

Significance
We performed a screen for extrachromosomal circular DNAs containing segments of genomic yeast DNA. We found 1,756 such extrachromosomal circular DNAs containing about 23% of the total yeast genomic information. The abundance of these circular forms of genomic DNA suggests that eccDNA formation might be a common mutation that can arise in any part of the genome, and not in only a few special loci. We propose that eccDNAs may be precursors to the copy number variation in eukaryotic genomes characteristic of both the evolutionary process and cancer progression.
Keywords: circular DNA, chromosomal instability, gene deletion, homologous recombination, DNA replication origins
Abstract
Examples of extrachromosomal circular DNAs (eccDNAs) are found in many organisms, but their impact on genetic variation at the genome scale has not been investigated. We mapped 1,756 eccDNAs in the Saccharomyces cerevisiae genome using Circle-Seq, a highly sensitive eccDNA purification method. Yeast eccDNAs ranged from an arbitrary lower limit of 1 kb up to 38 kb and covered 23% of the genome, representing thousands of genes. EccDNA arose both from genomic regions with repetitive sequences ≥15 bases long and from regions with short or no repetitive sequences. Some eccDNAs were identified in several yeast populations. These eccDNAs contained ribosomal genes, transposon remnants, and tandemly repeated genes (HXT6/7, ENA1/2/5, and CUP1-1/-2) that were generally enriched on eccDNAs. EccDNAs seemed to be replicated and 80% contained consensus sequences for autonomous replication origins that could explain their maintenance. Our data suggest that eccDNAs are common in S. cerevisiae, where they might contribute substantially to genetic variation and evolution.
Copy number variants (CNVs) are alterations in number of copies of particular genes or other extensive DNA sequences in a genome. Gene deletions and amplifications are important sources of genetic variation that have proven important in the evolution of multicellular organisms. The high prevalence of families of paralogous genes, such as globins, protein kinases, and serine proteases, strongly supports the idea that gene duplication and divergence underlie much of evolution (1). In human somatic cells, CNVs have been implicated in cancer (2) and aging (3), as well as developmental and neurological diseases (4, 5). The best-studied types of CNVs are chromosomal gene amplifications and deletions that can easily be characterized by genome sequencing or tiling arrays (6, 7) or as cytological visible homogeneously staining regions (8).
Extrachromosomal circular DNAs (eccDNAs) represent a less studied form of CNVs although examples of eccDNAs have been detected in many organisms. In humans, eccDNAs are produced during Ig heavy-chain class switching (9). In higher plant genomes, repeat-derived eccDNAs are considered intermediates in the evolution of satellite repeats (10). In aging Saccharomyces cerevisiae cells, ribosomal rDNA circles accumulate (11). Also, in yeast, eccDNAs are generated from telomeric regions that contain many repeated sequences (12), and recombination between retrotransposon or their remnants in the form of solo long terminal repeats (LTRs) has been implicated in the formation of eccDNA (13, 14). Generally, eccDNAs are acentric and thus expected to missegregate during cell division, resulting in copy number variation (15). However, the abundance of eccDNAs and their role in evolution remains unknown.
Simple recombinational models have been proposed that connect duplications, deletions, and eccDNA with one another. In the simplest model, a gene deletion can be produced by intrachromatid ectopic recombination between tandem repeats that flank the same gene, simultaneously producing a circular DNA element (Fig. 1A) (14). CNVs are also produced by other mechanisms that do not require a circular intermediate, such as replication slippage (16), interchromatid or interchromosome recombination between nonallelic homologs (17). However, DNA studies of several loci in human germ cells suggest that the frequency of intrachromatid ectopic recombination, which leads to eccDNA, could be just as high as other CNV events (18). Incorrect nonhomologous end joining of DNA ends can generate eccDNA (Fig. 1A) (2, 19),) in addition to other mechanisms (Fig. 1A). These mechanisms could include microhomology-mediated DNA repair processes at double-strand DNA breaks (20, 21), as well as processes independent of DNA breaks, such as replication errors near short inverted repeats (22) or small single-stranded DNA that prime formation of eccDNA (23).
