

Abstract
Parkinson disease (PD) is a late-onset neurodegenerative disorder. The mean age at onset is 61 years, but the disease can range from juvenile cases to cases in the 8th or 9th decade of life. The parkin gene on chromosome 6q and loci on chromosome 1p35-36 and 1p36 are responsible for some cases of autosomal recessive early-onset parkinsonism, but they do not appear to influence susceptibility or variability of age at onset for idiopathic PD. We have performed a genomewide linkage analysis using variance-component methodology to identify genes influencing age at onset of PD in a population of affected relatives (mainly affected sibling pairs) participating in the GenePD study. Four chromosomal loci showed suggestive evidence of linkage: chromosome 2p (maximum multipoint LOD [MaxLOD] = 2.08), chromosome 9q (MaxLOD = 2.00), chromosome 20 (MaxLOD = 1.82), and chromosome 21 (MaxLOD = 2.21). The 2p and 9q locations that we report here have previously been reported as loci influencing PD affection status. Association between PD age at onset and allele 174 of marker D2S1394, located on 2p13, was observed in the GenePD sample (P=.02). This 174 allele is common to the PD haplotype observed in two families that show linkage to PARK3 and have autosomal dominant PD, which suggests that this allele may be in linkage disequilibrium with a mutation influencing PD susceptibility or age at onset of PD.
Introduction
Parkinson disease (PD [MIM 168600]) is a late-onset neurodegenerative disorder characterized by resting tremor, rigidity, slowness of movement, and postural instability. The disorder results from degeneration of neurons within the substantia nigra, creating a deficiency in dopamine-mediated movement. Although dopamine replacement therapy mitigates symptoms and may increase longevity, it does not stop disease progression or restore full quality of life. The etiology of PD is unknown, although there is evidence to suggest that it is multifactorial, involving both genetic susceptibility and environmental agents or traumas.
To date, three genes causing familial forms of PD have been identified. These include the α-synuclein gene on 4q21-23/PARK1 (MIM 163890; Polymeropoulos et al. 1997) and ubiquitin C-terminal hydrolase on 4p14/PARK5 (MIM 191342; Leroy et al. 1998), both of which cause autosomal dominant forms of PD, as well as parkin on 6q25-27/PARK2 (MIM 602544; Kitada et al. 1998), which causes early-onset recessive PD. Linkage studies have identified four chromosomal regions with evidence for PD-related genes, at 2p13/PARK3 (MIM 602404; Gasser et al. 1998), 4p15/PARK4 (MIM 605543; Farrer et al. 1999), 1p35-p36/PARK6 (MIM 605909; Valente et al. 2001), and 1p36/PARK7 (MIM 606324; van Duijn et al. 2001), but genes have not been cloned at those locations.
Two genome scans evaluating linkage to affection status in multiplex families with idiopathic PD have been reported (DeStefano et al. 2001; Scott et al. 2001). Both studies yielded evidence of linkage to 9q and to several other regions that did not overlap between the two scans. Scott et al. (2001) observed significant evidence of linkage to 6q in the region of the parkin gene, in a subset of families with early-onset PD, but not to any of the other familial loci. No evidence of linkage to any familial PD loci was observed by DeStefano et al. (2001). Although stratification by age at onset was not performed in that sample, the families were screened for mutations in the parkin gene, and those with parkin mutations were excluded from the genome scan. On the basis of these analyses of linkage to affection status, there is no evidence that the genes implicated in the rare Mendelian forms of PD are major susceptibility genes for common idiopathic PD.
