868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Griendling K.K., FitzGerald G.A. Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation. 2003;108(17):2034–2040. doi: 10.1161/01.CIR.0000093661.90582.c4. 14581381 [DOI] [PubMed] [Google Scholar]
- 85.A. Daiber, M. Oelze, S. Daub, S. Steven, A. Schuff, S. Kroller-Schon, M. Hausding, P. Wenzel, E. Schulz, T. Gori, T. Munzel, Vascular redox signaling, redox switches in endothelial nitric oxide synthase and endothelial dysfunction, in: I. Laher (Ed.), Systems Biology of Free Radicals and Antioxidants, Springer-Verlag, Berlin Heidelberg, 2014, pp. 1177–1211.
- 86.Gori T., Münzel T. Oxidative stress and endothelial dysfunction: therapeutic implications. Ann. Med. 2011;43(4):259–272. doi: 10.3109/07853890.2010.543920. 21284528 [DOI] [PubMed] [Google Scholar]
- 87.Bjelakovic G., Nikolova D., Gluud L.L., Simonetti R.G., Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. J. Am. Med. Assoc. 2007;297(8):842–857. doi: 10.1001/jama.297.8.842. 17327526 [DOI] [PubMed] [Google Scholar]
- 88.Bjelakovic G., Nikolova D., Simonetti R.G., Gluud C. Antioxidant supplements for prevention of gastrointestinal cancers: a systematic review and meta-analysis. Lancet. 2004;364(9441):1219–1228. doi: 10.1016/S0140-6736(04)17138-9. 15464182 [DOI] [PubMed] [Google Scholar]
- 89.Lönn M.E., Dennis J.M., Stocker R. Actions of “antioxidants” in the protection against atherosclerosis. Free Radic. Biol. Med. 2012;53(4):863–884. doi: 10.1016/j.freeradbiomed.2012.05.027. 22664312 [DOI] [PubMed] [Google Scholar]
- 90.Rocha M., Apostolova N., Hernandez-Mijares A., Herance R., Victor V.M. Oxidative stress and endothelial dysfunction in cardiovascular disease: mitochondria-targeted therapeutics. Curr. Med. Chem. 2010;17(32):3827–3841. doi: 10.2174/092986710793205444. 20858217 [DOI] [PubMed] [Google Scholar]
- 91.Jazwa A., Cuadrado A. Targeting heme oxygenase-1 for neuroprotection and neuroinflammation in neurodegenerative diseases. Curr. Drug Targets. 2010;11(12):1517–1531. doi: 10.2174/1389450111009011517. 20704549 [DOI] [PubMed] [Google Scholar]
- 92.Wind S., Beuerlein K., Eucker T., Müller H., Scheurer P., Armitage M.E., Ho H., Schmidt H.H., Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br. J. Pharmacol. 2010;161(4):885–898. doi: 10.1111/j.1476-5381.2010.00920.x. 20860666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Adcock I.M., Cosio B., Tsaprouni L., Barnes P.J., Ito K. Redox regulation of histone deacetylases and glucocorticoid-mediated inhibition of the inflammatory response. Antioxid. Redox Signal. 2005;7(1–2):144–152. doi: 10.1089/ars.2005.7.144. 15650403 [DOI] [PubMed] [Google Scholar]
- 94.Haddad J.J. Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell Signal. 2002;14(11):879–897. doi: 10.1016/s0898-6568(02)00053-0. 12220615 [DOI] [PubMed] [Google Scholar]
- 95.Brigelius-Flohé R., Flohé L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011;15(8):2335–2381. doi: 10.1089/ars.2010.3534. 21194351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hausladen A., Privalle C.T., Keng T., DeAngelo J., Stamler J.S. Nitrosative stress: activation of the transcription factor OxyR. Cell. 1996;86(5):719–729. doi: 10.1016/s0092-8674(00)80147-6. 8797819 [DOI] [PubMed] [Google Scholar]
- 97.Fourquet S., Guerois R., Biard D., Toledano M.B. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J. Biol. Chem. 2010;285(11):8463–8471. doi: 10.1074/jbc.M109.051714. 20061377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wemmie J.A., Steggerda S.M., Moye-Rowley W.S. The Saccharomyces cerevisiae AP-1 protein discriminates between oxidative stress elicited by the oxidants H2O2 and diamide. J. Biol. Chem. 1997;272(12):7908–7914. doi: 10.1074/jbc.272.12.7908. 9065458 [DOI] [PubMed] [Google Scholar]
- 99.Dehne N., Brüne B. Sensors, transmitters, and targets in mitochondrial oxygen shortage-a hypoxia-inducible factor relay story. Antioxid. Redox Signal. 2014;20(2):339–352. doi: 10.1089/ars.2012.4776. 22794181 [DOI] [PubMed] [Google Scholar]
