

Abstract
Cyclooxygenase (COX) catalyzes the first step in prostanoid biosynthesis and exists as two isoforms. COX-1 is a constitutive enzyme involved in physiological processes, whereas COX-2 is induced by a variety of stimuli. MicroRNAs (miRNAs) are noncoding RNAs that function as key posttranscriptional regulators of gene expression. Although it is known that COX-2 expression is regulated by miRNAs, there are no data regarding COX-2 involvement in miRNA regulation. Considering our previous results showing that COX-2 expression in hepatocytes protects against insulin resistance, we evaluated the role of COX-2 in the regulation of a specific set of miRNAs implicated in insulin signaling in liver cells. Our results provide evidence of the molecular basis for a novel function of COX-2 in miRNA processing. COX-2 represses miRNA 23b (miR-23b), miR-146b, and miR-183 expression in liver cells by increasing the level of DEAD-box helicase p68 (DDX5) through phosphatidylinositol 3-kinase (PI3K)/p300 signaling and by modulating the enzymatic function of the Drosha (RNase type III) complex through its physical association with DDX5. The decrease of miR-183 expression promotes protection against insulin resistance by increasing insulin receptor substrate 1 (IRS1) levels. These results indicate that the modulation of miRNA processing by COX-2 is a key event in insulin signaling in liver and has potential clinical implications for the management of various hepatic dysfunctions.
INTRODUCTION
Cyclooxygenase 1 (COX-1) and 2 catalyze the first step in prostanoid biosynthesis. COX-1 (PTGS1) is constitutively expressed in many tissues, whereas COX-2 (PTGS2) expression is induced by a variety of stimuli such as growth factors, proinflammatory stimuli, hormones, and other cellular stresses (1–3). We and others have demonstrated that partial hepatectomy (PH) induced COX-2 expression in hepatocytes and contributed to the progression of the cell cycle during regeneration (4, 5). In addition to liver regeneration after PH or exposure to hepatotoxic agents, expression of COX-2 has been detected in animal models of cirrhosis (6), in human hepatoma cell lines (7, 8), in human hepatocellular carcinoma (HCC) (9), and after hepatitis B virus (HBV) and hepatitis C virus (HCV) infection (10, 11). Further, overexpression of COX-2 in liver exerts efficient protection against acute liver injury by an antiapoptotic/antinecrotic effect and by accelerated early hepatocyte proliferation (12, 13).
Insulin resistance (IR) plays a key role in the pathophysiology of obesity-related diseases such as type 2 diabetes and nonalcoholic fatty liver disease (NAFLD). Our previous results from studies performed in a model of transgenic mice constitutively expressing human COX-2 in hepatocytes (hCOX-2-Tg) indicate a protective role of COX-2 in a model of insulin resistance induced by a high-fat diet (HFD) (14) and in liver damage induced by hyperglycemia (15).
MicroRNAs (miRNAs) are small noncoding RNAs that negatively regulate their target genes primarily through RNA destabilization or translational repression. In mammalian miRNA biogenesis, the primary transcripts of miRNA (pri-miRNA) are cleaved into precursor miRNA (pre-miRNA) by the nuclear RNase III (ribonuclease type III) Drosha and further processed to mature miRNAs by cytosolic Dicer, another RNase III-related enzyme (16). The Drosha complex consists of Drosha, DGCR8 (DiGeorge syndrome critical region gene 8), DDX5 (RNA helicase p68), and DDX17 (RNA helicase p72). DDX5 and DDX17 are required for the maturation of some but not all miRNAs (17). Aberrant miRNA expression is associated with pathological conditions. In the context of liver diseases, previous studies revealed a role for miRNAs in acute liver injury, viral hepatitis, hepatocarcinogenesis, hepatic fibrogenesis, and NAFLD (18, 19). While many aspects of miRNA-induced protein regulation are known, there is a growing need to uncover the complex and incompletely understood regulatory mechanisms governing the activation and suppression of miRNA expression, considering that they can be controlled at multiple steps during RNA biogenesis: at the transcriptional level, during the multistep processing stage, and posttranscriptionally. The 3′ untranslated region (3′UTR) of COX-2 contains multiple copies of AU-rich elements (AREs) and microRNA response element (MRE) motifs which, when bound by specific ARE-binding factors or miRNAs, influence COX-2 stability and translational efficiency (20). A functional link between COX-2 and miRNA expression has been documented by some groups. Accordingly, Dey's group highlighted miRNA-mediated regulation of COX-2 by miRNA 101a (miR-101a) and miR-199a* during embryo implantation and in endometrial cancer cells (21, 22). Further, recent studies have reported that miR-101 and miR-16 downregulation is involved in COX-2 expression in human colon cancer cells (CRC) (23, 24). Our previous results revealed that miR-16 silences COX-2 expression in hepatoma cells by two mechanisms: by binding directly to the MRE motif in the COX-2 3′UTR and by decreasing the levels of HuR (25). However, less is known about the effects of COX-2 on miRNA expression patterns and their possible role in liver physiopathology.
In the present work, we have studied whether COX-2 regulates the expression profile of miRNAs in liver and examined the physiopathological properties related to the insulin signaling pathway. Here we provide the first demonstration that COX-2 decreases the levels of miR-23b, miR-146b, and miR-183 in liver by upregulating DDX5 expression through a phosphatidylinositol 3-kinase (PI3K)/p300-dependent mechanism. Moreover, COX-2 was shown to inhibit miRNA maturation by associating with the Drosha complex through DDX5 and preventing the conversion of pri-miRNAs into pre-miRNAs. In conclusion, our report highlights a novel miRNA-dependent mechanism through which COX-2 promotes protection against insulin resistance in the liver.
MATERIALS AND METHODS
Chemicals.
Antibodies were from Santa Cruz Biotech (Santa Cruz, CA), Sigma-Aldrich (St. Louis, MO), Cell Signaling Technology (Boston, MA), Abcam (Cambridge, United Kingdom), and Cayman Chemical (Ann Arbor, MI). Prostaglandin E2 (PGE2) and pharmacological inhibitors were from Cayman Chemical. C646 inhibitor was from Millipore (Calbiochem, Darmstadt, Germany). Reagents were from Merck, Roche Diagnostics (Mannheim, Germany), or Sigma. Reagents for electrophoresis were from Bio-Rad (Hercules, CA). Tissue culture dishes were from Falcon (Becton Dickinson Labware, Franklin Lakes, NJ). Tissue culture media were from Invitrogen (Grand Island, NY).
Patients.
This study included 10 nondiabetic patients with a clinical diagnosis of nonalcoholic steatosis (NAS), who underwent a liver biopsy specimen for diagnostic purposes. We further studied 10 subjects with histologically normal liver (NL). Informed consent in writing was obtained from each patient, and the study protocol conformed to ethical guidelines of the 1975 Declaration of Helsinki and was approved by the ethics committee of Princesa University Hospital (Madrid, Spain).
Animal experimentation.
hCOX-2-Tg mice (25 to 30 g body weight; 3 months of age) on a B6D2/OlaHsd background were used in this study together with corresponding age-matched wild-type (Wt) mice (12). The animals were housed on a 12-h-light/12-h-dark cycle in an air-conditioned room at 25°C with food and water available ad libitum and were treated according to the Institutional Care Instructions (Bioethical Commission from Consejo Superior de Investigaciones Científicas [CSIC], Spain). As indicated, mice were sacrificed and livers were removed, snap-frozen in liquid nitrogen, and stored at −80°C.
Primary culture of adult hepatocytes.
Hepatocytes were isolated from nonfasting male Wt and hCOX-2-Tg mice by perfusion with collagenase (2). Cells were plated in 6-cm-diameter dishes and cultured for 48 h in William's E medium supplemented with 20 ng/ml epidermal growth factor (EGF), 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (FBS).
Cell culture.
The CCL-13 human liver cell line (Chang liver [CHL]), an immortalized nontumor cell line derived from normal liver, was purchased from the American Type Culture Collection (Manassas, VA). The MIN6 pancreatic murine cell line was kindly provided by M. Vallejo (Madrid, Spain). Cells were grown in tissue culture dishes in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics (50 μg/ml each of penicillin, streptomycin, and gentamicin) at 37°C in a humidified air/5% CO2 atmosphere.
Construction of COX-2 expression vector.
pPyCAGIP, a cytomegalovirus (CMV) early enhancer/chicken beta actin (CAG) promoter-driven epiosomal vector, was a gift from I. Chambers (University of Edinburgh, Edinburgh, Scotland). Human COX-2 was PCR amplified from a human full-length COX-2 cDNA vector, a gift from S. Prescott (Huntsman Cancer Institute, Salt Lake City, UT), and cloned into the XhoI-NotI restriction site of pPyCAGIP. The correct orientation and integrity of the construct were confirmed by sequencing.
Generation of stable hCOX-2 cells.
Immortalized cell lines were derived from Wt (neonatal cell line [NCL] in vector [NCL-V]) and hCOX-2-Tg (NCL-C) liver by infection with simian virus 40 (SV40) large T antigen virus. To do this, primary hepatocytes from neonates (3 to 5 days old) were obtained from Wt and hCOX-2-Tg liver, dispersed with collagenase, and cultured as described previously (2). Primary hepatocytes were infected with Polybrene-supplemented virus for 48 h and maintained in culture medium for 72 h, followed by selection with puromycin for 3 weeks. Thereafter, immortalized cells were further cultured for 15 days in arginine-free medium supplemented with 10% FBS, to avoid growth of nonparenchymal cells. The cells displayed a typical hepatocyte phenotype, as assessed by albumin, carbamoyl phosphate synthase, and cytokeratine 18 expression (26), and were generously provided by A. M. Valverde (Madrid, Spain).
