

Abstract
In mammals the stress-inducible expression of genes encoding heat shock proteins is under the control of the heat shock transcription factor 1 (HSF1). Activation of HSF1 is a multistep process, involving trimerization, acquisition of DNA-binding and transcriptional activities, which coincide with several posttranslational modifications. Stress-inducible phosphorylation of HSF1, or hyperphosphorylation, which occurs mainly within the regulatory domain (RD), has been proposed as a requirement for HSF-driven transcription and is widely used for assessing HSF1 activation. Nonetheless, the contribution of hyperphosphorylation to the activity of HSF1 remains unknown. In this study, we generated a phosphorylation-deficient HSF1 mutant (HSF1Δ∼PRD), where the 15 known phosphorylation sites within the RD were disrupted. Our results show that the phosphorylation status of the RD does not affect the subcellular localization and DNA-binding activity of HSF1. Surprisingly, under stress conditions, HSF1Δ∼PRD is a potent transactivator of both endogenous targets and a reporter gene, and HSF1Δ∼PRD has a reduced activation threshold. Our results provide the first direct evidence for uncoupling stress-inducible phosphorylation of HSF1 from its activation, and we propose that the phosphorylation signature alone is not an appropriate marker for HSF1 activity.
INTRODUCTION
The heat shock response, as characterized by inducible expression of heat shock proteins (Hsps), is an ancient, evolutionarily conserved mechanism that protects cells from various proteotoxic insults, including exposures to elevated temperatures, heavy metals, proteasome inhibition, and oxidative stress (1). Hsps function as molecular chaperones, bind to misfolded proteins, facilitate their refolding or direct them to degradation, and block the formation of protein aggregates (2, 3). The heat shock response is controlled by heat shock transcription factors (HSFs) (4). In vertebrates, four HSFs (HSF1 to HSF4) have been found, whereas yeasts, flies, and nematodes have only a single HSF. HSFs bind DNA at evolutionarily well-conserved sequences, consisting of inverted nGAAn repeats, called heat shock elements (5–8). HSF1 is considered the master regulator of the heat shock response in mammals, since mice lacking HSF1 are unable to induce Hsp expression upon exposure to protein-damaging stress (9, 10). Besides controlling the stress-inducible expression of Hsps, HSF1 plays a role in development (10–12), life span regulation (13–15), immune responses (16), and the circadian cycle (17). In addition, HSF1 is a well-recognized transcriptional regulator in malignant human cancers (18–20).
The HSF1 protein is composed of five distinguishable functional domains (see Fig. 1A). The DNA-binding domain (DBD) is located at the N terminus (21), whereas the transactivation domain (TAD) resides in the C terminus (22). A unique requirement for HSF1 activation is the process of trimerization through an intermolecular interaction of leucine-zipper-like heptad repeat domains (HR-A/B) between HSF1 monomers (23, 24). Spontaneous trimerization under normal conditions is suppressed by another heptad repeat region (HR-C), which facilitates intramolecular interactions between HR-A/B and HR-C domains (25). A centrally located part of HSF1 is called the regulatory domain (RD). Deletion of the RD results in constitutive DNA-binding activity of HSF1 and induces expression of Hsps in the absence of stress (26–28). It has also been shown that the RD is self-sufficient in its heat-sensing capacity, since a chimeric transcription factor containing Gal4DBD-HSF1RD-VP16TAD is repressed under normal conditions but is capable of activating transcription in response to stress (22).
FIG 1.

Characterization of the HSF1 mutant that is phosphorylation-deficient within the RD, HSF1Δ∼PRD. (A) Schematic illustration of the HSF1 functional domains with the known phosphorylation sites. In HSF1Δ∼PRD, 15 phosphorylation sites in the regulatory domain (RD) were mutated from serine (S) and threonine (T) residues to alanines (A) as indicated. Additional HSF1 domains include the DNA-binding domain (DBD), heptad repeat domains (HR-A/B and HR-C), and transactivation domain (TAD). Note that the figure is not drawn to scale. (B) hsf1−/− MEFs were transfected with Mock plasmid [pcDNA3.1/myc-His(−)A], Myc-His-HSF1 WT, or Myc-His-HSF1Δ∼PRD. hsf1+/+ represents the endogenous levels of HSF1 in MEFs. Cells were either left untreated (−) or exposed to heat shock (+). HSF1 protein levels from cell lysates were detected by Western blotting with anti-HSF1 antibody. Hsc70 is shown as a loading control. An asterisk indicates an HSF1 protein that migrates slower on SDS-PAGE due to hyperphosphorylation (53). The difference in size between the endogenous HSF1 from hsf1+/+ MEFs and exogenous HSF1 WT is caused by the Myc-His tag on the human HSF1 WT construct. (C) hsf1−/− MEFs were transfected as in panel B. Cells were either left untreated (−) or heat shocked (+). Cell lysates were treated with lambda protein phosphatase (+λPP) or left untreated. Samples were analyzed by using Western blotting. α-Actin is shown as a loading control. An asterisk indicates the HSF1 protein that migrates slower on SDS-PAGE due to hyperphosphorylation (53). (D) hsf1−/− MEFs were transfected as in panel B and treated with cycloheximide (CHX) at 37°C for the indicated times. Cell lysates were analyzed with anti-HSF1 and anti-HSF2 antibodies. α-Actin is shown as a loading control.
Transient activation of HSF1 by various stresses includes accumulation in the nucleus, monomer-to-trimer transition, HSE-binding activity, and acquisition of transactivation capacity (1). During the activation-attenuation cycle, HSF1 is extensively posttranslationally modified (PTMs) and is subjected to, for example, phosphorylation, sumoylation, and acetylation (29–32). To date, 22 phosphorylation sites on serine and threonine residues have been identified within the HSF1 protein (33). Some sites, e.g., S303 and S307, appear to be constitutively phosphorylated (34–36), whereas other sites undergo inducible phosphorylation (37). Stress-inducible phosphorylation of HSF1, or hyperphosphorylation, is one of the most prominent modifications, coinciding with the acquisition of its transactivation capacity (30, 37–40). However, despite a wealth of studies on the role of single phosphorylation sites (29, 30, 34, 40–42), no direct link between hyperphosphorylation and HSF1 activation has been established.
Many pathological conditions, such as metabolic disorders, cancers, and neurodegenerative diseases, are associated with either increased or decreased activity of molecular chaperones (43, 44). Hence, modulating the heat shock response by altering HSF1 activity has been proposed as a potential therapeutic approach (44–46). Although a variety of compounds have been shown to affect HSF1 transactivation capacity (45–48), mechanistic understanding of how these compounds contribute to HSF1 activation is limited. Therefore, in development of drugs targeting specific phases in the HSF1 activation-attenuation cycle, emphasis should be placed on unraveling the functional impact of HSF1 PTMs. In order to investigate how the stress-inducible phosphorylation affects HSF1 activity, we generated a phosphorylation-deficient HSF1 mutant that lacks the known 15 phosphorylation sites within the RD (HSF1Δ∼PRD). Our results show that phosphorylation of HSF1 RD does not affect HSF1 nuclear localization and is not required for its DNA-binding activity, suggesting that the HSF1Δ∼PRD retains its properties to be accurately regulated upon exposure to stress. We conclude that the gain of HSF1 transactivation capacity is independent of the constitutive and stress-inducible phosphorylation of HSF1 within the RD, providing the first direct evidence for uncoupling HSF1 hyperphosphorylation from its activation.
MATERIALS AND METHODS
Plasmid constructs.
