

Abstract
Endosomal trafficking is a key mechanism to modulate signal propagation and cross talk. Ubiquitin adaptors, along with endosomal sorting complex required for transport (ESCRT) complexes, are also integrated to terminate ligand-receptor activation in late endosomes and multivesicular bodies (MVBs). Within these pathways, we recently demonstrated that the protein SIMPLE is a novel player in MVB regulation. SIMPLE is also clinically important and its mutation accounts for the Charcot-Marie-Tooth type 1C (CMT1C) disease. MVB defects of mutation and deletion of SIMPLE, however, are distinct. Here, we show that MVB defects found in mutation but not deletion of SIMPLE lead to impaired turnover and accumulation of ESCRT-0 protein Hrs puncta in late endosomes. We further uncover increased colocalization of ubiquitin ligase TRAF6 and Hrs in late endosomes. Upon stimulation with interkeukin-1 or transforming growth factor β, prolonged activation of p38 kinase/JNK is detected, while nuclear accumulation of NF-κB and phosphorylation of SMAD2 is reduced with CMT1C mutation. The aberrant kinetics we observed in inflammatory signaling may contribute to increased tumor susceptibility and changes in the levels of chemokines/cytokines that result from CMT1C mutation. We propose that altered endosomal trafficking due to malformations of MVBs and subsequent atypical signaling kinetic may account for a toxic gain of function in CMT1C pathogenesis.
INTRODUCTION
Receptor internalization is a fundamental cell signaling process that functions to limit ligand-receptor activation and to facilitate signaling cross talk (1–5). Internalized receptors are routed through endocytic endosomes and multivesicular bodies (MVBs). Ubiquitin adaptors, such as hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs/Hgs), signal transducing adaptor molecule (STAM), target of Myb protein 1 (Tom1) and arrestin proteins, function to selectively sort internalized membrane receptors (6–8). Ubiquitin adaptors also recruit multiprotein endosomal sorting complex required for transport (ESCRT) complexes to late endosomes/MVBs for the formation of intraluminal vesicles (ILVs) (9–12), which can be released extracellularly as exosomes (13–15). Thus, late endosomes/MVBs are critical hubs for sorting receptor signaling complexes for degradation, recycling, or secretion (16–18).
Current models indicate that Hrs, as a member of the ESCRT-0 complex, interacts with ligand-bound, ubiquitin-conjugated membrane receptors and facilitates their trafficking to the endosomal compartments (8–12). Ligand-bound, internalized receptors continue to signal downstream effectors in endosomes until dissociation of ligands from receptors with exposure to the low-pH environment of late endosomes/MVBs (3–5, 8). Ligand-free receptors are either recycled back to the plasma membrane or degraded through lysosomes to terminate the intracellular signaling. This degradation route is mediated by Hrs and additional ESCRT complexes. Hrs binds to ESCRT-1 protein Tsg101, which in turn recruits additional ESCRT-2 and ESCRT-3 components to initiate invagination on the endosomal membrane to generate ILVs inside the MVBs. Membrane receptors trafficked to ILVs are subjected to degradation once MVBs fuse with lysosomes. Disruption of MVBs can cause an imbalance in the turnover of membrane receptors, lead to aberrant cell signaling (19, 20), ultimately resulting in a range of pathological insults.
While membrane receptors are degraded, the turnover of Hrs requires its dissociation from the endosomal compartments and its return to the cytosol for recycling or degradation (21–23). Dissociation from the endosomal membrane is facilitated by Tyr-phosphorylation and ubiquitination of Hrs. Hence, endosomal localization and turnover of Hrs is dynamic, which is triggered by extracellular stimulation upon membrane receptor activation and is terminated by phosphorylation and ubiquitination of Hrs.
We recently demonstrated that the protein SIMPLE [small integral membrane protein of the lysosome/late endosome] is a novel player in MVB regulation (24). We showed that SIMPLE is located in late endosomes, MVBs, and lysosomes. Furthermore, deletion of SIMPLE [Simple−/−] reduces the formation of MVBs in primary embryonic fibroblasts (MEFs). Thus, SIMPLE is a previously unrecognized non-ESCRT protein in MVB biogenesis.
SIMPLE is also clinically important in that specific point mutations in one of the alleles causes autosomal-dominant Charcot-Marie-Tooth type 1C (CMT1C) demyelination (25–29). To better character CMT1C pathogenesis, we generated a physiological knock-in mouse model with one mutated SIMPLE allele (SimpleT115N/+) (24). In SimpleT115N/+ MEFs and SimpleT115N/+ primary Schwann cells, along with the CMT1C patient B cells, we found improper formation of MVBs. Malformations in the MVBs of SimpleT115N/+ MEFs, however, are distinct from Simple−/− MVBs (24). Multilamellar tubules are found in Simple−/− MEFs, while vacuolated MVBs are evident in SimpleT115N/+ MEFs. Although deletion or mutation of SIMPLE arrest cells at different stages of endosomal trafficking in MVB formation, both models (Simple−/− and SimpleT115N/+) showed a lack of ILVs and reduced exosome secretion. These data confirm SIMPLE as a novel player in endosomal trafficking and MVB formation.
Pathologically, SimpleT115N/+ mice developed modest neurological defects, while Simple−/− mice did not show any symptom (24). In particular, SimpleT115N/+ mice exhibited paralysis at an old age. The lack of neurological/locomotion defects in SIMPLE-null mice was also reported independently using another line of Simple−/− mice (30). These data indicate that a SIMPLE mutation exclusively elicits CMT1C demyelination, which is not seen in Simple−/− mice. Altogether, these data suggest the possibility that the mutation of SIMPLE imparts a toxic gain of function for CMT1C pathogenesis.
The purpose of this study is to elucidate the possible gain of function elicited by the mutation of SIMPLE to further understand CMT1C pathogenesis. Here, we report that mutation, but not deletion, of SIMPLE leads to accumulation of Hrs puncta in primary fibroblasts. Accumulation of Hrs puncta is also observed in CMT1C patient B cells and primary mouse Schwann cells. We further uncover increased colocalization of TRAF6 and Hrs in late endosomes. We also show that upon mutation of SIMPLE, the kinetics of p38 kinase and c-Jun N-terminal kinase (JNK) activation is altered, while nuclear accumulation of NF-κB is reduced in interleukin-1 (IL-1) challenge. Signaling kinetic elicited by transforming growth factor beta (TGF-β) is also altered in SimpleT115N/+ MEFs. The aberrant kinetics in inflammatory signaling correlate with increased tumor susceptibility and potentiated levels of chemokines/cytokines in SimpleT115N/+ mice. We propose that altered endosomal trafficking due to malformations of MVBs and subsequent atypical inflammatory signaling kinetics may account for the gain of function elicited by the SIMPLE mutation in CMT1C patients.
MATERIALS AND METHODS
Reagents.
Polyclonal antibody against SIMPLE was generated with COOH-terminal peptide DVDHYCPNCKALLGTYKRL as an antigen using standard techniques (24). The primary antibodies used were SIMPLE (Sigma; HPA006960), EEA1 (Cell Signaling; 3288), Rab7 (Cell Signaling; 9367), Rab11 (Cell Signaling; 5589), Hrs (Santa Cruz; SC271925), Tsg101 (Santa Cruz; SC22774), JNK (Santa Cruz; SC474), phospho-JNK (Cell Signaling; 9255), p38 kinase (Cell Signaling; 8690), phospho-p38 kinase (Cell Signaling; 9211), TAK1 (Cell Signaling; 5206), phospho-TAK1 (Cell Signaling; 4531), TRAF6 (Santa Cruz; SC7221 and SC8409), phospho-Tyr clone 4G10 (EMD Millipore; 05-321), ubiquitin (Santa Cruz; SC8017), IκB (Cell Signaling; 4814), NF-κB (Santa Cruz; SC372), SMAD2 (Cell Signaling; 5339), phospho-SMAD2 (Cell Signaling; 3108), V5 (Santa Cruz; SC81594), and HA (Santa Cruz; SC7392). Tubulin (E7) antibody was obtained from monoclonal antibody facility (University of Iowa). IL-1 and TGF-β were obtained from PreproTech. Cytokine/chemokine antibody arrays were obtained from RayBiotech.
Mice.
Animal experiments were performed in accordance with the guidelines of the Albert Einstein College of Medicine Institute of Animal Studies. Generation of SimpleT115N/+ mice with a C→A point mutation on codon 115 in exon 3 of the SIMPLE gene (T115N) was reported previously (24). The generation of Simple−/+ mice and mating to obtain Simple−/− mice has also been described (24). SimpleT115N/+ and Simple−/− mice were backcrossed into C57BL/6 more than 10 times before use. Subcutaneous implantation (2 × 106 cells/mouse) and tail vein injection (0.4 × 106 cells/mouse) of B16 melanoma cells into C57BL/6 backcrossed Simple+/+ and SimpleT115N/+ mice was carried out as described previously (31).
Cell culture.
Primary MEFs with Simple+/+, SimpleT115N/+, Simple−/−, and SimpleT115N/T115N genotypes were isolated from embryonic day 13.5 (E13.5) pups and cultured as described previously (32). Primary mouse Schwann cells were isolated from dorsal root ganglia of E13.5 pups and cultured as described previously (33). Epstein-Barr virus-transformed B cells from control and CMT1C patients were cultured as described previously (34). COS and B16F10 melanoma cells were cultured in Dulbecco modified Eagle medium. Transient transfection on COS cells were carried out using Lipofectamine according to the manufacturer. All media were supplemented with 10% fetal calf serum, 2 mM l-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml) (Invitrogen). Cells were transfected by using Lipofectamine (Invitrogen).
