

Abstract
It is hard to assess experimentally the importance of microbial diversity in soil for the functioning of terrestrial ecosystems. An approach that is often used to make such assessment is the so-called dilution method. This method is based on the assumption that the biodiversity of the microbial community is reduced after dilution of a soil suspension and that the reduced diversity persists after incubation of more or less diluted inocula in soil. However, little is known about how the communities develop in soil after inoculation. In this study, serial dilutions of a soil suspension were made and reinoculated into the original soil previously sterilized by gamma irradiation. We determined the structure of the microbial communities in the suspensions and in the inoculated soils using 454-pyrosequencing of 16S rRNA genes. Upon dilution, several diversity indices showed that, indeed, the diversity of the bacterial communities in the suspensions decreased dramatically, with Proteobacteria as the dominant phylum of bacteria detected in all dilutions. The structure of the microbial community was changed considerably in soil, with Proteobacteria, Bacteroidetes, and Verrucomicrobia as the dominant groups in most diluted samples, indicating the importance of soil-related mechanisms operating in the assembly of the communities. We found unique operational taxonomic units (OTUs) even in the highest dilution in both the suspensions and the incubated soil samples. We conclude that the dilution approach reduces the diversity of microbial communities in soil samples but that it does not allow accurate predictions of the community assemblage during incubation of (diluted) suspensions in soil.
INTRODUCTION
The significance of biodiversity for terrestrial ecosystem processes continues to be a matter of much debate (1–3). Compared to the importance of plants and animals, the role of microbial biodiversity is still poorly understood. This lack of knowledge is of great concern as soil microbes, particularly bacteria, represent the major source of biodiversity in terrestrial ecosystems and are known to carry out numerous essential ecosystem functions, including nutrient cycling and facilitating plant nutrition (4).
The biggest obstacle to a better understanding of the importance of microbial biodiversity for the functioning of terrestrial ecosystems is the lack of sound experimental approaches to make directed and predictable changes in the diversity of microbial communities in soil. One of the most interesting approaches so far is the so-called dilution method. This method involves the inoculation of sterilized soils with more or less diluted inocula derived from suspensions of the same soil (4–13). However, previous studies were often limited by the depth and extent of the samples used and focused only on the structure of the microbial community after regrowth in the soil. As a consequence, they do not provide information about the community from which the different communities after incubation originated and the process of community assemblage. Therefore, these studies do not allow testing of the assumption that dilution mainly influences diversity through the reduction of the number of the less abundant, rare species. In reality, rare species in the original community may have become common after incubation or vice versa.
High-throughput next-generation sequencing technologies have allowed researchers to use deeper sampling depths by providing large numbers of reads by cost-effective means to detect microbial phylogenetic diversity (14). This has provided new insights into the details of microbial communities in natural ecosystems (15–17) and in the human body (18). One of the exciting possibilities provided by this technology is the ability to estimate accurately the assembly processes and structures of microbial communities, including the long tail of less-abundant microbes that is evident in graphs of relative abundances of microbial species, which may lead to a better understanding of functional biodiversity in soil.
The major aim of this study was to determine the changes and the associated variation in the composition of a soil microbial community brought about by inoculation of serial dilutions of suspensions of that soil and to detect how the microbial community structure develops during regrowth in soil. This analysis will allow evaluation of the suitability of the dilution approach as a tool for the manipulation of microbial biodiversity and for the separation of rare from abundant species. It will also lead to a better understanding of the selective pressure of the soil environment on the assembly of microbial communities. We addressed three basic questions: (i) Does the dilution procedure reduce the diversity of the microbial community after inoculation and subsequent incubation of soil suspensions in soil? (ii) Does the composition of the microbial community change during incubation in soil? (iii) Is the dilution procedure effective in separating more and less abundant species so as to allow an assessment of their specific roles? In order to answer these questions, we established a range of microbial communities through the inoculation of serial dilutions of microbial suspensions from nonsterilized soil samples into the same soil after sterilization.
MATERIALS AND METHODS
Soil sampling and treatment.
Thirty liters of soil was collected at a depth of around 15 cm from dune sandy soil in Meijendel, The Netherlands (52°9′N, 4°22′E). Soil organic matter content was 1.70% ± 0.13% (n = 7), soil pH was 7.4 ± 0.06 (n = 7), soil nitrogen content was 1.19% ± 0.10% (n = 5), and soil phosphorus content was 0.12% ± 0.01% (n = 5); all soil had a sandy texture, with more than 99% of the grains greater than 75 μm. The soil was sieved and homogenized, and aliquots of 500 g were stored in plastic bags (Whirl-Pak sampling bag, 720 ml; Sigma-Aldrich). The bags containing soil were gamma irradiation sterilized (>25 kGy; Isotron, Ede, The Netherlands). One bag of soil was kept separately to serve as an inoculum. Sterility was checked by spreading 0.5 g of sterilized soil from the inoculum bag onto tryptic soy broth (TSB) and potato-dextrose agar (PDA) media. No bacterial and fungal growth on agar plates for six replicates was observed in the sterilized soil samples after 6 days. Three gamma irradiation-sterilized soil bags were inoculated with autoclaved demineralized water to be used as controls. A subsample of the fresh soil was taken to determine soil moisture (24 h, 105°C).