Although previous genetic studies have focused on affection status as the outcome of interest, there is also evidence of a genetic influence on age at onset of PD. A segregation analysis based on 948 consecutively ascertained PD index cases and a total of 4,351 individuals found the best-fit model to be a major gene with an additive effect influencing age at onset (Maher et al., in press). The estimated gene frequency of this onset-age or penetrance gene was .02, indicating a rare gene in the population. There was also evidence for a very rare (i.e., estimated frequency = 0.008) dominant gene influencing PD susceptibility. Evidence of a gene influencing age at onset of PD is important because identification of such a gene would contribute to the understanding of disease development and progression and could provide new targets for therapy. The ability to delay the onset of PD by 10 years, for example, could significantly reduce the impact of this disease. Recently, increasing attention has been paid to genetic influences on age at onset and methods to identify such genes (e.g., Horvath et al. 2001), particularly for adult-onset neurological disorders (Cardno et al. 2001; Rosenblatt et al. 2001).
To identify genes influencing age at onset of PD, we performed a genomewide linkage analysis of age at onset in a set of 103 multiplex families. Regions showing evidence of linkage were further examined using association tests and by genotyping of additional markers in a larger set of individuals. Follow-up analyses strengthened the evidence for linkage at 2p.
Subjects and Methods
Subjects
Relative pairs with PD were ascertained through index cases at 13 clinical sites. Neurologists from the participating sites examined and confirmed the PD diagnosis for each of the index patients and their affected relatives, with the exception of two individuals, for whom confirmation of diagnosis is ongoing. Diagnostic criteria for PD were based on the United Kingdom PD Society Brain Bank Criteria (Gibb and Lees 1988), but we removed “more than one affected relative” and “repeated head injury” as exclusionary criteria. In their original form, these criteria have a positive predictive value of 82% (Hughes et al. 1992); hence, we considered these modifications unlikely to degrade the positive predictive value. All neurologists participated in video-based interrater reliability training to ensure diagnostic consistency. All participants signed a consent form approved by the human-subjects committee of each participating institution.
A total of 103 families with multiple individuals with PD were included in the genome scan. There were 98 sibships of size 2, 3 sibships of size 3, and 1 sibship of size 4, giving a total of 113 affected full-sib pairs (see DeStefano et al. 2001). Included in these counts is an extended pedigree consisting of an affected sib pair with an unaffected sibling who had three offspring with PD, creating affected avuncular pairs within the family. Two families consisted of affected half-sib pairs. In addition, one affected parent and nine unaffected siblings were genotyped.
Follow-up studies were conducted in the original scan families, 49 additional sib pairs, and 1 additional half-sib pair with onset-age data. A total of 22 parent-offspring pairs were genotyped in the expanded sample.
Genotyping
Genomic DNA was extracted from lymphocytes through use of a Nucleon II DNA extraction kit. The scan consisted of 339 markers, with an average intermarker distance of 11 cM, from the Weber set 8, obtained from Research Genetics. The sib_kin program in the ASPEX package (The ASPEX Linkage Analysis ftp site) was used to verify sibling relationships. Pairs reported as full sibs but not confirmed as such were deleted from subsequent analyses. Mendelian inconsistencies in the genotype data were identified using Genehunter (Kruglyak et al. 1996). Genotypes for the entire nuclear family were deleted for the particular marker that yielded errors.
Analysis
Linkage analysis of PD affection status in these families, through use of the identical genome-scan genotype data, has been published elsewhere (DeStefano et al. 2001). The present article presents a novel analysis of these data through use of age at onset as the phenotype of interest.
Variance-component linkage analysis of age at onset was performed using Genehunter 2.1 (Pratt et al. 2000). Age at onset was known for all individuals with PD. Unaffected individuals are censored subjects with respect to age at onset; age at onset was coded as “missing” for all unaffected individuals. However, genotype information was included for nine unaffected siblings, to improve identity-by-descent estimation.