The plasmids encoding Myc-His-HSF1 WT [in pcDNA3.1/myc-His(−)A], Gal4-VP16, β-galacatosidase, and Gal4-driven luciferase have been described earlier (31, 40, 49). The phosphorylation-deficient HSF1 mutant (HSF1Δ∼PRD) was generated by replacing 15 phosphorylatable serine and threonine residues within the Myc-His-HSF1 wild-type (WT) RD with alanines (Fig. 1A). Fourteen sites (S230, S292, S303, S307, S314, S319, S320, T323, S326, S338, S344, S363, S368, and T369) were mutated by sequential rounds of site-directed mutagenesis using a QuikChange site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's instructions. Mutation 15, T367A, was performed by DNA Express, Inc. Gal4-VP16-HSF1 WT and Gal4-VP16-HSF1Δ∼PRD were generated by cloning the regulatory domain (amino acids [aa] 220 to 389) of Myc-His-HSF1 WT or Myc-His-HSF1Δ∼PRD into EcoRI-linearized pSGVP plasmid (pSGVP was kindly provided by Richard I. Morimoto, Northwestern University, Evanston, IL) by using an In-Fusion HD cloning kit (Clontech). The constructs were confirmed by sequencing.
Cell culture, treatments, and transfections.
hsf1−/− and hsf1+/+ mouse embryonic fibroblasts (MEFs) and human cervical cancer HeLa cells were cultured in high glucose Dulbecco modified Eagle medium (Sigma) containing 10% fetal calf serum (Gibco), 2 mM l-glutamine (Sigma), and streptomycin (100 μg/ml) and penicillin (100 U/ml) (both from VWR). Culture media for MEFs were supplemented with 1× MEM nonessential amino acid solution (Sigma). Heat shock treatments were conducted in a water bath at 39, 40, 41, 42, and 43°C for the indicated times. To induce heavy metal stress, CdSO4 (Sigma), dissolved in sterile water, was used at a concentration of 40 and 60 μM for the indicated times. For transfections, 6 × 106 HeLa or hsf1−/− MEFs were suspended in 0.4 ml of Opti-MEM (Gibco). Cells were subjected to a single electric pulse (220 V, 975 μF for HeLa cells; 280 V, 975 μF for MEFs) in 0.4-cm gap electroporation cuvettes (BTX) using a Bio-Rad Gene Pulser II electroporator. Transfected cells were left to recover in culture medium for 48 h prior to further treatments.
Western blot.
Cells were lysed in radioimmunoprecipitation assay lysis buffer (1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 0.15 M NaCl, 0.01 M sodium phosphate [pH 7.2], and 2 mM EDTA [pH 8.0]) supplemented with 0.5 mM phenylmethylsulfonyl fluoride and 1× Complete Mini-Protease inhibitor cocktail (Roche). Cell lysates, cleared by centrifugation (15,000 × g for 10 min at 4°C), were boiled in Laemmli sample buffer, resolved on an 8% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE), and transferred to nitrocellulose membrane (Pierce). The antibodies used for Western blotting were anti-HSF1 (AB-4; Thermo Scientific), anti-HSF2 (3E2; Millipore), anti-α-actin (AC-40; Sigma-Aldrich), anti-Hsc70 (SPA-815; Enzo Life Sciences), and anti-VP16 (V4388; Sigma-Aldrich). Horseradish peroxidase-conjugated secondary antibodies were purchased from Promega, Abcam, and GE Healthcare Life Sciences, and immunocomplexes were detected by enhanced chemiluminescence (GE Healthcare Life Sciences).
Protein dephosphorylation and protein turnover analyses.
For protein dephosphorylation analysis, lambda protein phosphatase (λPP; New England BioLabs) was used according to the manufacturer's instructions. Briefly, transfected MEFs were subjected to a 30-min heat shock at 43°C and lysed in buffer C (25% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, and 20 mM HEPES [pH 8]), and λPP was used at a concentration of 50 U/μg for whole-cell lysates. Samples were incubated at 30°C for 30 min, and the reaction was stopped by boiling in Laemmli sample buffer. To measure the protein turnover, transfected MEFs were treated for up to 15 h with cycloheximide (CHX; Sigma), which was added to culture medium at a concentration of 20 μg/ml.
ChIP.
Chromatin immunoprecipitation (ChIP) was performed as described by Vihervaara and coworkers (8) with minor changes to the protocol. A total of 5 × 107 transfected MEFs were cross-linked immediately after treatment for 10 min with a final concentration of 1% formaldehyde, followed by quenching in 125 mM glycine. After lysis in Joost lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl [pH 8.1]), chromatin was sonicated using a Bioruptor (Diagenode), and 1 mg of whole-cell extracts was used for each immunoprecipitation. Samples were precleared using a 50% slurry of protein G-Sepharose beads (GE Healthcare Life Sciences). Immunoprecipitation was performed overnight at 4°C using antibodies against HSF1 (SPA-901; Enzo Life Sciences). Normal rabbit serum (Jackson ImmunoResearch Laboratories) was used as a nonspecific antibody. After washing of the immunocomplexes, the remaining proteins and RNA were digested by using proteinase K and RNase A. Cross-links were reversed by incubating the samples overnight at 65°C. DNA was purified with phenol-chloroform. Samples were analyzed by quantitative PCR using StepOnePlus or QuantStudio 12K Flex Real-Time PCR Systems (both from Applied Biosystems). The following forward (f) and reverse (r) SYBR green primers were used: fHsp25 promoter, 5′-TGGGAATCGCTCCAGCTACCG-3′; rHsp25 promoter, 5′-AAGCTTGCAAAGGGGGCGGG-3′; fHsp70 promoter, 5′-CACCAGCACGTTCCCCA-3′; and rHsp70 promoter, 5′-CGCCCTGCGCCTTTAAG-3′. Immunoprecipitation samples were normalized to values obtained for input before fold enrichment was determined by setting the HSF1 WT control sample to value 1.
EMSA.
Electromobility shift assay (EMSA) was performed as described previously (50). Briefly, cell pellets from transfected hsf1−/− MEFs were lysed in buffer C, and the whole-cell extracts were incubated with 32P-labeled oligonucleotide representing the proximal HSE of the human Hsp70 promoter (forward, 5′-GAGGCGAAAACCCTGGAATATTCCCGACCTGGCAG-3′; reverse, 5′-CTGCCAGGTCGGGAATATTCCAGGGTTTTCGCCTC-3′). Samples were resolved on a 4% native polyacrylamide gel, and the protein-DNA complexes were visualized by autoradiography.
Immunofluorescence and confocal microscopy.
Transfected HeLa cells were cultured on coverslips for 48 h before treatments. Treated and untreated cells were fixed in 3.7% paraformaldehyde and permeabilized with 0.5% Triton-X in phosphate-buffered saline (PBS) for 12 min, followed by blocking with 5% bovine serum albumin (BSA) in PBS for 1 h at room temperature. The cells were incubated with rabbit anti-HSF1 (51) or mouse anti-myc (M4439; Sigma) antibodies overnight at 4°C, after which the unbound primary antibodies were washed off with PBS containing 0.1% Tween 20. After the washing step, the cells were incubated with secondary antibodies diluted 1:400 in 5% BSA-PBS for 1 h (donkey anti-rabbit antibody–Alexa Fluor 568 for anti-HSF1 and goat anti-mouse antibody–Alexa Fluor 488 for anti-myc, both from Life Technologies). Coverslips were mounted in Vectashield mounting medium with DAPI (4′,6′-diamidino-2-phenylindole; Vector Laboratories) for DNA staining. Immunofluorescence was performed with LSM 780 confocal microscope (Carl Zeiss, Inc.), and image analysis was performed using Fiji software (52).
Quantitative RT-PCR (qRT-PCR).