- 100.Abdelmohsen K., Kuwano Y., Kim H.H., Gorospe M. Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress: implications for cellular senescence. Biol. Chem. 2008;389(3):243–255. doi: 10.1515/BC.2008.022. 18177264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rodríguez-Pascual F., Redondo-Horcajo M., Magán-Marchal N., Lagares D., Martínez-Ruiz A., Kleinert H., Lamas S. Glyceraldehyde-3-phosphate dehydrogenase regulates endothelin-1 expression by a novel, redox-sensitive mechanism involving mRNA stability. Mol. Cell. Biol. 2008;28(23):7139–7155. doi: 10.1128/MCB.01145-08. 18809573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Klöss S., Srivastava R., Mülsch A. Down-regulation of soluble guanylyl cyclase expression by cyclic AMP is mediated by mRNA-stabilizing protein HuR. Mol. Pharmacol. 2004;65(6):1440–1451. doi: 10.1124/mol.65.6.1440. 15155837 [DOI] [PubMed] [Google Scholar]
- 103.Kuwano Y., Rabinovic A., Srikantan S., Gorospe M., Demple B. Analysis of nitric oxide-stabilized mRNAs in human fibroblasts reveals HuR-dependent heme oxygenase 1 upregulation. Mol. Cell. Biol. 2009;29(10):2622–2635. doi: 10.1128/MCB.01495-08. 19289500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stoecklin G., Stubbs T., Kedersha N., Wax S., Rigby W.F., Blackwell T.K., Anderson P. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 2004;23(6):1313–1324. doi: 10.1038/sj.emboj.7600163. 15014438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Go Y.M., Jones D.P. Redox control systems in the nucleus: mechanisms and functions. Antioxid. Redox Signal. 2010;13(4):489–509. doi: 10.1089/ars.2009.3021. 20210649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Luo M., He H., Kelley M.R., Georgiadis M.M. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid. Redox Signal. 2010;12(11):1247–1269. doi: 10.1089/ars.2009.2698. 19764832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Suthanthiran M., Anderson M.E., Sharma V.K., Meister A. Glutathione regulates activation-dependent DNA synthesis in highly purified normal human T lymphocytes stimulated via the CD2 and CD3 antigens. Proc. Natl. Acad. Sci. USA. 1990;87(9):3343–3347. doi: 10.1073/pnas.87.9.3343. 1970635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000;2(4):811–820. doi: 10.1089/ars.2000.2.4-811. 11213485 [DOI] [PubMed] [Google Scholar]
- 109.Lillig C.H., Berndt C., Holmgren A. Glutaredoxin systems. Biochim. Biophys. Acta. 2008;1780(11):1304–1317. doi: 10.1016/j.bbagen.2008.06.003. 18621099 [DOI] [PubMed] [Google Scholar]
- 110.Holmgren A., Johansson C., Berndt C., Lönn M.E., Hudemann C., Lillig C.H. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem. Soc. Trans. 2005;33(6):1375–1377. doi: 10.1042/BST0331375. 16246122 [DOI] [PubMed] [Google Scholar]
- 111.Hansen J.M., Zhang H., Jones D.P. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol. Sci. Off. J. Soc. Toxicol. 2006;91:643–650. doi: 10.1093/toxsci/kfj175. [DOI] [PubMed] [Google Scholar]
- 112.Yamawaki H., Berk B.C. Thioredoxin: a multifunctional antioxidant enzyme in kidney, heart and vessels. Curr. Opin. Nephrol. Hypertens. 2005;14(2):149–153. doi: 10.1097/00041552-200503000-00010. 15687841 [DOI] [PubMed] [Google Scholar]
- 113.Hirota K., Matsui M., Iwata S., Nishiyama A., Mori K., Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc. Natl. Acad. Sci. USA. 1997;94(8):3633–3638. doi: 10.1073/pnas.94.8.3633. 9108029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xanthoudakis S., Miao G.G., Curran T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl. Acad. Sci. USA. 1994;91(1):23–27. doi: 10.1073/pnas.91.1.23. 7506414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Azam S., Jouvet N., Jilani A., Vongsamphanh R., Yang X., Yang S., Ramotar D. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J. Biol. Chem. 2008;283(45):30632–30641. doi: 10.1074/jbc.M801401200. 18776186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gaiddon C., Moorthy N.C., Prives C. Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J. 1999;18(20):5609–5621. doi: 10.1093/emboj/18.20.5609. 10523305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Loboda A., Stachurska A., Dorosz J., Zurawski M., Wegrzyn J., Kozakowska M., Jozkowicz A., Dulak J. HIF-1 attenuates Ref-1 expression in endothelial cells: reversal by siRNA and inhibition of geranylgeranylation. Vascul. Pharmacol. 2009;51(2–3):133–139. doi: 10.1016/j.vph.2009.05.005. 19524065 [DOI] [PubMed] [Google Scholar]