Soil suspensions for inoculation were made by mixing 20 g of fresh soil and 190 ml of autoclaved demineralized water with a blender for 2 min. This procedure was repeated three times, and in between the blender was cooled down on ice for 2 min. This was called the 10−1 dilution. One hundred milliliters of the 10−1 dilution was transferred to a bottle containing 900 ml of autoclaved demineralized water, and the bottle was shaken by hand for 1 min. This procedure was repeated several times until 10−6 and 10−9 dilutions were made. Subsequently, 25 ml of each dilution was added to 500 g of soil in the bags, and additional autoclaved demineralized water was added to bring the moisture level of the inoculated soil to around 20%, which is roughly similar to the average level of the prevailing climatic conditions at the site from where the soil was taken. In total, 39 bags of soil (i.e., six replicate samples of the three dilutions in duplicate plus three controls) were used. We kept the six replicate samples (and the duplicates) per dilution separated throughout the experiment in order to be able to assess the variance caused by the dilution procedure. The remaining suspensions were centrifuged at 3,000 × g for 10 min at 4°C, and the pellets were stored at −20°C for further analysis. After inoculation, the soil bags were incubated at 20°C using sterilized cotton plug caps to ensure gas exchange. The soils were turned over regularly once a week to homogenize microbial growth. The aim was to reach similar microbial abundances with the different dilution treatments. After 8 weeks of incubation under laminar flow conditions, soil samples were taken to determine the microbial abundance in all treatments by quantitative real-time PCR (qPCR) using the Eub 338 (19) and Eub 518 (20) primer set for the 16S rRNA gene. Total DNA was extracted from the incubated soil using a MoBio Power Soil Extraction kit according to the supplier's instructions. Each 25-μl reaction mixture consisted of 12.5 μl of Sybr green mix (Bioline; GC-Biotech) with 4 mg/ml bovine serum albumin (BSA) in a total volume of 25 μl, 5 μM each primer, and 5 μl of template DNA (5 ng/μl). For bacteria, the standard curves were generated using a 10-fold dilution series from 108 to 103 of plasmid DNA. PCRs were run on a Rotor-Gene 3000 (Qiagen) and started with 15 min at 95°C, followed by 40 amplification cycles each of 95°C for 60 s, 53°C 50 s, and 72°C 60 s. A subsample of soil from each bag was stored at −20°C for further analysis. Triplicate reaction mixtures per DNA sample and the appropriate set of standards were used. For qPCR assays, there was a linear relationship between the log of the plasmid DNA copy number and the calculated threshold cycle (CT value). PCR efficiencies were 99%, and correlation coefficients (R2) for standard curves were 0.99. Bacterial abundance was similar for all dilution treatments after 8 weeks of incubation as determined by quantitative real-time PCR (see Fig. S1 in the supplemental material). We also measured fungal abundance by quantitative real-time PCR using the primers for the 5.8S and internal transcribed spacer 1 (ITS1) genes. For fungi, the standard curves were generated using a 10-fold dilution series from 108 to 103 of plasmid DNA obtained from fungi, but the fungal biomass appeared to be low in these soil samples; and because of the difficulty in assessing fungal abundance by quantitative real-time PCR due to heterogeneity in ribosomal operon number per fungal species/phylum, we decided to ignore the fungal community in the rest of our analyses. The primers we used for pyrosequencing target both bacteria and archaea. There were no significant numbers of archaeal sequences; therefore, we did not include archaea in our analyses.
DNA extraction, PCR, and 16S rRNA gene fragment pyrosequencing.
Total DNA was extracted from the soil suspensions and from incubated soil to determine the composition of the respective microbial communities by 454-pyrosequencing. DNA was extracted using a MoBio Power Soil Extraction kit according to the supplier's instructions (MoBio Laboratories, Carlsbad, CA, USA). Total DNA concentration was qualified on an ND-1000 spectrophotometer (NanoDrop Technology, Wilmington, DE). For DNA concentrations below 5 ng/μl, i.e., five samples of 10−6 and four samples of 10−9 suspensions, nested PCR was performed. The general bacterial primers 27F and 1492R (19) were used for the first amplification, and then 2 μl of the amplified products from the first round was used as the template for the second round PCR using bar-coded primers 515F and 806R (21). Five nanograms/microliter of DNA/sample of the diluted samples was used as the template for the first round of nested PCR with a PCR program of 95°C for 5 min, followed by 25 cycles each of 95°C for 30 s, 55°C 1 min, and 72°C 10 min. PCRs using bar-coded primers were performed using 5 μM each forward (515F) and reverse (806R) primer, 5 mM deoxynucleoside triphosphate (dNTP) mix (Invitrogen, Carlsbad, CA), 1 unit of Taq polymerase (Roche, Indianapolis, IN), and 5 ng/μl of sample DNA as the template in a total volume of 25 μl. The PCR conditions for the bar-coded primer were similar to those for the first-round PCR except that 25 cycles at an annealing temperature of 52°C were added. To control for contamination during PCR preparation, one negative control (water in place of DNA) was included for all PCRs. PCR products of each subsample from the bar-coded primers were generated in six replicates and purified using a Wizard SV Gel and PCR Clean-Up System (Promega). Equimolar purified PCR products that were quantified by PicoGreen assays were mixed and sequenced using a Roche Genome Sequencer FLX Titanium 454 sequencing platform (Macrogen, Seoul, South Korea).
Data analysis.
The raw sequence data were processed using the QIIME pipeline, version 1.6.0 (22). Low-quality sequences of less than 150 bp in length or with an average quality score of less than 25 were removed. After denoising the sequences using Denoiser, version 0.91 (23), and checking for chimeras using USEARCH, operational taxonomic units (OTUs) were identified using the UCLUST, version 1.2.21, algorithm (24) with a phylotype defined at the 97% sequence similarity level. The resulting OTUs were aligned against the Ribosomal Database Project (RDP) database (25).
Alpha diversity calculation was performed based on the rarefied OTU table to compare the diversity among samples at the given level of a sampling effort (26). The OTU table was rarefied to 1,535 reads by single rarefaction QIIME script since this number was the lowest number of reads for all samples. The average reads from the three sterilized controls were used as the baseline that was subtracted from the reads of the other 36 samples. The OTU table after subtraction of the control was used for further statistical analysis. Chao1 richness and Simpson and Shannon diversity and evenness indices were determined with the vegan package (27) in R (The R Foundation for Statistical Computing, Vienna, Austria). The percentage of coverage was calculated by Good's method using the following formula: % coverage = (1 − n/N) × 100, where n is the number of phylotypes represented by singletons and N is the total number of sequences (28).
