

Abstract
The small GTPase Rap1 controls the actin cytoskeleton by regulating Rho GTPase signaling. We recently established that the Rap1 effectors Radil and Rasip1, together with the Rho GTPase activating protein ArhGAP29, mediate Rap1-induced inhibition of Rho signaling in the processes of epithelial cell spreading and endothelial barrier function. Here, we show that Rap1 induces the independent translocations of Rasip1 and a Radil-ArhGAP29 complex to the plasma membrane. This results in the formation of a multimeric protein complex required for Rap1-induced inhibition of Rho signaling and increased endothelial barrier function. Together with the previously reported spatiotemporal control of the Rap guanine nucleotide exchange factor Epac1, these findings elucidate a signaling pathway for spatiotemporal control of Rho signaling that operates by successive protein translocations to and complex formation at the plasma membrane.
INTRODUCTION
The small GTPase Rap1 controls multiple processes linked to actin cytoskeletal dynamics, including integrin-mediated and cadherin-mediated cell adhesion. To this end, it translates spatial and temporal information into actin cytoskeleton modulation. This spatiotemporal information is received by Rap1 guanine nucleotide exchange factors (GEFs), such as Epac1 and PDZ-GEF. For instance, Epac1 responds to cyclic AMP (cAMP) by catalytic activation and translocation to specific sites at the plasma membrane resulting in the local activation of Rap1. Subsequently, actin cytoskeletal rearrangements occur, as exemplified in endothelial barrier regulation (1).
The endothelial barrier is under tight control to allow passage of fluids, solutes, and immune cells, without losing tissue integrity and barrier function (2–4). Rap1 plays an important role in this by enhancing the endothelial barrier function (5–9). It does so by impinging on the family of Rho GTPases, the master regulators of the actin cytoskeleton. Once activated, Rap1 inhibits the small GTPase Rho, resulting in the relaxation and disappearance of stress fibers (1, 10–13), and activates the small GTPase Cdc42, resulting in the formation of junctional actin bundles (1, 10). As a result, barrier function is enhanced. Indeed, Rho GTPases are important players in adherens junction formation and maintenance, which is necessary to sustain the barrier integrity of both epithelial and endothelial monolayers (14–16).
Recently, we have elucidated that one way for Rap1 to regulate Rho activity in endothelial barrier function is through the RhoGAP ArhGAP29. Rap1 does so through the highly related effectors Radil and Rasip1 (11, 17, 18), which have both been reported to interact with ArhGAP29 (19, 20). Activation of this pathway by active Rap1 reduces radial stress fibers, which exert tension on the adherens junctions, resulting in increased endothelial barrier function. Radil, Rasip1, and ArhGAP29 also control cell spreading of epithelial cells, indicating that the pathway is not restricted to endothelial barrier regulation but functions in a more universal manner (11). The interplay between Rasip1, Radil, and ArhGAP29, however, remained elusive. We demonstrate here that activation of Rap1 induces the translocation to the plasma membrane of Rasip1 and a complex of Radil and ArhGAP29 and the subsequent formation of a multimeric complex of Rap1, Rasip1, Radil, and ArhGAP29. We propose that through this translocation and complex formation Rap1 spatially restricts Rho-mediated signaling, which is necessary for endothelial barrier potentiation.
MATERIALS AND METHODS
Reagents and antibodies.
The reagent 007-AM (8-pCPT-2′-O-Me-cAMP-AM) was obtained from Biolog Life Sciences (Bremen, Germany) and used at the concentration of 1 μM (21). Prostacyclin (PGI2) was obtained from Sigma-Aldrich and used at the concentration of 10 μM. Antibodies were obtained from Invitrogen (V5, fluorescently labeled phalloidin and secondary antibodies), Santa Cruz Biotechnology (Ras-related protein 1 [Rap1]), Roche (green fluorescent protein [GFP]), Chemicon (α-tubulin), Novus Biologicals (Rho GTPase-activating protein 29 [ArhGAP29]), BD Biosciences (β-catenin), Sigma-Aldrich (Flag; M2), and Covance (hemagglutinin [HA]; HA11). All knockdowns were performed using ON-Target Plus siRNA (Dharmacon). Small interfering RNA (siRNA) SMARTpools were used for Rap1A, Rap1B, Rasip1, and Radil.
DNA constructs.
Rasip1, Radil, and ArhGAP29 were cloned into the Gateway system (Invitrogen) as described previously (11). Using this system, constructs were N-terminally cloned to a Flag-His, V5, cyan fluorescent protein (CFP), or citrine (yellow) fluorescent protein (YFP) tag using a pcDNA3 vector or an HA tag using a pMT2 vector or an mCherry or GFP tag using a modified pLV-CMVbc vector. Site-directed mutagenesis was used to create Rasip1 K163A, Radil K79A, and a premature stop-codon for ArhGAP29ΔC (P1258*). RadilΔPDZ(1–965) and RadilΔC(1–1058) were cloned into the Gateway system and subsequently N-terminally tagged.
Cell culture and transfections.
HEK293T cells were cultured at 37°C and 6% CO2 in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and antibiotics. Human umbilical vein endothelial cells (HUVECs; Lonza) were grown at 37°C and 6% CO2 on tissue culture dishes coated with 0.5% gelatin in EBM-2 culture medium (Lonza) supplemented with EGM-2 SingleQuots (epidermal growth factor [EGF], hydrocortisone, fetal bovine serum, vascular endothelial growth factor [VEGF], fibroblast growth factor B [FGF-B], R3-IGF-1, ascorbic acid, GA-100, and heparin; Lonza). The HUVECs were cultured maximally for 14 days before the experiments. siRNA transfections were performed 72 h before experiments with 40 to 50 nM ON-TARGETplus SMARTpools (Dharmacon, Inc.) targeting the indicated proteins using HiPerfect (Qiagen) or Dharmafect-1 (Dharmacon, Inc.), according to manufacturer's protocol. HEK293T cells were transfected with expression plasmids 24 to 48 h before performing experiments using X-tremeGENE 9 (Roche) according to the manufacturer's protocol. Overexpression of proteins in HUVECs was established by lentiviral transduction. For lentivirus production, the growth medium of HEK293T cells was replaced by EBM-2 growth medium, upon which these cells were transfected with the appropriate expression vector, together with third-generation packaging constructs using X-tremeGENE 9. HUVECs were infected 48 to 72 h before experiments using the undiluted growth medium of virus-producing HEK293T cells supplemented with Polybrene at 8 mg/liter. siRNA transfections occurred 16 h prior to transfection or infection of the expression plasmid.
Cell imaging.