- 118.Hirota K., Murata M., Sachi Y., Nakamura H., Takeuchi J., Mori K., Yodoi J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 1999;274(39):27891–27897. doi: 10.1074/jbc.274.39.27891. 10488136 [DOI] [PubMed] [Google Scholar]
- 119.Grösch S., Kaina B. Transcriptional activation of apurinic/apyrimidinic endonuclease (ape, Ref-1) by oxidative stress requires CREB. Biochem. Biophys. Res. Commun. 1999;261(3):859–863. doi: 10.1006/bbrc.1999.1125. 10441516 [DOI] [PubMed] [Google Scholar]
- 120.Walker L.J., Robson C.N., Black E., Gillespie D., Hickson I.D. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol. Cell. Biol. 1993;13(9):5370–5376. doi: 10.1128/mcb.13.9.5370. 8355688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kelley M.R., Parsons S.H. Redox regulation of the DNA repair function of the human AP endonuclease Ape1/ref-1. Antioxid. Redox Signal. 2001;3(4):671–683. doi: 10.1089/15230860152543014. 11554453 [DOI] [PubMed] [Google Scholar]
- 122.Bennett S.M., Neher T.M., Shatilla A., Turchi J.J. Molecular analysis of Ku redox regulation. BMC Mol. Biol. 2009;10:86. doi: 10.1186/1471-2199-10-86. 19715578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yoo S., Dynan W.S. Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res. 1999;27(24):4679–4686. doi: 10.1093/nar/27.24.4679. 10572166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Andrews B.J., Lehman J.A., Turchi J.J. Kinetic analysis of the Ku-DNA binding activity reveals a redox-dependent alteration in protein structure that stimulates dissociation of the Ku-DNA complex. J. Biol. Chem. 2006;281(19):13596–13603. doi: 10.1074/jbc.M512787200. 16537541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wei W., Yang Z., Tang C.H., Liu L. Targeted deletion of GSNOR in hepatocytes of mice causes nitrosative inactivation of O6-alkylguanine-DNA alkyltransferase and increased sensitivity to genotoxic diethylnitrosamine. Carcinogenesis. 2011;32(7):973–977. doi: 10.1093/carcin/bgr041. 21385828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cyr A.R., Domann F.E. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid. Redox Signal. 2011;15(2):551–589. doi: 10.1089/ars.2010.3492. 20919933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.A.M. Baird, K.J. O’Byrne, S.G. Gray, Reactive oxygen species and reactive nitrogen species in epigenetic modifications, in: I. Laher (Ed.). Systems Biology of Free Radicals and Antioxidants, Springer-Verlag, Berlin Heidelberg, 2014, pp. 437–455.
- 128.Hitchler M.J., Domann F.E. Redox regulation of the epigenetic landscape in cancer: a role for metabolic reprogramming in remodeling the epigenome. Free Radic. Biol. Med. 2012;53(11):2178–2187. doi: 10.1016/j.freeradbiomed.2012.09.028. 23022407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kim G.H., Ryan J.J., Archer S.L. The role of redox signaling in epigenetics and cardiovascular disease. Antioxid. Redox Signal. 2013;18(15):1920–1936. doi: 10.1089/ars.2012.4926. 23480168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Heyn H., Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat. Rev. Genet. 2012;13(10):679–692. doi: 10.1038/nrg3270. 22945394 [DOI] [PubMed] [Google Scholar]
- 131.Rahman I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. J. Biochem. Mol. Biol. 2003;36(1):95–109. doi: 10.5483/bmbrep.2003.36.1.095. 12542980 [DOI] [PubMed] [Google Scholar]