To compare the communities from the different dilution treatments, nonmetric multidimensional scaling (NMDS) plots were used to visualize the structure among samples at genus level. The NMDS plots were generated from Bray-Curtis similarity index matrices of all samples and created using the PAST software program (29).
RESULTS
Effect of dilution and incubation on bacterial community diversity.
Several indices were used to assess the diversity in the soil suspension dilutions and in the associated soil communities after incubation on the basis of OTU detection (Table 1). Remarkably, all indices for the diluted inocula of 10−6 and 10−9 were significantly higher after incubation than the indices of the associated suspensions, while the indices were lower for the 10−1 dilution after incubation in soil. Good's estimator of coverage increased with increasing dilution, indicating that microbial species were lost through dilution.
TABLE 1.
Estimators of sequence library diversity, evenness, and coverage in soil suspensions at three dilutions and the related samples after incubation in soila
| Dilution and treatment | Sobs (no. of OTUs)b | SChao1c | Shannon index | Simpson index | Evenness | Good's estimator of coverage |
|---|---|---|---|---|---|---|
| 10−1 | ||||||
| Soil suspension | 131.00 ± 3.27 | 169.15 ± 7.80 | 3.986 ± 0.036 | 0.966 ± 0.002 | 0.41 ± 0.01 | 97.61 ± 0.12 |
| Incubated soil | 107.20 ± 1.27 | 134.37 ± 2.96 | 3.719 ± 0.019 | 0.954 ± 0.002 | 0.38 ± 0.01 | 97.56 ± 0.11 |
| P value | * | * | * | * | * | |
| 10−6 | ||||||
| Soil suspension | 44.80 ± 7.98 | 53.09 ± 10.33 | 2.383 ± 0.416 | 0.774 ± 0.124 | 0.24 ± 0.07 | 99.32 ± 0.18 |
| Incubated soil | 70.09 ± 2.13 | 89.64 ± 4.46 | 3.208 ± 0.040 | 0.934 ± 0.004 | 0.35 ± 0.01 | 97.95 ± 0.21 |
| P value | * | * | * | NSd | * | |
| 10−9 | ||||||
| Soil suspension | 17.00 ± 2.17 | 19.54 ± 2.46 | 1.462 ± 0.293 | 0.623 ± 0.128 | 0.25 ± 0.05 | 99.77 ± 0.03 |
| Incubated soil | 55.83 ± 1.14 | 81.82 ± 3.37 | 2.633 ± 0.042 | 0.867 ± 0.006 | 0.25 ± 0.01 | 97.27 ± 0.24 |
| P value | * | * | * | * | NS |
Estimators were calculated for each dilution treatment of the soil suspensions (n = 5 to 6) and incubated soil samples (n = 11 to 12). Significance was determined at the species level (*, P < 0.05).
Observed number of species.
Estimated number of species with the Chao1 estimator.
NS, not significant.
Effect of dilution and incubation on bacterial community composition.
After the OTUs were classified according to the RDP database, the soil microbial community consisted of 18 phyla (Fig. 1). Phylum-level taxonomic assignments indicated that Proteobacteria, followed by Actinobacteria, Bacteroidetes, Acidobacteria, Verrucomicrobia, Planctomycetes, and Firmicutes dominated the microbial communities in the original nondiluted (10−1) soil suspension (>90% of all sequences). The variance in the abundances of the seven dominating phyla among the replicate suspension samples increased from the low-dilution treatments to the high-dilution treatments (Table 2; see also Fig. S2 in the supplemental material). The same was true for the incubated samples, while in general the variance among the replicates of the incubated samples (Table 2; see also Fig. S3) was lower than the variance among the replicate samples of the soil suspensions.
FIG 1.
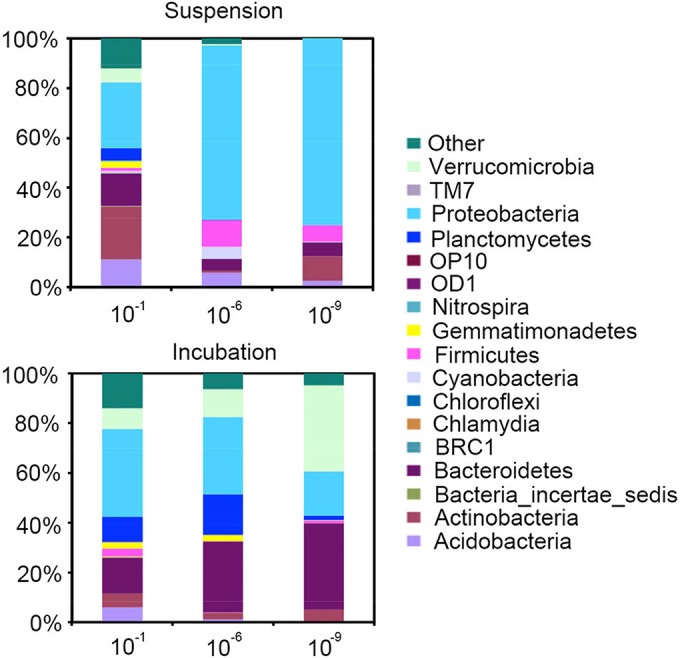
Bacterial community composition at the phylum level of soil suspensions and incubated soil samples in relative abundances.
TABLE 2.