RNA from transfected hsf1−/− MEFs was isolated using an RNeasy minikit (Qiagen) according to the manufacturer's instructions and quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). Then, 1 μg of total RNA was reverse transcribed with an iScript kit (Bio-Rad). A KAPA Probe Fast ABI Prism qPCR kit (KAPA Biosystems) and SensiFAST SYBR Hi-ROX kit (Bioline Reagents) were used for qRT-PCRs that were performed with StepOnePlus or QuantStudio 12K Flex real-time PCR systems (both from Applied Biosystems). Primers and probes were purchased from Oligomer. The following forward (f), reverse (r), and probe (pr) oligonucleotides were used in TaqMan assays: fRNA18S5, 5′-GCAATTATTCCCCATGAACG-3′; rRNA18S5, 5′-GGGACTTAATCAACGCAAGC-3′; prRNA18S5, 5′-FAM-TTCCCAGTAAGTGCGGGTC-BHQ-3′; fHSPA1A/B, 5′-AGGTGCTGGACAAGTGCCAG-3′; rHSPA1A/B, 5′-AACTCCTCCTTGTCGGCCA-3′; prHSPA1A/B, 5′-FAM-CATCTCCTGGCTGGACTCCAACACG-BHQ-3′; fHSPB1, 5′-CACTGGCAAGCACGAAGAAAG-3′; rHSPB1, 5′-GCGTGTATTTCCGGGTGAAG-3′; and prHSPB1, 5′-FAM-ACCGAGAGATGTAGCCATGTTCGTCCTG-BHQ-3′. The relative quantities of the target gene mRNAs were normalized against their respective 18S RNA (RNA18S5), and the fold induction was calculated against the respective mRNA levels in nontreated mock-transfected cells. All reactions were run in triplicate from samples derived from at least three biological replicates.
Luciferase assay.
Transfected HeLa cells were snap-frozen and lysed in Passive lysis buffer (Promega) according to the manufacturer's instructions. Cell lysates were cleared by centrifugation (15,000 × g for 10 min at 4°C), and the firefly luciferase activity, produced by the Gal4-driven luciferase plasmid, was measured by using a Luminoskan Ascent microplate luminometer (Thermo Scientific) with luciferase assay reagent (Promega) as a substrate. The luciferase activity was normalized using Rous sarcoma virus promoter-driven β-galactosidase as an internal control by incubating the cell lysates in 100 mM phosphate buffer (pH 7.0) with 0.67 mg of o-nitrophenyl β-d-galactoside (ONPG; Sigma)/ml, 1 mM MgCl2, and 45 mM β-mercaptoethanol at 37°C for 1 h. The absorbance was measured by a Multiskan MCC/340 (Labsystems) at 420 nm.
Statistical analysis.
Statistical analyses of the data were performed in GraphPad Prism 6. The data were analyzed within each time point using independent two-way analysis of variance and corrected for multiple comparisons using the Holm-Sidak post hoc test, and the significance level was set to 0.05.
RESULTS
HSF1Δ∼PRD and HSF1 WT display similar turnover and subcellular localization.
To study the impact of hyperphosphorylation on HSF1 activity, we generated a mutant construct of HSF1, where the known 15 phosphorylation sites residing within the RD (aa 220 to 389) were replaced with nonphosphorylatable alanines, that we designated HSF1Δ∼PRD (Fig. 1A). The RD harbors ∼70% of the known HSF1 phosphorylation sites (33) and is capable of repressing HSF1 TAD in the absence of stress, rendering HSF1 inactive under normal conditions (22, 27, 28), and we therefore mutated the phosphorylation sites within this domain.
We examined the expression, turnover, subcellular localization, and DNA-binding activity of HSF1Δ∼PRD and compared the properties to those of HSF1 WT before proceeding to the functional studies. To study specifically the properties of the mutant protein, without any interference from the endogenous HSF1, we expressed HSF1Δ∼PRD in hsf1 knockout (hsf1−/−) MEFs, derived from an hsf1−/− mouse (9, 10). To avoid generating a constitutively active HSF1, which has been observed as a result of HSF1 overexpression (26, 53), we titrated the exogenous HSF1 levels in hsf1−/− MEFs to mimic the endogenous levels in hsf1+/+ MEFs (Fig. 1B). Both endogenous and exogenous HSF1 WT from stressed cells migrated more slowly on SDS-PAGE than that from untreated cells (Fig. 1B, lane 1 versus lane 2; Fig. 1C, lane 3 versus lane 4), and previous studies have shown that this effect is caused by HSF1 hyperphosphorylation (39, 40, 53). In hsf1−/− MEFs where HSF1Δ∼PRD was expressed, the retarded migration of HSF1 mutant under heat shock conditions was greatly reduced compared to the HSF1 WT (Fig. 1C, lane 4 versus lane 6), indicating that the stress-inducible phosphorylation was diminished. The residual stress-inducible phosphorylation in HSF1Δ∼PRD (Fig. 1C, lane 5 versus lane 6) was assessed with lambda protein phosphatase (λPP) treatment (54). Since the retarded migration of HSF1Δ∼PRD upon heat stress was eliminated in the presence of λPP (Fig. 1C, lane 6 versus lane 12), it is plausible that HSF1Δ∼PRD undergoes stress-inducible phosphorylation beyond the 15 phosphorylation acceptor sites that were mutated (Fig. 1A).
Multisite phosphorylation has been shown to regulate the turnover of many transcriptional regulators (37). For example, under normal conditions, p53 is targeted for rapid degradation by the E3 ubiquitin ligase Mdm2 (55), whereas stress-inducible phosphorylation of the p53 N-terminal region impairs p53-Mdm2 interaction, resulting in p53 stabilization (56, 57). In contrast, EP300 undergoes phosphorylation-mediated degradation, where hyperphosphorylation precedes its proteasomal degradation (58). To address whether phosphorylation in the RD affects HSF1 turnover, we analyzed HSF1 protein levels in hsf1−/− MEFs, expressing either HSF1 WT or HSF1Δ∼PRD, treated with the eukaryotic translation inhibitor CHX (59). The protein levels of both HSF1 WT and HSF1Δ∼PRD remained constant throughout a 15-h CHX treatment (Fig. 1D), suggesting that lack of the phosphorylation within the RD does not alter the stability of the HSF1 protein. This finding is in agreement with a recent study showing that HSF1 phosphorylation does not affect its turnover (32). In order to validate that protein translation was inhibited by CHX, we analyzed the protein levels of HSF2 which is known to have a fast turnover rate (60, 61). As expected, HSF2 was rapidly degraded and not detectable after a 3-h CHX treatment (Fig. 1D).
HSF1 accumulates in the nucleus upon exposure to stress stimuli, while under nonstress conditions it is localized both in the nucleus and in the cytoplasm (53, 62, 63). Previously, it was proposed that phosphorylation of specific serine residues in the RD affects HSF1 cellular localization (64, 65). Accordingly, phosphorylation of S320 by protein kinase A would retain HSF1 in the nucleus (65), and phosphorylation of S303 and S307 would facilitate 14-3-3ε-mediated nuclear exclusion of HSF1 (64). In primate cells exposed to various proteotoxic stresses, HSF1 forms unique subnuclear granules, called nuclear stress bodies (nSBs) (66), the formation of which requires DNA-binding competent HSF1 and coincides with HSF1 hyperphosphorylation (53, 67). Using indirect immunofluorescence and confocal microscopy, we examined the subcellular localization of Myc-His-tagged HSF1Δ∼PRD and the formation of nSBs in HeLa cells. Under control conditions, the exogenously expressed HSF1 WT and HSF1Δ∼PRD, as well as the endogenous HSF1 protein, were diffusely distributed in the nucleus (Fig. 2). In response to heat stress, both HSF1Δ∼PRD and HSF1 WT were located in the nucleus and concentrated in nSBs. These results indicate that the phosphorylation within the RD has no effect on HSF1 localization under control or stress conditions and that the formation of nSBs is independent of HSF1 hyperphosphorylation.
FIG 2.

HSF1Δ∼PRD localizes to the same subcellular compartments as HSF1 WT under normal and stress conditions. HeLa cells were transfected with Mock plasmid [pcDNA3.1/myc-His(−)A], Myc-His-HSF1 WT, or Myc-His-HSF1Δ∼PRD, left untreated (C) or exposed to heat stress (HS; 1 h at 42°C), and analyzed by immunofluorescence microscopy. A monoclonal antibody against myc was used to detect exogenously expressed HSF1 protein, whereas an anti-HSF1 antibody was used to detect both endo- and exogenously expressed HSF1. DNA was stained with DAPI. The merge figure is an overlay of myc, HSF1, and DAPI signals. Scale bars, 25 μm.
HSF1Δ∼PRD binds to DNA in a stress-inducible manner.