- 132.Rahman I. Dietary polyphenols mediated regulation of oxidative stress and chromatin remodeling in inflammation. Nutr. Rev. 2008;66(Suppl. 1):S42–S45. doi: 10.1111/j.1753-4887.2008.00067.x. 18673489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rajendrasozhan S., Yang S.R., Edirisinghe I., Yao H., Adenuga D., Rahman I. Deacetylases and NF-kappaB in redox regulation of cigarette smoke-induced lung inflammation: epigenetics in pathogenesis of COPD. Antioxid. Redox Signal. 2008;10(4):799–811. doi: 10.1089/ars.2007.1938. 18220485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yang I.A., Relan V., Wright C.M., Davidson M.R., Sriram K.B., Savarimuthu Francis S.M., Clarke B.E., Duhig E.E., Bowman R.V., Fong K.M. Common pathogenic mechanisms and pathways in the development of COPD and lung cancer. Expert Opin. Ther. Targets. 2011;15(4):439–456. doi: 10.1517/14728222.2011.555400. 21284573 [DOI] [PubMed] [Google Scholar]
- 135.Cheng X., Ku C.H., Siow R.C. Regulation of the Nrf2 antioxidant pathway by microRNAs: new players in micromanaging redox homeostasis. Free Radic. Biol. Med. 2013;64:4–11. doi: 10.1016/j.freeradbiomed.2013.07.025. 23880293 [DOI] [PubMed] [Google Scholar]
- 136.Magenta A., Cencioni C., Fasanaro P., Zaccagnini G., Greco S., Sarra-Ferraris G., Antonini A., Martelli F., Capogrossi M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011;18(10):1628–1639. doi: 10.1038/cdd.2011.42. 21527937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mateescu B., Batista L., Cardon M., Gruosso T., de Feraudy Y., Mariani O., Nicolas A., Meyniel J.P., Cottu P., Sastre-Garau X., Mechta-Grigoriou F. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat. Med. 2011;17(12):1627–1635. doi: 10.1038/nm.2512. 22101765 [DOI] [PubMed] [Google Scholar]
- 138.O’Byrne K.J., Barr M.P., Gray S.G. The role of epigenetics in resistance to cisplatin chemotherapy in lung cancer. Cancers (Basel) 2011;3(1):1426–1453. doi: 10.3390/cancers3011426. 24212667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Thulasingam S., Massilamany C., Gangaplara A., Dai H., Yarbaeva S., Subramaniam S., Riethoven J.J., Eudy J., Lou M., Reddy J. miR-27b*, an oxidative stress-responsive microRNA modulates nuclear factor-kB pathway in RAW 264.7 cells. Mol. Cell. Biochem. 2011;352(1–2):181–188. doi: 10.1007/s11010-011-0752-2. 21350856 [DOI] [PubMed] [Google Scholar]
- 140.Espinosa-Diez C., Fierro-Fernández M., Sánchez-Gómez F., Rodríguez-Pascual F., Alique M., Ruiz-Ortega M., Beraza N., Martínez-Chantar M.L., Fernández-Hernando C., Lamas S. Targeting of gamma-glutamyl-cysteine ligase by miR-433 reduces glutathione biosynthesis and promotes TGF-beta-dependent fibrogenesis. Antioxid. Redox Signal. 2014 doi: 10.1089/ars.2014.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hayes P., Knaus U.G. Balancing reactive oxygen species in the epigenome: NADPH oxidases as target and perpetrator. Antioxid. Redox Signal. 2013;18(15):1937–1945. doi: 10.1089/ars.2012.4895. 23126619 [DOI] [PubMed] [Google Scholar]
- 142.Kang K.A., Zhang R., Kim G.Y., Bae S.C., Hyun J.W. Epigenetic changes induced by oxidative stress in colorectal cancer cells: methylation of tumor suppressor RUNX3. Tumour Biol. 2012;33(2):403–412. doi: 10.1007/s13277-012-0322-6. 22274925 [DOI] [PubMed] [Google Scholar]
- 143.Sanders Y.Y., Liu H., Liu G., Thannickal V.J. Epigenetic mechanisms regulate NADPH oxidase-4 expression in cellular senescence. Free Radic. Biol. Med. 2015;79:197–205. doi: 10.1016/j.freeradbiomed.2014.12.008. 25526894 [DOI] [PubMed] [Google Scholar]
- 144.Lim S.O., Gu J.M., Kim M.S., Kim H.S., Park Y.N., Park C.K., Cho J.W., Park Y.M., Jung G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology. 2008;135(6):2128–2140. doi: 10.1053/j.gastro.2008.07.027. 18801366 [DOI] [PubMed] [Google Scholar]
- 145.Hong J., Resnick M., Behar J., Wang L.J., Wands J., DeLellis R.A., Souza R.F., Spechler S.J., Cao W. Acid-induced p16 hypermethylation contributes to development of esophageal adenocarcinoma via activation of NADPH oxidase NOX5-S. Am. J. Physiol. Gastrointest. Liver Physiol. 2010;299(3):G697–G706. doi: 10.1152/ajpgi.00186.2010. 20576920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bartling T.R., Subbaram S., Clark R.R., Chandrasekaran A., Kar S., Melendez J.A. Redox-sensitive gene-regulatory events controlling aberrant matrix metalloproteinase-1 expression. Free Radic. Biol. Med. 2014;74:99–107. doi: 10.1016/j.freeradbiomed.2014.06.017. 