Coefficient of variation for each phylum measured in soil suspensions at the three dilution levels and in related soil samples after incubation
| Phylum | CV (%) by treatment and dilutiona |
|||||
|---|---|---|---|---|---|---|
| Soil suspension |
Incubated soil |
|||||
| 10−1 | 10−6 | 10−9 | 10−1 | 10−6 | 10−9 | |
| Proteobacteria | 13.94 | 46.66 | 26.66 | 6.05 | 20.73 | 50.69 |
| Actinobacteria | 24.95 | 146.26 | 205.76 | 10.97 | 60.29 | 77.59 |
| Bacteroidetes | 10.03 | 107.73 | 192.35 | 20.30 | 20.24 | 31.30 |
| Acidobacteria | 29.84 | 67.97 | 222.38 | 13.31 | 43.77 | 65.31 |
| Verrucomicrobia | 39.95 | 188.99 | 223.61 | 26.50 | 28.35 | 43.43 |
| Planctomycetes | 39.94 | 121.61 | 24.56 | 32.27 | 107.52 | |
| Firmicutes | 14.97 | 64.43 | 135.96 | 31.49 | 109.87 | 96.17 |
Coefficient of variation (CV) of each phylum based on absolute reads in soil suspensions and incubated soil samples was calculated as follows: CV = (standard deviation/mean) × 100.
To test the selective power of soil further, we analyzed the major phyla at the family level. Visually, we noticed that the diversity of the communities in the incubated soil samples and, in particular, those which were incubated with the highest, i.e., 10−9 dilution, suspensions differed strongly from the diversity of the inoculated suspensions (see Fig. S4 in the supplemental material). Thus, we compared the diversity of the communities in both the suspensions and the inoculated soil samples. Remarkably, for most phyla we found that the Shannon diversity index was significantly higher in the incubated soil samples than in the corresponding suspensions of the 10−6 and 10−9 dilutions (Table 3), while they were lower in the soil samples than in the associated suspensions at the 10−1 dilution.
TABLE 3.
Shannon diversity at the phylum level of three dilutions of a soil suspension and related soil samples after incubation
| Phylum | Shannon diversity index by dilution and treatmenta |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 10−1 |
P value | 10−6 |
P value | 10−9 |
P value | ||||
| Suspension | Incubation | Suspension | Incubation | Suspension | Incubation | ||||
| Acidobacteria | 1.60 ± 0.04 | 1.16 ± 0.04 | * | 0.73 ± 0.17 | 0.85 ± 0.07 | NS | 0.00 ± 0.00 | 0.54 ± 0.15 | * |
| Actinobacteria | 2.49 ± 0.03 | 2.34 ± 0.03 | * | 0.96 ± 0.25 | 1.78 ± 0.07 | * | 0.01 ± 0.01 | 1.46 ± 0.16 | * |
| Bacteroidetes | 1.49 ± 0.05 | 1.29 ± 0.04 | * | 0.61 ± 0.18 | 1.27 ± 0.06 | * | 0.00 ± 0.00 | 1.16 ± 0.07 | * |
| Firmicutes | 1.17 ± 0.05 | 1.04 ± 0.04 | NS | 0.94 ± 0.27 | 0.23 ± 0.12 | * | 0.22 ± 0.19 | 0.52 ± 0.11 | NS |
| Verrucomicrobia | 1.15 ± 0.07 | 1.23 ± 0.03 | NS | 0.07 ± 0.07 | 0.96 ± 0.06 | * | 0.00 ± 0.00 | 0.81 ± 0.07 | * |
| Alphaproteobacteria | 1.95 ± 0.01 | 1.88 ± 0.02 | NS | 1.05 ± 0.19 | 1.69 ± 0.04 | * | 0.66 ± 0.07 | 1.37 ± 0.12 | * |
| Betaproteobacteria | 1.39 ± 0.02 | 1.50 ± 0.03 | NS | 0.65 ± 0.21 | 0.75 ± 0.14 | NS | 0.41 ± 0.12 | 0.91 ± 0.08 | * |
| Deltaproteobacteria | 1.16 ± 0.03 | 1.31 ± 0.08 | NS | 0.02 ± 0.02 | 0.78 ± 0.13 | * | 0.00 ± 0.00 | 0.74 ± 0.09 | * |
| Gammaproteobacteria | 1.18 ± 0.02 | 0.94 ± 0.04 | * | 0.79 ± 0.09 | 0.95 ± 0.07 | NS | 0.33 ± 0.16 | 0.47 ± 0.11 | NS |
Diversity was calculated for each dilution of the soil suspensions (n = 5 to 6) and incubated soil samples (n = 11 to 12); six replicates of each were performed. Significance was determined at the family level (*, P < 0.05; NS, not significant).
To compare the overall community structure of the different dilution treatments and differences before and after incubation, the taxonomic abundance profiles were used to compute a Bray-Curtis similarity matrix, coordinated into two dimensions by using NMDS (Fig. 2). Samples were grouped according to before/after incubation. This analysis revealed clear differences in the microbial community structures before and after incubation. The community structures of the soil samples after incubation were more similar to each other than to the associated suspension samples. This may hint at selective processes in the soil leading to more equal communities. One-way analysis of similarities (ANOSIM) showed that the dilution treatment had a significant (R = 0.28, P < 0.001) overall effect on the structure of the bacterial community in the suspension and the soil samples after incubation.
FIG 2.
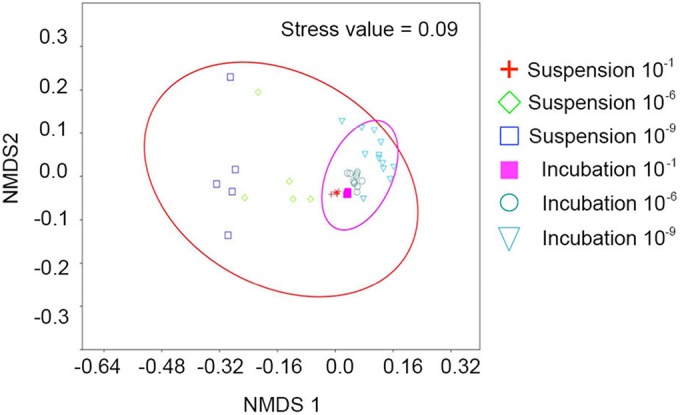
NMDS plot of the Bray-Curtis similarity matrix among six replicate samples of the three dilutions of the soil suspension and the related replicates (in duplicate) after incubation in soil.