For confocal live imaging, HEK293T cells were seeded overnight in glass-bottom wells (WillCo Wells). For the HUVECs, glass-bottom wells (WillCo Wells) were coated with fibronectin prior to seeding. The HUVECs were grown to confluence prior to imaging. Cells were imaged in L-15 Leibovitz medium (Invitrogen) at 37°C on an inverted Zeiss LSM510 confocal microscope equipped with ×63 magnification objective lens (numerical aperture, 1.4; Leica). Postacquisition image adjustments for brightness and contrast enhancement were performed using ImageJ software (National Institutes of Health). Images of HUVECs were taken prior to and after stimulation with 007-AM (15 min) or prostacyclin (5 min). For postacquisition analysis of the translocations of fluorescently labeled ArhGAP29, Rasip, and Radil, we used an analysis macro (ImageJ), custom designed to exclusively measure signal in the plasma membrane and cytosol, respectively, for each time point of the time series. The macro combines user-defined region of interest (ROI) drawing with (adaptable) intensity-based thresholding.
Immunofluorescence.
HUVECs were plated onto fibronectin-coated glass coverslips either 48 h after transfection and grown to confluence for another 24 h. After 15 min stimulation with 1 μM 007-AM, the cells were fixed with 4% formaldehyde for 15 min, permeabilized with 0.1% TX-100 for 5 min, and blocked with 1% bovine serum albumin for at least 30 min. Next, the cells were incubated with the indicated primary antibodies for 1 h and the indicated secondary antibody for 30 min and then mounted onto glass slides, which were subsequently examined on an Axioskop 2 mot plus microscope (Zeiss) with a 40× or 100× immersion oil objective lens and an Axiocam camera. Original images were used to create line scans in ImageJ. The graphs show profiles of signal intensities along the line scan. To allow easy visual comparison between channels, signal intensities were normalized to the mean.
Coimmunoprecipitations.
HEK293T cells were transfected using X-tremeGENE 9 and lysed 48 h after transfection using a buffer containing 0.5% NP-40, 20 mM Tris (pH 7.5), 150 mM NaCl, 20 mM MgCl2, 10% glycerol, and protease and phosphatase inhibitors. Cell lysates were cleared by centrifugation, and lysates were incubated with protein A-agarose beads (Pharmacia) coupled to the appropriate antibody. After extensive washing with lysis buffer, bound proteins were eluted in Laemmli buffer and analyzed by SDS-PAGE.
Endothelial barrier measurements.
Endothelial barrier was assessed by electrical cell impedance sensing (ECIS) measurements. At 48 h after siRNA transfection and/or 24 h after lentiviral infection, HUVECs were plated onto l-cysteine-reduced, fibronectin-coated 8W10E electrodes (Applied Biophysics) at a density of 105 cells/well and grown to confluence for another 24 h. The impedance was measured at multiple frequencies within the range of 62.5 to 16,000 Hz at 37°C and 6% CO2 using a 1600R ECIS system (Applied Biophysics). These frequency scans were used to calculate the endothelial barrier (Rb) with ECIS software (v1.2.135.0 PC) from Applied Biophysics. The graphs show multiple independent experiments represented by different colors. P values were calculated by using a Student t test (two-tailed, paired).
Real-time quantitative PCR (Q-PCR).
A total of 8 × 105 HUVECs were plated 48 h after siRNA transfection onto fibronectin-coated 6-cm dishes and grown for another 24 h. Total RNA was isolated using the RNeasy minikit (Qiagen) and transcribed into cDNA using the iScript cDNA synthesis kit (Bio-Rad). cDNA levels were quantified by SYBR green real-time PCR on a C1000 thermal cycler (Bio-Rad) using the following primers: CTGGACATCACAGGCTCGAA (Radil-forward), TTGGAGACATAGTAGACGCAC (Radil-reverse), TCAGCCAGAAAACCACCCTGA (Rasip1-forward), and CAGCACCTTCTCCTGCACAAA (Rasip1-reverse). Nonspecific signals were excluded based on nontemplate control samples. Amplification of the HPRT1 gene was used as a control for sample loading. Expression levels were normalized to the siScrambled control. Error bars indicate the standard deviations (n = 3).
RESULTS
ArhGAP29 is recruited to the plasma membrane upon activation of Epac1 and Rap1.
To investigate how ArhGAP29 is regulated downstream of Epac1-Rap1 signaling, we first determined the subcellular localization of ArhGAP29 by expressing GFP-ArhGAP29 in HUVECs. GFP-ArhGAP29 localized predominantly to the cytoplasmic compartment under resting conditions, with some localization to the plasma membrane. Upon activation of Epac1 by stimulating cells with an Epac-selective activator 8-pCPT-2′-O-Me-cAMP-AM (007-AM) (21), ca. 50% of the cells showed a clear enrichment of GFP-ArhGAP29 at nonspecific patches within the plasma membrane (Fig. 1A), which was not observed in HUVECs expressing GFP alone (Fig. 1B). This translocation was abrogated by depletion of Rap1 (Fig. 1C) and could also be observed when endogenous cAMP production was stimulated using the endothelial agonist prostacyclin (Fig. 1D), indicating that cAMP-activated Rap1 mediates the translocation of ArhGAP29 to the plasma membrane.
FIG 1.