Family-based association tests (FBATs) evaluating association between markers and age at onset were conducted using the program FBAT (Laird et al. 2000; FBAT Web Page). A brief review of the tests implemented in FBAT is given below. These tests are described in detail elsewhere (Rabinowitz and Laird 2000; Horvath et al. 2001). A general form of an FBAT statistic for family i (with ni offspring) is
 |
where Xij is a function of the genotype data of offspring j in family i and Tij is a function of the phenotype data of that offspring. For a biallelic marker, a score statistic based on Si can be defined as
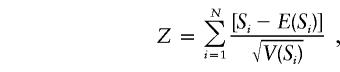 |
where E(Si) and V(Si) are the mean and variance, respectively, of Si under the null hypothesis of no linkage, and N is the total number of families. If the coding of Xij specifies an additive model (i.e., Xij = the number of alleles of interest [0, 1, or 2] carried by offspring j in family i) and Tij is specified as 0 for unaffected and 1 for affected, then this statistic is equivalent to the transmission/disequilibrium test for genotyped parent-offspring trios (Lunetta et al. 2000).
When parental genotypes are available, E(Si) can be computed by conditioning on the observed traits and parental marker genotypes and is based on Mendelian transmission probabilities (see Horvath et al. [2001] for details). For late-onset diseases such as PD, parental genotype information is often not available and computation of the null distribution is not straightforward. Rabinowitz and Laird (2000) invoke the statistical method of conditioning on sufficient statistics for the null hypothesis to construct a test of association when parental genotypes are not available. In this case, the offspring genotype distribution is defined by conditioning on the observed traits, the partially observed parental genotypes, and the offspring configuration. Tables presenting the conditional probabilities when partial or no parental genotype information is available are given in the FBAT technical report portion of the FBAT documentation. At least two distinct offspring genotypes must be observed for a family to contribute to the FBAT statistic when parental genotypes are not available. The statistical theory of conditioning on the sufficient statistics results in correct P values (type I error rate) regardless of the population admixture, patterns of missing genotypes, or genetic model (Rabinowitz and Laird 2000).
In the present study, both multiallelic and biallelic tests were performed. For the biallelic tests, an additive genetic model was assumed, with Xij coded as described above. Coding of Xij for multiallelic tests are described elsewhere (Horvath et al. 2001). The unknown underlying genetic model may determine which test, biallelic or multiallelic, is more powerful; hence, both were considered here. Two definitions of the trait were employed. In the first definition, Tij = age at onset for offspring j in family i. In the second definition, Tij=(onsetage-μ), where μ is a constant that is chosen to minimize the variance of the test statistic (Horvath et al. 2001). Age at onset was coded as “missing” for all unaffected individuals. For these trait definitions, a positive Z statistic indicates that the allele is associated with later onset of PD. In addition, the empirical variance option in FBAT was applied, to test the null hypothesis of no association in the presence of linkage, because association was tested only in those regions showing evidence of linkage (LOD>1.5) in the genome scan.
Results
Mean age at onset for the individuals with PD who were included in the genome scan was 60.4±11.79 years (range 26–88). The skewness and kurtosis for age at onset were −.45 and −.09, respectively. These values are consistent with a normal distribution and indicate that the normality assumptions underlying the variance-component linkage analysis are not being violated. Although age at onset was slightly higher for males (61.2±11.16; n=126) than females (59.2±12.59; n=89), this difference was not significant (P=.23).
Variance-component linkage analysis revealed four chromosomal regions that yielded multipoint LOD scores >1.5 (fig. 1). Maximum multipoint LOD scores (MaxLOD) >2.0 were observed for three of these regions, on chromosomes 2 (MaxLOD=2.08 at 99 cM), 9 (MaxLOD=2.00 at 116 cM), and 21 (MaxLOD=2.21 at 32 cM). In addition, a MaxLOD of 1.82 was observed at 54 cM on chromosome 20. Table 1 gives two-point LOD scores for the marker yielding the maximum two-point LOD score and for its flanking markers in these peak regions.
Figure 1.

Multipoint LOD-score curves for variance-component analysis of linkage to PD age at onset for chromosomes 2, 9, 20, and 21. The X-axes represent location in centimorgans, and the Y-axes represent LOD score.