Confocal microscopy.
Cells were plated on coverslips 24 h before the experiment. Cells were washed three times in cold phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 15 min. Paraformaldehyde was quenched by 0.1 M glycine in PBS for 10 min. After incubation in blocking solution (0.5% bovine serum albumin, 0.1% saponin, and 1% fetal bovine serum in PBS), the cells were incubated with primary antibodies at 4°C overnight, followed by secondary antibodies for 1 h at room temperature. All images were taken with Leica AOBS SP2 confocal microscope with a 63× objective lens, and primary representative images were shown. Processing and the brightness of images were uniformly adjusted to enhance contrast using Adobe Photoshop.
Image and statistical analysis.
Colocalization was analyzed in merged images using ImageJ software, and the relative numbers of colocalized puncta were counted and presented. Densitometry was performed to determine intensity in coimmunoprecipitation. The data are shown as means ± the standard errors of the mean. Two-way analysis of variance (ANOVA) was performed, and P values of <0.05 were considered significant.
RESULTS
Accumulation of Hrs puncta upon CMT1C mutation of SIMPLE.
ESCRT protein complexes play a key role in endosomal membrane invagination, which generates ILVs inside MVBs (9–12). Indeed, deletion of the ESCRT protein Hrs (ESCRT-0) or Tsg101 (ESCRT-1) (35, 36) leads to empty vacuoles, which are highly resemble to those found in SimpleT115N/+ MEFs (24). The similar vacuolated morphology in MVB defects suggested that regulation and function of ESCRT proteins might be affected upon the CMT1C mutation of SIMPLE. To investigate the possibility of a toxic gain-of-function mechanism upon CMT1C mutation of SIMPLE, we examined the subcellular localization of Tsg101 and Hrs. We performed confocal microscopy and found that endogenous Tsg101 exhibited perinuclear staining and that its distribution was indistinguishable among Simple+/+, SimpleT115N/+, and Simple−/− MEFs (Fig. 1A).
FIG 1.

Accumulation of Hrs puncta upon CMT1C mutation of SIMPLE. Confocal microscopy was performed and primary MEFs obtained from Simple+/+, Simple−/−, and SimpleT115N/+ mice were costained for endogenous Hrs and Tsg101 (A), Rab7 (B), EEA1 (C), and Rab11 (D). Costaining of Hrs-Rab7 (B) and Hrs-Rab11 (D) in SimpleT115N/T115N MEFs is also shown. DNA in nuclei was visualized using DAPI (blue). Representative images were shown. (E) Colocalization of Hrs-Rab7 puncta in Simple+/+, Simple−/−, SimpleT115N/+, and SimpleT115N/T115N MEFs was quantified. *, P < 0.05; **, P < 0.01.
Next, we examined the subcellular distribution of Hrs. In Simple+/+ and Simple−/− MEFs, Hrs exhibited similar diffused staining with occasional tiny speckles around the perinuclear region (Fig. 1A). In SimpleT115N/+ MEFs, however, we found the accumulation of Hrs puncta that were larger in size (Fig. 1A). Among the punctated staining pattern in SimpleT115N/+ MEFs, we found partial colocalization of Hrs and Rab7, a marker for late endosomes (Fig. 1B). Minimal colocalizations in the puncta, however, were detected by Hrs and early endosome antigen 1 (EEA1) (Fig. 1C), or Hrs and Rab11, a marker for recycling endosomes (Fig. 1D). Together, these data indicate that mutation, but not deletion, of SIMPLE leads to accumulation of Hrs in the late endosome compartments.
Given that SIMPLE is widely expressed, we further ascertained accumulation of Hrs puncta in CMT1C patient B cells and mouse primary SimpleT115N/+ Schwann cells (Fig. 2). Notably, the Schwann cells are suggested to account for the CMT1C neuropathy based upon increased sensitivity of this cell type. Confocal microscopy indicated increased colocalization of Hrs-Rab7 puncta in the CMT1C patient B cells (Fig. 2A) and SimpleT115N/+ Schwann cells (Fig. 2B). In SimpleT115N/+ Schwann cells, the Hrs-Rab7 puncta were localized along the cell periphery and were also obvious in the spindle extensions (Fig. 2B). Thus, these data indicate that CMT1C mutation of SIMPLE leads to the accumulation of Hrs in the late endosome compartments in multiple cell types.
FIG 2.
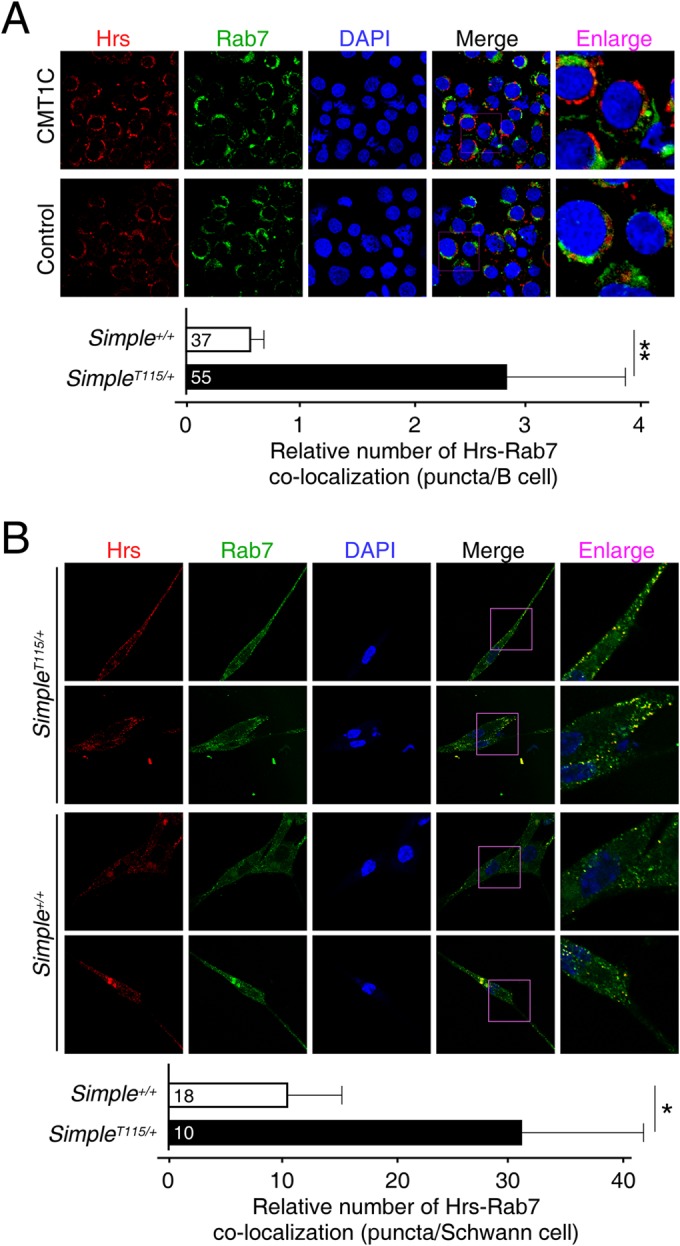
Accumulation of Hrs puncta in CMT1C patient B cells and in primary Schwann cells. Confocal microscopy was performed and costained for endogenous Hrs and Rab7 on control and CMT1C patient B cells (A) or Simple+/+ and SimpleT115N/+ mouse Schwann cells (B). DNA in nuclei was visualized using DAPI (blue). Representative images are shown. Colocalization of Hrs-Rab7 puncta was quantified. *, P < 0.05; **, P < 0.01.
CMT1C is an autosomal-dominant neuropathy and mutation of one copy of the SIMPLE allele is sufficient to cause demyelination. The possible gain of function elicited by the mutation of SIMPLE would suggest that the normal SIMPLE allele might not contribute to the pathogenesis. Indeed, we observed vacuolated appearances in MVBs in SimpleT115N/+ and SimpleT115N/T115N MEFs but not Simple−/− MEFs (24). All three genetic models, however, showed reduced exosome production. Thus, we examined whether Hrs puncta was accumulated in SimpleT115N/T115N MEFs. Similar to SimpleT115N/+ MEFs, SimpleT115N/T115N MEFs showed the accumulation of Hrs puncta which were colocalized with Rab7 (Fig. 1B) but not Rab11 (Fig. 1D). The accumulation of Hrs-Rab7 puncta in SimpleT115N/+ and SimpleT115N/T115N MEFs, therefore, was independent of the normal SIMPLE allele and was only observed following the CMT1C mutation of SIMPLE (Fig. 1E).
Regulation of Hrs upon CMT1C mutation of SIMPLE.
Endosomal localization and turnover of Hrs is dynamic, which is triggered by extracellular stimulation (e.g., serum) upon membrane receptor activation (3–5, 8) and is terminated by phosphorylation and ubiquitination of Hrs (21–23). To investigate whether the dynamics of endosomal localization accounted for the accumulation of Hrs puncta in SimpleT115N/+ MEFs, we examined the effect of extracellular stimulation using serum. Serum stimulation increased the accumulation of Hrs puncta in SimpleT115N/+ MEFs (Fig. 3A). Hrs puncta formed under serum stimulation in SimpleT115N/+ MEFs were more readily visualized by their increased size and number. Serum treatment also led to lesser diffused Hrs staining and more formation of tiny speckles in Simple+/+ and Simple−/− MEFs (Fig. 3A). The tiny speckles in Simple+/+ and Simple−/− MEFs, however, were not as pronounced as the distinctive Hrs puncta in SimpleT115N/+ MEFs.