For plasmid transfections, attached CHL cells at 70% confluence were transfected for 6 to 16 h with pPyCAGIP-hCOX-2 or control pPyCAGIP vector using Lipofectamine 2000 reagent (Invitrogen). At the end of this period, cells were replenished with fresh medium containing 10% FBS, and 3 μg/ml puromycin was added 24 h later. After approximately 12 days, single colonies of resistant cells were picked and cultured for a further 30 days in the presence of puromycin. Several clones were tested, with similar COX-2 expression levels seen. CHL and NCL cells stably expressing hCOX-2 or empty vector were termed CHL-C and NCL-C or CHL-V and NCL-V, respectively. In some experiments, cells were starved for 4 h in 2% FBS–1% bovine serum albumin (BSA) before addition of palmitate (P; 400 μM) for the indicated time periods. Then, 50 nM insulin was added for 15 min and cells were collected for analysis of insulin signaling.
miRNA microarray analysis.
miRNA from eight Wt and eight COX-2-Tg liver samples was extracted using Qiazol (Qiagen, Valencia, CA) and purified with an miRNeasy minikit (Qiagen). The quality and integrity of the microRNAs were assessed with an Agilent Bioanalyzer. For miRNA array analysis, we used a Mouse Genome V2.0 RT2 miRNA PCR array system (SA Biosciences, Frederick, MD), which included 440 unique sequences of miRNAs. For cDNA synthesis, purified miRNA was reverse transcribed (RT) using an RT2 miRNA first-strand kit (SA Biosciences). miRNA-specific cDNAs and RT2 SYBR green PCR master mix were diluted and loaded into each well of a PCR array consisting of six 96-well plates specific for a MyiQ PCR detection system (Bio-Rad). Each well contained a universal primer and a primer specific for 1 of 88 individual mouse miRNAs. Real-time PCR was performed using the following thermocycling parameters: 1 cycle of 95°C for 10 min followed by 40 cycles of 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s. Fold change in miRNA expression level was calculated with RT2 miRNA PCR array data analysis Web-based software (SA Biosciences) (27). The fold change in expression level of each miRNA was determined by comparing the expression levels of miRNAs in COX-2-Tg cells to those in Wt cells. Microarray data were validated by RT-PCR analysis of individual miRNAs.
Analysis of pathways and networks was performed using the “Database for Annotation, Visualization and Integrated Discovery” (DAVID) platform, allowing the identification of several candidate pathways for the miRNA target genes.
RNA extraction and quantitative real-time PCR analysis.
Total RNA from liver, cells, or human samples was extracted with TRIzol reagent (Invitrogen, Grand Island, NY). RNA (1 μg) was reverse transcribed using a Transcriptor First Strand cDNA synthesis kit (Roche Applied Science, Penzberg, Germany). cDNA was used as a template for real-time PCR with FastStart Universal SYBR green Master (Roche). Forward and reverse primers are described in Table 1.
TABLE 1.
The oligonucleotide sequences
| Gene | Orientation | Primer sequence (5′→3′) |
|---|---|---|
| Akt2 | Forward | ACGTGGTGAATACATCAAGACC |
| Reverse | GCTACAGAGAAATTGTTCAGGGG | |
| Insig1 | Forward | CTAGTGCTCTTCTCATTTGGCG |
| Reverse | AGGGATACAGTAAACCGACAACA | |
| Insig2 | Forward | GGAGTCACCTCGGCCTAAAAA |
| Reverse | CAAGTTCAACACTAATGCCAGGA | |
| Irs1 | Forward | CTCCTGCTAACATCCACCTTG |
| Reverse | AGCTCGCTAACTGAGATAGTCAT | |
| Irs2 | Forward | ACCGACTTGGTCAGCGAAG |
| Reverse | CACGAGCCCGTAGTTGTCAT | |
| Prkca | Forward | AGAGGTGCCATGAGTTCGTTA |
| Reverse | GGCTTCCGTATGTGTGGATTTT | |
| Rplp0 | Forward | ACTGGTCTAGGACCCGAGAAG |
| Reverse | TCCCACCTTGTCTCCAGTCT | |
| Tsc1 | Forward | CAGCCGGTATTGCACATCCT |
| Reverse | GGATAAACGGGTAGCAGCTTTG | |
| Pck1 | Forward | GGGCCGCTGGATGTCGGAAG |
| Reverse | GGTGCGGCCTTTCATGCACC | |
| Gck | Forward | TGAGCCGGATGCAGAAGGA |
| Reverse | GCAACATCTTTACACTGGCCT | |
| AKT2 | Forward | ACCACAGTCATCGAGAGGACC |
| Reverse | GGAGCCACACTTGTAGTCCA | |
| INSIG1 | Forward | GCCTACTGTACCCCTGTATCG |
| Reverse | TGGTTAATGCCAACAAAAACTGC | |
| INSIG2 | Forward | TAATGCGGTGTGTAGCAGTCT |
| Reverse | GTCCAATGGATAGTGCAGCCA | |
| IRS1 | Forward | CCCAGGACCCGCATTCAAA |
| Reverse | GGCGGTAGATACCAATCAGGT | |
| IRS2 | Forward | CAGTGCTGAGCGTCTTCTTTT |
| Reverse | ACCTACGCCAGCATTGACTT | |
| PRKCA | Forward | ATGTCACAGTACGAGATGCAAAA |
| Reverse | GCTTTCATTCTTGGGATCAGGAA | |
| RPLP0 | Forward | AGATGCAGCAGATCCGCAT |
| Reverse | GTTCTTGCCCATCAGCACC | |
| TSC1 | Forward | GTGGCGGAAGTCTATCTCGTC |
| Reverse | GCAAGGGTACATTCCATAAAGGC |
Real-time PCR was performed on a MyiQ detection system (Bio-Rad), and the thermocycling parameters were 95°C for 3 min and 40 cycles of 95°C for 30 s followed by 60°C for 30 s. Each sample was run in triplicate and was normalized to 36b4 mRNA. The replicates were then averaged, and fold induction was determined by a threshold cycle (ΔΔCT)-based fold change calculation.
For quantification of mature miRNAs, RNA (500 ng) was polyadenylated and reverse transcribed to cDNA using an NCode miRNA First-Strand cDNA synthesis kit (Invitrogen). cDNA was used as the template for real-time PCR FastStart Universal SYBR green Master (Roche) with the universal reverse primer provided in the kit and the following forward primers: for Let-7a, 5′-GGGTGAGGTAGTAGGTTGTATAGTT-3′; for miR-23b, 5′-ATCACATTGCCAGGGATTACC-3′; for miR-146b, 5′-GTGAGAACTGAATTCCATAGGCT-3′; and for miR-183, 5′-GGTATGGCACTGGTAGAATTCACT-3′. The following primers were used for quantification of pri- and pre-miRNAs: for pri-miR-23b, 5′-TGCTCCTCTATACAGTGCAG-3′ (forward) and 5′-AAATCAGCATGCCAGGAACC-3′ (reverse); for pre-miR-23b, 5′-TGCTTGGGTTCCTGGCAT-3′ (forward) and 5′-GGTTGCGTGGTAATCCCT-3′ (reverse); for pri-miR-146b, 5′-CAAGGCACACACTGAATAAATG-3′ (forward) and 5′-ACCAGAACTGAGTCCCTAG-3′ (reverse); for pre-miR-146b, 5′-CTGAGAACTGAATTCCATAGG-3′ (forward) and 5′-GCACCAGAAGTGAGTCC-3′ (reverse); for pri-miR-183, 5′-AGGAGCAGAGGAGGTCTTT-3′ (forward) and 5′-TATGGCACTGGTAGAATTCACT-3′ (reverse); and for pre-miR-183, 5′-CTGTGTATGGCACTGGTAG-3′ (forward) and 5′-CTGTTTATGGCCCTTCGGT-3′ (reverse). Real-time PCR was performed on a MyiQ detection system (Bio-Rad), and the thermocycling parameters were 95°C for 3 min and 40 cycles of 95°C for 15 s followed by 60°C for 30 s. Each sample was run in triplicate and was normalized to U6 snRNA levels (U6 primers 5′-CTTCGGCAGCACATATACT-3′ [forward] and 5′-AAAATATGGAACGCTTCACG-3′ [reverse]) or 36b4. Melting curve analysis was performed to confirm the specificity of the PCR products. The replicates were then averaged, and fold induction was determined by a ΔΔCT-based fold change calculation.
Construction and transfection of miRNA and small interfering RNA (siRNA) vectors and luciferase reporter assay.
Precursors for miR-23b, miR-146b, and miR-183 were generated in green fluorescent protein (GFP)-puromycin (pEGP)-miR cloning vector (Cell Biolabs, San Diego, CA). Briefly, microRNA stem-loop sequences were identified using the Sanger Center miRNA database (http://microrna.sanger.ac.uk/sequences) and were amplified by PCR from genomic DNA. PCR products were cloned into the BamHI-NheI restriction site of pEGP-miR vector, and the correct orientation and integrity of the construct were confirmed by sequencing.
siRNAs against hCOX-2 (siCOX-2) (5′-GGGCUGUCCCUUUACUUCATT-3′ [forward] and 5′-UGAAGUAAAGGGACAGCCCTT-3′ [reverse]) and mDDX5 (siDDX5) (5′-GAAGUCUACUUGCAUCUAUTT-3′ [forward] and 5′-AUAGAUGCAAGUAGACUUCCAA-3′ [reverse]) were purchased from Ambion (Austin, TX). For silencing experiments, CHL and NCL cells were seeded in a 6-well plate (3 × 105cells/well) at 70% confluence. After 24 h, cells were transfected with 4 μg of pEGP miR, 50 nM small interfering COX-2 (siCOX-2), or 30 nM siDDX5 using Lipofectamine 2000. After 16 h of incubation at 37°C, transfection medium was replaced with 2 ml of complete medium containing 10% FBS. After a further 48 h, cells were collected for Western blot and RT-PCR analyses.