Table 1.
Maximum Multipoint and Two-Point LOD Scores and Flanking Markers for Regions with Multipoint LOD >1.5[Note]
|
Two-Point LOD |
Multipoint LOD |
||||
| Chromosome | Locationa(cM) | Marker | LOD Score | Locationa(cM) | LOD Score |
| 2 | 91 | D2S1394 | .50 | ||
| 2 |
99 |
D2S1777 |
2.40 |
99 | 2.08 |
| 2 | 114 | D2S2972 | .36 | ||
| 9 | 111 | D9S938 | .46 | ||
| 9 |
120 |
D9S930 |
1.86 |
116 | 2.00 |
| 9 | 128 | D9S934 | .48 | ||
| 20 | 48 | D20S477 | .14 | ||
| 20 |
54 |
D20S478 |
.95 |
54 | 1.82 |
| 20 | 62 | D20S481 | .89 | ||
| 21 | 13 | D21S1437 | .39 | ||
| 21 |
24 |
D21S2052 |
.94 |
32 | 2.21 |
| 21 | 37 | D21S1440 | .73 | ||
Note.— Underlining indicates markers with maximum two-point LOD in regions with multipoint LOD >1.5.
Location is in relation to p-ter.
Tests of association with age at onset were performed for the markers in the four regions described in table 1. No significant association was detected using the global multiallelic test. Diallelic tests were also performed, to evaluate association with specific alleles. There were three markers, shown in table 2, for which specific alleles showed significant evidence of association consistently across the two tests considered (see the “Methods” section). As shown in table 2, the number of families that were informative for a particular test was small, because of the low frequency of a specific allele in a highly polymorphic microsatellite marker.
Table 2.
Diallelic Family-Based Association Tests Yielding a Nominal P < .05.
|
P Value |
||||
| Marker | Allele | EmpiricalVariance | MinimizedVariance | No. of Families |
| D2S1394 | 174 | .045 | .017 | 18 |
| D20S477 | 242 | .049 | .049 | 10 |
| D21S2052 | 120 | .027 | .023 | 18 |
Genotyping in a larger set of affected relative pairs was performed for the marker D2S1394. The two-point LOD score for D2S1394 increased from 0.50 to 0.58 with the addition of 49 affected sibling pairs. In the larger sample, there were 34 families that were informative for evaluating association between the 174 allele and age at onset of PD. Significant association was observed in this larger sample when either the empirical variance test (P=.017) or the minimized variance test (P=.036) was used. The test statistic computed by FBAT (Z=2.40, using empirical variance) was positive, indicating that later age at onset was associated with the 174 allele in the family-based test. Examination of age at onset among all individuals with PD revealed that mean age at onset was similar for individuals without a 174 allele (61.1±12.03; n=307) compared with those with one 174 allele (60.6±12.28; n=70). Age at onset was greater for 174 homozygotes (69.8±8.75), although the frequency of these individuals was low (n=5).
Discussion
Segregation analysis in PD has implicated the presence of a gene influencing age at onset of PD (Maher et al., in press). We performed a genomewide scan using 103 multiplex families from the GenePD study and identified four regions with suggestive evidence of linkage to age at onset. Two of these regions, on chromosomes 2p13 and 9q, have previously been implicated in linkage studies examining PD affection status (Gasser et al. 1998; Scott et al. 2001). Given the limited support for additional studies, we chose to concentrate on chromosome 2p13 for follow-up in a larger sample.
The strongest evidence of linkage to age at onset (two-point LOD=2.4) in our original sample was observed at 99 cM on chromosome 2, which coincides with the location of PARK3. Affected individuals from families that show linkage to PARK3 appear clinically similar to individuals with idiopathic PD, although dementia was observed in several affected individuals (Gasser et al. 1998). Mean age at onset for PD in these families that show linkage to PARK3 was 59 years (Gasser et al. 1998), which is comparable to the mean age reported here. Two North American families that show linkage to PARK3 trace their ancestry to a common region of southern Denmark and northern Germany and share a common haplotype over a 2.5-Mb region, which narrows the location of the PARK3 gene (West et al. 2001).