FIG 3.

Regulation of Hrs upon CMT1C mutation of SIMPLE. (A) Confocal microscopy was performed, and primary MEFs obtained from Simple+/+, Simple−/−, and SimpleT115N/+ mice were serum starved for 2 h prior to stimulation or not with 20% serum for 30 min. Cells were costained for endogenous Hrs and Rab7, and representative images are shown. (B) Confocal microscopy was performed, and primary MEFs were cultured in serum or serum withdrawal was carried out for 1 or 4 h. Cells were costained for endogenous Hrs and Rab7, and representative images are shown. (C) Simple+/+ (WT) and SimpleT115N/+ (MT) MEFs were treated or not treated with 20% serum for 30 min. Cell lysate prepared was immunoprecipitated (IP) with an antibody against Hrs. Modification of Hrs by phospho-Tyr or ubiquitin (Ub) was examined by immunoblotting (IB). Antibody against Hrs (Ab) was loaded in reference to the IgG in the immunoprecipitates. The levels of pTyr and Ub modification of Hrs were quantified. *, P < 0.05.
We also examined the effect of serum withdrawal on the dynamics of the Hrs puncta (Fig. 3B). Serum withdrawal led to a time-dependent reduction of Hrs puncta in SimpleT115N/+ MEFs. By 1 h after serum withdrawal, the size and number of Hrs puncta were less evident in SimpleT115N/+ MEFs. By 4 h after serum withdrawal, tiny speckles along with a diffused pattern of Hrs were observed in SimpleT115N/+ MEFs. For Simple+/+ MEFs under serum culture, a tiny speckled pattern of Hrs was observed, which became diffused upon serum withdrawal. Together, these data indicate that the accumulation of Hrs puncta in SimpleT115N/+ MEFs is dynamic and is regulated by extracellular stimulation.
Given that the turnover of Hrs on the endosomal membrane is regulated by ubiquitination and Tyr phosphorylation (21–23), we further examined posttranslational modifications of endogenous Hrs in Simple+/+ and SimpleT115N/+ MEFs. Under both basal and serum-treated conditions, the extent of ubiquitination of Hrs in SimpleT115N/+ MEFs remained lower than in Simple+/+ MEFs (Fig. 3C). Under the basal condition, the extent of the Tyr phosphorylation of Hrs in SimpleT115N/+ MEFs was reduced compared to Simple+/+ MEFs (Fig. 3C). Upon serum stimulation, Tyr phosphorylation of Hrs was increased, as reported previously (21–23). The extent of Tyr phosphorylation of Hrs under serum stimulation, however, was indistinguishable in Simple+/+ and SimpleT115N/+ MEFs. Together, these data indicate that reduced ubiquitination may in part contribute to the impaired turnover of Hrs in SimpleT115N/+ MEFs.
Dysregulated TRAF6-TAK1 axis upon CMT1C mutation of SIMPLE.
The accumulation of Hrs puncta and its dynamic nature suggested a role for SIMPLE in modulating cell signaling. A complex protein-protein interaction network is critical for receptor signaling and subsequent internalization (2–5, 7, 10–12). Thus, we performed binding assays to identify protein interactors to further investigate the possible role of SIMPLE in endosomal signaling. We found that SIMPLE interacted with Tsg101 and all members of the Nedd4 E3 ligase family (Fig. 4A). Indeed, previous reports demonstrated that the PTAP and PPXY motifs are required for the interaction of SIMPLE with Tsg101 and Nedd4 E3 ligase, respectively (34, 37).
FIG 4.

Activation of the RAF6-TAK1 axis upon CMT1C mutation of SIMPLE. (A) COS cells were transiently transfected with HA-SIMPLE and V5-tagged Tsg101. The presence of Tsg101 in SIMPLE immunoprecipitates (IP) was determined by immunoblotting (IB). Similar coimmunoprecipitations of SIMPLE and different members of the V5-tagged Nedd4 E3 ligase family (Nedd4.1, Nedd4.2, Itch, WWP1, and WWP2) were performed. (B) COS cells were transiently transfected with HA-SIMPLE and V5-tagged Tom1 and Tab2. The presence of Tom1 and Tab2 in SIMPLE immunoprecipitates (IP) was determined by immunoblotting (IB). (C and D) Cell lysate prepared from Simple+/+ (WT) and SimpleT115N/+ (MT) MEFs was immunoprecipitated (IP) with an antibody against TRAF6 or TAK1. The ubiquitination (Ub) of TRAF6 or TAK1 is shown in panel C. Association and phosphorylation (P) of TAK1 in the immunoprecipitates was examined by immunoblotting (IB) as shown in panel D. Antibody against TRAF6 or TAK1 (Ab) was loaded in reference to the IgG in the immunoprecipitates. The levels of ubiquitinated TRAF6 or TAK1 were quantified. *, P < 0.05; **, P < 0.01. (E and F) Confocal microscopy was performed, and primary MEFs obtained from Simple+/+ and SimpleT115N/+ mice were costained for endogenous TRAF6 and Hrs (E) or TRAF6 and Rab7 (F). DNA in nuclei was visualized by using DAPI (blue). Representative images are shown. The colocalization of Hrs-TRAF6 and TRAF6-Rab7 puncta was quantified. *, P < 0.05; **, P < 0.01.
In addition, we found that SIMPLE interacts with an ESCRT-0 protein Tom1 and a signaling adaptor Tab2 (Fig. 4B). Tom1 and Tab2 play an important role in the internalization and signal propagation upon stimulation with inflammatory cytokines, such as IL-1 and TGF-β (38–44). Tom1 is an ubiquitin adaptor and interacts with receptor adaptor SARA and Tollip for its role in TGF-β and IL-1 signaling, respectively (8, 33, 63). Tab2 provides an ubiquitin platform and recruits TGF-β associated kinase (TAK1) in TGF-β and IL-1 signaling (41, 45–48). Furthermore, distinct types of ubiquitin conjugation mediated by different ubiquitin E3 ligases are required for activation, propagation, and termination of the inflammatory signaling (42, 49–53). Some of these ubiquitin ligases, such as Nedd4 and Itch, are known SIMPLE interacting proteins (Fig. 4A) (34, 37). Lastly, SIMPLE is also known as LITAF, which is induced upon inflammation and modulates cytokine gene induction (54–56). Together, these observations strongly suggested that SIMPLE might play a role in ubiquitin-mediated inflammatory signaling. The accumulation of Hrs puncta further suggested a possible alteration in inflammatory signaling in SimpleT115N/+ MEFs.
Next, we examined ubiquitination of key signaling components in the inflammatory pathway in Simple+/+ and SimpleT115N/+ MEFs. Among them, the TRAF family of ubiquitin ligases serves as a critical node for recruiting upstream kinases, such as TAK1, for activation of downstream signaling effectors (46, 57, 58). Coimmunoprecipitations indicated increased ubiquitination of the TRAF6 adaptor in SimpleT115N/+ MEFs (Fig. 4C). In SimpleT115N/+ MEFs, the ubiquitinated TRAF6 also showed increased recruitment of TAK1 kinase (Fig. 4D). Indeed, TAK1 was phosphorylated and ubiquitinated more definitively in SimpleT115N/+ MEFs than in Simple+/+ MEFs (Fig. 4C and D). These data indicate increased formation of a TRAF6-TAK1 complex and potentiated phosphorylation of TAK1 in SimpleT115N/+ MEFs.
We further investigated the localization of TRAF6 by confocal microscopy. We found partial colocalization of TRAF6 with the Hrs puncta in SimpleT115N/+ MEFs (Fig. 4E). TRAF6 was also partially colocalized with the late endosome marker Rab7 in SimpleT115N/+ MEFs (Fig. 4F). In Simple+/+ MEFs, TRAF6 exhibited a diffused cytoplasmic pattern. These data indicate that mutation of SIMPLE leads to the colocalization of TRAF6 in the Hrs puncta in SimpleT115N/+ MEFs. Collectively, accumulation of Hrs puncta and their colocalization with TRAF6 in the late endosomes may account for the increased ubiquitination and phosphorylation of TAK1.
Altered kinetics of p38 kinase and JNK phosphorylation upon CMT1C mutation of SIMPLE.
The TRAF6-TAK1 axis mediates phosphorylation of mitogen-activated protein kinases (MAPKs), including p38 kinase and JNK (46, 59–61). The presence of TRAF6 in the accumulated Hrs puncta and the increased phosphorylation of TAK1 suggested that phosphorylation of the p38 kinase and JNK might be altered in SimpleT115N/+ MEFs. We examined the phosphorylation of p38 kinase and JNK under basal conditions and upon stimulation with IL-1 for different times in SimpleT115N/+, Simple+/+, and Simple−/− MEFs (Fig. 5). We found that optimal phosphorylation of p38 kinase and JNK occurred at about 5 to 15 min after IL-1 stimulation. The kinetics of p38 kinase and JNK phosphorylation were indistinguishable in Simple+/+ and Simple−/− MEFs (Fig. 5A). The kinetics of p38 kinase and JNK phosphorylation in SimpleT115N/+ MEFs, however, were different (Fig. 5B). In particular, the phosphorylation at the basal level and late stage of p38 kinase and JNK was elevated in SimpleT115N/+ MEFs. These data indicate that mutation of SIMPLE leads to aberrant activation kinetics in p38 kinase and JNK.
FIG 5.