For DDX5 overexpression, vector pcDNA3.1-DDX5 (GenScript USA Inc., Piscataway, NJ) was used. For C646 treatment, NCL cells were seeded in 6-well plates at 5 × 105 cells/well. C646 was added in the dose range of 2.5 to 20 μM in serum-free DMEM for 24 h. Cells were then harvested for Western blot analysis.
Using several computational tools for miRNA prediction (RNAhybrid, PITA, and TargetScan), miR-183 was predicted to associate with different MRE motifs in the 3′UTR region of insulin receptor substrate 1 (IRS1). Using RNAhybrid, we found one predicted MRE in IRS1 at position 190, where position 1 was defined as the commencement of the 3′UTR region. The 3′UTR sequences of IRS1 were retrieved using Ensembl Data base (http://www.ensembl.org). Human and mouse miRNA sequences were downloaded from the miRBase website (http://www.mirbase.org). A fragment of the 3′UTR of IRS1 mRNA (region 4750, from NM_010570) that included the MRE binding site for miR-183 and a mutant (mut) variant were cloned into the pGL3-Promoter vector (Promega) downstream of the firefly luciferase gene (SacI and NheI sites) to obtain reporter constructs pGL3-seed and pGL3-mut. The mouse sequences cloned were 5′-CTCAGGAGTTCATTGACTGAACTGCACGTTCTACATTGTGCCAAGCAACAAGAAAGCAC-3′ (seed) and 5′-CTCAGGAGTTCATTGACAGGACTGCACTCGCCGTATTTCGGTCAGCAACAAGAAAGCAC-3′ (mut). Cells (3 × 104 cells/well) were seeded in a 24-well plate and transfected for 18 h with pGL3-empty (750 ng), pGL3-seed (750 ng), pGL3-mut (750 ng), pEGP-miR-183 (800 ng), or pRL (50 ng), or different combinations thereof, using Lipofectamine 2000. Cells were harvested 48 h after transfection, and lysates were assayed with the dual-luciferase reporter assay (Promega, USA).
Analysis of insulin release.
MIN6 cells were transfected with 4 μg of pEGP miR or pPyCAGIP-hCOX-2 using Lipofectamine 2000. Cells were serum starved for 1 h in Krebs Ringer HEPES (KRH) buffer containing 0.1% (wt/vol) BSA and 2 mM glucose prior to the addition of KRH buffer containing 2 or 30 mM glucose for a further hour. After this time, insulin released into the medium was measured with using a mouse insulin enzyme-linked immunosorbent assay (ELISA) kit (Crystal Chem Inc., Downers Grove, IL).
Immunoprecipitation assay.
The binding of COX-2 to the Drosha complex was analyzed by immunoprecipitation and Western blotting. Immunoprecipitation was carried out in a buffer containing 10 mM Tris-HCl (pH 7.5), 1 mM MgCl2, 1 mM EGTA, 10% glycerol, 0.5% CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}, 1 mM β-mercaptoethanol, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and a protease and phosphatase inhibitor cocktail (Sigma-Aldrich). Total crude extracts (500 μg) from NCL-C cells were immunoprecipitated with Drosha or COX-2 antibody (1/50) and mixed overnight at 4°C. An equal volume of protein A/G-Sepharose was then added, and mixing was continued for 4 h at 4°C. Protein A/G-Sepharose was then pelleted at 12,000 rpm for 5 min at 4°C, and beads were resuspended in 20 μl of Laemmli sample buffer and heated at 100°C for 5 min. For RNase A digestion, the immunoprecipitated proteins were incubated at 37°C with 20 μg/ml RNase A for 30 min.
In vitro pri-miRNA processing assay.
In vitro pri-miRNA processing was performed as described previously (28). In brief, biotin-labeled pri-miRNAs were prepared by standard in vitro transcription with T7 RNA polymerase, using 150-bp pri-miRNAs cloned into pGEM-T Easy as the templates. The processing reaction was carried out in a 30-μl volume containing 10 μl of immunoprecipitation beads (Drosha complex), immunoprecipitated or recombinant COX-2 (Sigma), where indicated, 6.4 mM MgCl2, 1 mM ATP, 1 U/μl RNase inhibitor, and 3 μl of the in vitro-transcribed pri-miRNA. The reaction mixture was incubated at 37°C for 90 min. RNA was then extracted by the use of phenol. For the analysis of COX-2 dose-dependent inhibition of pri-miRNA processing, the in vitro pri-miRNA processing assay was performed with different concentrations (1, 2, and 4 μg) of recombinant COX-2. RNA was extracted and subjected to reverse transcription-quantitative PCR (RT-qPCR) for analysis of pri- and pre-miRNAs.
Western blot analysis.
Extracts from cells (2 × 106 to 3 × 106) or from liver tissue were obtained as described previously (12). For Western blot analysis, whole-cell extracts were boiled for 5 min in Laemmli sample buffer, and equal amounts (20 to 30 μg) of protein were separated on 10% to 15% SDS-polyacrylamide electrophoresis gels (SDS-PAGE). The relative amounts of each protein were determined with the following polyclonal or monoclonal antibodies: COX-2 (Cayman 160107 and Santa Cruz sc-1747); α-tubulin (Sigma T9026); antiacetyllysine, p-IR, IRS1, and IRS2 (Millipore 06-933, 07-841, 06-248, and 06-506, respectively); DGCR8 (Proteintech 10996-1-AP); DDX5, Drosha, p-AKT (Ser473), AKT, and IR (Cell Signaling Technology 9877, 3364, 4060, 9272, and 3020, respectively); DDX17 (Abcam ab70184); and Dicer (dicer 1, RNase type III; Santa Cruz sc-30226). After incubation with the corresponding anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibody, blots were developed by the use of the ECL protocol (GE Healthcare, Chalfont St Giles, United Kingdom). Protein band densities were normalized to α-tubulin. Densitometric analysis was carried out with Image Gauge 4.0 software (Fujifilm), and the results were expressed in arbitrary units.
Data analysis.
Data are expressed as means ± standard deviations (SD) (n ranged from three to five independent experiments). Statistical significance was evaluated with Student's two-tailed t test for unpaired observations, using the statistical software GraphPad Prism 5. A P value of <0.05 was considered significant.
RESULTS
COX-2 regulates the expression of a specific set of miRNAs in liver cells.
To identify hepatic miRNAs regulated by COX-2, we isolated total RNA from liver extracts of Wt and COX-2-Tg mice and performed an RT2 miRNA PCR array. Of 440 murine mature miRNAs analyzed, 30 were differentially expressed in COX-2-Tg liver relative to the Wt (Fig. 1A) (P values, <0.05; fold change cutoff value, >2). Of these miRNAs, 21 were downregulated and 9 were upregulated. Analysis of pathways and networks using the DAVID platform identified several candidate pathways for these miRNA target genes. Among them, the insulin signaling pathway was of interest because, in accordance with our previous results, COX-2 is implicated in protection against hepatic insulin resistance in mice on a high-fat diet (14). Only eight of these miRNAs (miR-146b, Let-7b, miR-183, miR-181a1*, miR-23b, miR-184, miR-204, and Let-7a) have target genes involved in the insulin signaling pathway, and their data were confirmed by real-time RT-PCR in liver extracts from Wt and COX-2-Tg mice. Additionally, we attempted to confirm the array data in different cell models in isolated hepatocytes from Wt and COX-2-Tg mice and in NCL and CHL cells stably transfected with a COX-2 expression vector (CHL-C and NCL-C cells), in comparison with the parental cells (CHL-V and NCL-V cells). From these analyses, we confirmed that one miRNA was upregulated (Let-7a) and three miRNAs were downregulated (miR-23b, miR-146b, and miR-183) in the presence of COX-2 (Fig. 1B to E). To test the specificity of COX-2 with respect to expression of these miRNAs, NCL-V cells were treated with PGE2 and NCL-C cells were transfected with siCOX-2 mRNA. As shown in Fig. 2, PGE2 reproduced the effect of COX-2 overexpression, and miR-23b, miR-183, and miR-146b were significantly downregulated. Conversely, siCOX-2 treatment led to an increase in the levels of these miRNAs in NCL-C cells. However, neither PGE2 treatment nor siCOX-2 treatment modified Let-7a expression as expected (Fig. 2), suggesting an indirect regulation of this miRNA by COX-2. Thus, Let-7a was excluded from further analysis.
FIG 1.

Identification of miRNAs regulated by COX-2 in liver. (A) Identification of 30 differentially expressed miRNAs in Wt and hCOX-2-Tg liver, as detected by qPCR array. Values represent fold change relative to liver of Wt mice. (B to E) Validation of the upregulated expression of Let-7a and the downregulated expression of miR-23b, miR-146b, and miR-183 in Wt and hCOX-2-Tg liver, primary hepatocytes, and NCL-V (V) and NCL-C (C) and CHL-V (V) and CHL-C (C) cells by real-time PCR. miRNA expression was normalized to U6 RNA expression. Data are reported as means ± SD of the results of three independent experiments. *, P < 0.05 (versus absence of COX-2).