Fig. 1.

Genome-wide eccDNA measurement by Circle-Seq. (A) Models of extrachromosomal circular DNA (eccDNA) formation: intrachromatid ectopic homologous recombination (HR), nonhomologous end-joining (NHEJ), and other circularization mechanisms. (B) Circle-Seq procedure. From disrupted S. cerevisiae cells, eccDNA was purified by (1) column purification for circular DNA, (2) digestion of remaining linear DNA by plasmid-safe ATP-dependent DNase facilitated by NotI endonuclease, and (3) rolling-circle amplification by ϕ29 DNA polymerase and subsequent high-throughput DNA sequencing. (C) Sequenced 141-nucleotide reads were mapped to the S288C S. cerevisiae reference genome. Shown are data from reference sample R3 for part of chromosome IV. Green, paralogous genes; black, unique genes or ORFs; blue, read coverage mapped to the chromosome; gray boxes, individual reads with homology to the Watson (+) (light gray) and Crick (−) (dark gray) strands. (D) EccDNA was identified from regions covered by contiguous mapped reads >1 kb that were found to be significantly overrepresented by Monte Carlo simulation. Black bars, chromosomal regions recorded as eccDNAs; white bars, chromosomal regions excluded from the analysis (contiguous reads <1 kb).
There is likely an intimate connection between deletions, duplications, and circular DNA forms: reintegration of a circular copy of a gene by homologous recombination results in a duplication, just as recombination between duplicates can generate circular DNA copies and deletions. Chromosomal CNVs have been generally detected well after their establishment. Thus, the mechanism for formation of copy number variable regions can be inferred only from their structure (24, 25). Detection of new, potentially transient chromosomal duplications is challenging because it involves detection of alterations in single DNA molecules within large cell populations. We reasoned that detection and recovery of eccDNAs might be more tractable than other CNVs because of the unique and well-studied biophysical and biochemical properties of circular DNA molecules.
We developed a sensitive, genome-scale enrichment and detection method for eccDNA (Circle-Seq), based on well-established prokaryotic plasmid purification (26, 27) and deep sequencing technology. With Circle-Seq, we were able to purify, sequence, and map eccDNAs that, among them, cover nearly a quarter of the whole genome sequence of S. cerevisiae. We infer from the numerous eccDNA findings that circularization of genomic sequences is common enough to account for the generation of much of the variation in gene copy numbers observed in cancer and other human genetic diseases, as well as the process of evolution by duplication, deletion, and divergence.
Results
Circle-Seq, A Method for Genome-Scale Detection of eccDNA.
To obtain knowledge about the abundance of eccDNA in the S288C S. cerevisiae genome, we developed a method for genome-scale detection of eccDNA (Circle-Seq). EccDNAs were purified from 10 independent cultures of haploid S. cerevisiae cells grown in complete nutrient medium for 10 generations (1010 cells per population). After disrupting cells, the Circle-Seq method consists of three steps that exploit differences in the structural and chemical properties of circular and linear chromosomal DNA. The first step is denaturation and fast neutralization of DNA with alkali, followed by column chromatography to separate the fast reannealing circular DNA from linear DNA (Fig. 1B). The second step is treatment with the rare cutting endonuclease NotI and subsequent digestion with exonuclease to remove remaining linear single-stranded and double-stranded DNA. Elimination of linear DNA was confirmed by quantitative PCR for the actin encoding gene ACT1 (Fig. S1). The third step is enrichment of circular DNA by rolling circle amplification, using the highly processive ϕ29 DNA polymerase (Fig. 1B) (28).