24973648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Case A.J., Madsen J.M., Motto D.G., Meyerholz D.K., Domann F.E. Manganese superoxide dismutase depletion in murine hematopoietic stem cells perturbs iron homeostasis, globin switching, and epigenetic control in erythrocyte precursor cells. Free Radic. Biol. Med. 2013;56:17–27. doi: 10.1016/j.freeradbiomed.2012.11.018. 23219873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang X., Vatamaniuk M.Z., Roneker C.A., Pepper M.P., Hu L.G., Simmons R.A., Lei X.G. Knockouts of SOD1 and GPX1 exert different impacts on murine islet function and pancreatic integrity. Antioxid. Redox Signal. 2011;14(3):391–401. doi: 10.1089/ars.2010.3302. 20586612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Verdin E., Ott M. 50 Years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015;16(4):258–264. doi: 10.1038/nrm3931. 25549891 [DOI] [PubMed] [Google Scholar]
- 150.Falkenberg K.J., Johnstone R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014;13(9):673–691. doi: 10.1038/nrd4360. 25131830 [DOI] [PubMed] [Google Scholar]
- 151.Radak Z., Koltai E., Taylor A.W., Higuchi M., Kumagai S., Ohno H., Goto S., Boldogh I. Redox-regulating sirtuins in aging, caloric restriction, and exercise. Free Radic. Biol. Med. 2013;58:87–97. doi: 10.1016/j.freeradbiomed.2013.01.004. 23339850 [DOI] [PubMed] [Google Scholar]
- 152.Escobar J., Pereda J., López-Rodas G., Sastre J. Redox signaling and histone acetylation in acute pancreatitis. Free Radic. Biol. Med. 2012;52(5):819–837. doi: 10.1016/j.freeradbiomed.2011.11.009. 22178977 [DOI] [PubMed] [Google Scholar]
- 153.Zee R.S., Yoo C.B., Pimentel D.R., Perlman D.H., Burgoyne J.R., Hou X., McComb M.E., Costello C.E., Cohen R.A., Bachschmid M.M. Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid. Redox Signal. 2010;13(7):1023–1032. doi: 10.1089/ars.2010.3251. 20392170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bräutigam L., Jensen L.D., Poschmann G., Nyström S., Bannenberg S., Dreij K., Lepka K., Prozorovski T., Montano S.J., Aktas O., Uhlén P., Stühler K., Cao Y., Holmgren A., Berndt C. Glutaredoxin regulates vascular development by reversible glutathionylation of sirtuin 1. Proc. Natl. Acad. Sci. USA. 2013;110(50):20057–20062. doi: 10.1073/pnas.1313753110. 24277839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shao D., Fry J.L., Han J., Hou X., Pimentel D.R., Matsui R., Cohen R.A., Bachschmid M.M. A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress. J. Biol. Chem. 2014;289(11):7293–7306. doi: 10.1074/jbc.M113.520403. 24451382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Caito S., Rajendrasozhan S., Cook S., Chung S., Yao H., Friedman A.E., Brookes P.S., Rahman I. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010;24(9):3145–3159. doi: 10.1096/fj.09-151308. 20385619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Doyle K., Fitzpatrick F.A. Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J. Biol. Chem. 2010;285(23):17417–17424. doi: 10.1074/jbc.M109.089250. 20385560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fritz K.S., Galligan J.J., Smathers R.L., Roede J.R., Shearn C.T., Reigan P., Petersen D.R. 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chem. Res. Toxicol. 2011;24(5):651–662. doi: 10.1021/tx100355a. 21449565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Vatsyayan R., Kothari H., Pendurthi U.R., Rao L.V. 4-Hydroxy-2-nonenal enhances tissue factor activity in human monocytic cells via p38 mitogen-activated protein kinase activation-dependent phosphatidylserine exposure. Arterioscler. Thromb. Vasc. Biol. 2013;33(7):1601–1611. doi: 10.1161/ATVBAHA.113.300972. 23640483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Monfort A., Wutz A. Breathing-in epigenetic change with vitamin C. EMBO Rep. 2013;14(4):337–346. doi: 10.1038/embor.2013.29. 23492828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang T., Chen K., Zeng X., Yang J., Wu Y., Shi X., Qin B., Zeng L., Esteban M.A., Pan G., Pei D. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell. 2011;9(6):575–587. doi: 10.1016/j.stem.2011.10.005. 22100412 [DOI] [PubMed] [Google Scholar]