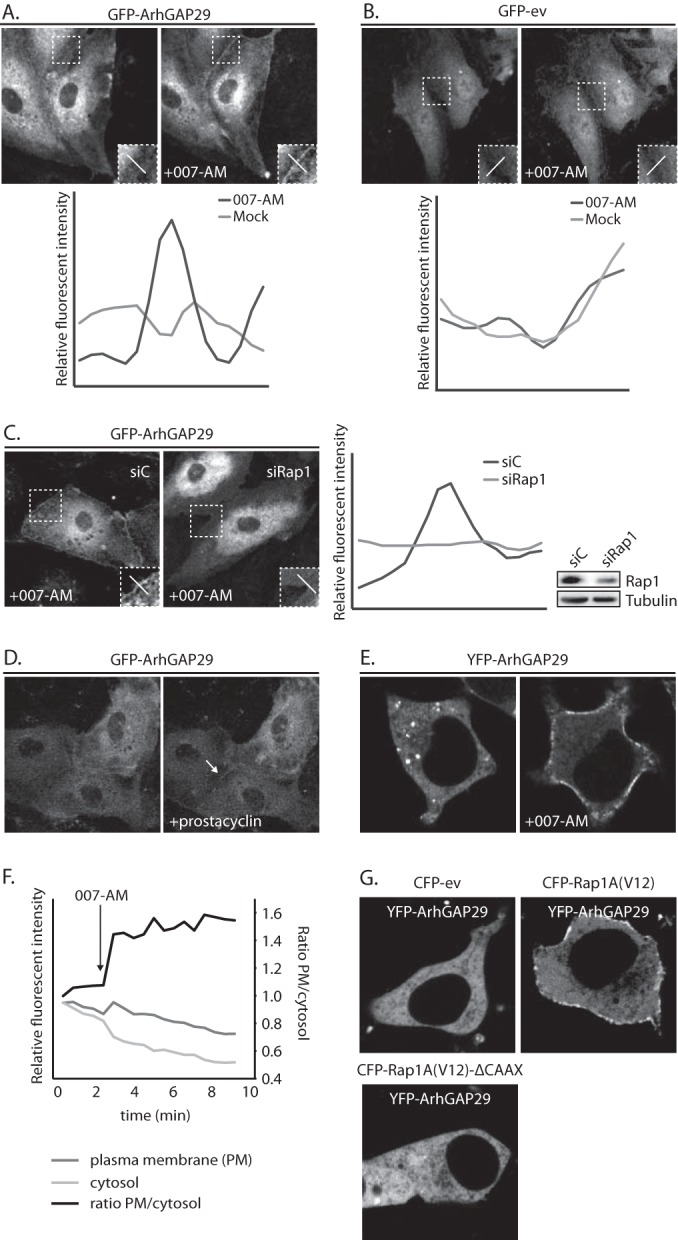
Active Rap1 recruits ArhGAP29 to the plasma membrane. (A) The upper panel shows live imaging of confluent monolayers of HUVECs infected with a lentivirus transducing GFP-ArhGAP29. The cells were imaged prior to and 15 min after stimulation with 007-AM. The boxed areas of cell-cell contacts are enlarged in the insets. In the lower panel, a graph shows the relative intensity profiles of fluorescent signal intensities along the line scans depicted in the boxed area. (B) The upper panel shows live imaging of confluent monolayers of HUVECs infected with a lentivirus transducing GFP-ev. The cells were imaged prior to and 15 min after stimulation with 007-AM. The boxed areas of cell-cell contacts are enlarged in the insets. In the lower panel, a graph shows the relative intensity profiles of fluorescent signal intensities along the line scans depicted in the boxed area. (C) The left panel shows live imaging of GFP-ArhGAP29 expressing HUVECs, treated with control siRNA (siC) or siRNA targeting Rap1A and Rap1B (siRap1). The cells were grown to confluence and stimulated with 007-AM 15 min prior to imaging. The boxed areas are enlarged in insets. In the right panel, a graph shows the relative intensity profiles of fluorescent signal intensities along the line scans depicted in the boxed area. The knockdown efficiency was assessed by Western blotting. (D) Live imaging of confluent monolayers of HUVECs infected with a lentivirus transducing GFP-ArhGAP29. The cells were imaged prior to (left image) and 5 min after (right image) stimulation with prostacyclin. Enrichment of ArhGAP29 at cell-cell contacts is indicated by the arrow. (E) Live imaging of HEK293T cells transiently transfected with HA-Epac1, HA-Rap1A, V5-Radil, Flag-Rasip1, and YFP-ArhGAP29, imaged prior to and 10 min after stimulation with 007-AM. (F) Quantitative analysis of a representative ArhGAP29 translocation experiment in HEK293T cells (transfected as in panel E; see Movie S1 in the supplemental material). A custom-made analysis macro (ImageJ), which detects and measures signal in the plasma membrane (PM) and cytosol, respectively. (G) Imaging of YFP-ArhGAP29 in HEK293T cells transfected with V5-Radil, HA-Rasip1, or YFP-ArhGAP29 and with either CFP-ev, CFP-Rap1A(V12), or CFP-Rap1A(V12)ΔCAAX. Experiments were repeated at least three times, and representative images were chosen.
Similar results were obtained upon reconstituting the signaling cascade in HEK293T cells by coexpressing HA-Epac1, HA-Rap1, and YFP-ArhGAP29, together with V5-Radil and Flag-Rasip1, the Rap1 effectors involved in this process (11). Stimulation with 007-AM readily induced the translocation of YFP-ArhGAP29 from the cytosol to the plasma membrane (Fig. 1E and F, showing the analysis of Movie S1 in the supplemental material).
To confirm that the translocation of ArhGAP29 to the plasma membrane was induced by activated Rap1, we transfected HEK293T cells with YFP-ArhGAP29, V5-Radil, HA-Rasip1, and CFP-Rap1A(V12), an active Rap1A mutant. Indeed, expression of CFP-Rap1(V12) resulted in plasma membrane localized YFP-ArhGAP29 (Fig. 1G). Transfection of a CFP-Rap1A(V12)-ΔCAAX mutant (unable to associate with the plasma membrane) did not result in plasma membrane localization of YFP-ArhGAP29 (Fig. 1G), indicating that plasma membrane recruitment of ArhGAP29 requires active Rap1 to be membrane localized. We conclude that activation of Rap1 results in the translocation of ArhGAP29 to the plasma membrane in both HUVECs and HEK293T cells.
Radil and Rasip1 translocate to the plasma membrane by binding active Rap1.
Since Rap1 regulates ArhGAP29 through Radil and Rasip1 (11), we next investigated the localizations of Radil and Rasip1 upon activation of Epac1-Rap1 signaling. As we observed for ArhGAP29, Radil and Rasip1 localize predominantly to the cytoplasmic compartment in HUVECs (Fig. 2A and B). Upon stimulation with 007-AM, both GFP-Radil and GFP-Rasip1 largely redistributed to the plasma membrane. Similarly, reconstitution of this pathway in HEK293T cells, by coexpressing HA-Epac1, HA-Rap1A, and HA-ArhGAP29, together with mCherry-Radil and HA-Rasip1 (Fig. 2C and D; see also Movie S2 in the supplemental material) or YFP-Rasip1 and V5-Radil (Fig. 2E and F; see also Movie S3 in the supplemental material) revealed translocation to the plasma membrane of both Radil and Rasip1 upon stimulation with 007-AM. As for ArhGAP29, depletion of Rap1 abrogated this translocation in HUVECs (Fig. 2A and B), whereas overexpression of CFP-Rap1A(V12) resulted in plasma membrane localization of mCherry-Radil (Fig. 2G) and YFP-Rasip1 (Fig. 2H) in HEK293T cells. Furthermore, the CFP-Rap1A(V12)-ΔCAAX mutant could not recruit Radil (Fig. 2G) or Rasip1 (Fig. 2H) to the plasma membrane, demonstrating that both Radil and Rasip1 are recruited to an active pool of Rap1 at the plasma membrane.
FIG 2.