Amplified circular DNA was sequenced as 141-nucleotide single-end reads on an Illumina Hi-Seq. We obtained 40–100 million reads from each of 10 independent populations. Virtually all (99.7–99.8%) reads mapped to the S. cerevisiae S288C reference genome (example in Fig. 1C). We considered mapped reads to indicate amplification of the circular DNA. To exclude residual noise from potential minor linear DNA fragments, we focused on sequence data that included a minimum of seven contiguous mapped reads corresponding to DNA regions >1 kb (Fig. 1D and Fig. S2). The chosen cut-off means that small eccDNAs were excluded from further analysis (example in Fig. 1D). Regions mapped with contiguous reads >1 kb were finally tested for the likelihood of occurring by chance using Monte Carlo simulations (Fig. S3). Regions that contained contiguous reads >1 kb and had a depth coverage significantly greater than expected by chance (P < 0.1) were considered as eccDNAs.
The number of >1-kb-long regions covered by contiguous reads increased as a function of sequenced DNA up to 30–45 million reads (Fig. 2). Sequencing beyond 45 million reads did not substantially increase the number of >1-kb-long regions covered by contiguous reads (Fig. 2), indicating that saturation was reached for most samples.
Fig. 2.

Contiguous reads >1 kb as function of sequence depth. Recorded eccDNA from 1 × 1010 cells increased as a function of sequence depth (in millions of mapped reads). Shown are quadruplicate reference samples (R1, R2, R3, R4) from mixed populations of ∼4,000 congenic single-gene deletion mutants from haploid S288C S. cerevisiae reference genome; an identical set of quadruplicate reference samples grown with 0.03 mM Zeocin (Z1, Z2, Z3, Z4); and duplicate samples of clonal isogenic haploid S288C populations (S1, S2). G1, G2, two populations that contain [GAP1circles] (14).
The Circle-Seq method identified a total of 1,756 different eccDNAs (Fig. 3 and Dataset S1). Combining and merging eccDNA regions from all 10 samples covered 23% of the S. cerevisiae genome, suggesting that a substantial fraction of the yeast genome is prone to circularization (Fig. S4). A large subgroup of the eccDNAs (38%, 669 eccDNAs) covered regions of DNA that were not unique in the reference genome. The homologous sequences included telomeric regions, gene duplications, and retrotransposons. The majority of eccDNAs were less than 5 kb whereas the longest recorded eccDNA was nearly 40 kb (Dataset S1). Most recorded eccDNAs (97.3%) carried at least a partial gene fragment (example in Fig. 1C), and only 2.7% were formed entirely from intergenic regions. A chromosomal overview of all 1,756 detected eccDNAs (Fig. 3) showed that eccDNAs formed from all chromosomes. Specific eccDNAs were annotated according to the nomenclature for non-Mendelian elements and superscripted to describe the allele type: [genecircle].
Fig. 3.

Genomic overview of eccDNA types detected in S. cerevisiae. Chromosomal map of S. cerevisiae eccDNA from Circle-Seq of 10 populations (R, Z, and S samples; see Fig. 2 legend). Highly conserved genes on eccDNAs are annotated, including genes that encode cell cycle regulators (CDC8 and CDC20), phosphoglucomutase (PGM2), a recombination repair protein (RAD57), and a helicase complex component (MCM7). Annotated also are genes encoding glucose, sodium, proline, and allantoin transporters HXT6, HXT7, ENA1, ENA2, ENA5, PUT4, DAL2, DAL4, and DAL7, as well as the asparagine synthetase gene ASN2, some retrotransposons (Ty elements) and telomeres with Y′ genes (see Dataset S1 for a list of all genes on eccDNAs). x axis, chromosomal coordinates for chromosomes I–XVI. (Scale bar, upper right.) y axis, logarithmic representation of normalization of each eccDNA to the number of mapped read fragments per kilobase from a million mapped reads (FPKM). Circles, unique eccDNA; triangles, nonunique eccDNA; black, eccDNAs with proposed replication origins or ARS sites; gray, eccDNAs with the ARS 17-bp consensus sequence (ACS); open circles and open triangles, eccDNAs without origin, ARS, and ACS; gene with two punctuation marks, more than one gene on the eccDNA. EccDNAs were annotated as unique if they included at least one uniquely mapped read to the reference genome and were annotated as nonunique if all included reads mapped equally well to several loci.