Effect of dilution on rare/abundant OTUs.
A possible way to determine if the dilution approach is appropriate to separate rare species from abundant ones is to make Venn diagrams to assess the shared and unique OTUs between dilution treatments in the soil suspensions (Fig. 3A) and the incubated soil samples (Fig. 3B). We found 954, 77, and 10 unique OTUs in the 10−1, 10−6, and 10−9 dilution samples of the soil suspensions, respectively, and 386, 96, and 88 unique OTUs in the respective dilution treatments of the incubated soil samples. To identify the unique OTUs in the different treatments, the phylogenetic affiliation was done at the genus level. From the unique OTUs that were assigned to a genus, a total of 158, 38, and 10 unique genera were detected in the 10−1, 10−6, and 10−9 dilutions of soil suspensions, respectively (see Table S1 in the supplemental material), and 84, 33, and 34 unique genera were detected in the 10−1, 10−6, and 10−9 dilutions of the incubated soil samples, respectively (see Table S2).
FIG 3.
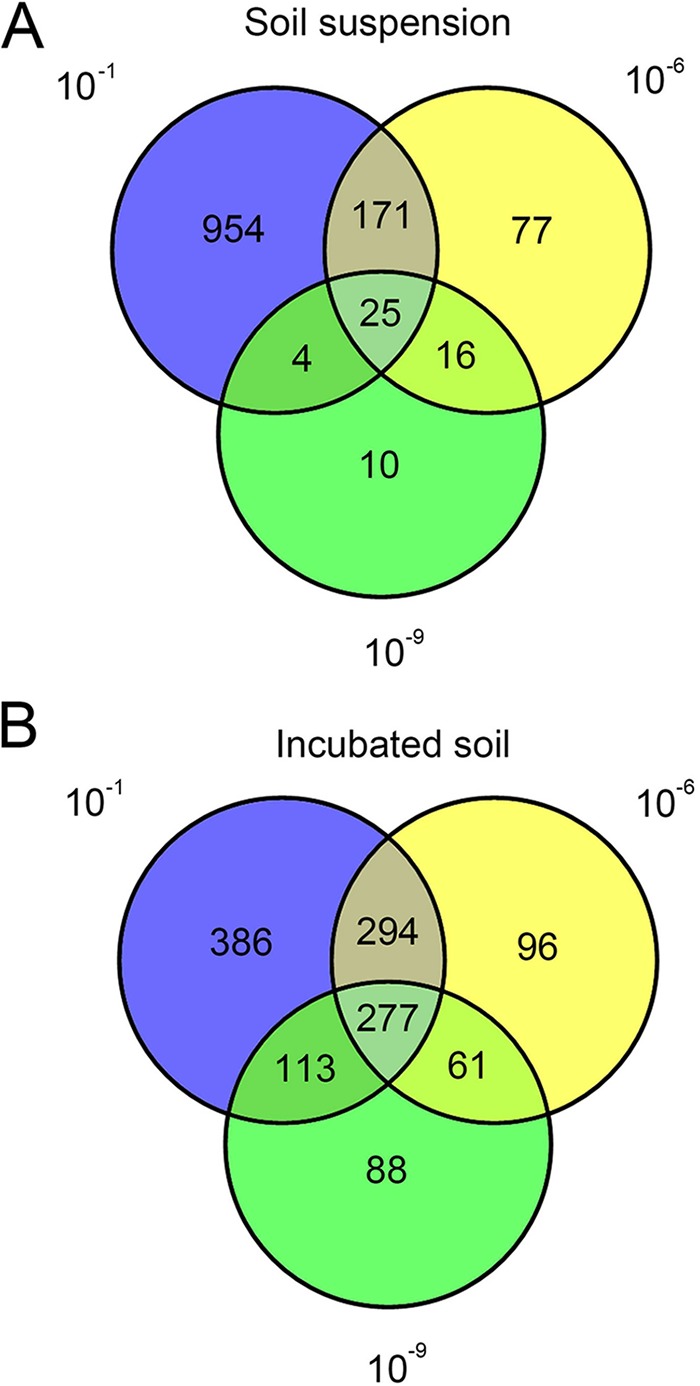
Venn diagram of shared and unique OTUs in each dilution of the soil suspensions and incubated soil samples.
DISCUSSION
A number of studies have used the dilution method approach to artificially change microbial diversity (4–13). This approach is one of the few available methods to manipulate the microbial biodiversity of complex natural ecosystems such as the soil. And, indeed, our results show that dilution reduces the microbial biodiversity in the soil suspension and the soil after incubation of more or less diluted suspensions. Previous studies mostly based their conclusions on community measurements with limited resolution, detecting only the more abundant species since those species can be detected in the easiest way. However, compared to the rare biosphere, the abundant members represent only a small fraction of microbial diversity (15), and, thus, in this way, the real microbial biodiversity in these ecosystems may not be accounted for. Furthermore, none of those studies focused on changes in the community structure from the original more or less diluted inocula into different communities after incubation in soil or on the degree of variation in the suspensions after dilution and the consequences of this variation for the variances in the incubated soils. We suspected that the variance among the replicate samples would be considerable, and therefore we determined this variation in the suspension samples and how the variant communities developed during incubation and regrowth in soil. We were especially interested in the possibilities created by the dilution approach to separate abundant and rare species and thus allow experimental studies on the importance of rare (and abundant) microbes in soil ecosystems.