FIG 2.
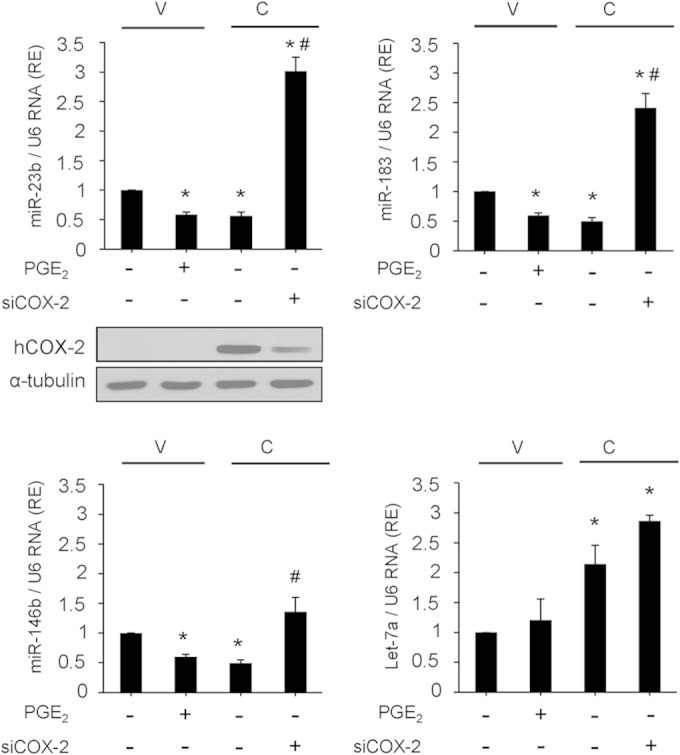
COX-2 downregulates miR-23b, miR-146b, and miR-183 in NCL cells. NCL-V and NCL-C cells were transfected with 50 nM scrambled siRNA or 50 nM siCOX-2 or were stimulated with 5 μM PGE2. COX-2 protein levels are shown by Western blotting. After 48 h, total RNA was extracted and miRNA expression was analyzed by real-time PCR and normalized to U6 RNA expression. The expression level was determined relative to that in NCL-V cells (arbitrarily assigned a value of 1). Data are reported as means ± SD of the results of three independent experiments.*, P < 0.05 (versus NCL-V cells); #, P < 0.05 (versus NCL-C cells).
COX-2 decreases miRNA expression through a DDX5- and PI3K/p300-dependent mechanism.
To gain insight into mechanisms linking COX-2 with regulation of these specific miRNAs, we used Western blotting to analyze proteins involved in the biosynthesis of miRNAs. Interestingly, compared with the levels seen with DGCR8, Drosha, DDX17, and Dicer proteins, the level of DDX5 was significantly increased in liver from COX-2-Tg mice (1.86-fold, P < 0.05) and also in NCL-C and CHL-C cells (Fig. 3A), suggesting that COX-2 regulates DDX5. To determine the possible signaling pathways related to COX-2 and involved in miRNA biosynthesis, NCL-C cells were treated with pharmacological inhibitors of p38 mitogen-activated protein kinase (MAPK) kinase (MEK) (BRIB796), MEK/extracellular signal-regulated kinase (MEK/ERK) (PD98059), PI3K (LY294002), and protein kinase C (PKC) (GO6983). As shown in Fig. 3B, only PI3K inhibition with LY294002 decreased the expression of DDX5. Moreover, treatment of NCL-C cells with LY294002 also increased the levels of miR-23b, miR-146b, and miR-183 (Fig. 3C). Previous work has shown that hCOX-2 overexpression produces an activation of PI3K/Akt in vivo (15). In our study, we measured the activity of PI3K by an in vitro activity assay (Millipore catalog no. 17-493), and PI3K activity increased significantly in the NCL-V cells treated with PGE2 and NCL-C cells. However, when NCL-C was treated with LY294002, the PI3K activity was reduced (data not shown). It has been shown previously that AKT phosphorylates p300, leading to an increase in its histone acetyltransferase (HAT) activity (29); also, DDX5 is acetylated by p300, stabilizing its expression (30). Using immunoprecipitation with an acetyllysine antibody, we confirmed that DDX5 is acetylated. Moreover, the acetylation is decreased in the presence of siCOX-2 (Fig. 3D). Furthermore, the expression of DDX5 could be decreased in NCL-C cells by treatment with C646, a selective inhibitor of p300 (Fig. 3E), indicating that DDX5 stability is regulated by p300 acetylation in this cells.
FIG 3.

COX-2 increases DDX5 protein levels largely through PI3K signaling. (A) Representative Western blot showing expression of COX-2 and proteins involved in miRNA processing (DGCR8, Drosha, DDX5, DDX17, and Dicer). Total cellular extracts were prepared from NCL and CHL cells and from Wt and COX-2-Tg mouse liver, and protein levels were analyzed by Western blotting. For densitometric analysis of DDX5 protein in liver extracts, the relative level of Wt expression was defined as 1. α-Tubulin served as a loading control. (B) NCL cells were serum starved for 6 h prior to treatment with different pharmacological inhibitors for 12 h (0.5 μM BRIB796, 50 μM PD98059, 20 μM LY294002, and 1 μM GO6983). Protein levels were analyzed by Western blotting. Ctrl, control; DMSO, dimethyl sulfoxide. (C) Analysis of miRNA expression by real-time PCR after treatment with 20 μM LY294002 (LY29) for 12 h. The expression level was determined relative to that in NCL-V cells (arbitrarily assigned a value of 1). Data are reported as means ± SD of the results of three independent experiments.*, P < 0.05 (versus NCL-V cells); #, P < 0.05 (versus NCL-C cells). (D) NCL-C cells were subjected to immunoprecipitation (IP) with an acetyllysine antibody (α-Acetyl Lys) after treatment with siCOX-2 (50 nM), followed by immunostaining with anti-DDX5 antibody. IB, immunoblot. The input panel shows that comparable amounts of total DDX5 were present in all immunoprecipitation experiments. p85 served as a loading control. (E) NCL cells were cultured without or with 2.5 μM and 5 μM C646, as indicated. After 24 h, cells were harvested and COX-2 and DDX5 expression was determined by Western blotting. α-Tubulin served as a loading control.
DDX5 inhibition restores the expression of the miRNAs downregulated by COX-2 in liver.
To establish a direct relationship between DDX5 and COX-2, DDX5 protein was analyzed after treatment of NCL-V cells with PGE2 and after COX-2 silencing in NCL-C cells. PGE2 treatment resulted in a significant increase of DDX5 in NCL-V cells (Fig. 4A). Conversely, NCL-C cells treated with siCOX-2 had significantly less DDX5 protein (Fig. 4A). No significant changes were observed in DGCR8 or Drosha proteins upon treatment. To determine whether DDX5 was responsible for the decreased expression of miR23b, miR-146b, and miR-183 after COX-2 overexpression, DDX5 was silenced with a specific siRNA in NCL-C cells. As shown in Fig. 4B, the miRNA levels increased in DDX5 knockdown NCL-C cells. Moreover, we transfected NCL-V cells with pcDNA3.1-DDX5 and the results showed that DDX5 overexpression reduced the miR-23b, miR-146b, and miR-183 levels significantly. All these data indicated a role for DDX5 in the COX-2-mediated downregulation of these miRNAs in liver. This led us to hypothesize that COX-2 modulates the enzymatic function of the Drosha complex through its physical association with DDX5. Indeed, endogenous DDX5 and Drosha coimmunoprecipitate with COX-2 in NCL-C cells (data not shown). Further, the association between Drosha and COX-2 was markedly decreased when DDX5 was silenced, indicating that COX-2 interacts with the Drosha complex through DDX5. Furthermore, the association of COX-2 with Drosha and DDX5 was not decreased by treatment with RNase A (single-stranded RNA nuclease), indicating that COX-2 interaction with the Drosha complex is RNA independent (Fig. 4C). Moreover, RNA immunoprecipitation (RIP) analysis showed no interaction of COX-2 with pri-miR-23b, pri-miR-146b, and pri-miR-183 (data not shown).
FIG 4.

DDX5 is implicated in COX-2-dependent regulation of miRNAs. (A) Analysis of protein levels after transfection with 50 nM scrambled siRNA (Scr) or 50 nM siCOX-2 or stimulation with 5 μM PGE2 for 48 h. Protein expression was normalized to that of α-tubulin. Results of densitometric analysis of DDX5 protein were quantified relative to the level in NCL-V cells (assigned a value of 1), and data are represented as relative expression (RE) levels. (B) Analysis of miRNA expression by real-time PCR after transfection with 4 μg pcDNA3.1-DDX5 (pDDX5) and 30 nM siDDX5. DDX5 protein levels are shown by Western blotting. Data are reported as means ± SD of the results of three independent experiments. *, P < 0.05 (versus NCL-V cells); #, P < 0.05 (versus NCL-C cells). (C) COX-2 and Drosha interaction is DDX5 dependent and RNA independent. NCL-C cells were transfected with Scr or siDDX5 and digested with RNase A and then subjected to immunoprecipitation (IP) with anti-Drosha, followed by immunostaining with anti-COX-2 or anti-DDX5 antibody. IB, immunoblot. (D) Expression levels of the primary (pri), precursor (pre), and mature (mat) forms of the indicated miRNAs were examined by real-time PCR after transfection with 50 nM scrambled siRNA or 50 nM siCOX-2 in NCL-V and NCL-C cells. Pri- and pre-miRNAs were normalized to 36b4 RNA, and mature miRNA was normalized to U6 RNA. (E) COX-2 dose-dependent inhibition of pri-miRNA processing. The in vitro pri-miRNA processing assay was performed with different concentrations of recombinant COX-2, and RNA was extracted and subjected to RT-qPCR for analysis of pri- and pre-miRNAs. The pre-miRNA/pri-miRNA ratio is represented. Data are reported as means ± SD of the results of three independent experiments.*, P < 0.05 (versus NCL-V cells); #, P < 0.05 (versus NCL-C cells).