In addition to evidence of linkage, we also observed association between the 174 allele of marker D2S1394 and age at onset. Given this association, we focused on the D2S1394 marker when typing additional individuals and observed both an increase in the LOD score and a decrease in the P value for the test of association to allele 174 in the augmented sample. Both the family-based association tests and examination of mean age at onset by genotype among all individuals with PD suggest that the 174 allele is associated with a later age at onset in this study population. Marker D2S1394 is within the 2.5-Mb region defining PARK3, and the 174 allele is present in the PD haplotype shared by the two families that show linkage to PARK3 (West et al. 2001). Association of the same allele of the D2S1394 marker in two independent studies provides strong evidence that this allele is in linkage disequilibrium with a PD-susceptibility gene or a gene influencing age at onset of PD. Further studies are needed to determine the specific gene influencing PD, variants within such a gene, and effects of these variants. Candidate genes in this region include transforming growth factor-α (TGFA), cytochrome P450 retinoid metabolizing protein (P450RAI-2), sepiapterin reductase (SPR, or 7,8-dihydrobiopterin:NADP+ oxidoreductase), and the putative N-acetyltransferase Camello 2 (CML2). In addition, a number of as-yet-undiscovered genes are predicted in this region of linkage.
The linkage peak for age at onset on chromosome 9 overlaps with the previously reported linkage for affection status in the GenePD sample (DeStefano et al. 2001), although the peaks do not coincide, as shown in figure 2. There are no obvious candidate genes at this location.
Figure 2.

Overlay of multipoint LOD-score curve assessing linkage to PD affection status and to PD age at onset for chromosome 9.
Evidence of linkage to PD age at onset was also observed on chromosomes 20 (multipoint LOD=1.82) and 21 (multipoint LOD=2.21) in regions that have not been implicated in other PD linkage studies. Candidate genes in the region of linkage on chromosome 20 include induced transcription factor 2-β (TGIF2), neuronatin, and protein tyrosine phosphatase receptor T. Candidate genes on chromosome 21 include amyloid β (A4) precursor protein and ubiquitin-specific protease 16. Evidence of association was observed for one allele in each region. Additional studies are planned, to examine these regions more closely.
Susceptibility to a disease and age at onset of a disease are two distinct but related phenotypes. It is possible that separate genes may contribute to each of these aspects of genetic vulnerability to disease or that a gene may influence both risk for disease and age at onset. We have identified four regions that show suggestive evidence of linkage to age at onset of PD. Follow-up analysis on chromosome 2 strengthened evidence for linkage. This region overlaps with a locus previously implicated in studies of linkage with affection status. Association with a specific allele on 2p13 suggests close proximity to the gene, and additional work is ongoing, in the GenePD and other studies, to identify the gene at this location.
Acknowledgment
The present study was supported by Public Health Service grant RO-1 NS36711-04 (Genetic Linkage Study in Parkinson’s Disease).