Replication Origins in eccDNA.
The considerable read coverage of eccDNAs indicated that at least some eccDNA replicated at mitosis. Previous studies showed that eccDNAs replicate and propagate in populations of yeast cells when they carry a replication origin (11, 13, 14). In S. cerevisiae, replication is controlled by autonomously replicating sequences (ARSs) that bind the origin recognition complex (ORC). More than 10,000 copies of the 17-bp ARS “core consensus sequence” (ACS) are found in the S. cerevisiae reference genome (29) although only 803 sites have been validated or proposed as replication origins (29–32). Of the 1,756 eccDNAs, 18.7% contained one of the 803 putative origins or ARS sites (Fig. 3). The ARS and origins were not found to be overrepresented on eccDNAs. However, when we included the ACS, we found that 80% of all eccDNAs included either an origin of replication, ARS, or ACS (Fig. 3 and Dataset S1).
Validating the Circle-Seq Procedure.
The Circle-Seq method was validated using three distinguishable exogenous plasmids spiked into lysates just before eccDNA purification at different ratios: specifically, 1:1 (plasmid:cell) for pBR322; 1:50 for pUC19; and 1:2,500 for pUG72. Each of the plasmids was detected by Circle-Seq (Fig. 4A and Fig. S5A), and plasmids were confirmed by size-specific DNA bands amplified by inverse PCR (Fig. S5B). Based on these findings, the detection sensitivity was at least 1 eccDNA per 2,500 cells (Fig. 4A and Fig. S5C).
Fig. 4.

Detection of known circular DNA elements. (A) Scatter plot shows fraction of uniquely mapped reads for plasmids spiked into samples after cell lysis and before column purification. Plasmids added in ratios per cell: pBR322 (crosses) 1:1, pUC19 (circles) 1:50, and pUG72 (triangles) 1:2,500. (B and C) Plots show fraction of uniquely mapped reads corresponding to 2µ (B) and mitochondrial DNA (mtDNA) (C). See Fig. 2 legend for description of the 10 populations (R, Z, and S samples). (D) Read coverage across the GAP1 locus on chromosome XI after Circle-Seq of G1 and G2 populations containing [GAP1circles]. (E) Analysis of [GAP1circle] junctions using single nucleotide polymorphisms (SNPs) in the LTRs YKRδ11 and YKRδ12. Recombination occurred between nucleotides in YKRδ11/12 as illustrated by a representative recombination junction identified in both G samples. The recombination site is shown by the square box supported by at least 50 junction reads. Vertical lines indicate homology between YKRδ11 and YKRδ12.
We identified the very abundant and naturally occurring circular DNA species, the mitochondrial DNA (mtDNA), and the 2µ plasmid (Fig. 4 B and C). The Circle-Seq method also recorded reads at chromosomal loci previously reported to circularize and form eccDNA. These eccDNAs included the ribosomal RNA genes (11), the paralogous metallothionein genes CUP1-1 and CUP1-2 (33), and telomeres (12). The [rDNAcircle] (Fig. 3 and Fig. S6A), the [CUP1/2circle] (Fig. 3 and Fig. S6B), and circularized telomeric Y′ elements (Fig. 3; example in Fig. S6C) were found in all samples tested by Circle-Seq.
To further verify that Circle-Seq recovered genomic sequences known to circularize and amplify under appropriate selection, biological replicates of a [GAP1circle] mutant (14) were grown in l-glutamine as a sole nitrogen source. The GAP1 locus, encoding the general amino acid permease 1, was extensively represented in reads obtained after Circle-Seq of the two cultures (G1 and G2) (Fig. 4D). Presence of [GAP1circles] was confirmed by inverse PCR (Fig. S7), and, as expected, [GAP1circles] were not recorded in yeast populations cultivated in complete media (Fig. S7). We previously showed that [GAP1circles] form by recombination between two homologous long-terminal repeats (LTRs), YKRδ11 and YKRδ12, that flank the GAP1 gene (14). We confirmed the recombination using distinguishable single-nucleotide polymorphisms in YKRδ11 and YKRδ12 as fingerprints for reads mapping across the recombination site (Fig. 4E).