- 162.Chervona Y., Costa M. The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic. Biol. Med. 2012;53(5):1041–1047. doi: 10.1016/j.freeradbiomed.2012.07.020. 22841757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Polytarchou C., Pfau R., Hatziapostolou M., Tsichlis P.N. The JmjC domain histone demethylase Ndy1 regulates redox homeostasis and protects cells from oxidative stress. Mol. Cell. Biol. 2008;28(24):7451–7464. doi: 10.1128/MCB.00688-08. 18838535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li J., Braganza A., Sobol R.W. Base excision repair facilitates a functional relationship between guanine oxidation and histone demethylation. Antioxid. Redox Signal. 2013;18(18):2429–2443. doi: 10.1089/ars.2012.5107. 23311711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Perillo B., Ombra M.N., Bertoni A., Cuozzo C., Sacchetti S., Sasso A., Chiariotti L., Malorni A., Abbondanza C., Avvedimento E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319(5860):202–206. doi: 10.1126/science.1147674. 18187655 [DOI] [PubMed] [Google Scholar]
- 166.Chia N., Wang L., Lu X., Senut M.C., Brenner C., Ruden D.M. Hypothesis: environmental regulation of 5-hydroxymethylcytosine by oxidative stress. Epigenetics. 2011;6(7):853–856. doi: 10.4161/epi.6.7.16461. 21617369 [DOI] [PubMed] [Google Scholar]
- 167.Valinluck V., Tsai H.H., Rogstad D.K., Burdzy A., Bird A., Sowers L.C. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Res. 2004;32(14):4100–4108. doi: 10.1093/nar/gkh739. 15302911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Valinluck V., Sowers L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67(3):946–950. doi: 10.1158/0008-5472.CAN-06-3123. 17283125 [DOI] [PubMed] [Google Scholar]
- 169.Valinluck V., Sowers L.C. Inflammation-mediated cytosine damage: a mechanistic link between inflammation and the epigenetic alterations in human cancers. Cancer Res. 2007;67(12):5583–5586. doi: 10.1158/0008-5472.CAN-07-0846. 17575120 [DOI] [PubMed] [Google Scholar]
- 170.Lao V.V., Herring J.L., Kim C.H., Darwanto A., Soto U., Sowers L.C. Incorporation of 5-chlorocytosine into mammalian DNA results in heritable gene silencing and altered cytosine methylation patterns. Carcinogenesis. 2009;30(5):886–893. doi: 10.1093/carcin/bgp060. 19279184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Azad N., Zahnow C.A., Rudin C.M., Baylin S.B. The future of epigenetic therapy in solid tumours − lessons from the past. Nat. Rev. Clin. Oncol. 2013;10(5):256–266. doi: 10.1038/nrclinonc.2013.42. 23546521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lupo B., Trusolino L. Inhibition of poly(ADP-ribosyl)ation in cancer: old and new paradigms revisited. Biochim. Biophys. Acta. 2014;1846(1):201–215. doi: 10.1016/j.bbcan.2014.07.004. 25026313 [DOI] [PubMed] [Google Scholar]
- 173.Curtin N.J. PARP inhibitors for cancer therapy. Expert Rev. Mol. Med. 2005;7(04):1–20. doi: 10.1017/S146239940500904X. [DOI] [PubMed] [Google Scholar]
- 174.Delhommeau F., Dupont S., Della Valle V., James C., Trannoy S., Massé A., Kosmider O., Le Couedic J.P., Robert F., Alberdi A., Lécluse Y., Plo I., Dreyfus F.J., Marzac C., Casadevall N., Lacombe C., Romana S.P., Dessen P., Soulier J., Viguié F., Fontenay M., Vainchenker W., Bernard O.A. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009;360(22):2289–2301. doi: 10.1056/NEJMoa0810069. 19474426 [DOI] [PubMed] [Google Scholar]
- 175.Kantarjian H.M., Thomas X.G., Dmoszynska A., Wierzbowska A., Mazur G., Mayer J., Gau J.P., Chou W.C., Buckstein R., Cermak J., Kuo C.Y., Oriol A., Ravandi F., Faderl S., Delaunay J., Lysak D., Minden M., Arthur C. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J. Clin. Oncol. 2012;30(21):2670–2677. doi: 10.1200/JCO.2011.38.9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fenaux P., Mufti G.J., Hellstrom-Lindberg E., Santini V., Finelli C., Giagounidis A., Schoch R., Gattermann N., Sanz G., List A., Gore S.D., Seymour J.F., Bennett J.M., Byrd J., Backstrom J., Zimmerman L., McKenzie D., Beach C., Silverman L.R., International Vidaza High-Risk MDS Survival Study Group Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–232. doi: 10.1016/S1470-2045(09)70003-8. 19230772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nass N., Kalinski T. Tamoxifen resistance: from cell culture experiments towards novel biomarkers. Pathol. Res. Pract. 2015;211(3):189–197. doi: 10.1016/j.prp.2015.01.004. 25666016 [DOI] [PubMed] [Google Scholar]