Dysregulated IL-1 signaling upon CMT1C mutation of SIMPLE. Primary MEFs prepared from Simple+/+ and Simple−/− mice (A and C) or Simple+/+ and SimpleT115N/+ mice (B and D) were treated or not treated with IL-1 for the times indicated. A cell lysate prepared was immunoblotted for phosphorylated (indicated by a “P-” prefix) and total p38 kinase and JNK, and the results are shown in panels A and B. The expression levels of IκB and tubulin are also shown in panels C and D. The relative levels of phosphorylation of p38 kinase and IκB expression were also quantified and graphed. *, P < 0.05.
We further performed confocal microscopy to investigate the kinetics of p38 kinase and JNK phosphorylation in SimpleT115N/+ MEFs. Under the basal condition, partial staining of phospho-p38 kinase and phospho-JNK was found in SimpleT115N/+ nuclei but not in Simple+/+ nuclei (Fig. 6). Optimal phosphorylation and nuclear localization of p38 kinase and JNK were evident after 10 min of IL-1 stimulation in both SimpleT115N/+ and Simple+/+ nuclei. After 30 and 45 min of IL-1 stimulation, staining of phospho-p38 kinase and phospho-JNK was minimal in Simple+/+ nuclei. Staining of phospho-p38 kinase and phospho-JNK, however, remained evident in many of the SimpleT115N/+ nuclei after 30 or 45 min of IL-1 stimulation. These data confirm that the mutation of SIMPLE elicits aberrant signaling kinetics with prolonged activation of p38 kinase and JNK.
FIG 6.

Increased nuclear localization of p38 kinase and JNK upon CMT1C mutation of SIMPLE. Primary MEFs prepared from Simple+/+ and SimpleT115N/+ mice were treated (10, 30, and 45 min) or not treated with IL-1. Confocal microscopy was performed to detect the phosphorylation of p38 kinase (P-p38) (A) or JNK (P-JNK) (B). DNA in nuclei was visualized using DAPI, and this is represented in the red channel. The percentage of cells with nuclear accumulated P-p38 kinase or P-JNK was quantified. *, P < 0.05; **, P < 0.01.
Altered kinetics of NF-κB activation upon CMT1C mutation of SIMPLE.
The TRAF6-TAK1 axis also mediates nuclear accumulation of NF-κB through the activation of the IκB protein kinase (IKK) pathway (46, 59, 61, 62). We examined the kinetics of NF-κB nuclear accumulation upon IL-1 stimulation by confocal microscopy (Fig. 7A). At 10, 30, and 45 min after IL-1 stimulation, nuclear accumulation of NF-κB was found in Simple+/+ MEFs (Fig. 7A). IL-1 also led to nuclear accumulation of NF-κB in SimpleT115N/+ MEFs after 10 or 30 min of stimulation. By 45 min, however, many SimpleT115N/+ MEFs did not show nuclear accumulation of NF-κB. These data further indicate altered kinetics of NF-κB activation upon SIMPLE mutation.
FIG 7.

Reduced activation of NF-κB upon CMT1C mutation of SIMPLE. Primary MEFs prepared from Simple+/+ and SimpleT115N/+ mice were treated (10, 30, and 45 min) or not treated with IL-1. (A) Confocal microscopy was performed to investigate the localization of NF-κB. (B) The expression of IκB was also examined. DNA in the nuclei was visualized using DAPI, and this is represented in the red channel. The percentage of cells with nuclear accumulated NF-κB or expression of IκB was quantified. *, P < 0.05.
Nuclear accumulation of NF-κB is controlled by the level of IκB protein, which is degraded upon phosphorylation by the IKK pathway (48). Activation of the NF-κB pathway, however, provides a negative-feedback regulation and re-expression of IκB protein suppresses and terminates nuclear accumulated NF-κB. Thus, we examined the levels of IκB protein upon IL-1 stimulation by immunoblot analysis (Fig. 5C and D) and confocal microscopy (Fig. 7B). We found that the levels of IκB protein in Simple+/+ MEFs were abrogated after 10 min of IL-1 stimulation (Fig. 5C and D). By 25 min after IL-1 stimulation, the expression of IκB protein had reappeared, and the levels of IκB protein were further accumulated in Simple+/+ MEFs by 45 min. The kinetics of IκB protein expression was indistinguishable between Simple+/+ and Simple−/− MEFs (Fig. 5C). In SimpleT115N/+ MEFs, however, the degradation of IκB protein was incomplete after 10 to 20 min of IL-1 stimulation (Fig. 5D). At 45 min after IL-1 stimulation, the expression levels of IκB protein in SimpleT115N/+ MEFs were lower than in Simple+/+ MEFs. Confocal microscopy also indicated similar altered expression of the IκB protein in SimpleT115N/+ MEFs compared to Simple+/+ MEFs (Fig. 7B). These data confirm that mutation of SIMPLE leads to distorted kinetics in the activation of NF-κB, which is partly due to incomplete degradation of IκB.
Dysregulated TGF-β signaling upon CMT1C mutation of SIMPLE.
In addition to the IL-1 signaling, Tom1, Tab2, TRAF6, and TAK1 also play a role in TGF-β signal transduction (63–66). TGF-β signaling leads to the activation of MAPKs, as well as the phosphorylation of transcription factor SMAD2 (67–69). Thus, we examined the phosphorylation of p38 kinase and SMAD2 upon stimulation with TGF-β for different times in SimpleT115N/+, Simple+/+, and Simple−/− MEFs (Fig. 8). Similar to the IL-1 stimulation, we found that optimal phosphorylation of p38 kinase occurred at about 5 to 15 min after TGF-β stimulation. Phosphorylation of SMAD2 also showed similar activation kinetics after TGF-β stimulation. The kinetics of p38 kinase and SMAD2 phosphorylation were similar in Simple+/+ and Simple−/− MEFs (Fig. 8A). In parallel to IL-1 stimulation, administration of TGF-β also altered the kinetics of p38 kinase phosphorylation in SimpleT115N/+ MEFs (Fig. 8B). In particular, phosphorylation of p38 kinase at the basal level and the late stage were elevated in SimpleT115N/+ MEFs upon TGF-β stimulation. Phosphorylation kinetics of SMAD2 in SimpleT115N/+ MEFs, however, was reduced compared to Simple+/+ MEFs. These data indicate that mutation, but not deletion, of SIMPLE also leads to aberrant signaling kinetics with prolonged activation of p38 kinase but diminished SMAD2 phosphorylation upon TGF-β stimulation.
FIG 8.

Dysregulated TGF-β signaling upon CMT1C mutation of SIMPLE. Primary MEFs prepared from Simple+/+ and Simple−/− mice (A) or Simple+/+ and SimpleT115N/+ mice (B) were treated or not treated with TGF-β for the times indicated. A cell lysate was prepared and immunoblotted for phosphorylated (indicated by a “P-” prefix) and total p38 kinase and SMAD2. The expression levels of tubulin are also shown. The relative levels of phosphorylation of p38 kinase and SMAD2 were quantified. *, P < 0.05.
Increased tumor susceptibility upon CMT1C mutation of SIMPLE.
Given the dysregulated activation kinetics in the IL-1 and TGF-β pathways, we sought to determine whether CMT1C mutation would exert in vivo pathological consequence. In addition to peripheral neuropathy, dysregulation of SIMPLE is also associated with arrhythmias and sudden cardiac death in patients with idiopathic prolonged QT interval duration (LQT syndrome) (70, 71). SIMPLE also contributes to tumorigenesis (72–74), in which inflammatory pathways such as those utilized by IL-1 and TGF-β are frequently altered.
We first tested whether CMT1C mutation would affect tumor susceptibility in a syngeneic xenograft model using B16 melanoma cells. Subcutaneous implantation of B16 melanoma cells led to formation of tumor mass at the injected site. The tumor masses from SimpleT115N/+ mice, however, were larger than those from Simple+/+ mice (Fig. 9A). These data indicate increased tumor susceptibility upon CMT1C mutation of SIMPLE.
FIG 9.

Increased tumor susceptibility upon CMT1C mutation of SIMPLE. (A) Simple+/+ (n = 7) and SimpleT115N/+ (n = 6) mice were subcutaneously implanted with B16 melanoma cells (2 × 106 cells/mouse) in the upper right chest. A tumor mass on day 10 postimplantation was isolated and weighed. *, P < 0.05. (B and C) Simple+/+ (n = 21) and SimpleT115N/+ (n = 12) mice were injected with B16 melanoma cells (0.4 × 106 cells/mouse) via tail vein. Tumor cells that colonized to form dark nodules in the lung were visualized at day 10 or day 22 postinjection as shown in panel B. The numbers and sizes of dark nodules that colonized in the liver and kidney at day 10 or day 22 postinjection were also determined, as shown in panel C. *, P < 0.05; ND, not detected. (D) Simple+/+ and SimpleT115N/+ mice were intravenously inoculated (B16) or not inoculated (control, CTL) with B16 melanoma cells. Serum samples were incubated with antibody arrays to determine the relative abundance of different cytokines/chemokines. The level of cytokine/chemokine detected in control Simple+/+ serum was used as a reference for normalization. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
We further ascertained tumor susceptibility in SimpleT115N/+ mice by tail vein injection of B16 melanoma cells, which led to the deposits of melanin and the formation of dark nodules with primary colonization in the lung (Fig. 9B). At day 10 postinjection, the numbers and sizes of dark nodules formed in SimpleT115N/+ lungs were elevated compared to Simple+/+ lungs. We also observed increased numbers and sizes of dark nodules in SimpleT115N/+ livers (Fig. 9C), which is a common secondary site for tumor cell colonization. By day 22 postinjection, the increased numbers and sizes of dark nodules formed in SimpleT115N/+ lungs and livers were more evident (Fig. 9B and C). Furthermore, additional dark nodules were detected in SimpleT115N/+ kidneys (Fig. 9C), which were not found in Simple+/+ kidneys, by day 22 postinjection. These data confirm increased tumor susceptibility upon CMT1C mutation of SIMPLE.