When the effect of COX-2 expression on maturation of miRNAs was evaluated using real-time RT-PCR in NCL-V and NCL-C cells, we found that COX-2 decreased the levels of precursor and mature forms of miR-23b, miR-146b, and miR-183; however, expression of their primary transcripts was slightly increased (Fig. 4D). Furthermore, COX-2 knockdown abolished the decrease in the expression of these miRNAs at the precursor and mature levels (Fig. 4D). To confirm these findings, we performed an in vitro pri-miRNA processing assay by incubating pri-miR183 substrate with the immunoprecipitated Drosha complex from NCL-V cells, in the presence or absence of COX-2. The processing activity for the pri-miR183 primary transcript was significantly reduced in the presence of COX-2 (immunoprecipitated and recombinant), and the effect was dose dependent and more pronounced with recombinant COX-2 (Fig. 4E). These results indicated that COX-2 associates with the Drosha Microprocessor complex through interaction with DDX5, ultimately promoting the inhibition of miRNA processing.
Relationship between COX-2-regulated miRNAs and the insulin signaling pathway.
Analysis of pathways and networks for miR-23b, miR-146b, and miR-183 using the DAVID platform identified several target genes related to the insulin signaling pathway. Therefore, some of these target genes were measured by real-time RT-PCR in different hepatic cell models. A significant increase in the expression of several genes, including IRS1, was observed in NCL-C and CHL-C cells and also in COX-2-Tg liver (Fig. 5A to C). Indeed, several programs (RNAhybrid, PITA, and TargetScan) predicted an association of miR183 with the 3′UTR region of IRS1 at different MRE motifs, and we found one predicted MRE for miR183 at position 190 (where position 1 was defined as the beginning of the 3′UTR region). To confirm that miR183 could bind to this predicted region, we performed a luciferase reporter gene assay in NCL cells. We cloned the 3′UTR region of IRS1 containing the miR-183 putative binding site (seed region) and constructed a mutant variant. These regions were cloned downstream of the luciferase gene in the pGL3 vector (pGL3-seed and pGL3-mut, respectively). Compared with control cells (pGL3-seed only), luciferase activity was significantly decreased after cotransfection of pGL3-seed and miR-183. Additionally, no differences on luciferase activity were found when cells were cotransfected with pGL3-mut and miR-183 relative to cells transfected with pGL3-mut only (Fig. 5D). These results suggested that miR-183 binds specifically to the 3′UTR region of IRS1, inhibiting its expression.
FIG 5.

Analysis of miR-23b, miR-183, and miR-146b predicted target genes. (A to C) Expression of the identified target genes involved in insulin signaling (Akt2 [v-Akt murine thymoma viral oncogene homolog 2], Irs1 [insulin receptor substrate 1], Irs2 [insulin receptor substrate 2], Tsc1 [tuberous sclerosis 1], Prkca [protein kinase C alpha], Insig1 [insulin-induced gene 1], and Insig2 [insulin-induced gene 2]) in NCL-C and CHL-C cells and hCOX-2-Tg liver extracts determined by real-time PCR. Expression is shown relative to that seen with NCL-V, CHL-V, or Wt mice. *, P < 0.05 (versus absence of COX-2). (D) A luciferase assay was carried out on NCL-C cells for IRS 3′UTR, using pGL3-seed and pGL3-mut reporter vectors. Firefly luciferase activity was evaluated 48 h after cotransfection with pGL3-empty/seed/mut (750 ng), miR-183 (50 nM), and miR-NC (50 nM), as indicated. Data were normalized to renilla luciferase activity (all samples were cotransfected with 50 ng pRL vector and refer to the positive control, pGL3 empty vector). Data are reported as means ± SD of the results of three independent experiments. *, P < 0.05 (versus the pGL3 empty vector).
COX-2-expressing liver cells are protected against IR induced by palmitate.
Given the results presented above, we next examined the insulin signaling pathway in COX-2-Tg liver and in NCL-C and CHL-C cells under basal conditions and after treatment with 400 μM palmitate (P) to induce insulin resistance (IR). As shown in Fig. 6A, p-IR, IRS1, IRS2, and p-AKT protein levels were increased in COX-2-Tg liver, and also in NCL-C and CHL-C cells, indicating that the insulin signaling pathway is activated in the presence of COX-2. To confirm that DDX5 was implicated in the miRNA downregulation by COX-2, and thus was responsible for the increase in insulin signaling, we inhibited DDX5 activity with siRNA. As shown in Fig. 6B, DDX5 silencing abrogated the COX-2-mediated increase in insulin signaling components in NCL-C cells. Similar results were obtained after silencing of COX-2. When palmitate was used to induce insulin resistance (IR), NCL cells expressing COX-2 exhibited lower insulin resistance after challenge with insulin, as illustrated by higher expression of IRS1 and p-AKT in the presence of palmitate than in NCL-V cells, indicating a COX-2-mediated protective effect (Fig. 6C).
FIG 6.

COX-2 regulates insulin signaling through DDX5 and protects against insulin resistance. (A) Western blot analysis of COX-2 and proteins involved in insulin signaling in NCL and CHL cells and Wt and hCOX-2-Tg liver. (B) Analysis of protein expression in NCL cells after transfection with 30 nM Scr, 30 nM siDDX5, or 50 nM siCOX-2. The expression of target proteins was normalized to that of α-tubulin. (C) NCL cells were serum starved for 6 h prior to treatment with 400 μM palmitate in medium supplemented with 1% BSA for 24 h and then stimulated with 50 nM insulin for 10 min. Protein levels were measured by Western blotting. Results of densitometric analysis of IRS1 and p-AKT/AKT ratio protein levels are expressed relative to those of NCL-V cells (assigned a value of 1) and are represented as relative expression (RE) levels. Data are reported as means ± SD of the results of three independent experiments. *, P < 0.05 (versus NCL-V); #, P < 0.05 (versus absence of palmitate).
To confirm that the activation of insulin signaling was mediated by COX-2-dependent downregulation of miRNAs, NCL-C cells were transiently transfected with a vector expressing miR-23b, miR-146b, and miR-183 separately or in combination. As anticipated, expression of individual miRNAs reduced the effect of COX-2 in the activation of insulin signaling, and this abrogation was more pronounced when all three miRNAs were transfected in combination (Fig. 7A). Moreover, ectopic miR-183 expression could reverse the protective effect of COX-2 against palmitate-induced IR in liver cells, as measured by IRS1 expression and AKT phosphorylation (Fig. 7B). Additionally, we analyzed the expression of phosphoenolpyruvate carboxykinase (encoded by Pck1) and glucokinase (encoded by Gck), bona fide targets of insulin signaling and glucose homeostasis, respectively (31, 32). A significant reduction in Pck1 mRNA expression was observed in NCL-C cells relative to NCL-V cells; however, when NCL-C cells were transiently transfected with a vector encoding miR-23b, miR-146b, or miR-183, the decrease in Pck1 mRNA levels was reversed. Again, this effect was more pronounced when all three miRNAs were overexpressed simultaneously. Conversely, COX-2 increased the expression of Gck, and this was reversed in the presence of miR-23b, miR-146b, and miR-183 (Fig. 7C). Together, these results indicate that the downregulation of miRNAs by COX-2 enhances insulin signaling, partly through the inhibition of gluconeogenesis and the activation of glycolysis. In addition to testing the insulin signaling pathway, we performed a glucose-stimulated insulin secretion (GSIS) functional assay. Our results showed that GSIS in MIN6 cells was significantly reduced after transfection with a vector encoding miR-183 (Fig. 7D). Further, COX-2-transfected MIN6 cells exhibited increased insulin secretion, and this was abrogated by cotransfection with miR-183 (Fig. 7D).
FIG 7.

Ectopic miR-183 expression counteracts the protective effect of COX-2 against insulin resistance. (A) Western blot analysis of proteins after transfection with 4 μg of pEGP-miRNA expression vectors. Results of densitometric analysis of COX-2, DDX5, p-IR/IR ratio, IRS1, IRS2, and the p-AKT/AKT ratio are reported relative to NCL-V cells (assigned a value of 1) and is represented as relative expression (RE) levels. (B) Western blot analysis of proteins after transfection with pEGP-miR183 expression vector and treatment with 400 μM palmitate and/or 50 nM insulin. Results of densitometric analysis of IRS1 and p-AKT/AKT ratio are shown. (C) Real-time PCR measurement of Pck1 and Gck mRNA in NCL-C cells after transfection with 4 μg of pEGP-miRNA. Data are reported as means ± SD of the results of three independent experiments. *, P < 0.05 (versus NCL-V cells); #, P < 0.05 (versus NCL-C cells). (D) MIN6 β cells were transfected with pPyCAGIP-COX-2 and/or pEGP-miR-183 for 48 h prior to incubation with glucose (2 or 30 mM) for 1 h. Glucose-stimulated insulin secretion (GSIS) was then assessed as described in Materials and Methods. Data are reported as means ± SD of the results of three independent experiments. *, P < 0.05 (versus 2 mM glucose); #, P < 0.05 (versus 30 mM glucose).