EccDNAs Represent Diverse Genomic Loci.
To explore the number and types of eccDNAs in a population of yeast cells, we analyzed eccDNA from four reference populations that were mixtures of single-gene deletion mutants (R1–R4). We compared the reference samples to clonal isogenic WT populations (S1 and S2). We also examined the effect of DNA damage on eccDNAs by growing the pooled, single-gene deletion mutants in the presence of the DNA-damaging agent Zeocin (Z1–Z4) (Fig. 2). The reference R1–R4 samples had an average of 147 eccDNAs per sample (181, 42, 248, and 118 eccDNAs); the clonal S1 and S2 populations had a similar average of 146 eccDNAs per sample (144 and 148 eccDNAs); whereas the Z1–Z4 samples had an average of 219 eccDNAs (159, 210, 218, and 288 eccDNAs). Populations treated with the DNA-damaging agent Zeocin showed a tendency to more eccDNA types than the untreated reference (R1–R4) (Fig. 2 and Fig. S4). This result suggested that single-strand and double-strand DNA breaks contributed to formation of eccDNAs, in agreement with studies in S. cerevisiae (34), Drosophila melanogaster (35, 36), and human carcinoma KB-V1 cells (37).
A majority (72%) of the genes on eccDNAs in the reference R1–R4 samples were unique to a particular sample (74% in Z1–Z4 samples); only 15.4% of the genes were common to all four R-samples (Fig. 5A; 12.9% in Z-samples). In addition to sequences from 2µ, mtDNA, [rDNAcircle], and [Y′-TELcircles], several genes were found on eccDNAs in all four reference samples. These genes included HXT6, HXT7, encoding high-affinity plasma membrane hexose transporters; ENA1, ENA2, and ENA5, encoding sodium pump ATPases; and CUP1-1 and CUP1-2, encoding metallothioneins (Fig. 5A). Almost all genes common for the reference samples were also present in other samples (Fig. 5 B and C and Dataset S2). The orthologous hexose transporter genes HXT6 and HXT7 (38) are 99.7% identical, suggesting that the [HXT6/7circle] most likely formed by homologous recombination between the two genes (Fig. 5C). We confirmed the existence of [HXT6/7circle] in all samples using inverse PCR on DNA enriched for eccDNA and in samples without linear DNA (Fig. 5D). In addition to multiple reads covering the HXT6 and HXT7 locus, reads also covered the intergenic region next to HXT6 and the hexose transporter ortholog HXT3 (38). These mapped reads suggested the existence of other [HXTcircles] formed by recombination between HXT3 and HXT7 or HXT3 and HXT6. Inverse PCR confirmed the [HXT3/6circle] in samples, R2, Z2, and S2 (Fig. 5D).
Fig. 5.
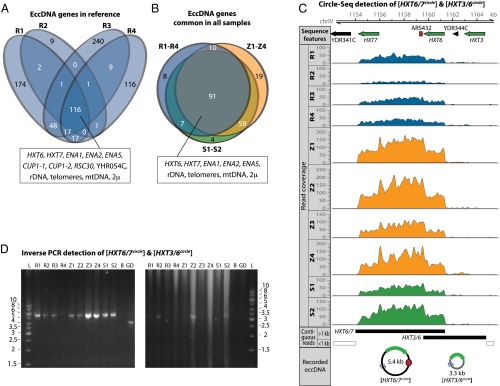
Common eccDNA elements. (A) Venn diagram displaying overlap among 747 genes on 612 eccDNA elements in four reference samples (R1, R2, R3, R4). (B) Venn diagram of the 116 redundant genes present in all reference samples (R1–R4) compared with eccDNA genes present in all samples of Z1–Z4, and S1 and S2, respectively. (C) Read coverage of the HXT7, HXT6, and HXT3 locus on chromosome IV, recording the [HXT6/7circle] and the [HXT6/3circle]. Blue arrows on recorded eccDNA indicate the position of inverse PCR primers for D. (D) Confirmation of [HXT6/7circle] and [HXT6/3circle] by inverse PCR and agarose gel electrophoresis. Samples R1–R4, Z1–Z4, and S1 and S2 as in Fig. 2 legend. Confirming DNA bands are found at 5.4 kb and 3.3 kb. L, molecular weight marker; B, no DNA template; GD, genomic DNA from S288C.