Previous reports showed that SIMPLE plays a key role in cytokine production upon lipopolysaccharide (LPS) challenge (54, 56). Increased tumor susceptibility upon CMT1C mutation of SIMPLE suggested that the levels of cytokine and chemokines might be altered. Thus, we collected serum from both SimpleT115N/+ and Simple+/+ mice upon intravenous inoculation or not with B16 melanoma cells. Cytokine/chemokine analysis indicated the level of CCL1, CCL12, CCL17, CCL19, CCL20, and CCL25 chemokines were specifically elevated in SimpleT115N/+ serum samples upon B16 challenge (Fig. 9D). We also found that the levels of several chemokines and immune cell mitogens were elevated in SimpleT115N/+ sera compared to Simple+/+ sera. The induction of these cytokines/chemokines was independent of B16 injection. These included IL-10, IL-12, XCL1 TPO, sTNFRII, and CXCL1. There were also some cytokines/chemokines, such as CXCL16, macrophage colony-stimulating factor (M-CSF), vascular endothelial growth factor (VEGF), and CXCL4, in which the levels were comparable in both SimpleT115N/+ and Simple+/+ sera. Collectively, these data indicate that changes in the profiles of cytokines/chemokines may be associated with increased tumor susceptibility and aberrant kinetics in inflammatory signaling upon CMT1C mutation of SIMPLE.
DISCUSSION
In this report, we show that mutation, but not deletion, of protein SIMPLE leads to accumulation of Hrs puncta. We also show altered signaling kinetics in SimpleT115N/+ MEFs upon IL-1 or TGF-β challenges. Dysregulated inflammatory signaling may contribute to the increased tumor susceptibility and abnormal levels of chemokines/cytokines in SimpleT115N/+ mice. These findings, therefore, demonstrate altered signaling kinetics upon mutation of SIMPLE, which exhibit distinct defects in MVBs. These data also for the first time provide a possible understanding of the toxic gain of function in CMT1C pathogenesis.
Protein SIMPLE and receptor trafficking.
We have uncovered the accumulation of Hrs puncta in SimpleT115N/+ MEFs. We found that the accumulation of Hrs puncta is dynamic and is caused, in part, by its impaired turnover after receptor signaling. Reduced ubiquitination and Tyr phosphorylation support the impaired turnover and the accumulation of Hrs in SimpleT115N/+ late endosomes. Ubiquitination is a result of opposing actions of ubiquitin ligases and deubiquitinases (50, 75). Similarly, Tyr phosphorylation is mediated by an overall balance of kinases and phosphatases. Perhaps, recruitment of ubiquitin ligases and/or deubiquitinases to late endosomes is altered upon CMT1C mutation. By the same token, the binding of kinases and/or phosphatases may be affected upon CMT1C mutation. Thus, the changes in the SIMPLE binding partners affect turnover of inflammatory receptors and/or ESCRT proteins in late endosomes.
The role of SIMPLE in inflammatory signaling is highly similar to that of the arrestin protein family (α-arrestins and β-arrestins) in modulating G protein-coupled receptor (GPCR) signaling (2, 76, 77). In particular, similar signature binding motifs that facilitate receptor endocytosis and endosomal trafficking are found in both SIMPLE and arrestin proteins. First, both SIMPLE and β-arrestin proteins encode a di-Leu type AP2 binding motif to facilitate clathrin-mediated receptor internalization (24, 76, 78). Second, SIMPLE interacts with Nedd4 E3 ubiquitin ligases via the PPXY motifs (34, 37). PPXY motifs are also found in all of the α-arrestin proteins except the Arrdc5 member (79, 80). Third, SIMPLE encodes a PTAP motif for its interaction with ESCRT-1 protein Tsg101 (34, 37). Interaction with Tsg101 is unique for Arrdc1 among the six members of the α-arrestin family (81). Homo- and heteromerization among α-arrestin and β-arrestin proteins (82) may further assemble a composite complex that resembles SIMPLE, encoding all three signature binding motifs.
In addition to endosomal trafficking in the intracellular compartments, SIMPLE also functions highly similarly to the α-arrestin protein Arrdc1 in the production of extracellular nanovesicles. Extracellular nanovesicles include microvesicles budded from plasma membrane and exosomes derived from MVBs (83–85). Extracellular nanovesicles have been shown to mediate intercellular signaling (86, 87). Previous reports indicated that Arrdc1 plays a key role in the budding of microvesicles from the plasma membrane (88, 89). Budding of microvesicles mediated by Arrdc1 is analogous to the invagination of the endosomal membrane for the generation of ILVs and exosomes: our initial findings of SIMPLE in MVB biogenesis (24). Collectively, we surmise that SIMPLE acts, in parallel to the arrestin proteins in the GPCR pathway and budding of microvesicles, as a ubiquitin adaptor to downregulate inflammatory receptor signaling and participate in exosome production in MVBs.
Protein SIMPLE and inflammatory signaling.
Our data indicate a role for SIMPLE in inflammatory signaling mediated by IL-1 and TGF-β. The possible involvement of SIMPLE in other inflammatory pathways, such as the Toll-like receptor (TLR) complex and tumor necrosis factor alpha (TNF-α) signaling, however, cannot be excluded. In particular, the TLR complex and the TNF-α receptor elicit a similar ubiquitin platform for the recruitment of various signaling adaptors and upstream kinases (50, 90). Many of these adaptors and upstream kinases (e.g., Tollip and TAK1) are also associated with the IL-1 and TGF-β signaling pathways. In addition, Tom1, which interacts with SIMPLE and was proposed as an ancestral type ESCRT-0 (91, 92), facilitates the internalization and termination of the activated TLR complex and the TNF-α receptor (93). Furthermore, previous reports showed that SIMPLE-null mice are less sensitive to challenge by a lethal dose of LPS, in part due to altered cytokine levels (54, 56, 94). Thus, we expect a similar role for SIMPLE in the TLR complex and the TNF-α receptor signaling pathway.
We have uncovered aberrant signaling kinetics upon IL-1 or TGF-β stimulation in SimpleT115N/+ MEFs. We found prolonged activation of p38 kinase and JNK, whereas the nuclear accumulation of NF-κB is reduced upon IL-1 stimulation. Similarly, upon TGF-β challenge, phosphorylation of p38 kinase is extended, whereas the activation of SMAD2 is diminished in SimpleT115N/+ MEFs. The differential effects displayed in SimpleT115N/+ MEFs are highly reminiscent of the “biased” signaling reported in the GPCR pathway (95–97).
Current models indicate that activation of the GPCR pathway elicits protein kinase A (PKA) activation via accumulation of second messenger cyclic AMP. Ligand-bound GPCR also leads to increased phosphorylation of MAPKs via β-arrestin scaffolds. Deletion of β-arrestin proteins or administration of modified ligands can preferentially elicit either a PKA response or a MAPK activation (95–97). The “biased” GPCR signaling seems to exert a beneficial therapeutic response in a context-dependent manner toward specific disease types (98–101). Given the differential effects on p38 kinase/JNK and NF-κB/SMAD2 in SimpleT115N/+ MEFs, it is tempting to speculate that similar “biased” signaling mechanisms are present in the inflammatory pathways. Thus, in parallel to the synthetic ligands that selectively activate “biased” GPCR pathways, molecular engineering to produce recombinant IL-1 or TGF-β ligands/biologics may be carried out. Such “biased” reagents may elicit a preferential signaling and achieve a differential therapeutic response, allowing future exploitations that target the beneficial side of the inflammatory pathways.
CMT1C pathogenesis.
Previously, we showed that the CMT1C mutation elicits distinctive MVB morphology that is not found in the SIMPLE-null mice (24). Here, we extend the exclusive changes and show the accumulation of Hrs, along with abnormal IL-1 and TGF-β signaling kinetics, in SimpleT115N/+ MEFs but not in Simple−/− MEFs. These selective defects in MVB structure, Hrs accumulation, and aberrant inflammatory signaling kinetics may contribute to locomotion defects and paralysis that are found in SimpleT115N/+ mice but not in Simple−/− mice (24, 30). Further investigation to uncover the exclusive molecular and pathological changes upon SIMPLE mutation will expose and provide new understanding of this toxic gain-of-function CMT1C pathogenesis.
The molecular basis for onset in CMT1C patients is a pressing question in the CMT1C pathogenesis. Given the dysregulated inflammatory responses, we propose that an injury-repair imbalance could be a trigger for the CMT1C onset. Increased inflammation and a subsequent aberrant response due to the SIMPLE mutation will alter the injury-repair balance. This model suggests that a yet-to-be-detected repair signal may reset the balance of injury to repair to maintain nerve function during development or before disease onset in asymptomatic CMT1C carriers. Nonetheless, fortuitous exposure to damaging agents that elicit inflammatory injury (or reduce repair compensation) may account for the onset in CMT1C by tilting the injury-repair balance.