Although less abundant microbes should be more prone to being lost from the original microbial community at increasing dilutions, our results show that unique OTUs still show up in the highest-dilution treatment in the suspensions (Fig. 3). Most likely, certain microbial species are suppressed or masked for amplicon measurements in the low-dilution samples and only show up in the higher-dilution treatments. An issue that may have played an important role in the preparation of the diluted soil suspensions is the adsorption of cells on soil particles. Bakken (30) claimed that a satisfactory separation of microorganisms and soil particles is not possible, and thus this could have influenced the structure of the microbial communities in the suspensions and, in particular, the large variation therein. Moreover, methodological errors may also have played a role in the failure of the sequencing approach to detect all species in the suspensions. For instance, the nested PCR could be a possible source of bias; therefore, the patterns from nested PCRs between samples were compared with the ones from direct PCRs. The patterns obtained from the nested PCRs were similar to those from the direct PCRs of soil suspensions. Only minor variations were observed. In this experiment, the PCR products were purified before sequencing to exclude the nonincorporated primers. Thus, we concluded that the nested PCR approach may not have significantly influenced the results.
Similarly, our results indicate that, most likely, rare species that were suppressed in the low-dilution samples may have acquired an opportunity to develop in the higher-dilution samples because the cellular densities were low in those samples after dilution.
The data shown in Table 3 and the diagrams of Fig. S4 in the supplemental material clearly indicate that the present methodology, i.e., 454 pyrosequencing, does not allow a complete view of the species present even in a dilute suspension as these data show that the diversity of the communities of the diluted samples increased during incubation in soil. We do not know what the precise detection limit is of the 454 pyrosequencing technique for observing microbial species in a suspension, but it is fair to assume that the bacterial species that are detected in soil after incubation were present but not detected in the soil suspensions, most probably because of their low abundances. As mentioned, the data of the Venn diagram (Fig. 3) also clearly indicate the presence of species in all suspensions, including the 10−9 dilutions, which were not detected by our method.
The fact that these organisms were detected in soil but not in suspensions may be because these organisms were better adapted to the prevailing conditions of the soil environment (31) than other organisms that were detected in the suspensions but not in soils. These other taxa may have been lost during incubation since they might have had special requirements not available in soil. It is not possible to conclude that these hidden species are rare species, and, thus, the conclusion is warranted that the dilution approach does not guarantee the identification of rare or less abundant versus abundant species.
Although all inoculated organisms were returned into the same environment where they came from originally, the actual conditions for the individual organisms could have changed dramatically due to the difference in spatial arrangements and the large degree of heterogeneity in soil. The factors that are responsible for the selection of microbes in soil resulting in the differences between the communities found in soil and those in the suspensions are not clear. Previous studies have indicated that soil microbial communities are largely influenced by soil moisture (32, 33). In our study, moisture availability after incubation could be a potential clue for the structuring of the community by selecting for individual microbial species with relatively high moisture stress resistance. Other factors are also said to be key to the shaping of bacterial communities in soil (34–37), but the relevance of these factors for the assemblage of the communities from various inocula, as in this study, is not known.
In this study, we have considered several taxonomic diversity indicators. All indicated that the dilution procedure has a strong reducing effect on microbial diversity (Table 1). We have used these different diversity indices because they give different insights into the diversity of complex communities such as soil microbial communities. In contrast to the richness index (Chao estimator), the diversity indices (Shannon and Simpson) focus on both the richness and evenness of a community. Shannon diversity is often sensitive to the presence of rare species, while the Simpson index emphasizes the dominant members (38). Haegeman et al. (39) suggest that community diversity is best estimated by Shannon and Simpson indices, whereas the Chao estimator is not a reliable estimator of richness in the presence of rare species. Despite the differences in the focus of the diversity indices used here, all indices showed similar trends. This strongly suggests that the alpha diversity decreases in response to the dilution of microbial communities and that this decrease is reflected in the diversity of the communities after incubation in soil.
Interestingly, when we compared the diversity of the different phyla in suspension and in soil after incubation, we observed that the Shannon diversity index of most phyla decreased from suspension to soil sample for the undiluted (10−1) samples but increased for the most-diluted (10−9) samples (Table 3). Obviously, there are strong selection mechanisms operating in soil that lead to a certain homogenization in the communities that are formed after regrowth of the suspensions. That observation is confirmed by the data shown in Fig. 2 and Table 2, both of which show that the variances in the communities formed in the replicate samples diminish. We are not aware of similar observations presented in literature, but the findings are in line with the wealth of information that indicates that the soil is a strong factor shaping the structure of the microbial community inhabiting the soil.
Analysis of the overall microbial community revealed that the community changed through dilution treatment of the soil suspensions and incubated soil samples at both the phylum (Fig. 1) and OTU levels (Fig. 2). A detailed look at the microbial communities in the original nondiluted (10−1) soil suspension revealed that the core groups comprised the well-known soil microbial phyla of Proteobacteria, Actinobacteria, Bacteroidetes, Acidobacteria, Verrucomicrobia, Planctomycetes, and Firmicutes (40, 41). During incubation, the same core groups were observed again, but the relative abundances of each group changed substantially. The largest changes in the occurrences of specific phyla were detected for the phylum Proteobacteria, which was highly dominant especially in the higher dilutions but less dominant in the incubated soil samples, for the phylum Bacteroidetes, which decreased slightly with increasing dilution in the soil suspensions but outgrew and increased significantly in the samples after incubation, and for the phylum Verrucomicrobia, which was not detected in the higher soil suspension dilutions but showed up in high numbers after incubation. At the family level, we detected high proportions of Betaproteobacteria represented by Alcaligenaceae, Burkholderiaceae, and Comamonadaceae in the highest suspension dilution. Remarkably, their relative abundances decreased during incubation in soil. That was unexpected as Proteobacteria are dominant members in various soils, and as they are mostly fast-growing r-strategists, we expected them to be abundantly present in the incubated soil samples. The result may have been caused by the oligotrophic conditions prevailing in our test soil. However, the same observations after incubation in soil were made for Acidobacteria, which are generally considered to be soil-adapted oligotrophic organisms (34), and for other well-known soil inhabitants such as Actinobacteria. It is interesting that groups such as Verrucomicrobia and Sphingobacteriaceae and Chitinophagaceae families of Bacteroidetes grew out significantly in all dilution treatments during incubation in soil. This contradicts what is known about Verrucomicrobia, which is usually considered a low-abundance phylum in soil (40). Verrucomicrobia may highly depend on C availability due to their slow-growing life strategy (21); that, in combination with the observed results, may indicate that Verrucomicrobia are a potential indicator of the response of these taxa to environmental factors (42).