Active Rap1 recruits Radil and Rasip1 to the plasma membrane. (A) The left panel shows live imaging of confluent monolayers of HUVECs infected with a lentivirus transducing GFP-Radil and treated with control siRNA (siC) or siRNA targeting Rap1A and Rap1B (siRap1). The cells were imaged prior to or 15 min after stimulation with 007-AM. The boxed areas of cell-cell contacts are enlarged in the insets. In the right panel, a graph shows the relative intensity profiles of fluorescent signal intensities along the line scans depicted in the boxed area. The knockdown efficiency was assessed by Western blotting. (B) The left panel shows live imaging of confluent monolayers of HUVECs infected with a lentivirus transducing GFP-Rasip1 and treated with control siRNA (siC) or siRNA targeting Rap1A and Rap1B (siRap1). The cells were imaged prior to or 15 min after stimulation with 007-AM. The boxed areas of cell-cell contacts are enlarged in the insets. In the right panel, a graph shows the relative intensity profiles of fluorescent signal intensities along the line scans depicted in the boxed area. Knockdown efficient was assessed by Western blotting. (C) Still images (taken prior to and 3 min after 007-AM stimulation) corresponding to the live imaging (see Movie S2 in the supplemental material) of HEK293T cells transiently transfected with HA-Epac1, HA-Rap1A, mCherry-Radil, HA-Rasip1, and HA-ArhGAP29. (D) Quantitative plasma membrane (PM)/cytosol analysis (as in Fig. 1F) of mCherry-Radil translocation in Movie 2 in the supplemental material. (E) Still images (taken prior to and 2 min after 007-AM stimulation) corresponding to the live imaging (see Movie S3 in the supplemental material) of HEK293T cells transiently transfected with HA-Epac1, HA-Rap1A, V5-Radil, YFP-Rasip1, and HA-ArhGAP29. (F) Quantitative plasma membrane (PM)/cytosol analysis (as in Fig. 1F) of the YFP-Rasip1 translocation shown in Movie S3 in the supplemental material. (G) Live imaging of mCherry-Radil in HEK293T cells transfected with mCherry-Radil, HA-Rasip1, and HA-ArhGAP29 and with either CFP-ev, CFP-Rap1A(V12) or CFP-Rap1A(V12)ΔCAAX. (H) Live imaging of YFP-Rasip1 in HEK293T cells transfected with YFP-Rasip1, V5-Radil, and HA-ArhGAP29 and with either CFP-ev, CFP-Rap1A(V12), or CFP-Rap1A(V12)ΔCAAX. (I) Alignment of the RA domains of Radil, Rasip1, and the second RA domain of AF6. Highlighted with gray is the conserved lysine, which is essential for interaction with GTP-loaded Rap1. (J) Live imaging of wild-type versus RA mutant mCherry-Radil (upper panels) and wild-type versus RA mutant YFP-Rasip1 (lower panels) in HEK293T cells transfected with HA-Rasip1, V5-Radil, HA-ArhGAP29, and HA-Rap1A(V12), together with mCherry-Radil, mCherry-Radil K79A, YFP-Rasip1, or YFP-Rasip1 K163A. The experiments were repeated at least three times; representative images are shown.
Previously, it was shown that both Radil and Rasip1 can directly bind to Rap1, most likely through their respective RA domains (17, 22). To test whether Radil and Rasip1 are recruited to the plasma membrane by directly binding active Rap1 at the plasma membrane, we mutated a conserved lysine (Fig. 2I) to an alanine in the RA-domains of Radil and Rasip1, since a similar mutation in the Rap1-effector AF6 was shown to abolish the interaction with Rap1 (23). As expected, Radil K79A and Rasip1 K163A did not localize to the plasma membrane in the presence of HA-Rap1A(V12) (Fig. 2J). These data strongly suggest that activation of Rap1 results in the translocation of Radil and Rasip1 to the plasma membrane by directly interacting with their RA domains.
Rasip1 and Radil translocate to the plasma membrane independently.
Next, we investigated whether the translocations of Radil and Rasip1 to the plasma membrane depend on each other. Rap1-induced plasma membrane localization of Radil was unaffected by siRNA-mediated depletion of Rasip1 in HUVECs (Fig. 3A), whereas in the reconstituted system in HEK293T cells the overexpression of Rasip1 is not required (Fig. 3C). Similarly, Rap1-induced plasma membrane localization of Rasip1 was unaffected by siRNA-mediated depletion of Radil in HUVECs or the absence of Radil overexpression in HEK293Ts (Fig. 3B and C). Moreover, the Rap1-binding mutant of Rasip1 did not prevent the translocation of wild-type Radil to the plasma membrane in HEK293Ts and vice versa (Fig. 3D and E), indicating that both effectors must bind active Rap1 at the plasma membrane through their own RA domain. These results indicate that Rasip1 and Radil translocate to active Rap1 at the plasma membrane independently of each other.
FIG 3.
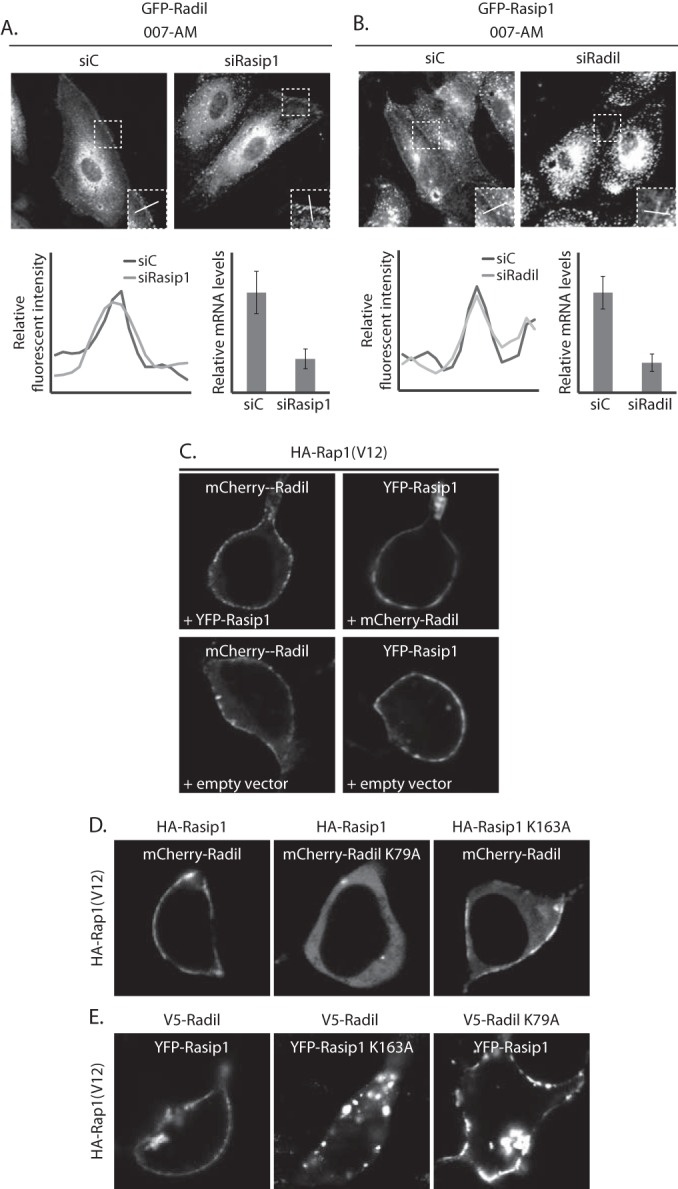
Rasip1 and Radil translocate to the plasma membrane independently. (A) The upper panel shows live imaging of GFP-Radil-expressing HUVECs, treated with control siRNA (siC), or siRNA targeting Rasip1 (siRasip1). Cells were grown to confluence and stimulated with 007-AM 15 min prior to imaging. Boxed areas are enlarged in insets. In the lower panel, the left graph shows the relative intensity profiles of fluorescent signal intensities along the line scans depicted in the boxed area. The right graph shows the knockdown efficiency as assessed by Q-PCR. (B) The upper panel shows live imaging of GFP-Rasip1-expressing HUVECs, treated with control siRNA (siC) or siRNA targeting Radil (siRadil). The cells were grown to confluence and stimulated with 007-AM for 15 min prior to imaging. The boxed areas are enlarged in insets. In the lower panel, the left graph shows the relative intensity profiles of fluorescent signal intensities along the line scans depicted in the boxed area. The right graph shows the knockdown efficiency as assessed by Q-PCR. (C) Live imaging of mCherry-Radil or YFP-Rasip1 in HEK293T cells transfected with HA-Rap1A(V12) and both mCherry-Radil and YFP-Rasip1 (upper panels), only mCherry-Radil (lower left panel), or only YFP-Rasip1 (lower right panel). (D) Live imaging of wild or mutant mCherry-Radil (as indicated) in HEK293T cells transfected with HA-Rap1A(V12) and either mCherry-Radil or mCherry-Radil K79A, together with either HA-Rasip1 or HA-Rasip1 K163A. (E) Live imaging of wild or mutant YFP-Rasip1 (as indicated) in HEK293T cells transfected with HA-Rap1A(V12) and either YFP-Rasip1 or YFP-Rasip1 K163A, together with either V5-Radil or V5-Radil K79A.