The injury imbalance is not limited to inflammatory signaling per se. In addition to IL-1, TGF-β, TLR, and TNF-α signaling as prominent inflammatory pathways, SIMPLE interactors Tom1 and Tab2 are also important for bone morphogenic protein (BMP) signaling (8, 102). BMP signaling employs a cascade similar to that for the TGF-β pathway, including receptor Ser kinases and SMAD transcription factors (103). BMP signaling plays a critical role in growth, differentiation, maintenance, and regeneration (104). Given that most CMT diseases are symmetric and lead to injury in both legs (105), an improper maintenance with aberrant signaling kinetics may also contribute to the onset of demyelination.
Peripheral demyelination is the only known symptom in CMT1C patients thus far. SIMPLE, however, is widely expressed in many different cell types. Other defects in CMT1C patients are emerging (28, 70–72, 74, 106). For example, nucleotide polymorphism at the SIMPLE locus shows strong correlation with patients diagnosed with idiopathic LQT syndrome. SIMPLE expression is also associated with tumorigenesis. Given the ubiquitous expression of SIMPLE and its basic function in endosomal signaling and MVB regulation, additional defects may be dependent on cellular states and specific pathological triggers. Thus, a defined pathological challenge will reveal additional biological roles for SIMPLE in future studies.
Although we have shown that SimpleT115N/+ mice exhibit increased tumor susceptibility, it is currently not known whether CMT1C patients are more prone to tumorigenesis. A challenging impediment is the low prevalence of CMT1C (1:∼400,000) in humans (25, 107, 108). Further examination of our SimpleT115N/+ mice and their derived primary cells, the most physiological relevant model with one mutated SIMPLE allele, may reveal in CMT1C patients broader pathophysiological defects that have thus far remained undetected.
Conclusion.
We have demonstrated here that mutation, but not deletion, of SIMPLE affects the turnover of the ESCRT-0 Hrs protein and elicits aberrant kinetics in inflammatory signaling. The aberrant kinetics likely contributes to the increased tumor susceptibility in SimpleT115N/+ mice. Thus, dysregulated signaling kinetics could account for a toxic gain of function and the onset of CMT1C pathogenesis.
ACKNOWLEDGMENTS
We thank members of our laboratories for their suggestions and critical reading of the manuscript.
This research is supported, in part, by a grant from the National Institutes of Health (R01-DK904881 [C.-W.C.]).
REFERENCES
- 1.Magalhaes AC, Dunn H, Ferguson SS. 2011. Regulation of G protein-coupled receptor activity, trafficking, and localization by Gpcr-interacting proteins. Br J Pharmacol 165:1717–1736. doi: 10.1111/j.1476-5381.2011.01552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marchese A, Paing MM, Temple BR, Trejo J. 2008. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol 48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Platta HW, Stenmark H. 2011. Endocytosis and signaling. Curr Opin Cell Biol 23:393–403. doi: 10.1016/j.ceb.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Sorkin A, Goh LK. 2009. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res 315:683–696. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 5.Zwang Y, Yarden Y. 2009. Systems biology of growth factor-induced receptor endocytosis. Traffic 10:349–363. doi: 10.1111/j.1600-0854.2008.00870.x. [DOI] [PubMed] [Google Scholar]
- 6.Katoh Y, Shiba Y, Mitsuhashi H, Yanagida Y, Takatsu H, Nakayama K. 2004. Tollip and Tom1 form a complex and recruit ubiquitin-conjugated proteins onto early endosomes. J Biol Chem 279:24435–24443. doi: 10.1074/jbc.M400059200. [DOI] [PubMed] [Google Scholar]
- 7.Premont RT, Gainetdinov RR. 2007. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol 69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- 8.Wang T, Liu NS, Seet LF, Hong W. 2010. The emerging role of VHS domain-containing Tom1, Tom1L1, and Tom1L2 in membrane trafficking. Traffic 11:1119–1128. doi: 10.1111/j.1600-0854.2010.01098.x. [DOI] [PubMed] [Google Scholar]
- 9.Babst M. 2011. MVB vesicle formation: ESCRT-dependent, ESCRT-independent, and everything in between. Curr Opin Cell Biol 23:452–457. doi: 10.1016/j.ceb.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henne WM, Buchkovich NJ, Emr SD. 2011. The ESCRT pathway. Dev Cell 21:77–91. doi: 10.1016/j.devcel.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 11.Hurley JH. 2010. The ESCRT complexes. Crit Rev Biochem Mol Biol 45:463–487. doi: 10.3109/10409238.2010.502516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rusten TE, Vaccari T, Stenmark H. 2012. Shaping development with ESCRTs. Nat Cell Biol 14:38–45. doi: 10.1038/nrm3495. [DOI] [PubMed] [Google Scholar]
- 13.Keller S, Sanderson MP, Stoeck A, Altevogt P. 2006. Exosomes: from biogenesis and secretion to biological function. Immunol Lett 107:102–108. doi: 10.1016/j.imlet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 14.van Niel G, Porto-Carreiro I, Simoes S, Raposo G. 2006. Exosomes: a common pathway for a specialized function. J Biochem 140:13–21. doi: 10.1093/jb/mvj128. [DOI] [PubMed] [Google Scholar]
- 15.Vella LJ, Sharples RA, Nisbet RM, Cappai R, Hill AF. 2008. The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur Biophysics J 37:323–332. doi: 10.1007/s00249-007-0246-z. [DOI] [PubMed] [Google Scholar]
- 16.Brankatschk B, Wichert SP, Johnson SD, Schaad O, Rossner MJ, Gruenberg J. 2012. Regulation of the EGF transcriptional response by endocytic sorting. Sci Signal 5:ra21. doi: 10.1126/scisignal.2002351. [DOI] [PubMed] [Google Scholar]
- 17.Falguières T, Luyet P-P, Gruenberg J. 2009. Molecular assemblies and membrane domains in multivesicular endosome dynamics. Exp Cell Res 315:1567–1573. doi: 10.1016/j.yexcr.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 18.Lobert VH, Stenmark H. 2011. Cell polarity and migration: emerging role for the endosomal sorting machinery. Physiology (Bethesda) 26:171–180. doi: 10.1152/physiol.00054.2010. [DOI] [PubMed] [Google Scholar]
- 19.Saksena S, Emr SD. 2009. ESCRTs and human disease. Biochem Soc Trans 037:167–172. doi: 10.1042/BST0370167. [DOI] [PubMed] [Google Scholar]
- 20.Stuffers S, Brech A, Stenmark H. 2009. ESCRT proteins in physiology and disease. Exp Cell Res 315:1619–1626. doi: 10.1016/j.yexcr.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 21.Gasparrini F, Molfetta R, Quatrini L, Frati L, Santoni A, Paolini R. 2012. Syk-dependent regulation of Hrs phosphorylation and ubiquitination upon FεRI engagement: impact on Hrs membrane/cytosol localization. Eur J Immunol 42:2744–2753. doi: 10.1002/eji.201142278. [DOI] [PubMed] [Google Scholar]
- 22.Row PE, Clague MJ, Urbe S. 2005. Growth factors induce differential phosphorylation profiles of the Hrs-STAM complex: a common node in signaling networks with signal-specific properties. Biochem J 389:629–636. doi: 10.1042/BJ20050067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stern KA, Visser Smit GD, Place TL, Winistorfer S, Piper RC, Lill NL. 2007. Epidermal growth factor receptor fate is controlled by Hrs tyrosine phosphorylation sites that regulate Hrs degradation. Mol Cell Biol 27:888–898. doi: 10.1128/MCB.02356-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu H, Guariglia S, Yu RY, Li W, Brancho D, Peinado H, Lyden D, Salzer J, Bennett C, Chow CW. 2013. Mutation of SIMPLE in Charcot-Marie-Tooth 1C alters production of exosomes. Mol Biol Cell 24:1619–1637. doi: 10.1091/mbc.E12-07-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett CL, Shirk AJ, Huynh HM, Street VA, Nelis E, Van Maldergem L, De Jonghe P, Jordanova A, Guergueltcheva V, Tournev I, Van Den Bergh P, Seeman P, Mazanec R, Prochazka T, Kremensky I, Haberlova J, Weiss MD, Timmerman V, Bird TD, Chance PF. 2004. SIMPLE mutation in demyelinating neuropathy and distribution in sciatic nerve. Ann Neurol 55:713–720. doi: 10.1002/ana.20094. [DOI] [PubMed] [Google Scholar]
- 26.Gerding WM, Koetting J, Epplen JT, Neusch C. 2009. Hereditary motor and sensory neuropathy caused by a novel mutation in LITAF. Neuromuscular Disorders 19:701–703. doi: 10.1016/j.nmd.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Latour P, Gonnaud PM, Ollagnon E, Chan V, Perelman S, Stojkovic T, Stoll C, Vial C, Ziegler F, Vandenberghe A, Maire I. 2006. SIMPLE mutation analysis in dominant demyelinating Charcot-Marie-Tooth disease: three novel mutations. J Peripher Nerv Syst 11:148–155. doi: 10.1111/j.1085-9489.2006.00080.x. [DOI] [PubMed] [Google Scholar]
- 28.Saifi GM, Szigeti K, Wiszniewski W, Shy ME, Krajewski K, Hausmanowa-Petrusewicz I, Kochanski A, Reeser S, Mancias P, Butler I, Lupski JR. 2005. SIMPLE mutations in Charcot-Marie-Tooth disease and the potential role of its protein product in protein degradation. Hum Mutat 25:372–383. doi: 10.1002/humu.20153. [DOI] [PubMed] [Google Scholar]