- 178.Olm E., Jönsson-Videsäter K., Ribera-Cortada I., Fernandes A.P., Eriksson L.C., Lehmann S., Rundlöf A.K., Paul C., Björnstedt M. Selenite is a potent cytotoxic agent for human primary AML cells. Cancer Lett. 2009;282(1):116–123. doi: 10.1016/j.canlet.2009.03.010. 19345479 [DOI] [PubMed] [Google Scholar]
- 179.Goto S., Kamada K., Soh Y., Ihara Y., Kondo T. Significance of nuclear glutathione S-transferase pi in resistance to anti-cancer drugs. Jpn. J. Cancer Res. 2002;93(9):1047–1056. doi: 10.1111/j.1349-7006.2002.tb02482.x. 12359059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cyr A.R., Hitchler M.J., Domann F.E. Regulation of SOD2 in cancer by histone modifications and CpG methylation: closing the loop between redox biology and epigenetics. Antioxid. Redox Signal. 2013;18(15):1946–1955. doi: 10.1089/ars.2012.4850. 22946823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wang R., An J., Ji F., Jiao H., Sun H., Zhou D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem. Biophys. Res. Commun. 2008;373(1):151–154. doi: 10.1016/j.bbrc.2008.06.004. 18555005 [DOI] [PubMed] [Google Scholar]
- 182.Archer S.L., Marsboom G., Kim G.H., Zhang H.J., Toth P.T., Svensson E.C., Dyck J.R., Gomberg-Maitland M., Thébaud B., Husain A.N., Cipriani N., Rehman J. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121(24):2661–2671. doi: 10.1161/CIRCULATIONAHA.109.916098. 20529999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kamiya T., Machiura M., Makino J., Hara H., Hozumi I., Adachi T. Epigenetic regulation of extracellular-superoxide dismutase in human monocytes. Free Radic. Biol. Med. 2013;61:197–205. doi: 10.1016/j.freeradbiomed.2013.04.013. 23602908 [DOI] [PubMed] [Google Scholar]
- 184.Zelko I.N., Folz R.J. Regulation of Oxidative Stress in Pulmonary Artery Endothelium: modulation of EC-SOD and NOX4 expression using HDAC Class I. Inhibitors. Am. J. Respir. Cell Mol. Biol. 2015 doi: 10.1165/rcmb.2014-0260OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Teoh-Fitzgerald M.L., Fitzgerald M.P., Zhong W., Askeland R.W., Domann F.E. Epigenetic reprogramming governs EcSOD expression during human mammary epithelial cell differentiation, tumorigenesis and metastasis. Oncogene. 2014;33(3):358–368. doi: 10.1038/onc.2012.582. 23318435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ito K., Lim S., Caramori G., Cosio B., Chung K.F., Adcock I.M., Barnes P.J. A molecular mechanism of action of theophylline: induction of histone deacetylase activity to decrease inflammatory gene expression. Proc. Natl. Acad. Sci. USA. 2002;99(13):8921–8926. doi: 10.1073/pnas.132556899. 12070353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ford P.A., Durham A.L., Russell R.E., Gordon F., Adcock I.M., Barnes P.J. Treatment effects of low-dose theophylline combined with an inhaled corticosteroid in COPD. Chest. 2010;137(6):1338–1344. doi: 10.1378/chest.09-2363. 20299628 [DOI] [PubMed] [Google Scholar]
- 188.Mukhopadhyay P., Pacher P., Das D.K. MicroRNA signatures of resveratrol in the ischemic heart. Ann. N. Y. Acad. Sci. 2011;1215:109–116. doi: 10.1111/j.1749-6632.2010.05866.x. 21261648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Farghali H., Kutinová Canová N., Lekić N. Resveratrol and related compounds as antioxidants with an allosteric mechanism of action in epigenetic drug targets. Physiol. Res. 2013;62(1):1–13. doi: 10.33549/physiolres.932434. 23173686 [DOI] [PubMed] [Google Scholar]
- 190.Xia N., Forstermann U., Li H. Resveratrol as a gene regulator in the vasculature. Curr. Pharm. Biotechnol. 2014;15(4):401–408. doi: 10.2174/1389201015666140711114450. 25022271 [DOI] [PubMed] [Google Scholar]
- 191.Gatz S.A., Wiesmüller L. Take a break – resveratrol in action on DNA. Carcinogenesis. 2008;29(2):321–332. doi: 10.1093/carcin/bgm276. 18174251 [DOI] [PubMed] [Google Scholar]