Inverse correlation between COX-2 expression and miRNA expression in nonalcoholic steatosis.
Since COX-2-mediated inflammation is implicated in fatty liver disease, we examined whether there was a relationship between COX-2 expression and miRNA expression in nonalcoholic steatosis (NAS). Results from RT-PCR analysis of human biopsy samples showed that, compared with normal liver (NL) levels, COX-2 expression was significantly increased in NAS samples, whereas expression of miR-23b, miR-146b, and miR-183 was significantly decreased (Fig. 8A). Furthermore, expression of DDX5 and IRS1 was also found to be increased in NAS (Fig. 8B).
FIG 8.
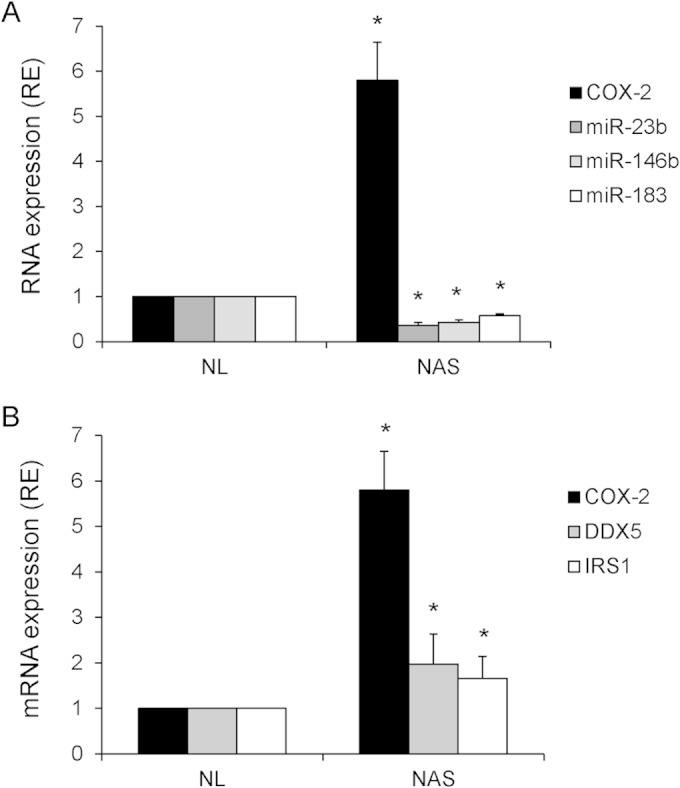
COX-2 correlates inversely to miR-23b, miR-146b, and miR-183 and directly to DDX5 and IRS1 in NAS human biopsy specimens. The expression of COX-2, DDX5, and IRS1 mRNA and also of miR-23b, miR-146b, and miR-183 was analyzed using real-time PCR in NL and NAS samples. *, P < 0.05 (versus NL samples). Data were normalized to 36b4 mRNA and U6 RNA levels, respectively.
DISCUSSION
The current report proposes a novel miRNA-dependent mechanism through which COX-2 modulates insulin signaling in liver. COX-2 represses miR-23b, miR-146b, and miR-183 expression in hepatic cell lines by associating with the Drosha complex through DDX5 acetylation and modulates the efficiency of Drosha-mediated pri-miRNA cleavage. Specifically, we and others have shown that IRS1 is a direct target of miR-183 (33). Thus, the regulation of miR-183 expression allows COX-2 to contribute to protection against IR by increasing IRS1 protein levels.
miRNA processing can be regulated at multiple steps during biogenesis and leads to either elevated or decreased miRNA levels. The Microprocessor complex, consisting of Drosha, DGCR8, DDX5, and DDX17, drives the maturation of pri-miRNA to pre-miRNA (34). The DDX5/DDX17 complex has been demonstrated to alter the miRNA processing efficiency differently for specific miRNAs, decreasing or increasing expression (28, 35). In addition to their playing a role in miRNA processing, it has been recently reported that DDX5 and DDX17 control miRNA production at the transcriptional level (36). It has been previously reported that in the absence of DDX5, the expression of only 35% of miRNAs (and pre-miRNAs) was reduced without concomitant changes in the levels of the corresponding pri-miRNAs, while the expression of other miRNAs was increased or unaltered (17). Alternatively, as DDX5 and DDX17 are known to interact with a variety of proteins, they may serve as a scaffold for the recruitment of multiple factors to the Drosha complex (36, 37). Considering these data, we propose that the mechanism by which COX-2 selectively inhibits miRNA processing is by stabilizing DDX5 through PI3K/p300, thus modulating its interaction with the Drosha complex and altering its correct positioning on the miRNA hairpin structure.
Previous studies have shown that PI3K enhances the metabolic stability of endogenous p300 protein (38). Indeed, AKT, activated in a PI3K-dependent manner, translocates to the nucleus, where it binds to and phosphorylates p300 at S1834, leading to an increase in its histone acetyltransferase (HAT) activity (29). Moreover, DDX5 is a substrate for p300 in vitro and in vivo, and its acetylation results in stabilization (30). In the present study, we demonstrated that the inhibition of PI3K (LY294002) and p300 (C646) leads to a decrease of DDX5 protein levels in NCL-C cells, along with the acetylated status of DDX5, indicating that the increase in the DDX5 levels mediated by COX-2 is PI3K/p300 dependent.
While the positive regulatory machinery of miRNA processing is relatively well understood (28, 39), negative regulation of the pathway is largely uncharacterized. Our results demonstrate a negative regulation of miRNA processing exerted by COX-2 through DDX5. The best-studied negative regulator of miRNA biogenesis is the Lin-28 RNA binding protein, which is able to repress the processing of Let-7 family members by Dicer (40). Also, the predominant consequence of activation of the Myc oncogenic transcription factor is a widespread repression of miRNA expression as a result of Myc binding to miRNA promoters (41). Although the inhibition of miRNA processing by COX-2 is due to its interaction with the Drosha complex, we demonstrated that treatment of NCL-V cells with PGE2 mimics the effect of COX-2, downregulating miR-23b, miR-183, and miR-146b. This could be explained by the fact that we and others have shown that PGE2 induces COX-2 transcription through the PI3K/AKT signaling pathway (42, 43) and also stabilizes DDX5 expression, thus favoring the binding of COX-2 with the Drosha complex.
We analyzed the enriched signaling pathways among the predicted gene targets of the miRNAs regulated by COX-2 and focused on the insulin signaling pathway based on our previous results that indicated that COX-2 reduces liver damage induced by hyperglycemia (15) and protects against adiposity, inflammation, and hepatic insulin resistance in mice challenged with a high-fat diet (14). These miRNA target genes were validated by real-time RT-PCR and presented increased expression in COX-2-Tg liver and in both CHL and NCL cells expressing COX-2.
Recent studies have suggested that differential miRNA expression levels may play a role in the regulation of metabolic pathways, including those involved in insulin signaling and lipid and glucose metabolism, which are all events generally associated with pathogenesis and development of nonalcoholic fatty liver disease (NAFLD) (44). For example, Dávalos et al. reported that miR-33a/b contributes to the regulation of fatty acid and glucose metabolism and insulin signaling, by targeting key enzymes involved in fatty acid oxidation and IRS2 (45). Moreover, miR-155 has been shown to have a protective role in the development of nonalcoholic hepatosteatosis in mice, by regulating lipid metabolism through liver X receptor alpha (LXRα) (46).
Regarding COX-2 participation in metabolism, some studies have indicated that prostaglandins might favor fat accumulation in hepatocytes and hence the development of hepatic steatosis. Other studies provide evidence that PGE2 might suppress de novo lipogenesis. The work of Hsieh et al. (47–49) showed that rats fed with a fructose or high-fat diet and treated with COX-2 inhibitors improve muscle and fat insulin resistance. However, contradictory results were obtained by Coll et al., who showed that COX-2 inhibition exacerbated palmitate-induced inflammation and IR in skeletal muscle (50).
Previous results obtained using hepatocytes from our COX-2-Tg model indicated that, although a diabetic state decreased PI3K activity, the activity still remained significantly higher in COX-2-Tg diabetic mice than in Wt mice, suggesting an activation of the PI3K/AKT pathway by COX-2 (15). Similar results were obtained in the present study, where p-AKT levels per se were increased in NCL-C and CHL-C cells and in COX-2-Tg liver relative to Wt levels and were further increased by insulin. Hepatic insulin resistance may stem from compromised signaling through the insulin receptor substrate (IRS1and IRS2) proteins, a family of docking molecules that connect insulin receptor activation to essential downstream kinase cascades, such as the PI3K or mitogen-activated protein kinase (MAPK) pathways (51). In this context, there is probably positive feedback leading to increased PI3K activation. IRS proteins are a critical link in hepatic insulin signaling, and it has been previously shown that the decrease in expression of IRS proteins in liver may represent a key molecular lesion of hepatic insulin resistance (52). In addition, and in accordance with the results of this report, previous work has shown that a reduction in IRS1 expression resulted in a significant increase in the mRNA abundance of Pck1, encoding the essential gluconeogenic enzyme, and a decrease in Gck expression (51). All these data agree with our results, since COX-2 upregulates IRS1 by reducing miR-183 expression, thus protecting against insulin resistance in liver cells. Moreover, miR-183 expression leads to metabolic dysfunction, illustrated by a reduction in GSIS, suggesting a functional consequence of COX-2-mediated miRNA regulation. Our analysis revealed that, while expression of miR-183 impairs GSIS in MIN6 β cells, COX-2 has the opposite effect and is able to reverse the defect in insulin signaling.