- 29.Street VA, Goldy JD, Golden AS, Tempel BL, Bird TD, Chance PF. 2002. Mapping of Charcot-Marie-Tooth disease type 1C to chromosome 16p identifies a novel locus for demyelinating neuropathies. Am J Hum Genet 70:244–250. doi: 10.1086/337943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Somandin C, Gerber D, Pereira JA, Horn M, Suter U. 2012. LITAF (SIMPLE) regulates Wallerian degeneration after injury but is not essential for peripheral nerve development and maintenance: implications for Charcot-Marie-Tooth disease. Glia 60:1518–1528. doi: 10.1002/glia.22371. [DOI] [PubMed] [Google Scholar]
- 31.Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, Lang RA, Pollard JW. 2009. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment, and growth. PLoS One 4:e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang TT, Suk HY, Yang X, Olabisi O, Yu RY, Durand J, Jelicks LA, Kim JY, Scherer PE, Wang Y, Feng Y, Rossetti L, Graef IA, Crabtree GR, Chow CW. 2006. Role of transcription factor NFAT in glucose and insulin homeostasis. Mol Cell Biol 26:7372–7387. doi: 10.1128/MCB.00580-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang DP, Kim J, Syed N, Tung YJ, Bhaskaran A, Mindos T, Mirsky R, Jessen KR, Maurel P, Parkinson DB, Kim HA. 2012. p38 MAPK activation promotes denervated Schwann cell phenotype and functions as a negative regulator of Schwann cell differentiation and myelination. J Neurosci 32:7158–7168. doi: 10.1523/JNEUROSCI.5812-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shirk AJ, Anderson SK, Hashemi SH, Chance PF, Bennett CL. 2005. SIMPLE interacts with NEDD4 and TSG101: evidence for a role in lysosomal sorting and implications for Charcot-Marie-Tooth disease. J Neurosci Res 82:43–50. doi: 10.1002/jnr.20628. [DOI] [PubMed] [Google Scholar]
- 35.Bache KG, Brech A, Mehlum A, Stenmark H. 2003. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol 162:435–442. doi: 10.1083/jcb.200302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Razi M, Futter CE. 2006. Distinct roles for Tsg101 and Hrs in multivesicular body formation and inward vesiculation. Mol Biol Cell 17:3469–3483. doi: 10.1091/mbc.E05-11-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eaton HE, Desrochers G, Drory SB, Metcalf J, Angers A, Brunetti CR. 2011. SIMPLE/LITAF expression induces the translocation of the ubiquitin ligase itch towards the lysosomal compartments. PLoS One 6:e16873. doi: 10.1371/journal.pone.0016873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen YG. 2009. Endocytic regulation of TGF-beta signaling. Cell Res 19:58–70. doi: 10.1038/cr.2008.315. [DOI] [PubMed] [Google Scholar]
- 39.De Boeck M, ten Dijke P. 2012. Key role for ubiquitin protein modification in TGFβ signal transduction. Uppsala J Med Sci 117:153–165. doi: 10.3109/03009734.2012.654858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heldin CH, Landstrom M, Moustakas A. 2009. Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol 21:166–176. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 41.Mu Y, Gudey SK, Landstrom M. 2012. Non-Smad signaling pathways. Cell Tissue Res 347:11–20. doi: 10.1007/s00441-011-1201-y. [DOI] [PubMed] [Google Scholar]
- 42.Verstrepen L, Verhelst K, Carpentier I, Beyaert R. 2011. TAX1BP1, a ubiquitin-binding adaptor protein in innate immunity and beyond. Trends Biochem Sci 36:347–354. doi: 10.1016/j.tibs.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 43.Wuerzberger-Davis SM, Miyamoto S. 2010. TAK-ling IKK activation: “Ub” the judge. Sci Signal 3:pe3. doi: 10.1126/scisignal.3105pe3. [DOI] [PubMed] [Google Scholar]
- 44.Xu P, Liu J, Derynck R. 2012. Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Lett 586:1871–1884. doi: 10.1016/j.febslet.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishitani T, Takaesu G, Ninomiya-Tsuji J, Shibuya H, Gaynor RB, Matsumoto K. 2003. Role of the TAB2-related protein TAB3 in IL-1 and TNF signaling. EMBO J 22:6277–6288. doi: 10.1093/emboj/cdg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Landstrom M. 2010. The TAK1-TRAF6 signaling pathway. Int J Biochem Cell Biol 42:585–589. doi: 10.1016/j.biocel.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 47.Sakurai H. 2012. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol Sci 33:522–530. doi: 10.1016/j.tips.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 48.Skaug B, Jiang X, Chen ZJ. 2009. The role of ubiquitin in NF-κB regulatory pathways. Annu Rev Biochem 78:769–796. doi: 10.1146/annurev.biochem.78.070907.102750. [DOI] [PubMed] [Google Scholar]
- 49.Clague MJ, Liu H, Urbe S. 2012. Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev Cell 23:457–467. doi: 10.1016/j.devcel.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 50.Corn JE, Vucic D. 2014. Ubiquitin in inflammation: the right linkage makes all the difference. Nat Struct Mol Biol 21:297–300. doi: 10.1038/nsmb.2808. [DOI] [PubMed] [Google Scholar]
- 51.MacGurn JA, Hsu PC, Emr SD. 2012. Ubiquitin and membrane protein turnover: from cradle to grave. Annu Rev Biochem 81:231–259. doi: 10.1146/annurev-biochem-060210-093619. [DOI] [PubMed] [Google Scholar]
- 52.Polo S. 2012. Signaling-mediated control of ubiquitin ligases in endocytosis. BMC Biol 10:25. doi: 10.1186/1741-7007-10-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raiborg C, Stenmark H. 2009. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- 54.Merrill JC, You J, Constable C, Leeman SE, Amar S. 2011. Whole-body deletion of LPS-induced TNF-α factor (LITAF) markedly improves experimental endotoxic shock and inflammatory arthritis. Proc Natl Acad Sci U S A 108:21247–21252. doi: 10.1073/pnas.1111492108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moriwaki Y, Begum NA, Kobayashi M, Matsumoto M, Toyoshima K, Seya T. 2001. Mycobacterium bovis Bacillus Calmette-Guerin and its cell wall complex induce a novel lysosomal membrane protein, SIMPLE, that bridges the missing link between lipopolysaccharide and p53-inducible gene, LITAF(PIG7), and estrogen-inducible gene, EET-1. J Biol Chem 276:23065–23076. doi: 10.1074/jbc.M011660200. [DOI] [PubMed] [Google Scholar]
- 56.Srinivasan S, Leeman SE, Amar S. 2010. Beneficial dysregulation of the time course of inflammatory mediators in lipopolysaccharide-induced tumor necrosis factor alpha factor-deficient mice. Clin Vaccine Immunol 17:699–704. doi: 10.1128/CVI.00510-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chung JY, Park YC, Ye H, Wu H. 2002. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci 115:679–688. [DOI] [PubMed] [Google Scholar]
- 58.Xie P. 2013. TRAF molecules in cell signaling and in human diseases. J Mol Signal 8:7. doi: 10.1186/1750-2187-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang Z, Ninomiya-Tsuji J, Qian Y, Matsumoto K, Li X. 2002. Interleukin-1 (IL-1) receptor-associated kinase-dependent IL-1-induced signaling complexes phosphorylate TAK1 and TAB2 at the plasma membrane and activate TAK1 in the cytosol. Mol Cell Biol 22:7158–7167. doi: 10.1128/MCB.22.20.7158-7167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, Ninomiya-Tsuji J, Matsumoto K. 2000. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell 5:649–658. doi: 10.1016/S1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 61.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 62.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. 1999. The kinase TAK1 can activate the NIK-I κB as well as the MAP kinase cascade in the IL-1 signaling pathway. Nature 398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 63.Seet LF, Liu N, Hanson BJ, Hong W. 2004. Endofin recruits TOM1 to endosomes. J Biol Chem 279:4670–4679. [DOI] [PubMed] [Google Scholar]
- 64.Shibuya H, Yamaguchi K, Shirakabe K, Tonegawa A, Gotoh Y, Ueno N, Irie K, Nishida E, Matsumoto K. 1996. TAB1: an activator of the TAK1 MAPKKK in TGF-β signal transduction. Science 272:1179–1182. doi: 10.1126/science.272.5265.1179. [DOI] [PubMed] [Google Scholar]
- 65.Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH, Landstrom M. 2008. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol 10:1199–1207. doi: 10.1038/ncb1780. [DOI] [PubMed] [Google Scholar]