HSF1 activation can be divided into two separate steps. First, HSF1 forms trimers, accumulates in the nucleus, and acquires DNA-binding activity (1). Second, HSF1 acquires transactivating capacity, an event that coincides with the stress-inducible phosphorylation of HSF1 (39). To study whether phosphorylation within the RD alters the DNA-binding activity of HSF1, we used ChIP to compare the occupancy of HSF1 WT and HSF1Δ∼PRD at Hsp70 (HSPA1A and HSPA1B; HSPA1A/B) and Hsp25 (HSPB1) promoters. hsf1−/− MEFs, expressing HSF1 WT or HSF1Δ∼PRD, were either left untreated or exposed to a 30-min heat shock at 43°C, followed by immunoprecipitation with HSF1 antibody or normal rabbit serum as a nonspecific antibody. Under control conditions, the signal for the occupancy of HSF1 WT and HSF1Δ∼PRD at the Hsp70 and Hsp25 promoters was below that of the nonspecific antibody (Fig. 3A), showing that removal of the basal phosphorylation from the RD does not spontaneously induce the DNA-binding activity of HSF1. Upon heat stress, the occupancy of HSF1 WT and HSF1Δ∼PRD increased similarly at the Hsp70 and Hsp25 promoters. Next we examined if the phosphorylation within the RD affects the DNA-binding activity of HSF1 under prolonged stress. hsf1−/− MEFs, expressing either HSF1 WT or HSF1Δ∼PRD, were exposed to cadmium sulfate (60 μM CdSO4), which in addition to promoting expression of metallothioneins induces HSF1-dependent Hsp expression (68). Whole-cell extracts were incubated with a 32P-labeled oligonucleotide containing the proximal HSE of the Hsp70 promoter and binding was studied by EMSA. During prolonged exposure to CdSO4 and during recovery from stress, we did not detect any difference between HSF1 WT and HSF1Δ∼PRD DNA-binding activities (Fig. 3B). Taken together, we conclude that neither basal nor stress-inducible phosphorylation within the RD is involved in the regulation of HSF1 DNA-binding activity.
FIG 3.

HSF1Δ∼PRD binds to DNA in a stress-inducible manner. hsf1−/− MEFs were transfected with Mock plasmid [pcDNA3.1/myc-His(−)A], Myc-His-HSF1 WT, or Myc-His-HSF1Δ∼PRD, and left either untreated (C) or exposed to a 30-min heat shock at 43°C (A) or heavy metal stress (B). (A) The occupancy of HSF1 at the HSPA1A/B (Hsp70) and HSPB1 (Hsp25) promoters was analyzed by ChIP, followed by qPCR. The qPCR values of the immunoprecipitations were normalized to the input values and related to the HSF1 WT control sample, which was set to value 1. The data are presented as mean values from three independent experiments plus the standard errors of the mean (SEM). The values obtained for the nonspecific antibody (normal rabbit serum) are 1.07 for HSPA1A/B and 2.92 for HSPB1. (B) For assessing HSF1Δ∼PRD DNA-binding activity during prolonged stress, the cells were treated with 60 μM CdSO4 for the indicated times (3+R: 3 h CdSO4, followed by a 3-h recovery in fresh culture medium). The HSE-HSF complex (HSF-HSE) was analyzed by EMSA. Expression of HSF1 constructs was detected by Western blotting with anti-HSF1 antibody. α-Actin was used as a loading control. The pound sign indicates nonspecific HSE interactions, and the asterisk indicates HSF1 protein that migrates more slowly on SDS-PAGE due to hyperphosphorylation (53).
Phosphorylation in the regulatory domain suppresses HSF1 transactivating capacity.
To investigate the effect of phosphorylation on HSF1 transactivating capacity, we transfected HSF1 WT and HSF1Δ∼PRD into hsf1−/− MEFs, exposed the cells to stress and measured the steady-state mRNA levels of Hsp70 (HSPA1A and HSPA1B; HSPA1A/B), Hsp25 (HSPB1), and Hsp40 (DnaJB1) by qRT-PCR. Under control conditions, neither HSF1Δ∼PRD nor HSF1 WT was spontaneously activated, since the levels of Hsps were equal to those in cells transfected with an empty plasmid (Mock) (Fig. 4A and B). Upon a 30-min exposure to heat stress at 43°C, we observed an HSF1-dependent increase in HSPA1A/B, HSPB1, and DnaJB1 mRNAs (Fig. 4A and data not shown). Surprisingly, not only was HSF1Δ∼PRD activated upon heat stress, but it exceeded the HSF1 WT in transactivating capacity, since steady-state mRNA levels of HSPA1A/B and HSPB1 were 2-fold higher in the HSF1Δ∼PRD-expressing cells than in HSF1 WT-expressing cells. The 2-fold difference in Hsp mRNA levels between HSF1 WT and HSF1Δ∼PRD was observed also after 1 h exposure to heat stress. Our observation that HSF1Δ∼PRD is capable of driving transcription provides the first evidence for uncoupling the stress-inducible phosphorylation from HSF1 activation.
FIG 4.

Phosphorylation in the regulatory domain suppresses HSF1 transactivating capacity. (A) hsf1−/− MEFs were transfected with Mock plasmid [pcDNA3.1/myc-His(−)A], Myc-His-HSF1 WT, or Myc-His-HSF1Δ∼PRD and left either untreated (C) or exposed to heat stress at 43°C up to 60 min. The mRNA levels of HSPA1A/B (Hsp70) and HSPB1 (Hsp25) were quantified with qRT-PCR and normalized against RNA18S5. The values are shown relative to the respective mRNA levels in the Mock-transfected cells in control conditions (C), which was arbitrarily set to value 1. (B) hsf1−/− MEFs were transfected as in panel A and either left untreated (C), treated with 60 μM CdSO4 for 3 h (3 h), or treated for 3 h and left to recover in fresh culture medium for 3 h (3 h + R3h). mRNA quantification and data analysis were performed as in panel A. The data are presented as mean values from at least three independent experiments plus the SEM. *, P ≤ 0.05; ***, P ≤ 0.001; ****, P ≤ 0.0001.
Next, we examined whether the increased levels of Hsps in cells expressing HSF1Δ∼PRD were specific for heat stress only. For this purpose, we measured HSPA1A/B and HSPB1 mRNA levels from cells exposed to heavy metals. We treated hsf1−/− MEFs, transfected with either HSF1 WT or HSF1Δ∼PRD, with CdSO4, and found that after a 3-h exposure the mRNA levels of HSPA1A/B and HSPB1 were higher in HSF1Δ∼PRD-expressing cells than in HSF1 WT-expressing cells, and the difference was maintained also after 3 h of recovery (Fig. 4B). These results demonstrate that the phosphorylation-mediated repression of HSF1 transactivating capacity is not specific for a particular type of stress.
Elevated stress-inducible Hsp mRNAs in cells expressing HSF1Δ∼PRD could be due to a lowered threshold of stress stimuli. To address this possibility, we exposed hsf1−/− MEFs transfected with HSF1 WT or HSF1Δ∼PRD to heat shock temperatures at 39, 40, and 41°C (Fig. 5A). Moderate heat stress can activate the heat shock response, albeit less efficiently than an exposure to 43°C (69, 70). We did not detect HSF1-mediated induction of HSPA1A/B mRNA within 1 h at 39°C, and at 40°C only cells expressing HSF1Δ∼PRD displayed elevated levels of HSPA1A/B mRNA, whereas at 41°C both HSF1 WT and HSF1Δ∼PRD were capable of inducing HSPA1A/B mRNA. Importantly, upon exposure to 41°C, only cells expressing HSF1Δ∼PRD displayed elevated levels of HSPA1A/B mRNA as early as at a 30-min time point (Fig. 5A).
FIG 5.