In summary, our results indicate that the dilution procedure leads to a reduction of bacterial diversity, but the assembly of the microbial community during incubation in soil cannot be predicted on the basis of the composition of the inoculum. Obviously, soil has a strong selective power in shaping the microbial community, which leads to more uniform structures of the communities even after inoculation of much more variable suspensions. Also the deep-sequencing approach applied here did not allow a complete view of the microbial species present in even highly diluted suspensions. This also hinders the assessment and identification of rare species in a soil sample as even undetected species in the suspensions could develop into abundant populations after only 8 weeks of incubation. In future studies we hope to be able to learn more about the functional responses of more or less diverse samples and the consequences of these changes for the functioning of the soil ecosystem.
Supplementary Material
ACKNOWLEDGMENTS
We thank K. van der Veen-van Wijk and A. Pijl for their technical assistance and G. Hol and M. de Hollander for data analysis assistance.
Footnotes
This article is publication number 5604 of the Netherlands Institute of Ecology (NIOO-KNAW).
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00958-15.
REFERENCES
- 1.Sala OE, Chapin FS, Armesto JJ, Berlow E, Bloomfield J, Dirzo R, Huber-Sanwald E, Huenneke LF, Jackson RB, Kinzig A, Leemans R, Lodge DM, Mooney HA, Oesterheld M, Poff NL, Sykes MT, Walker BH, Walker M, Wall DH. 2000. Biodiversity-global biodiversity scenarios for the year 2100. Science 287:1770–1774. doi: 10.1126/science.287.5459.1770. [DOI] [PubMed] [Google Scholar]
- 2.Magurran AE, Henderson PA. 2003. Explaining the excess of rare species in natural species abundance distributions. Nature 422:714–716. doi: 10.1038/nature01547. [DOI] [PubMed] [Google Scholar]
- 3.Butchart SH, Walpole M, Collen B, van Strien A, Scharlemann JP, Almond RE, Baillie JE, Bomhard B, Brown C, Bruno J, Carpenter KE, Carr GM, Chanson J, Chenery AM, Csirke J, Davidson NC, Dentener F, Foster M, Galli A, Galloway JN, Genovesi P, Gregory RD, Hockings M, Kapos V, Lamarque JF, Leverington F, Loh J, McGeoch MA, McRae L, Minasyan A, Morcillo MH, Oldfield TE, Pauly D, Quader S, Revenga C, Sauer JR, Skolnik B, Spear D, Stanwell-Smith D, Stuart SN, Symes A, Tierney M, Tyrrell TD, Vie JC, Watson R. 2010. Global biodiversity: indicators of recent declines. Science 328:1164–1168. doi: 10.1126/science.1187512. [DOI] [PubMed] [Google Scholar]
- 4.Philippot L, Spor A, Henault C, Bru D, Bizouard F, Jones CM, Sarr A, Maron PA. 2013. Loss in microbial diversity affects nitrogen cycling in soil. ISME J 7:1609–1619. doi: 10.1038/ismej.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salonius PO. 1981. Metabolic capabilities of forest soil microbial-populations with reduced species diversity. Soil Biol Biochem 13:1–10. doi: 10.1016/0038-0717(81)90094-8. [DOI] [Google Scholar]
- 6.Garland JL, Lehman RM. 1999. Dilution/extinction of community phenotypic characters to estimate relative structural diversity in mixed communities. FEMS Microbiol Ecol 30:333–343. doi: 10.1111/j.1574-6941.1999.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 7.Griffiths BS, Ritz K, Wheatley R, Kuan HL, Boag B, Christensen S, Ekelund F, Sorensen SJ, Muller S, Bloem J. 2001. An examination of the biodiversity-ecosystem function relationship in arable soil microbial communities. Soil Biol Biochem 33:1713–1722. doi: 10.1016/S0038-0717(01)00094-3. [DOI] [Google Scholar]
- 8.Franklin RB, Garland JL, Bolster CH, Mills AL. 2001. Impact of dilution on microbial community structure and functional potential: comparison of numerical simulations and batch culture experiments. Appl Environ Microbiol 67:702–712. doi: 10.1128/AEM.67.2.702-712.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matos A, Kerkhof L, Garland JL. 2005. Effects of microbial community diversity on the survival of Pseudomonas aeruginosa in the wheat rhizosphere. Microb Ecol 49:257–264. doi: 10.1007/s00248-004-0179-3. [DOI] [PubMed] [Google Scholar]
- 10.Franklin RB, Mills AL. 2006. Structural and functional responses of a sewage microbial community to dilution-induced reductions in diversity. Microb Ecol 52:280–288. doi: 10.1007/s00248-006-9033-0. [DOI] [PubMed] [Google Scholar]
- 11.Pedrós-Alió C. 2006. Marine microbial diversity: can it be determined? Trends Microbiol 14:257–263. doi: 10.1016/j.tim.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 12.Hol WHG, de Boer W, Termorshuizen AJ, Meyer KM, Schneider JHM, van Dam NM, van Veen JA, van der Putten WH. 2010. Reduction of rare soil microbes modifies plant-herbivore interactions. Ecol Lett 13:292–301. doi: 10.1111/j.1461-0248.2009.01424.x. [DOI] [PubMed] [Google Scholar]