- 66.Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. 2008. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell 31:918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kang JS, Liu C, Derynck R. 2009. New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol 19:385–394. doi: 10.1016/j.tcb.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 68.Massague J. 2012. TGFβ signaling in context. Nat Rev Mol Cell Biol 13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moustakas A, Heldin CH. 2009. The regulation of TGFβ signal transduction. Development 136:3699–3714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- 70.Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, Bis JC, Marciante K, Rivadeneira F, Noseworthy PA, Sotoodehnia N, Smith NL, Rotter JI, Kors JA, Witteman JC, Hofman A, Heckbert SR, O'Donnell AG, Uitterlinden CJ, Psaty BM, Lumley T, Larson MG, Stricker BH. 2009. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet 41:399–406. doi: 10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pfeufer A, Sanna S, Arking DE, Muller M, Gateva V, Fuchsberger C, Ehret GB, Orru M, Pattaro C, Kottgen A, Perz S, Usala G, Barbalic M, Li M, Putz B, Scuteri A, Prineas RJ, Sinner MF, Gieger C, Najjar SS, Kao WH, Muhleisen TW, Dei M, Happle C, Mohlenkamp S, Crisponi L, Erbel R, Jockel KH, Naitza S, Steinbeck G, Marroni F, Hicks AA, Lakatta E, Muller-Myhsok B, Pramstaller PP, Wichmann HE, Schlessinger D, Boerwinkle E, Meitinger T, Uda M, Coresh J, Kaab S, Abecasis GR, Chakravarti A. 2009. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet 41:407–414. doi: 10.1038/ng.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bertolo C, Roa S, Sagardoy A, Mena-Varas M, Robles EF, Martinez-Ferrandis JI, Sagaert X, Tousseyn T, Orta A, Lossos IS, Amar S, Natkunam Y, Briones J, Melnick A, Malumbres R, Martinez-Climent JA. 2013. LITAF, a BCL6 target gene, regulates autophagy in mature B-cell lymphomas. Br J Haematol 162:621–630. doi: 10.1111/bjh.12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang D, Liu J, Tang K, Xu Z, Xiong X, Rao Q, Wang M, Wang J. 2009. Expression of pig7 gene in acute leukemia and its potential to modulate the chemosensitivity of leukemic cells. Leuk Res 33:28–38. doi: 10.1016/j.leukres.2008.06.034. [DOI] [PubMed] [Google Scholar]
- 74.Zhou J, Yang Z, Tsuji T, Gong J, Xie J, Chen C, Li W, Amar S, Luo Z. 2011. LITAF and TNFSF15, two downstream targets of AMPK, exert inhibitory effects on tumor growth. Oncogene 30:1892–1900. doi: 10.1038/onc.2010.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Behrends C, Harper JW. 2011. Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol 18:520–528. doi: 10.1038/nsmb.2066. [DOI] [PubMed] [Google Scholar]
- 76.Kommaddi RP, Shenoy SK. 2013. Arrestins and protein ubiquitination. Prog Mol Biol Transl Sci 118:175–204. doi: 10.1016/B978-0-12-394440-5.00007-3. [DOI] [PubMed] [Google Scholar]
- 77.Shukla AK, Xiao K, Lefkowitz RJ. 2011. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci 36:457–469. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tian X, Kang DS, Benovic JL. 2014. beta-arrestins and G protein-coupled receptor trafficking. Handb Exp Pharmacol 219:173–186. doi: 10.1007/978-3-642-41199-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Becuwe M, Herrador A, Haguenauer-Tsapis R, Vincent O, Leon S. 2012. Ubiquitin-mediated regulation of endocytosis by proteins of the arrestin family. Biochem Res Int 2012:242764. doi: 10.1155/2012/242764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Puca L, Brou C. 2014. Alpha-arrestins: new players in Notch and GPCR signaling pathways in mammals. J Cell Sci 127:1359–1367. doi: 10.1242/jcs.142539. [DOI] [PubMed] [Google Scholar]
- 81.Rauch S, Martin-Serrano J. 2011. Multiple interactions between the ESCRT machinery and arrestin-related proteins: implications for PPXY-dependent budding. J Virol 85:3546–3556. doi: 10.1128/JVI.02045-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Puca L, Chastagner P, Meas-Yedid V, Israel A, Brou C. 2013. α-Arrestin 1 (ARRDC1) and β-arrestins cooperate to mediate Notch degradation in mammals. J Cell Sci 126:4457–4468. doi: 10.1242/jcs.130500. [DOI] [PubMed] [Google Scholar]
- 83.Huotari J, Helenius A. 2011. Endosome maturation. EMBO J 30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mittelbrunn M, Sanchez-Madrid F. 2012. Intercellular communication: diverse structures for exchange of genetic information. Nat Rev Mol Cell Biol 13:328–335. doi: 10.1038/nrm3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Simons M, Raposo G. 2009. Exosomes–vesicular carriers for intercellular communication. Curr Opin Cell Biol 21:575–581. doi: 10.1016/j.ceb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 86.Katoh M. 2013. Therapeutics targeting angiogenesis: genetics and epigenetics, extracellular miRNAs and signaling networks. Int J Mol Med 32:763–767. doi: 10.3892/ijmm.2013.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sheldon H, Heikamp E, Turley H, Dragovic R, Thomas P, Oon CE, Leek R, Edelmann M, Kessler B, Sainson RC, Sargent I, Li JL, Harris AL. 2010. New mechanism for Notch signaling to endothelium at a distance by Delta-like 4 incorporation into exosomes. Blood 116:2385–2394. doi: 10.1182/blood-2009-08-239228. [DOI] [PubMed] [Google Scholar]
- 88.Kuo L, Freed EO. 2012. ARRDC1 as a mediator of microvesicle budding. Proc Natl Acad Sci U S A 109:4025–4026. doi: 10.1073/pnas.1201441109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nabhan JF, Hu R, Oh RS, Cohen SN, Lu Q. 2012. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc Natl Acad Sci U S A 109:4146–4151. doi: 10.1073/pnas.1200448109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bhoj VG, Chen ZJ. 2009. Ubiquitylation in innate and adaptive immunity. Nature 458:430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- 91.Blanc C, Charette SJ, Mattei S, Aubry L, Smith EW, Cosson P, Letourneur F. 2009. Dictyostelium Tom1 participates to an ancestral ESCRT-0 complex. Traffic 10:161–171. doi: 10.1111/j.1600-0854.2008.00855.x. [DOI] [PubMed] [Google Scholar]
- 92.Herman EK, Walker G, van der Giezen M, Dacks JB. 2011. Multivesicular bodies in the enigmatic amoeboflagellate Breviata anathema and the evolution of ESCRT 0. J Cell Sci 124:613–621. doi: 10.1242/jcs.078436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamakami M, Yoshimori T, Yokosawa H. 2003. Tom1, a VHS domain-containing protein, interacts with Tollip, ubiquitin, and clathrin. J Biol Chem 278:52865–52872. doi: 10.1074/jbc.M306740200. [DOI] [PubMed] [Google Scholar]
- 94.Tang X, Metzger D, Leeman S, Amar S. 2006. LPS-induced TNF-alpha factor (LITAF)-deficient mice express reduced LPS-induced cytokine: evidence for LITAF-dependent LPS signaling pathways. Proc Natl Acad Sci U S A 103:13777–13782. doi: 10.1073/pnas.0605988103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Luttrell LM. 2014. Minireview: more than just a hammer: ligand “bias” and pharmaceutical discovery. Mol Endocrinol 28:281–294. doi: 10.1210/me.2013-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shukla AK. 2014. Biasing GPCR signaling from inside. Sci Signal 7:pe3. doi: 10.1126/scisignal.2005021. [DOI] [PubMed] [Google Scholar]
- 97.Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. 2014. Recent developments in biased agonism. Curr Opin Cell Biol 27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carr R III, Du Y, Quoyer J, Panettieri RA Jr, Janz JM, Bouvier M, Kobilka BK, Benovic JL. 2014. Development and characterization of pepducins as Gs-biased allosteric agonists. J Biol Chem 289:35668–35684. doi: 10.1074/jbc.M114.618819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chakir K, Depry C, Dimaano VL, Zhu WZ, Vanderheyden M, Bartunek J, Abraham TP, Tomaselli GF, Liu SB, Xiang YK, Zhang M, Takimoto E, Dulin N, Xiao RP, Zhang J, Kass DA. 2011. Gαs-biased β2-adrenergic receptor signaling from restoring synchronous contraction in the failing heart. Sci Transl Med 3:100ra188. doi: 10.1126/scitranslmed.3001909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, Shenoy SK, Gygi SP, Lefkowitz RJ. 2011. Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal 4:ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Violin JD, Crombie AL, Soergel DG, Lark MW. 2014. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci 35:308–316. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 102.Nohe A, Keating E, Knaus P, Petersen NO. 2004. Signal transduction of bone morphogenetic protein receptors. Cell Signal 16:291–299. doi: 10.1016/j.cellsig.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 103.Wotton D, Massague J. 2001. Smad transcriptional corepressors in TGF beta family signaling. Curr Top Microbiol Immunol 254:145–164. [PubMed] [Google Scholar]
- 104.Kitisin K, Saha T, Blake T, Golestaneh N, Deng M, Kim C, Tang Y, Shetty K, Mishra B, Mishra L. 2007. TGF-β signaling in development. Sci STKE 2007:cm1. [DOI] [PubMed] [Google Scholar]
- 105.Chance PF, Pleasure D. 1993. Charcot-Marie-Tooth syndrome. Arch Neurol 50:1180–1184. doi: 10.1001/archneur.1993.00540110060006. [DOI] [PubMed] [Google Scholar]
- 106.Potulska-Chromik A, Sinkiewicz-Darol E, Kostera-Pruszczyk A, Drac H, Kabzinska D, Zakrzewska-Pniewska B, Golebiowski M, Kochanski A. 2012. Charcot-Marie-Tooth type 1C disease coexisting with progressive multiple sclerosis: a study of an overlapping syndrome. Folia Neuropathol 50:369–374. [DOI] [PubMed] [Google Scholar]
- 107.Bird TD. 1998. Charcot-Marie-Tooth neuropathy type 1. In Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Fong C-T, Smith RJH (ed), GeneReviews. GeneReviews, Seattle, WA: http://www.ncbi.nlm.nih.gov/books/NBK1205/. [PubMed] [Google Scholar]
- 108.Chance PF. 2004. Genetic evaluation of inherited motor/sensory neuropathy. Suppl Clin Neurophysiol 57:228–242. doi: 10.1016/S1567-424X(09)70360-5. [DOI] [PubMed] [Google Scholar]