HSF1Δ∼PRD requires a lower threshold for activation than HSF1 WT. hsf1−/− MEFs were transfected with Mock plasmid [pcDNA3.1/myc-His(−)A], Myc-His-HSF1 WT, or Myc-His-HSF1Δ∼PRD and left either untreated (C) or exposed to heat stress (39, 40, and 41°C for 30 and 60 min) (A) or heavy metal stress (40 and 60 μM CdSO4 for 60, 120, and 180 min) (B). The mRNA levels of HSPA1A/B (Hsp70) were quantified using qRT-PCR and normalized against RNA18S5. The values are shown relative to the respective mRNA levels in the Mock-transfected cells in control conditions (C), which was arbitrarily set to value 1. The data are presented as mean values from at least three independent experiments plus the SEM. *, P ≤ 0.05; **, P ≤ 0.01; ****, P ≤ 0.0001.
To further study the activation threshold of HSF1 WT and HSF1Δ∼PRD, we treated cells with 40 μM and 60 μM CdSO4 and measured HSPA1A/B mRNA at 1-h intervals up to 3 h. After a 3-h exposure to 40 μM CdSO4, the heat shock response was activated only in cells expressing HSF1Δ∼PRD, while at 60 μM CdSO4, HSPA1A/B mRNA was induced both in HSF1 WT- and in HSF1Δ∼PRD-expressing cells (Fig. 5B). Taken together, our results revealed that cells expressing HSF1Δ∼PRD activated the heat shock response upon moderate stress, which indicates that the activation threshold of HSF1 is lowered when the RD is not phosphorylated.
Intrinsic capacity of the regulatory domain to control transactivation depends on its phosphorylation status.
The Gal4 DNA-binding domain fused to the herpes simplex virus 1 VP16 activation domain (AD) is a potent transcriptional activator capable of expressing eukaryotic genes under a promoter containing Gal4-binding sites (49). In earlier studies, where HSF1 RD was introduced into that chimeric protein, the transactivating capacity of VP16 AD became stress-responsive (22), providing evidence for HSF1 RD possessing an intrinsic ability to regulate transcription and sense heat stress. Here, we wanted to investigate whether the lack of phosphorylation within the RD contributes to the enhanced transactivating capacity of the heterologous VP16 AD. For this purpose, we cloned the regulatory domain (aa 220 to 389) of HSF1 WT and HSF1Δ∼PRD into the Gal4-VP16 chimeric construct (Fig. 6A). HeLa cells were cotransfected with the indicated chimeric constructs and Gal4-driven luciferase reporter gene. Luciferase activity was measured from untreated cells as well as from cells exposed to a 30-min heat shock at 42°C, followed by a 5-h recovery. To exclude the possibility that the obtained results were due to unequal expression of Gal4-VP16 chimeras, we analyzed their protein levels, and found them equally expressed (Fig. 6C). Cells expressing Gal4-VP16 displayed constitutive luciferase activity, whereas Gal4-VP16-HSF1 WT repressed the transcription of the reporter gene under nonstress conditions, reducing luciferase activity by 50% (Fig. 6B). In contrast, the luciferase activity in cells expressing Gal4-VP16-HSF1Δ∼PRD was equal to the cells transfected with Gal4-VP16. These results demonstrate that the lack of phosphorylation within the HSF1 RD reverses the repressed transactivating capacity of VP16 AD under control conditions.
FIG 6.
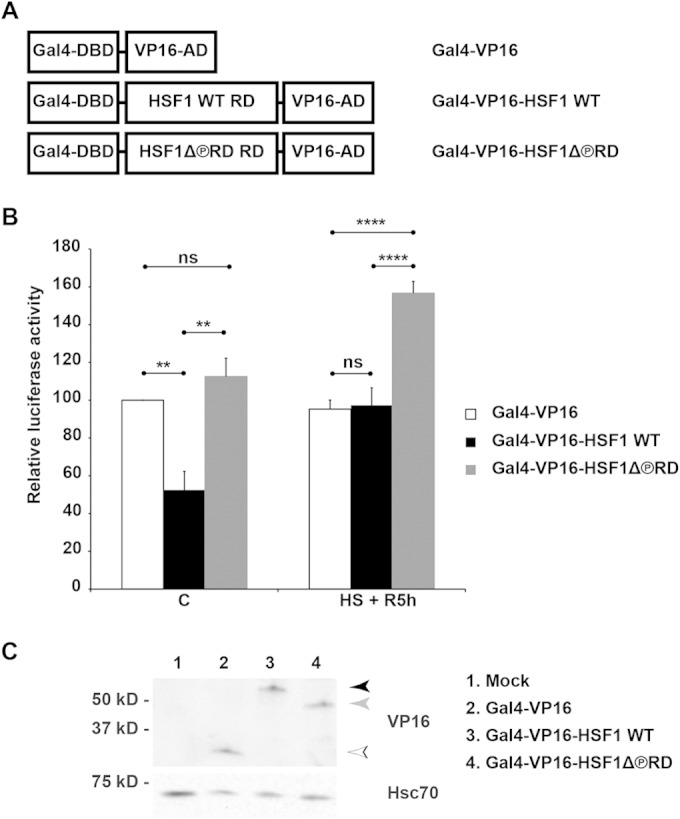
Phosphorylation defines capacity of the RD to control transactivation. (A) Schematic illustration of the chimeric proteins consisting of Gal4 DNA-binding domain (Gal4-DBD, aa 1 to 147), HSF1 WT or HSF1Δ∼PRD regulatory domain (HSF1 WT RD and HSF1Δ∼PRD RD, aa 220 to 389), and herpes simplex virus protein VP16 activation domain (VP16-AD, aa 413 to 490). Note that the figure is not drawn to scale. (B and C) HeLa cells were transfected with plasmids encoding Gal4-driven luciferase and β-galactosidase, together with indicated plasmids encoding chimeric proteins described in panel A or with an empty plasmid (Mock). Cells were left untreated (C) or heat shocked for 30 min at 42°C and let to recover for 5 h at 37°C (HS + R5h). In panel B, the relative luciferase activity was calculated against the activity in the Gal4-VP16 samples under control conditions, which was set to value 100. The data are presented as mean values from four independent experiments plus the SEM. ns, nonsignificant; **, P ≤ 0.01; ****, P ≤ 0.0001. In panel C, the protein levels of the Gal4-VP16 chimeras, under control conditions, were analyzed using Western blotting with anti-VP16 antibody. Hsc70 is shown as a loading control. The arrowheads indicate Gal4-VP16 (white), Gal4-VP16-HSF1 WT (black), and Gal4-VP16-HSF1Δ∼PRD (gray). The difference in the migration pattern between Gal4-VP16-HSF1 WT and Gal4-VP16-HSF1Δ∼PRD is likely due to phosphorylation.
Repression of Gal4-VP16-HSF1 WT was eliminated upon heat shock due to the intrinsic capacity of HSF1 RD to sense heat shock, and the luciferase activity corresponded to that observed in cells expressing Gal4-VP16 (Fig. 6B). Surprisingly, after heat shock, Gal4-VP16-HSF1Δ∼PRD was 60% more effective than Gal4-VP16 or Gal4-VP16-HSF1 WT, indicating that the phosphorylation-deficient RD is capable of further enhancing transactivation in a stress-dependent manner. Based on the obtained results, we conclude that the phosphorylation status defines the intrinsic capacity of HSF1 RD to control transactivation in response to heat stress.
DISCUSSION
Involvement of HSF1 in a plethora of cellular functions requires a sophisticated regulatory mechanism(s) that can accurately control the conditions under which HSF1 is activated. It has been suggested that phosphorylation of HSF1 would serve as an integrator of various signaling pathways triggering HSF1-driven transcription (37). The regulatory domain (RD) harbors ∼70% of the known HSF1 phosphorylation sites, and we hypothesized that HSF1 activation would primarily be controlled by phosphorylation within the RD. The results obtained here provide, to the best of our knowledge, the first direct evidence for uncoupling hyperphosphorylation from HSF1 activation. In contrast to previous studies, where disruption of the RD led to a constitutively active HSF1 (34, 35, 38), HSF1Δ∼PRD is not spontaneously active and is capable of inducing Hsp expression in a stress-dependent manner. These results are surprising, since phosphorylation has been regarded as an important hallmark of HSF1 activation (20, 30, 38). Although HSF1 is phosphorylated on several residues during its activation, it has been reported that only one of these residues, S326, substantially contributes to HSF1 transcriptional activity (30) and is widely used as a marker for activated HSF1 in carcinogenesis (20, 71). In agreement with the earlier report (30), the stress-inducible DNA-binding capacity of HSF1 is not dependent on S326 as an intact phosphorylation acceptor site, whereas our results show that HSF1 can be a potent transcriptional activator without being phosphorylated on multiple sites, including S326, within the RD. This discrepancy is presumably due to a different experimental approach, i.e., single-site versus multisite mutagenesis of HSF1.