- 192.Malireddy S., Kotha S.R., Secor J.D., Gurney T.O., Abbott J.L., Maulik G., Maddipati K.R., Parinandi N.L. Phytochemical antioxidants modulate mammalian cellular epigenome: implications in health and disease. Antioxid. Redox Signal. 2012;17(2):327–339. doi: 10.1089/ars.2012.4600. 22404530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kodach L.L., Jacobs R.J., Voorneveld P.W., Wildenberg M.E., Verspaget H.W., van Wezel T., Morreau H., Hommes D.W., Peppelenbosch M.P., van den Brink G.R., Hardwick J.C. Statins augment the chemosensitivity of colorectal cancer cells inducing epigenetic reprogramming and reducing colorectal cancer cell ‘stemness’ via the bone morphogenetic protein pathway. Gut. 2011;60(11):1544–1553. doi: 10.1136/gut.2011.237495. 21551187 [DOI] [PubMed] [Google Scholar]
- 194.Ishikawa S., Hayashi H., Kinoshita K., Abe M., Kuroki H., Tokunaga R., Tomiyasu S., Tanaka H., Sugita H., Arita T., Yagi Y., Watanabe M., Hirota M., Baba H. Statins inhibit tumor progression via an enhancer of zeste homolog 2-mediated epigenetic alteration in colorectal cancer. Int. J. Cancer. 2014;135(11):2528–2536. doi: 10.1002/ijc.28672. 24346863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tikoo K., Patel G., Kumar S., Karpe P.A., Sanghavi M., Malek V., Srinivasan K. Tissue specific up regulation of ACE2 in rabbit model of atherosclerosis by atorvastatin: role of epigenetic histone modifications. Biochem. Pharmacol. 2015;93(3):343–351. doi: 10.1016/j.bcp.2014.11.013. 25482567 [DOI] [PubMed] [Google Scholar]
- 196.Münzel T., Daiber A., Gori T. Nitrate therapy: new aspects concerning molecular action and tolerance. Circulation. 2011;123(19):2132–2144. doi: 10.1161/CIRCULATIONAHA.110.981407. 21576678 [DOI] [PubMed] [Google Scholar]
- 197.Schuhmacher S., Wenzel P., Schulz E., Oelze M., Mang C., Kamuf J., Gori T., Jansen T., Knorr M., Karbach S., Hortmann M., Mäthner F., Bhatnagar A., Förstermann U., Li H., Münzel T., Daiber A. Pentaerythritol tetranitrate improves angiotensin II-induced vascular dysfunction via induction of heme oxygenase-1. Hypertension. 2010;55(4):897–904. doi: 10.1161/HYPERTENSIONAHA.109.149542. 20157049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Schuhmacher S., Oelze M., Bollmann F., Kleinert H., Otto C., Heeren T., Steven S., Hausding M., Knorr M., Pautz A., Reifenberg K., Schulz E., Gori T., Wenzel P., Münzel T., Daiber A. Vascular dysfunction in experimental diabetes is improved by pentaerithrityl tetranitrate but not isosorbide-5-mononitrate therapy. Diabetes. 2011;60(10):2608–2616. doi: 10.2337/db10-1395. 21844097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pautz A., Rauschkolb P., Schmidt N., Art J., Oelze M., Wenzel P., Förstermann U., Daiber A., Kleinert H. Effects of nitroglycerin or pentaerithrityl tetranitrate treatment on the gene expression in rat hearts: evidence for cardiotoxic and cardioprotective effects. Physiol. Genomics. 2009;38(2):176–185. doi: 10.1152/physiolgenomics.00035.2009. 19417013 [DOI] [PubMed] [Google Scholar]
- 200.Wu Z., Siuda D., Xia N., Reifenberg G., Daiber A., Münzel T., Förstermann U., Li H. Maternal treatment of spontaneously hypertensive rats with pentaerythritol tetranitrate reduces blood pressure in female offspring. Hypertension. 2015;65(1):232–237. doi: 10.1161/HYPERTENSIONAHA.114.04416. 25385760 [DOI] [PubMed] [Google Scholar]
- 201.Colussi C., Scopece A., Vitale S., Spallotta F., Mattiussi S., Rosati J., Illi B., Mai A., Castellano S., Sbardella G., Farsetti A., Capogrossi M.C., Gaetano C. P300/CBP associated factor regulates nitroglycerin-dependent arterial relaxation by N{varepsilon}-lysine acetylation of contractile proteins. Arterioscler. Thromb. Vasc. Biol. 2012;32(10):2435–2443. doi: 10.1161/ATVBAHA.112.254011. 22859492 [DOI] [PubMed] [Google Scholar]
- 202.Lawless M.W., O’Byrne K.J., Gray S.G. Oxidative stress induced lung cancer and COPD: opportunities for epigenetic therapy. J. Cell. Mol. Med. 2009;13(9A):2800–2821. doi: 10.1111/j.1582-4934.2009.00845.x. 19602054 [DOI] [PMC free article] [PubMed] [Google Scholar]