Our results shed light on the mechanism through which COX-2 regulates miRNA processing by modulating the Drosha complex activity through the interaction with DDX5, thus downregulating miR-183 (Fig. 9). Importantly, we have demonstrated this inverse relationship between COX-2 and expression of miRNAs not only in transgenic animal and in vitro models but also in human hepatic biopsy specimens of NAS patients. Considering all these data, we propose that the decrease of miR-183 promotes protection against IR by increasing IRS1 levels in liver.
FIG 9.
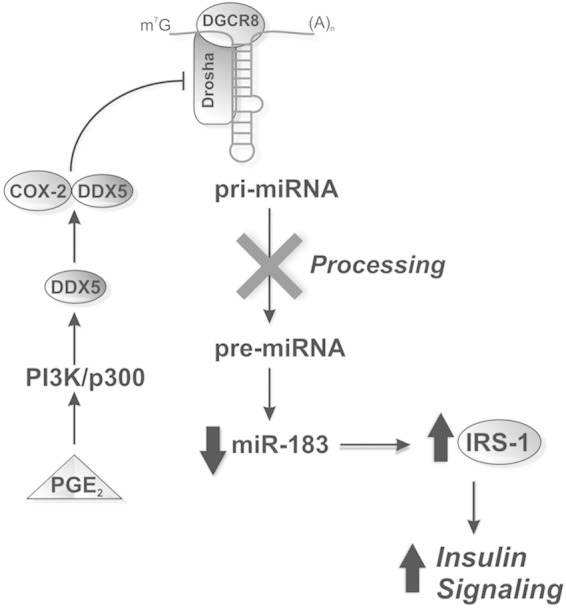
COX-2 increases insulin signaling by downregulation of miR-183. PGE2 stabilizes DDX5 expression through the PI3K/p300 signaling pathway. COX-2 associates with the Drosha complex through DDX5 and prevents the conversion of pri-miRNAs into pre-miRNAs. The association leads to a decrease of mature miR-183 levels and therefore to an increase in the level of its gene target, IRS1, a key protein in the insulin signaling pathway. DDX5, DEAD-box helicase p68; Drosha, RNase type III; DGCR8, DiGeorge syndrome critical region gene 8; m7G, 7-methylguanosine-cap.
miRNAs represent attractive therapeutic targets, and various chemically engineered oligonucleotides have been developed to safely inhibit miRNA function through sequestering mature miRNAs while preventing RNA-induced silencing complex (RISC) processing (53). Indeed, miravirsen, the first drug targeting a specific miRNA, has shown encouraging properties in clinical trials in patients infected with HCV (54). Our results suggest that antagonism of endogenous miR-183 might have utility as a therapeutic strategy for treating IR and also for treating metabolic syndrome and NAFLD.
ACKNOWLEDGMENTS
This work was supported by the Ministerio de Economía y Competitividad, Spain (SAF2010-16037 and SAF2013-43713-R to P.M.-S., BFU2011-24760 to L.B., and SAF2012-397329 and CIBERehd to M.C.), Comunidad de Madrid, Spain (S2010/BMD-2378 to L.B. and P.M.-S.), and Instituto de Salud Carlos III, Spain (RD12/0042/0019 and CIBERehd to L.B. and P.M.-S. and PI13/01299 to C.G.-M). N.A. is supported by Red de Investigación Cardiovascular (RD12/0042/0019). D.E.F. is supported by Financing Program for short stays abroad CONICET-Argentina. L.C.-S. is supported by a postdoctoral fellowship from CONACYT, Mexico.
We declare no conflict of interest.
REFERENCES
- 1.Ledwith BJ, Pauley CJ, Wagner LK, Rokos CL, Alberts DW, Manam S. 1997. Induction of cyclooxygenase-2 expression by peroxisome proliferators and non-tetradecanoylphorbol 12,13-myristate-type tumor promoters in immortalized mouse liver cells. J Biol Chem 272:3707–3714. doi: 10.1074/jbc.272.6.3707. [DOI] [PubMed] [Google Scholar]
- 2.Martín-Sanz P, Callejas NA, Casado M, Díaz-Guerra MJ, Boscá L. 1998. Expression of cyclooxygenase-2 in foetal rat hepatocytes stimulated with lipopolysaccharide and pro-inflammatory cytokines. Br J Pharmacol 125:1313–1319. doi: 10.1038/sj.bjp.0702196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simmons DL, Botting RM, Hla T. 2004. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev 56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 4.Casado M, Callejas NA, Rodrigo J, Zhao X, Dey SK, Bosca L, Martin-Sanz P. 2001. Contribution of cyclooxygenase 2 to liver regeneration after partial hepatectomy. FASEB J 15:2016–2018. doi: 10.1096/fj.01-0158fje. [DOI] [PubMed] [Google Scholar]
- 5.Rudnick DA, Perlmutter DH, Muglia LJ. 2001. Prostaglandins are required for CREB activation and cellular proliferation during liver regeneration. Proc Natl Acad Sci U S A 98:8885–8890. doi: 10.1073/pnas.151217998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto H, Kondo M, Nakamori S, Nagano H, Wakasa K, Sugita Y, Chang-De J, Kobayashi S, Damdinsuren B, Dono K, Umeshita K, Sekimoto M, Sakon M, Matsuura N, Monden M. 2003. JTE-522, a cyclooxygenase-2 inhibitor, is an effective chemopreventive agent against rat experimental liver fibrosis1. Gastroenterology 125:556–571. doi: 10.1016/S0016-5085(03)00904-1. [DOI] [PubMed] [Google Scholar]
- 7.Kern MA, Haugg AM, Koch AF, Schilling T, Breuhahn K, Walczak H, Fleischer B, Trautwein C, Michalski C, Schulze-Bergkamen H, Friess H, Stremmel W, Krammer PH, Schirmacher P, Muller M. 2006. Cyclooxygenase-2 inhibition induces apoptosis signaling via death receptors and mitochondria in hepatocellular carcinoma. Cancer Res 66:7059–7066. doi: 10.1158/0008-5472.CAN-06-0325. [DOI] [PubMed] [Google Scholar]
- 8.Mayoral R, Fernandez-Martinez A, Bosca L, Martin-Sanz P. 2005. Prostaglandin E2 promotes migration and adhesion in hepatocellular carcinoma cells. Carcinogenesis 26:753–761. doi: 10.1093/carcin/bgi022. [DOI] [PubMed] [Google Scholar]
- 9.Cusimano A, Fodera D, Lampiasi N, Azzolina A, Notarbartolo M, Giannitrapani L, D'Alessandro N, Montalto G, Cervello M. 2009. Prostaglandin E2 receptors and COX enzymes in human hepatocellular carcinoma: role in the regulation of cell growth. Ann N Y Acad Sci 1155:300–308. doi: 10.1111/j.1749-6632.2009.03701.x. [DOI] [PubMed] [Google Scholar]
- 10.Cheng AS, Yu J, Lai PB, Chan HL, Sung JJ. 2008. COX-2 mediates hepatitis B virus X protein abrogation of p53-induced apoptosis. Biochem Biophys Res Commun 374:175–180. doi: 10.1016/j.bbrc.2008.06.098. [DOI] [PubMed] [Google Scholar]
- 11.Núñez O, Fernández-Martínez A, Majano PL, Apolinario A, Gómez-Gonzalo M, Benedicto I, López-Cabrera M, Boscá L, Clemente G, García-Monzón C, Martín-Sanz P. 2004. Increased intrahepatic cyclooxygenase 2, matrix metalloproteinase 2, and matrix metalloproteinase 9 expression is associated with progressive liver disease in chronic hepatitis C virus infection: role of viral core and NS5A proteins. Gut 53:1665–1672. doi: 10.1136/gut.2003.038364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casado M, Molla B, Roy R, Fernandez-Martinez A, Cucarella C, Mayoral R, Bosca L, Martin-Sanz P. 2007. Protection against Fas-induced liver apoptosis in transgenic mice expressing cyclooxygenase 2 in hepatocytes. Hepatology 45:631–638. doi: 10.1002/hep.21556. [DOI] [PubMed] [Google Scholar]
- 13.Mayoral R, Molla B, Flores JM, Bosca L, Casado M, Martin-Sanz P. 2008. Constitutive expression of cyclo-oxygenase 2 transgene in hepatocytes protects against liver injury. Biochem J 416:337–346. doi: 10.1042/BJ20081224. [DOI] [PubMed] [Google Scholar]
- 14.Frances DE, Motino O, Agra N, Gonzalez-Rodriguez A, Fernandez-Alvarez A, Cucarella C, Mayoral R, Castro-Sanchez L, Garcia-Casarrubios E, Bosca L, Carnovale CE, Casado M, Valverde AM, Martin-Sanz P. 24 November 2014, posting date. Hepatic cyclooxygenase-2 expression protects against diet-induced steatosis, obesity and insulin resistance. Diabetes doi: 10.2337/db14-0979. [DOI] [PubMed] [Google Scholar]
- 15.Frances DE, Ingaramo PI, Mayoral R, Traves P, Casado M, Valverde AM, Martin-Sanz P, Carnovale CE. 2013. Cyclooxygenase-2 over-expression inhibits liver apoptosis induced by hyperglycemia. J Cell Biochem 114:669–680. doi: 10.1002/jcb.24409. [DOI] [PubMed] [Google Scholar]
- 16.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. 2009. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 17.Fukuda T, Yamagata K, Fujiyama S, Matsumoto T, Koshida I, Yoshimura K, Mihara M, Naitou M, Endoh H, Nakamura T, Akimoto C, Yamamoto Y, Katagiri T, Foulds C, Takezawa S, Kitagawa H, Takeyama K, O'Malley BW, Kato S. 2007. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol 9:604–611. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- 18.Ferreira DM, Simao AL, Rodrigues CM, Castro RE. 2014. Revisiting the metabolic syndrome and paving the way for microRNAs in non-alcoholic fatty liver disease. FEBS J 281:2503–2524. doi: 10.1111/febs.12806. [DOI] [PubMed] [Google Scholar]
- 19.Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, Janssen J, Koppe C, Knolle P, Castoldi M, Tacke F, Trautwein C, Luedde T. 2011. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53:209–218. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- 20.Harper KA, Tyson-Capper AJ. 2008. Complexity of COX-2 gene regulation. Biochem Soc Trans 36:543–545. doi: 10.1042/BST0360543. [DOI] [PubMed] [Google Scholar]