Our finding that HSF1 activity can be uncoupled from the phosphorylation events occurring in the RD, is supported by recently published studies. The results by Rossi et al. indicate that cells treated with the proteasome inhibitor bortezomib display inducible HSF1-dependent Hsp70 expression accompanied by only a modest increase in HSF1 phosphorylation (72). Another recent study revealed that ethanol exposure leads to transcriptional activation of HSF1, which lacks hyperphosphorylation (73). Taking all of these findings together, we conclude that activation of HSF1 can occur in the absence of hyperphosphorylation. Intriguingly, hyperphosphorylated HSF1 can also be transcriptionally incompetent under certain circumstances, which was recently observed in heat-stressed mitotic cells, where Hsps were not induced despite hyperphosphorylation of HSF1 (74). Thus, employing phosphorylation as a sole marker for HSF1 activation should be reconsidered.
Given that hyperphosphorylation is not required for HSF1 activation, the question of why HSF1 is hyperphosphorylated during its activation remains to be answered. The finding that HSF1Δ∼PRD can be activated by milder stress than HSF1 WT (Fig. 5) indicates that phosphorylation within the RD defines the activation threshold in response to distinct stress stimuli. Furthermore, the phosphorylation-deficient HSF1Δ∼PRD is a more potent transactivator than HSF1 WT (Fig. 4), suggesting that hyperphosphorylation limits the magnitude of the heat shock response. Based on these results, we propose that phosphorylation serves as a fine-tuning mechanism for regulating the transcriptionally competent HSF1. Phosphorylation can modulate transcription factor activity on at least three levels: subcellular localization, DNA-binding activity, and interaction with the transcriptional machinery (75). Since we did not observe any changes in HSF1 localization (Fig. 2) or DNA-binding activity (Fig. 3), it is likely that phosphorylation within the RD modulates the interaction between HSF1 and the transcriptional machinery both via conformational changes (76) and electrostatic effects (77). It has earlier been shown that HSF1 interacts with various protein complexes required for active transcription, such as the chromatin remodeling complex SWI/SNF (78), Mediator complex (79), components of the preinitiation complex (80), and the histone chaperone FACT (81). However, the role of HSF1 phosphorylation in these protein-protein interactions has not been reported and will be investigated in our forthcoming studies. Another mechanism by which phosphorylation modulates transcription factor activity is through interconnected posttranslational modifications (PTMs) (82). It is known that HSF1 is sumoylated in a phosphorylation-dependent manner (29). In addition, HSF1 is subjected to acetylation (31), ubiquitination (32), and glycosylation (83), and it is plausible that other PTMs modulate HSF1-driven gene expression in an orchestrated manner. Thus, to fully understand how transactivation capacity of HSF1 is regulated, emphasis should be placed on phosphorylation-dependent interactions within the RD.
Our results validate hyperphosphorylation as a fundamental regulator of HSF1 transactivation capacity. Phosphorylation, in cooperation with other PTMs, creates distinct PTM signatures, which can adequately modulate the transcriptional response based on the type and severity of the stimuli. Since HSF1 is involved in a multitude of physiological processes, such as the transcriptional control of development and life span, in addition to proteotoxic stress responses (4), a specific PTM signature would provide a mechanism enabling precise temporal, spatial, and environmental transcriptional programs to ensure that a cell is able to perform its designated function. Importantly, identification and regulation of the HSF1 PTM signatures provides new possibilities to counteract the actions of HSF1 in pathological conditions, such as cancer and neurodegenerative diseases (20, 46).
ACKNOWLEDGMENTS
We thank Julius Anckar for insightful discussion in the initial stage of the project, Alexandra Elsing for expert help with the statistical analysis, Samu Himanen for help with the ChIP assay, and Emine Lundsten and Cynthia Swan for assistance with writing of the manuscript. All of the members of the Sistonen laboratory are acknowledged for their constructive comments.
This study was financially supported by the Academy of Finland, the Sigrid Jusélius Foundation, the Magnus Ehrnrooth Foundation, Åbo Akademi University Foundation (L.S.), and the Turku Doctoral Program of Biomedical Sciences (M.A.B. and J.J.).
REFERENCES
- 1.Anckar J, Sistonen L. 2011. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem 80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 2.Richter K, Haslbeck M, Buchner J. 2010. The heat shock response: life on the verge of death. Mol Cell 40:253–266. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 3.Hartl FU, Bracher A, Hayer-Hartl M. 2011. Molecular chaperones in protein folding and proteostasis. Nature 475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 4.Åkerfelt M, Morimoto RI, Sistonen L. 2010. Heat shock factors: integrators of cell stress, development, and life span. Nat Rev Mol Cell Biol 11:545–555. doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amin J, Ananthan J, Voellmy R. 1988. Key features of heat shock regulatory elements. Mol Cell Biol 8:3761–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Littlefield O, Nelson HCM. 1999. A new use for the ‘wing’ of the ‘winged’ helix-turn-helix motif in the HSF–DNA cocrystal. Nat Struct Mol Biol 6:464–470. doi: 10.1038/8269. [DOI] [PubMed] [Google Scholar]
- 7.Trinklein ND, Murray JI, Hartman SJ, Botstein D, Myers RM. 2004. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol Biol Cell 15:1254–1261. doi: 10.1091/mbc.E03-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vihervaara A, Sergelius C, Vasara J, Blom MAH, Elsing AN, Roos-Mattjus P, Sistonen L. 2013. Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc Natl Acad Sci U S A 110:E3388–E3397. doi: 10.1073/pnas.1305275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ. 1998. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J Biol Chem 273:7523–7528. doi: 10.1074/jbc.273.13.7523. [DOI] [PubMed] [Google Scholar]
- 10.Xiao X, Zuo X, Davis AA, McMillan DR, Curry BB, Richardson JA, Benjamin IJ. 1999. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J 18:5943–5952. doi: 10.1093/emboj/18.21.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christians E, Davis A, Thomas S, Benjamin I. 2000. Embryonic development: maternal effect of Hsf1 on reproductive success. Nature 407:693–694. doi: 10.1038/35037669. [DOI] [PubMed] [Google Scholar]
- 12.Page TJ, Sikder D, Yang L, Pluta L, Wolfinger RD, Kodadek T, Thomas RS. 2006. Genome-wide analysis of human HSF1 signaling reveals a transcriptional program linked to cellular adaptation and survival. Mol Biosystems 2:627–639. doi: 10.1039/b606129j. [DOI] [PubMed] [Google Scholar]
- 13.Hsu AL, Murphy CT, Kenyon C. 2003. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- 14.Morley JF, Morimoto RI. 2004. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell 15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steele AD, Hutter G, Jackson WS, Heppner FL, Borkowski AW, King OD, Raymond GJ, Aguzzi A, Lindquist S. 2008. Heat shock factor 1 regulates life span as distinct from disease onset in prion disease. Proc Natl Acad Sci U S A 105:13626–13631. doi: 10.1073/pnas.0806319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inouye S, Izu H, Takaki E, Suzuki H, Shirai M, Yokota Y, Ichikawa H, Fujimoto M, Nakai A. 2004. Impaired IgG production in mice deficient for heat shock transcription factor 1. J Biol Chem 279:38701–38709. doi: 10.1074/jbc.M405986200. [DOI] [PubMed] [Google Scholar]