ArhGAP29 translocation to the plasma membrane is dependent on Radil but not Rasip1.
We next investigated whether the Rap1-induced translocation of ArhGAP29 is dependent on Radil and/or Rasip1. Simultaneous depletion of both Radil and Rasip1 abrogated Epac1-Rap1 induced translocation of ArhGAP29 to the plasma membrane in HUVECs (Fig. 4A). Interestingly, depletion of Radil alone but not Rasip1 alone was sufficient to abrogate ArhGAP29 translocation to the plasma membrane upon activation of Epac1 with 007-AM (Fig. 4A). Similarly, only the presence of Radil but not Rasip1 was required for ArhGAP29 translocation in the reconstituted HEK293T system (Fig. 4B). This suggests that the translocation of ArhGAP29 to the plasma membrane is solely dependent on Radil. Indeed, in HEK293T cells HA-Rap1A(V12) failed to induce plasma membrane localization of YFP-ArhGAP29 when coexpressed with of V5-Radil K79A and HA-Rasip1, whereas YFP-ArhGAP29 normally localized to the plasma membrane when coexpressed with HA-Rap1A(V12), V5-Radil and HA-Rasip1 K179A (Fig. 4C). We conclude that Radil, and not Rasip1, mediates Rap1-induced translocation of ArhGAP29 to the plasma membrane.
FIG 4.
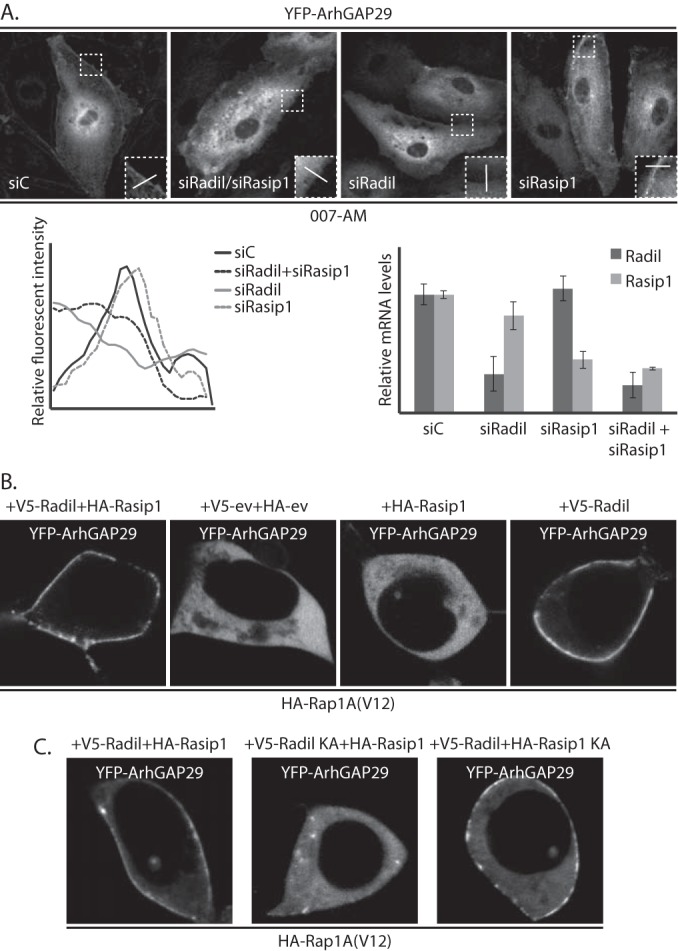
Rap1 recruits ArhGAP29 to the plasma membrane through Radil. (A) In the upper panel, GFP-ArhGAP29 expressing HUVECs were treated with control siRNA (siC), siRNA targeting Radil and Rasip1 (siRadil + siRasip1), and Radil (siRadil) or Rasip1 (siRasip1). The cells were grown to confluence and treated with 007-AM for 15 min. The boxed areas of cell-cell contacts are enlarged in the insets. In the lower panel, the left graph shows the relative intensity profiles of fluorescent signal intensities along the line scans depicted in the boxed area. The right graph shows the knockdown efficiency as assessed by Q-PCR. (B) Live imaging of YFP-ArhGAP29 in HEK293T cells transfected with HA-Rap1A(V12) and YFP-ArhGAP29 upon cotransfection with either V5-Radil and HA-Rasip1 (first panel), empty vector only (second panel), HA-Rasip1 supplemented with empty vector (third panel), or V5-Radil supplemented with empty vector (fourth panel). (C) Live imaging of YFP-ArhGAP29 in HEK293T cells transfected with HA-Rap1A(V12) and YFP-ArhGAP29 upon cotransfection with either V5-Radil and HA-Rasip1 (first panel), V5-Radil K79A (V5-Radil KA) and HA-Rasip1 (second panel), or V5-Radil and HA-Rasip1 K163A (HA-Rasip1 KA) (third panel). The experiments were repeated at least three times, and representative images were chosen.
ArhGAP29 directly interacts with the PDZ domain of Radil.