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- ASPEX Linkage Analysis Package, The, ftp://lahmed.stanford.edu/pub/aspex/index.html
- FBAT Web Page, http://www.biostat.harvard.edu/~fbat/default.html
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for PD [MIM 168600], PARK1 [MIM 163890], PARK2 [MIM 602544], PARK3 [MIM 602404], PARK4 [MIM 605543], PARK5 [MIM 191342], PARK6 [MIM 605909], and PARK7 [MIM 606324])
References
- Cardno AG, Holmans PA, Rees MI, Jones LA, McCarthy GM, Hamshere ML, Williams NM, Norton N, Williams HJ, Fenton I, Murphy KC, Sanders RD, Gray MY, O'Donovan MC, McGuffin P, Owen MJ (2001) A genomewide linkage study of age at onset in schizophrenia. Am J Med Genet 105:439–445 [DOI] [PubMed] [Google Scholar]
- DeStefano AL, Golbe LI, Mark MH, Lazzarini AM, Maher NE, Saint-Hilaire M, Feldman RG, et al (2001) Genome-wide scan for Parkinson's disease: the GenePD study. Neurology 57:1124–1126 [DOI] [PubMed] [Google Scholar]
- Farrer M, Gwinn-Hardy K, Muenter M, DeVrieze FW, Crook R, Perez-Tur J, Lincoln S, Maraganore D, Adler C, Newman S, MacElwee K, McCarthy P, Miller C, Waters C, Hardy J (1999) A chromosome 4p haplotype segregating with Parkinson's disease and postural tremor. Hum Mol Genet 8:81–85 [DOI] [PubMed] [Google Scholar]
- Gasser T, Muller-Myhsok B, Wszolek ZK, Oehlmann R, Calne DB, Bonifati V, Bereznai B, Fabrizio E, Vieregge P, Horstmann RD (1998) A susceptibility locus for Parkinson's disease maps to chromosome 2p13. Nat Genet 18:262–265 [DOI] [PubMed] [Google Scholar]
- Gibb WR, Lees AJ (1988) The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiat 51:745–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Xu X, Laird NM (2001) The family based association test method: strategies for studying general genotype-phenotype associations. Eur J Hum Genet 9:301–306 [DOI] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiat 55:181–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608 [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Laird N, Horvath S, Xu X (2000) Implementing a unified approach to family based tests of association Genetic Epi 19 Suppl 1:S36–S42 [DOI] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH (1998) The ubiquitin pathway in Parkinson's disease. Nature 395:451–452 [DOI] [PubMed] [Google Scholar]
- Lunetta KL, Faraone SV, Biederman J, Laird NM (2000) Family-based tests of association and linkage that use unaffected sibs, covariates, and interactions. Am J Hum Genet 66:605–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher NE, Currie LJ, Lazzarini AM, Wilk JB, Taylor CA, Saint-Hilaire MH, Feldman RG, Golbe LI, Wooten GF, Myers RH. A segregation analysis of Parkinson disease revealing evidence for a major causative gene. Am J Med Genet (in press) [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047 [DOI] [PubMed] [Google Scholar]
- Pratt SC, Daly MJ, Kruglyak L (2000) Exact multipoint quantitative-trait linkage analysis in pedigrees by variance components. Am J Hum Genet 66:1153–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz D, Laird N (2000) A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum Hered 50:211–223 [DOI] [PubMed] [Google Scholar]
- Rosenblatt A, Brinkman RR, Liang KY, Almqvist EW, Margolis RL, Huang CY, Sherr M, Franz ML, Abbott MH, Hayden MR, Ross CA (2001) Familial influence on age of onset among siblings with Huntington disease. Am J Med Genet 105:399–403 [PubMed] [Google Scholar]
- Scott WK, Nance MA, Watts RL, Hubble JP, Koller WC, Lyons K, Pahwa R, et al (2001) Complete genomic screen in Parkinson disease: evidence for multiple genes. JAMA 286:2239–2244 [DOI] [PubMed] [Google Scholar]
- Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, Albanese A, Wood NW (2001) Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet 68:895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJLM, Testers L, Breedveld GJ, Horstink M, Sandkuijl LA, van Swieten JC, Oostra BA, Heutink P (2001) PARK7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet 69:629–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Zimprich A, Lockhart PJ, Farrer M, Singleton A, Holtom B, Lincoln S, Hofer A, Hill L, Müller-Myhsok B, Wszolek ZK, Hardy J, Gasser T (2001) Refinement of the Park3 locus on chromosome 2p13 and the analysis of 14 candidate genes. Eur J Hum Genet 9:659–666 [DOI] [PubMed] [Google Scholar]