- 13.Vivant AL, Garmyn D, Maron PA, Nowak V, Piveteau P. 2013. Microbial diversity and structure are drivers of the biological barrier effect against Listeria monocytogenes in soil. PLoS One 8:e76991. doi: 10.1371/journal.pone.0076991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen ZT, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc Natl Acad Sci U S A 103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huber JA, Mark Welch D, Morrison HG, Huse SM, Neal PR, Butterfield DA, Sogin ML. 2007. Microbial population structures in the deep marine biosphere. Science 318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- 17.Neufeld JD, Li J, Mohn WW. 2008. Scratching the surface of the rare biosphere with ribosomal sequence tag primers. FEMS Microbiol Lett 283:146–153. doi: 10.1111/j.1574-6968.2008.01124.x. [DOI] [PubMed] [Google Scholar]
- 18.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. 2009. A core gut microbiome in obese and lean twins. Nature 457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175. In Stackebrandt E, Goodfellow M (ed), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Chichester, United Kingdom. [Google Scholar]
- 20.Muyzer G, de Waal EC, Uitterlinden AG. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergmann GT, Bates ST, Eilers KG, Lauber CL, Caporaso JG, Walters WA, Knight R, Fierer N. 2011. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol Biochem 43:1450–1455. doi: 10.1016/j.soilbio.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Tumbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reeder J, Knight R. 2010. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat Methods 7:668–669. doi: 10.1038/nmeth0910-668b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 25.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hughes JB, Hellmann JJ. 2005. The application of rarefaction techniques to molecular inventories of microbial diversity. Environ Microbiol 397:292–308. doi: 10.1016/S0076-6879(05)97017-1. [DOI] [PubMed] [Google Scholar]
- 27.Dixon P. 2003. VEGAN, a package of R functions for community ecology. J Veg Sci 14:927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x. [DOI] [Google Scholar]
- 28.Good IJ. 1953. The population frequencies of species and the estimation of population parameters. Biometrika 40:237–264. doi: 10.1093/biomet/40.3-4.237. [DOI] [Google Scholar]
- 29.Hammer HD, , Ryan PD et al. 2001. PAST: paleontological statistics software package for education and data analysis. Palaeontologia Electronica 4:1–9. [Google Scholar]
- 30.Bakken LR. 1985. Separation and purification of bacteria from soil. Appl Environ Microbiol 49:1482–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brazelton WJ, Ludwig KA, Sogin ML, Andreishcheva EN, Kelley DS, Shen CC, Edwards RL, Baross JA. 2010. Archaea and bacteria with surprising microdiversity show shifts in dominance over 1,000-year time scales in hydrothermal chimneys. Proc Natl Acad Sci U S A 107:1612–1617. doi: 10.1073/pnas.0905369107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brockett BFT, Prescott CE, Grayston SJ. 2012. Soil moisture is the major factor influencing microbial community structure and enzyme activities across seven biogeoclimatic zones in western Canada. Soil Biol Biochem 44:9–20. doi: 10.1016/j.soilbio.2011.09.003. [DOI] [Google Scholar]
- 33.Schimel JP, Gulledge JM, Clein-Curley JS, Lindstrom JE, Braddock JF. 1999. Moisture effects on microbial activity and community structure in decomposing birch litter in the Alaskan taiga. Soil Biol Biochem 31:831–838. doi: 10.1016/S0038-0717(98)00182-5. [DOI] [Google Scholar]
- 34.Eichorst SA, Breznak JA, Schmidt TM. 2007. Isolation and characterization of soil bacteria that define Teniglobus gen. nov., in the phylum Acidobacteria. Appl Environ Microbiol 73:2708–2717. doi: 10.1128/AEM.02140-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fierer N, Allen AS, Schimel JP, Holden PA. 2003. Controls on microbial CO2 production: a comparison of surface and subsurface soil horizons. Global Change Biol 9:1322–1332. doi: 10.1046/j.1365-2486.2003.00663.x. [DOI] [Google Scholar]
- 36.Navarrete AA, Kuramae EE, de Hollander M, Pijl AS, van Veen JA, Tsai SM. 2013. Acidobacterial community responses to agricultural management of soybean in Amazon forest soils. FEMS Microbiol Ecol 83:607–621. doi: 10.1111/1574-6941.12018. [DOI] [PubMed] [Google Scholar]
- 37.Kuramae EE, Gamper HA, Yergeau E, Piceno YM, Brodie EL, DeSantis TZ, Andersen GL, van Veen JA, Kowalchuk GA. 2010. Microbial secondary succession in a chronosequence of chalk grasslands. ISME J 4:711–715. doi: 10.1038/ismej.2010.11. [DOI] [PubMed] [Google Scholar]
- 38.Nagendra H. 2002. Opposite trends in response for the Shannon and Simpson indices of landscape diversity. Appl Geogr 22:175–186. doi: 10.1016/S0143-6228(02)00002-4. [DOI] [Google Scholar]
- 39.Haegeman B, Hamelin J, Moriarty J, Neal P, Dushoff J, Weitz JS. 2013. Robust estimation of microbial diversity in theory and in practice. ISME J 7:1092–1101. doi: 10.1038/ismej.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janssen PH. 2006. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol 72:1719–1728. doi: 10.1128/AEM.72.3.1719-1728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AKM, Kent AD, Daroub SH, Camargo FAO, Farmerie WG, Triplett EW. 2007. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1:283–290. doi: 10.1038/ismej.2007.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fierer N, Bradford MA, Jackson RB. 2007. Toward an ecological classification of soil bacteria. Ecology 88:1354–1364. doi: 10.1890/05-1839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.