- 17.Reinke H, Saini C, Fleury-Olela F, Dibner C, Benjamin IJ, Schibler U. 2008. Differential display of DNA-binding proteins reveals heat-shock factor 1 as a circadian transcription factor. Genes Dev 22:331–345. doi: 10.1101/gad.453808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai C, Whitesell L, Rogers AB, Lindquist S. 2007. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130:1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santagata S, Hu R, Lin NU, Mendillo ML, Collins LC, Hankinson SE, Schnitt SJ, Whitesell L, Tamimi RM, Lindquist S, Ince TA. 2011. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc Natl Acad Sci U S A 108:18378–18383. doi: 10.1073/pnas.1115031108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S. 2012. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150:549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vuister GW, Kim S, Orosz A, Marquardt J, Wu C, Bax A. 1994. Solution structure of the DNA-binding domain of Drosophila heat shock transcription factor. Nat Struct Biol 1:605–614. doi: 10.1038/nsb0994-605. [DOI] [PubMed] [Google Scholar]
- 22.Newton EM, Knauf U, Green M, Kingston RE. 1996. The regulatory domain of human heat shock factor 1 is sufficient to sense heat stress. Mol Cell Biol 16:839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorger PK, Nelson HCM. 1989. Trimerization of a yeast transcriptional activator via a coiled-coil motif. Cell 59:807–813. doi: 10.1016/0092-8674(89)90604-1. [DOI] [PubMed] [Google Scholar]
- 24.Peteranderl R, Nelson HCM. 1992. Trimerization of the heat shock transcription factor by a triple-stranded α-helical coiled coil. Biochemistry 31:12272–12276. doi: 10.1021/bi00163a042. [DOI] [PubMed] [Google Scholar]
- 25.Rabindran SK, Haroun RI, Clos J, Wisniewski J, Wu C. 1993. Regulation of heat shock factor trimer formation: role of a conserved leucine zipper. Science 259:230–234. doi: 10.1126/science.8421783. [DOI] [PubMed] [Google Scholar]
- 26.Zuo J, Rungger D, Voellmy R. 1995. Multiple layers of regulation of human heat shock transcription factor 1. Mol Cell Biol 15:4319–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green M, Schuetz TJ, Sullivan EK, Kingston RE. 1995. A heat shock-responsive domain of human HSF1 that regulates transcription activation domain function. Mol Cell Biol 15:3354–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takemori Y, Enoki Y, Yamamoto N, Fukai Y, Adachi K, Sakurai H. 2009. Mutational analysis of human heat-shock transcription factor 1 reveals a regulatory role for oligomerization in DNA-binding specificity. Biochem J 424:253–261. doi: 10.1042/BJ20090922. [DOI] [PubMed] [Google Scholar]
- 29.Hietakangas V, Ahlskog JK, Jakobsson AM, Hellesuo M, Sahlberg NM, Holmberg CI, Mikhailov A, Palvimo JJ, Pirkkala L, Sistonen L. 2003. Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol Cell Biol 23:2953–2968. doi: 10.1128/MCB.23.8.2953-2968.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guettouche T, Boellmann F, Lane WS, Voellmy R. 2005. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem 6:4. doi: 10.1186/1471-2091-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Westerheide SD, Anckar J, Stevens SM Jr, Sistonen L, Morimoto RI. 2009. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raychaudhuri S, Loew C, Körner R, Pinkert S, Theis M, Hayer-Hartl M, Buchholz F, Hartl FU. 2014. Interplay of acetyltransferase EP300 and the proteasome system in regulating heat shock transcription factor 1. Cell 156:975–985. doi: 10.1016/j.cell.2014.01.055. [DOI] [PubMed] [Google Scholar]
- 33.Xu YM, Huang DY, Chiu JF, Lau AT. 2012. Post-translational modification of human heat shock factors and their functions: a recent update by proteomic approach. J Proteome Res 11:2625–2634. doi: 10.1021/pr201151a. [DOI] [PubMed] [Google Scholar]
- 34.Knauf U, Newton EM, Kyriakis J, Kingston RE. 1996. Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev 10:2782–2793. doi: 10.1101/gad.10.21.2782. [DOI] [PubMed] [Google Scholar]
- 35.Kline MP, Morimoto RI. 1997. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol Cell Biol 17:2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu B, Zhong R, Soncin F, Stevenson MA, Calderwood SK. 1998. Transcriptional activity of heat shock factor 1 at 37°C is repressed through phosphorylation on two distinct serine residues by glycogen synthase kinase 3 and protein kinases Cα and Cζ. J Biol Chem 273:18640–18646. doi: 10.1074/jbc.273.29.18640. [DOI] [PubMed] [Google Scholar]
- 37.Holmberg CI, Tran SE, Eriksson JE, Sistonen L. 2002. Multisite phosphorylation provides sophisticated regulation of transcription factors. Trends Biochem Sci 27:619–627. doi: 10.1016/S0968-0004(02)02207-7. [DOI] [PubMed] [Google Scholar]
- 38.Xia W, Voellmy R. 1997. Hyperphosphorylation of heat shock transcription factor 1 is correlated with transcriptional competence and slow dissociation of active factor trimers. J Biol Chem 272:4094–4102. doi: 10.1074/jbc.272.7.4094. [DOI] [PubMed] [Google Scholar]
- 39.Cotto JJ, Kline M, Morimoto RI. 1996. Activation of heat shock factor 1 DNA binding precedes stress-induced serine phosphorylation. Evidence for a multistep pathway of regulation. J Biol Chem 271:3355–3358. [DOI] [PubMed] [Google Scholar]
- 40.Holmberg CI, Hietakangas V, Mikhailov A, Rantanen JO, Kallio M, Meinander A, Hellman J, Morrice N, MacKintosh C, Morimoto RI, Eriksson JE, Sistonen L. 2001. Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J 20:3800–3810. doi: 10.1093/emboj/20.14.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Yao M, Gu J, Sun L, Shen Y, Liu X. 2002. Blocking HSF1 by dominant-negative mutant to sensitize tumor cells to hyperthermia. Biochem Biophys Res Commun 290:1454–1461. doi: 10.1006/bbrc.2002.6373. [DOI] [PubMed] [Google Scholar]
- 42.Boellmann F, Guettouche T, Guo Y, Fenna M, Mnayer L, Voellmy R. 2004. DAXX interacts with heat shock factor 1 during stress activation and enhances its transcriptional activity. Proc Natl Acad Sci U S A 101:4100–4105. doi: 10.1073/pnas.0304768101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. 2009. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 44.Calamini B, Morimoto RI. 2012. Protein homeostasis as a therapeutic target for diseases of protein conformation. Curr Top Med Chem 12:2623–2640. doi: 10.2174/1568026611212220014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Westerheide SD, Morimoto RI. 2005. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem 280:33097–33100. doi: 10.1074/jbc.R500010200. [DOI] [PubMed] [Google Scholar]
- 46.Neef DW, Turski ML, Thiele DJ. 2010. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol 8:e1000291. doi: 10.1371/journal.pbio.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Calamini B, Silva MC, Madoux F, Hutt DM, Khanna S, Chalfant MA, Saldanha SA, Hodder P, Tait BD, Garza D, Balch WE, Morimoto RI. 2012. Small-molecule proteostasis regulators for protein conformational diseases. Nat Chem Biol 8:185–196. doi: 10.1038/nchembio.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.West JD, Wang Y, Morano KA. 2012. Small molecule activators of the heat shock response: chemical properties, molecular targets, and therapeutic promise. Chem Res Toxicol 25:2036–2053. doi: 10.1021/tx300264x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sadowski I, Ma J, Triezenberg S, Ptashne M. 1988. GAL4-VP16 is an unusually potent transcriptional activator. Nature 335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 50.Mosser DD, Theodorakis NG, Morimoto RI. 1988. Coordinate changes in heat shock element-binding activity and HSP70 gene transcription rates in human cells. Mol Cell Biol 8:4736–4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holmberg CI, Illman SA, Kallio M, Mikhailov A, Sistonen L. 2000. Formation of nuclear HSF1 granules varies depending on stress stimuli. Cell Stress Chaperones 5:219–228. doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarge KD, Murphy SP, Morimoto RI. 1993. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol 13:1392–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhuo S, Clemens JC, Hakes DJ, Barford D, Dixon JE. 1993. Expression, purification, crystallization, and biochemical characterization of a recombinant protein phosphatase. J Biol Chem 268:17754–17761. [PubMed] [Google Scholar]