- 21.Chakrabarty A, Tranguch S, Daikoku T, Jensen K, Furneaux H, Dey SK. 2007. MicroRNA regulation of cyclooxygenase-2 during embryo implantation. Proc Natl Acad Sci U S A 104:15144–15149. doi: 10.1073/pnas.0705917104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daikoku T, Hirota Y, Tranguch S, Joshi AR, DeMayo FJ, Lydon JP, Ellenson LH, Dey SK. 2008. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res 68:5619–5627. doi: 10.1158/0008-5472.CAN-08-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strillacci A, Griffoni C, Sansone P, Paterini P, Piazzi G, Lazzarini G, Spisni E, Pantaleo MA, Biasco G, Tomasi V. 2009. MiR-101 downregulation is involved in cyclooxygenase-2 overexpression in human colon cancer cells. Exp Cell Res 315:1439–1447. doi: 10.1016/j.yexcr.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 24.Young LE, Moore AE, Sokol L, Meisner-Kober N, Dixon DA. 2012. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol Cancer Res 10:167–180. doi: 10.1158/1541-7786.MCR-11-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agra Andrieu N, Motino O, Mayoral R, Llorente Izquierdo C, Fernandez-Alvarez A, Bosca L, Casado M, Martin-Sanz P. 2012. Cyclooxygenase-2 is a target of microRNA-16 in human hepatoma cells. PLoS One 7:e50935. doi: 10.1371/journal.pone.0050935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valverde AM, Burks DJ, Fabregat I, Fisher TL, Carretero J, White MF, Benito M. 2003. Molecular mechanisms of insulin resistance in IRS-2-deficient hepatocytes. Diabetes 52:2239–2248. doi: 10.2337/diabetes.52.9.2239. [DOI] [PubMed] [Google Scholar]
- 27.Pepini T, Gorbunova EE, Gavrilovskaya IN, Mackow JE, Mackow ER. 2010. Andes virus regulation of cellular microRNAs contributes to hantavirus-induced endothelial cell permeability. J Virol 84:11929–11936. doi: 10.1128/JVI.01658-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. 2009. Modulation of microRNA processing by p53. Nature 460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 29.Huang WC, Chen CC. 2005. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol 25:6592–6602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mooney SM, Goel A, D'Assoro AB, Salisbury JL, Janknecht R. 2010. Pleiotropic effects of p300-mediated acetylation on p68 and p72 RNA helicase. J Biol Chem 285:30443–30452. doi: 10.1074/jbc.M110.143792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang J, Liu C, Qiao A, Cui Y, Zhang H, Cui A, Zhang S, Yang Y, Xiao X, Chen Y, Fang F, Chang Y. 2013. MicroRNA-29a-c decrease fasting blood glucose levels by negatively regulating hepatic gluconeogenesis. J Hepatol 58:535–542. doi: 10.1016/j.jhep.2012.10.024. [DOI] [PubMed] [Google Scholar]
- 32.Iynedjian PB. 2009. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 66:27–42. doi: 10.1007/s00018-008-8322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J, Wong C. 2008. A computational screen for mouse signaling pathways targeted by microRNA clusters. RNA 14:1276–1283. doi: 10.1261/rna.997708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. 2004. The Microprocessor complex mediates the genesis of microRNAs. Nature 432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 35.Sakamoto S, Aoki K, Higuchi T, Todaka H, Morisawa K, Tamaki N, Hatano E, Fukushima A, Taniguchi T, Agata Y. 2009. The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol Cell Biol 29:3754–3769. doi: 10.1128/MCB.01836-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dardenne E, Polay Espinoza M, Fattet L, Germann S, Lambert MP, Neil H, Zonta E, Mortada H, Gratadou L, Deygas M, Chakrama FZ, Samaan S, Desmet FO, Tranchevent LC, Dutertre M, Rimokh R, Bourgeois CF, Auboeuf D. 2014. RNA helicases DDX5 and DDX17 dynamically orchestrate transcription, miRNA, and splicing programs in cell differentiation. Cell Rep 7:1900–1913. doi: 10.1016/j.celrep.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 37.Trabucchi M, Briata P, Filipowicz W, Rosenfeld MG, Ramos A, Gherzi R. 2009. How to control miRNA maturation? RNA Biol 6:536–540. doi: 10.4161/rna.6.5.10080. [DOI] [PubMed] [Google Scholar]
- 38.Chen J, Halappanavar SS, St-Germain JR, Tsang BK, Li Q. 2004. Role of Akt/protein kinase B in the activity of transcriptional coactivator p300. Cell Mol Life Sci 61:1675–1683. doi: 10.1007/s00018-004-4103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawai S, Amano A. 2012. BRCA1 regulates microRNA biogenesis via the DROSHA microprocessor complex. J Cell Biol 197:201–208. doi: 10.1083/jcb.201110008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, Wulczyn FG. 2008. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat Cell Biol 10:987–993. doi: 10.1038/ncb1759. [DOI] [PubMed] [Google Scholar]
- 41.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. 2008. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet 40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dey I, Lejeune M, Chadee K. 2006. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br J Pharmacol 149:611–623. doi: 10.1038/sj.bjp.0706923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Obermajer N, Kalinski P. 2012. Key role of the positive feedback between PGE(2) and COX2 in the biology of myeloid-derived suppressor cells. Oncoimmunology 1:762–764. doi: 10.4161/onci.19681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ceccarelli S, Panera N, Gnani D, Nobili V. 2013. Dual role of microRNAs in NAFLD. Int J Mol Sci 14:8437–8455. doi: 10.3390/ijms14048437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dávalos A, Goedeke L, Smibert P, Ramírez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, Esplugues E, Fisher EA, Penalva LO, Moore KJ, Suárez Y, Lai EC, Fernández-Hernando C. 2011. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci U S A 108:9232–9237. doi: 10.1073/pnas.1102281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller AM, Gilchrist DS, Nijjar J, Araldi E, Ramirez CM, Lavery CA, Fernandez-Hernando C, McInnes IB, Kurowska-Stolarska M. 2013. MiR-155 has a protective role in the development of non-alcoholic hepatosteatosis in mice. PLoS One 8:e72324. doi: 10.1371/journal.pone.0072324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsieh PS, Jin JS, Chiang CF, Chan PC, Chen CH, Shih KC. 2009. COX-2-mediated inflammation in fat is crucial for obesity-linked insulin resistance and fatty liver. Obesity (Silver Spring) 17:1150–1157. doi: 10.1038/oby.2008.674. [DOI] [PubMed] [Google Scholar]
- 48.Hsieh PS, Lu KC, Chiang CF, Chen CH. 2010. Suppressive effect of COX2 inhibitor on the progression of adipose inflammation in high-fat-induced obese rats. Eur J Clin Invest 40:164–171. doi: 10.1111/j.1365-2362.2009.02239.x. [DOI] [PubMed] [Google Scholar]
- 49.Tian YF, Hsia TL, Hsieh CH, Huang DW, Chen CH, Hsieh PS. 2011. The importance of cyclooxygenase 2-mediated oxidative stress in obesity-induced muscular insulin resistance in high-fat-fed rats. Life Sci 89:107–114. doi: 10.1016/j.lfs.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 50.Coll T, Palomer X, Blanco-Vaca F, Escola-Gil JC, Sanchez RM, Laguna JC, Vazquez-Carrera M. 2010. Cyclooxygenase 2 inhibition exacerbates palmitate-induced inflammation and insulin resistance in skeletal muscle cells. Endocrinology 151:537–548. doi: 10.1210/en.2009-0874. [DOI] [PubMed] [Google Scholar]
- 51.Taniguchi CM, Ueki K, Kahn R. 2005. Complementary roles of IRS-1 and IRS-2 in the hepatic regulation of metabolism. J Clin Invest 115:718–727. doi: 10.1172/JCI23187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. 2000. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell 6:77–86. doi: 10.1016/S1097-2765(05)00010-9. [DOI] [PubMed] [Google Scholar]
- 53.van Rooij E, Purcell AL, Levin AA. 2012. Developing microRNA therapeutics. Circ Res 110:496–507. doi: 10.1161/CIRCRESAHA.111.247916. [DOI] [PubMed] [Google Scholar]
- 54.Gebert LF, Rebhan MA, Crivelli SE, Denzler R, Stoffel M, Hall J. 2014. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res 42:609–621. doi: 10.1093/nar/gkt852. [DOI] [PMC free article] [PubMed] [Google Scholar]