Recently, ArhGAP29 was identified in a mass spectrometry screen for proteins interacting with the PDZ domain of Radil (19). Indeed, wild-type Radil coprecipitated with ArhGAP29, whereas a truncation mutant lacking the PDZ-domain and the neighboring C-terminal tail (RadilΔPDZ) did not (Fig. 5A and B). This lack of binding is fully attributed to the missing PDZ domain, since a truncation mutant lacking only the C terminus directly after the PDZ domain (RadilΔC) interacted normally with ArhGAP29 (Fig. 5B). Furthermore, plasma membrane localization of ArhGAP29 in HEK293T cells by CFP-Rap1A(V12) was abrogated when cells were cotransfected with V5-tagged RadilΔPDZ instead of wild-type Radil (Fig. 5C). PDZ-binding domains are usually harbored in the last C-terminal amino acids. ArhGAP29 contains an unconventional PDZ-binding motif at its C terminus, comprising P-Q-F-V. To determine whether this motif was responsible for the interaction with Radil, we created a truncation mutant of ArhGAP29 (ArhGAP29ΔC) lacking these last four residues. Whereas Radil readily coprecipitated with wild-type ArhGAP29 in HEK293T cells, far less Radil precipitated with ArhGAP29ΔC (Fig. 5D). As expected, this truncated ArhGAP29ΔC exhibited reduced plasma membrane localization upon Rap1A(V12)-Radil signaling in HEK293T cells (Fig. 5E). Altogether, these results indicate that Radil, through its PDZ domain, directly interacts with the C terminus of ArhGAP29 and that this interaction is required for efficient plasma membrane localization of ArhGAP29 downstream of Epac1-Rap1 signaling.
FIG 5.
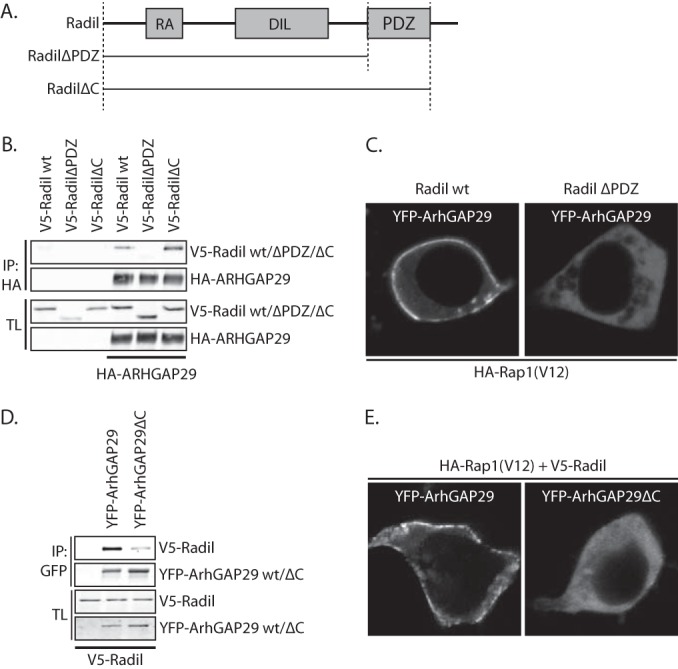
ArhGAP29 binds the PDZ-domain of Radil through its C terminus. (A) Domain architecture of Radil and the truncation mutants used for the assays depicted in panels B and C. RA, Ras association domain; DIL, dilute domain; PDZ, PSD-95/Dlg1/ZO-1. (B) Coimmunoprecipitation of V5-Radil truncation mutants with HA-ArhGAP29 expressed in HEK293T cells. The Western blot is representative of three independent experiments. (C) Live imaging of YFP-ArhGAP29 in HEK293T cells transfected with YFP-ArhGAP29 and HA-Rap1A(V12) in the presence of V5-Radil or V5-RadilΔPDZ. Experiments were repeated four times, and representative images were chosen. (D) Coimmunoprecipitation of V5-Radil with YFP-tagged full-length ArhGAP29 or ArhGAP29 lacking its last four amino acids (ArhGAP29ΔC) expressed in HEK293T. The Western blot is representative of two independent experiments. (E) Live imaging of YFP-ArhGAP29 in HEK293T cells transfected with YFP-ArhGAP29 or YFP-ArhGAP29ΔC with HA-Rap1A(V12) and V5-Radil. The experiments were repeated four times, and a representative image was chosen.
Rasip1 forms a ternary complex with Radil and ArhGAP29.
Since both Rasip1 and the Radil-ArhGAP29 complex translocate to the plasma membrane upon Rap1 activation, we addressed the question whether the Rasip1 and Radil-ArhGAP29 remain independent complexes or whether they form a Rap1-induced multimeric complex. To that end, we performed coimmunoprecipitations in HEK293T cells. Immunoprecipitation of ArhGAP29 coprecipitated Radil but not Rasip1. However, when Rasip1 and Radil were both expressed, Rasip1 is also coprecipitated with ArhGAP29, indicating the presence of a trimeric complex (Fig. 6A). Importantly, Rap1A(V12) increased the amount of Rasip1 that precipitated with ArhGAP29 and Radil, whereas the interaction between Radil and ArhGAP29 was not affected (Fig. 6B). These results indicate that upon translocation to the plasma membrane Rasip1 and the Radil-ArhGAP29 complex form a multimeric protein complex.
FIG 6.
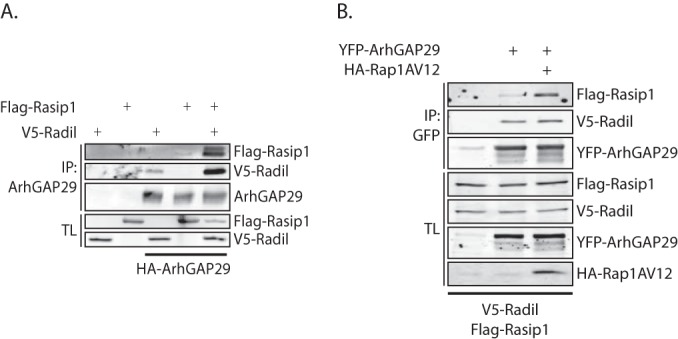
Rasip1 forms a multimeric complex with ArhGAP29 and Radil. (A) Coimmunoprecipitation of Flag-Rasip1 with YFP-ArhGAP29 (detected using an anti-GFP antibody) in the presence or absence of V5-Radil in HEK293T cells. The experiments were repeated at least four times. (B) Coimmunoprecipitation of Flag-Rasip1 and V5-Radil with YFP-ArhGAP29 (detected with an anti-GFP antibody) in the presence or absence of HA-Rap1A(V12) in HEK293T cells.
Both Rasip1 and Radil are required for Rap1-induced inhibition of stress fiber formation and endothelial barrier function.
The formation of a multimeric complex of Rasip1, Radil, and ArhGAP29 by Rap1 suggests that both Radil and Rasip1 are required for Rap1-induced inhibition of stress fibers and endothelial barrier function. Indeed, depletion of either Rasip1 or Radil inhibited Rap1A(V12)-induced endothelial barrier function (Fig. 7A). Furthermore, in HUVECs the reduction of radial stress fibers by 007-AM was abrogated by the depletion of either Rasip1 or Radil (Fig. 7B; also see Fig. S1D in the supplemental material). We conclude that both Rasip1 and Radil are required for active Rap1-induced inhibition of radial stress fibers and increase in barrier function. This result is compatible with our previous notion that Radil and Rasip1 are also both required for Rap1-ArhGAP29-mediated spreading of epithelial cells (11).