- 55.Moll UM, Petrenko O. 2003. The MDM2-p53 interaction. Mol Cancer Res 1:1001–1008. [PubMed] [Google Scholar]
- 56.Appella E, Anderson CW. 2001. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 268:2764–2772. doi: 10.1046/j.1432-1327.2001.02225.x. [DOI] [PubMed] [Google Scholar]
- 57.Vousden KH. 2002. Activation of the p53 tumor suppressor protein. Biochim Biophys Acta 1602:47–59. doi: 10.1016/S0304-419X(02)00035-5. [DOI] [PubMed] [Google Scholar]
- 58.Poizat C, Puri PL, Bai Y, Kedes L. 2005. Phosphorylation-dependent degradation of p300 by doxorubicin-activated p38 mitogen-activated protein kinase in cardiac cells. Mol Cell Biol 25:2673–2687. doi: 10.1128/MCB.25.7.2673-2687.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Obrig TG, Culp WJ, McKeehan WL, Hardesty B. 1971. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem 246:174–181. [PubMed] [Google Scholar]
- 60.Mathew A, Mathur SK, Morimoto RI. 1998. Heat shock response and protein degradation: regulation of HSF2 by the ubiquitin-proteasome pathway. Mol Cell Biol 18:5091–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahlskog JK, Björk JK, Elsing AN, Aspelin C, Kallio M, Roos-Mattjus P, Sistonen L. 2010. Anaphase-promoting complex/cyclosome participates in the acute response to protein-damaging stress. Mol Cell Biol 30:5608–5620. doi: 10.1128/MCB.01506-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cotto J, Fox S, Morimoto RI. 1997. HSF1 granules: a novel stress-induced nuclear compartment of human cells. J Cell Sci 110:2925–2934. [DOI] [PubMed] [Google Scholar]
- 63.Jolly C, Morimoto RI, Robert-Nicoud M, Vourc'h C. 1997. HSF1 transcription factor concentrates in nuclear foci during heat shock: relationship with transcription sites. J Cell Sci 110:2935–2941. [DOI] [PubMed] [Google Scholar]
- 64.Wang X, Grammatikakis N, Siganou A, Calderwood SK. 2003. Regulation of molecular chaperone gene transcription involves the serine phosphorylation, 14-3-3 epsilon binding, and cytoplasmic sequestration of heat shock factor 1. Mol Cell Biol 23:6013–6026. doi: 10.1128/MCB.23.17.6013-6026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murshid A, Chou S, Prince T, Zhang Y, Bharti A, Calderwood SK. 2010. Protein kinase A binds and activates heat shock factor 1. PLoS One 5:e13830. doi: 10.1371/journal.pone.0013830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Biamonti G, Vourc'h C. 2010. Nuclear stress bodies. Cold Spring Harb Perspect Biol 2:a000695. doi: 10.1101/cshperspect.a000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alastalo TP, Hellesuo M, Sandqvist A, Hietakangas V, Kallio M, Sistonen L. 2003. Formation of nuclear stress granules involves HSF2 and coincides with the nucleolar localization of Hsp70. J Cell Sci 116:3557–3570. doi: 10.1242/jcs.00671. [DOI] [PubMed] [Google Scholar]
- 68.Murata M, Gong P, Suzuki K, Koizumi S. 1999. Differential metal response and regulation of human heavy metal-inducible genes. J Cell Physiol 180:105–113. doi:. [DOI] [PubMed] [Google Scholar]
- 69.Wang S, Diller KR, Aggarwal SJ. 2003. Kinetics study of endogenous heat shock protein 70 expression. J Biomech Eng 125:794–797. doi: 10.1115/1.1632522. [DOI] [PubMed] [Google Scholar]
- 70.Shinkawa T, Tan K, Fujimoto M, Hayashida N, Yamamoto K, Takaki E, Takii R, Prakasam R, Inouye S, Mezger V, Nakai A. 2011. Heat shock factor 2 is required for maintaining proteostasis against febrile-range thermal stress and polyglutamine aggregation. Mol Biol Cell 22:3571–3583. doi: 10.1091/mbc.E11-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chou SD, Murshid A, Eguchi T, Gong J, Calderwood SK. 2015. HSF1 regulation of beta-catenin in mammary cancer cells through control of HuR/elavL1 expression. Oncogene 34:2178–2188. doi: 10.1038/onc.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rossi A, Riccio A, Coccia M, Trotta E, La Frazia S, Santoro MG. 2014. The proteasome inhibitor bortezomib is a potent inducer of zinc-finger AN1-type domain 2a gene expression: role of HSF1/HSF2 heterocomplexes. J Biol Chem 289:12705–12715. doi: 10.1074/jbc.M113.513242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.El Fatimy R, Miozzo F, Le Mouel A, Abane R, Schwendimann L, Saberan-Djoneidi D, de Thonel A, Massaoudi I, Paslaru L, Hashimoto-Torii K, Christians E, Rakic P, Gressens P, Mezger V. 2014. Heat shock factor 2 is a stress-responsive mediator of neuronal migration defects in models of fetal alcohol syndrome. EMBO Mol Med 6:1043–1061. doi: 10.15252/emmm.201303311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Elsing AN, Aspelin C, Björk JK, Bergman HA, Himanen SV, Kallio MJ, Roos-Mattjus P, Sistonen L. 2014. Expression of HSF2 decreases in mitosis to enable stress-inducible transcription and cell survival. J Cell Biol 206:735–749. doi: 10.1083/jcb.201402002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hunter T, Karin M. 1992. The regulation of transcription by phosphorylation. Cell 70:375–387. doi: 10.1016/0092-8674(92)90162-6. [DOI] [PubMed] [Google Scholar]
- 76.Sprang S, Acharya K, Goldsmith E, Stuart D, Varvill K, Fletterick R, Madsen N, Johnson L. 1988. Structural changes in glycogen phosphorylase induced by phosphorylation. Nature 336:215–221. doi: 10.1038/336215a0. [DOI] [PubMed] [Google Scholar]
- 77.Hurley JH, Dean AM, Sohl JL, Koshland DE Jr, Stroud RM. 1990. Regulation of an enzyme by phosphorylation at the active site. Science 249:1012–1016. doi: 10.1126/science.2204109. [DOI] [PubMed] [Google Scholar]
- 78.Corey LL, Weirich CS, Benjamin IJ, Kingston RE. 2003. Localized recruitment of a chromatin-remodeling activity by an activator in vivo drives transcriptional elongation. Genes Dev 17:1392–1401. doi: 10.1101/gad.1071803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park JM, Werner J, Kim JM, Lis JT, Kim Y. 2001. Mediator, not holoenzyme, is directly recruited to the heat shock promoter by HSF upon heat shock. Mol Cell 8:9–19. doi: 10.1016/S1097-2765(01)00296-9. [DOI] [PubMed] [Google Scholar]
- 80.Yuan C, Gurley WB. 2000. Potential targets for HSF1 within the preinitiation complex. Cell Stress Chaperones 5:229–242. doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fujimoto M, Takaki E, Takii R, Tan K, Prakasam R, Hayashida N, Iemura S, Natsume T, Nakai A. 2012. RPA assists HSF1 access to nucleosomal DNA by recruiting histone chaperone FACT. Mol Cell 48:182–194. doi: 10.1016/j.molcel.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 82.Filtz TM, Vogel WK, Leid M. 2014. Regulation of transcription factor activity by interconnected posttranslational modifications. Trends Pharmacol Sci 35:76–85. doi: 10.1016/j.tips.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Singleton KD, Wischmeyer PE. 2008. Glutamine induces heat shock protein expression via O-glycosylation and phosphorylation of HSF-1 and Sp1. J Parent Enteral Nutr 32:371–376. doi: 10.1177/0148607108320661. [DOI] [PubMed] [Google Scholar]