FIG 7.
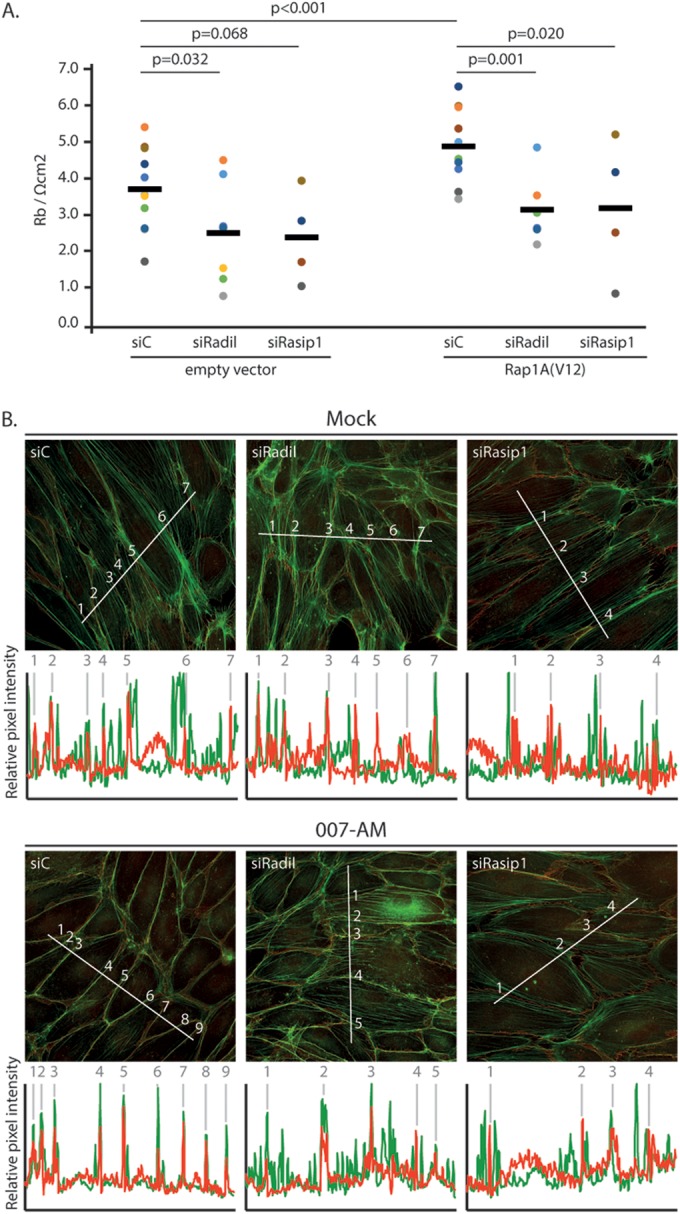
Rasip1 and Radil are required for Rap1-induced endothelial barrier function. (A) Endothelial barrier (Rb) of control HUVEC monolayers (siC) and HUVEC monolayers depleted of Radil (siRadil) or Rasip1 (siRasip1), transduced with control lentivirus or Rap1A(V12) containing lentivirus. Different colors represent independent experiments (n > 4). Averages are indicated by the black line. (For the knockdown efficiencies, see Fig. S1A and B in the supplemental material.) (B) Immunofluorescence of HUVECs transfected with control siRNA (siC), siRNA targeting Radil (siRadil), or siRNA targeting Rasip1 (siRasip1). Cells were grown to confluence and either not stimulated or stimulated with 007-AM 15 min prior to fixation. The cells were stained for β-catenin (red) and F-actin (phalloidin, green). The graphs show the relative intensity profiles of fluorescent signal intensities of phalloidin (green) and β-catenin (red) along the line scans depicted in the immunofluorescent images. Gray lines and numbers correlate to where the line scan crosses an adherens junction, as assessed by β-catenin staining. (For the knockdown efficiencies, see Fig. S1C in the supplemental material.)
DISCUSSION
We have previously shown that Rap1 induces cell spreading and endothelial barrier function by inhibiting Rho signaling through its effectors Rasip1 and Radil and their binding partner, the RhoGAP ArhGAP29 (11, 19, 20). Here, we show that active Rap1 achieves this by inducing the translocation of all three components—Radil, Rasip1, and ArhGAP29—to the plasma membrane. Our results confirm that the previously reported Rap1-dependent plasma membrane localization of Radil (24) and Rasip1 (18) are due to direct binding of the effectors to active Rap1 through their RA domains. Whereas the translocation of ArhGAP29 depends on Radil alone, a trimeric complex between Radil, Rasip1, and ArhGAP29 is formed upon Rap1 activation. Since all three proteins are required for Rap1-induced inhibition of stress fiber formation and endothelial barrier function (the present study and see also reference 11), we conclude that the formation of this complex results in local ArhGAP29 activity, inhibiting Rho and subsequently stress fiber formation.
It is currently unclear whether complex formation allosterically activates ArhGAP29 or whether local recruitment of the GAP is sufficient. Hence, Radil, which clearly controls the localization of ArhGAP29, might separately control the GAP activity of ArhGAP29. Also, the contribution of Rasip1 in the complex to regulate Rho requires further research. Rasip1 indirectly complexes with ArhGAP29 and is clearly required for the biological effect but not for the translocation of ArhGAP29. Presumably, Rasip1 controls the activity of ArhGAP29. Indeed, it was demonstrated that depletion of Rasip1 results in increased Rho-GTP levels in three-dimensional HUVEC cultures (18). However, this was not observed in two-dimensional HUVEC cultures (11, 18, 20), implying that Rasip1 could also function independently of ArhGAP29 in the control of endothelial barrier function.
Our results further our understanding of Rap1 signaling and, more specifically, its key role in spatiotemporal control of the actin cytoskeleton. Previously, we already showed that the Rap1 GEFs PDZ-GEF and the cAMP-inducible Epac1 are regulated by translocation to the plasma membrane (25, 26). Together with our current results, this reveals a signaling pathway, which operates by successive protein translocations to the plasma membrane, spatially restricting Rho activity and subsequent actin cytoskeletal dynamics. Parallel to this, Rap1 activates Cdc42 through the recruitment of the Cdc42GEF FGD5 to cell-cell junctions, resulting in cortical actin bundle formation (10).
Altogether, a picture emerges for endothelial barrier control in which Rap1 functions to translate cortical cues into the spatial regulation of Rho GTPase activity and subsequent actin cytoskeletal remodeling, through the dynamic spatiotemporal control of GEFs and GAPs for the various GTPases involved.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the department for continuous discussions and critical comments.
A.P., M.J.V., and W.J.P. were funded by grants from the Dutch Cancer Society, and B.P. was funded by a gravitation grant from Netherlands Organization for Scientific Research.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01453-14.
REFERENCES
- 1.Pannekoek WJ, Post A, Bos JL. 2014. Rap1 signaling in endothelial barrier control. Cell Adhesion Migration 8:100–107. doi: 10.4161/cam.27352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baluk P, Hashizume H, McDonald DM. 2005. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev 15:102–111. doi: 10.1016/j.gde.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Bazzoni G, Dejana E. 2004. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 4.Weis SM. 2008. Vascular permeability in cardiovascular disease and cancer. Curr Opin Hematol 15:243–249. doi: 10.1097/MOH.0b013e3282f97d86. [DOI] [PubMed] [Google Scholar]
- 5.Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. 2005. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 105:1950–1955. doi: 10.1182/blood-2004-05-1987. [DOI] [PubMed] [Google Scholar]
- 6.Kooistra MR, Corada M, Dejana E, Bos JL. 2005. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett 579:4966–4972. doi: 10.1016/j.febslet.2005.07.080. [DOI] [PubMed] [Google Scholar]
- 7.Pannekoek WJ, van Dijk JJG, Chan OYA, Huveneers S, Linnemann JR, Spanjaard E, Brouwer PM, van der Meer AJ, Zwartkruis FJT, Rehmann H, de Rooij J, Bos JL. 2011. Epac1 and PDZ-GEF cooperate in Rap1 mediated endothelial junction control. Cell Signal 23:2056–2064. doi: 10.1016/j.cellsig.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 8.Wittchen ES, Worthylake RA, Kelly P, Casey PJ, Quilliam LA, Burridge K. 2005. Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J Biol Chem 280:11675–11682. doi: 10.1074/jbc.M412595200. [DOI] [PubMed] [Google Scholar]
- 9.Wittchen ES, Aghajanian A, Burridge K. 2011. Isoform-specific differences between Rap1A and Rap1B GTPases in the formation of endothelial cell junctions. Small GTPases 2:65–76. doi: 10.4161/sgtp.2.2.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ando K, Fukuhara S, Moriya T, Obara Y, Nakahata N, Mochizuki N. 2013. Rap1 potentiates endothelial cell junctions by spatially controlling myosin II activity and actin organization. J Cell Biol 202:901–916. doi: 10.1083/jcb.201301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Post A, Pannekoek WJ, Ross SH, Verlaan I, Brouwer PM, Bos JL. 2013. Rasip1 mediates Rap1 regulation of Rho in endothelial barrier function through ArhGAP29. Proc Natl Acad Sci U S A 110:11427–11432. doi: 10.1073/pnas.1306595110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glading A, Han J, Stockton RA, Ginsberg MH. 2007. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell-cell junctions. J Cell Biol 179:247–254. doi: 10.1083/jcb.200705175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stockton RA, Shenkar R, Awad IA, Ginsberg MH. 2010. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med 207:881–896. doi: 10.1084/jem.20091258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. 2010. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost 103:40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- 15.Spindler V, Schlegel N, Waschke J. 2010. Role of GTPases in control of microvascular permeability. Cardiovasc Res 87:243–253. doi: 10.1093/cvr/cvq086. [DOI] [PubMed] [Google Scholar]
- 16.Szulcek R, Beckers CM, Hodzic J, de Wit J, Chen Z, Grob T, Musters RJ, Minshall RD, van Hinsbergh VW, van Nieuw Amerongen GP. 2013. Localized RhoA GTPase activity regulates dynamics of endothelial monolayer integrity. Cardiovasc Res 99:471–482. doi: 10.1093/cvr/cvt075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smolen GA, Schott BJ, Stewart RA, Diederichs S, Muir B, Provencher HL, Look AT, Sgroi DC, Peterson RT, Haber DA. 2007. A Rap GTPase interactor, RADIL, mediates migration of neural crest precursors. Genes Dev 21:2131–2136. doi: 10.1101/gad.1561507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson CW, Parker LH, Hall CJ, Smyczek T, Mak J, Crow A, Posthuma G, De Maziere A, Sagolla M, Chalouni C, Vitorino P, Roose-Girma M, Warming S, Klumperman J, Crosier PS, Ye W. 2013. Rasip1 regulates vertebrate vascular endothelial junction stability through Epac1-Rap1 signaling. Blood 122:3678–3690. doi: 10.1182/blood-2013-02-483156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmed SM, Theriault BL, Uppalapati M, Chiu CW, Gallie BL, Sidhu SS, Angers S. 2012. KIF14 negatively regulates Rap1a-Radil signaling during breast cancer progression. J Cell Biol 199:951–967. doi: 10.1083/jcb.201206051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu K, Sacharidou A, Fu S, Chong DC, Skaug B, Chen ZJ, Davis GE, Cleaver O. 2011. Blood vessel tubulogenesis requires Rasip1 regulation of GTPase signaling. Dev Cell 20:526–539. doi: 10.1016/j.devcel.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vliem MJ, Ponsioen B, Schwede F, Pannekoek WJ, Riedl J, Kooistra MR, Jalink K, Genieser HG, Bos JL, Rehmann H. 2008. 8-pCPT-2′-O-Me-cAMP-AM: an improved Epac-selective cAMP analogue. Chembiochem 9:2052–2054. doi: 10.1002/cbic.200800216. [DOI] [PubMed] [Google Scholar]
- 22.Mitin NY, Ramocki MB, Zullo AJ, Der CJ, Konieczny SF, Taparowsky EJ. 2004. Identification and characterization of rain, a novel Ras-interacting protein with a unique subcellular localization. J Biol Chem 279:22353–22361. doi: 10.1074/jbc.M312867200. [DOI] [PubMed] [Google Scholar]
- 23.Wohlgemuth S, Kiel C, Kramer A, Serrano L, Wittinghofer F, Herrmann C. 2005. Recognizing and defining true Ras binding domains I: biochemical analysis. J Mol Biol 348:741–758. doi: 10.1016/j.jmb.2005.02.048. [DOI] [PubMed] [Google Scholar]
- 24.Ahmed SM, Daulat AM, Meunier A, Angers S. 2010. G protein βγ subunits regulate cell adhesion through Rap1a and its effector Radil. J Biol Chem 285:6538–6551. doi: 10.1074/jbc.M109.069948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ponsioen B, Gloerich M, Ritsma L, Rehmann H, Bos JL, Jalink K. 2009. Direct spatial control of Epac1 by cyclic AMP. Mol Cell Biol 29:2521–2531. doi: 10.1128/MCB.01630-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consonni SV, Gloerich M, Spanjaard E, Bos JL. 2012. cAMP regulates DEP domain-mediated binding of the guanine nucleotide exchange factor Epac1 to phosphatidic acid at the plasma membrane. Proc Natl Acad Sci U S A 109:3814–3819. doi: 10.1073/pnas.1117599109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.