Mechanisms of eccDNA Formation.
As suggested for the [HXT6/7circle], homologous recombination between repeated sequences is both a simple and intuitive mechanism for eccDNA formation although there are alternative mechanisms, some examples of which have been documented in eukaryotes (2, 19–21, 23, 39). We explored the extent to which eccDNAs were products of recombination between homologous regions preexisting in chromosomes. Eight percent (140 eccDNAs) of all putative eccDNAs (1,756) had homologous sequences longer than 50 nucleotides (>50 nt), and another two percent (35 eccDNAs) contained shorter homologous sequences between 15 and 50 nucleotides (Dataset S1). Homologous regions >50 nt included intragenic repeats of FLO11 and paralogous genes such as HXT6/HXT7 (Fig. 5C), CUP1-1/CUP1-2 (Fig. S6B), and ENA1/ENA2/ENA5 (Fig. S8), as well as a large group of elements consisting of long terminal repeats (LTRs), retrotransposons, and solo LTRs (Dataset S1). One example was the [Ty1_NUP116_CSM3_ERB1_Ty1circle] that seemed to be formed by recombination between two Ty1 LTR elements flanking NUP116_CSM3_ERB1 that respectively encode a nucleoporin component, a replication fork associated factor, and a constituent of 66S pre-ribosomal particles. Inverse PCR confirmed that recombination had taken place between YMRCTy1-3 and YMRCTy1-4, leaving one chimeric Ty1 element on the eccDNA (Fig. S9). Overall, LTR elements were significantly overrepresented in the recombination site of unique eccDNAs compared with the LTRs in the genome (P < 0.001 by Monte Carlo simulation), suggesting that LTR elements are more likely to recombine and circularize than other elements in the genome.
Next, we searched for DNA circularization events that could involve microhomologies or end joining at the recombination sites. We searched for sequence reads that spanned the recombination junction and identified such reads at 130 eccDNAs (7.4%, excluding nonunique eccDNAs, rDNA region at chrXII, and plasmids) (Dataset S1). These reads strongly supported the existence of the corresponding eccDNAs and gave exact information about the chromosomal DNA that had served as substrate for DNA circularization (e.g., Fig. 6A and Fig. S10).
Fig. 6.

EccDNA read junctions and overall distribution. (A) Example of junction reads spanning the eccDNAs [CDC8circle], [CDC20circle], [MCM7circle], [PGM2circle], and [ASN2circle] compared with the genomic sequence. All junctions are based on at least two junctions reads each. Junction points are marked with a dashed line or a square box. (B) Pie chart of distribution of homologous sequences at putative eccDNA recombination sites based on BLAST+ analyses. Nt, nucleotides; N.A., none annotated.
Combined analyses of homologous recombination sites and junction reads revealed that ∼10% of the different eccDNAs (174 eccDNAs) arose between chromosomal regions with ≥15-nt homology (e.g., [HXT6/7circle]) (Fig. 5C) whereas 9% (161 eccDNAs) derived from regions with 7- to 14-nt homology (e.g., [MCM7circle] and [ASN2circle]) (Fig. 6A), and another 3% (60 eccDNAs) derived from regions with less than <7-nt homology (e.g., [CDC8circle], [CDC20circle], and [PGM2circle]) (Fig. 6A). The remaining 78% (1,361 eccDNAs) could not be annotated (Fig. 6B) either because junction reads were not located or because larger stretches of homology did not exist at the edges of chromosomal coordinates from which eccDNA likely arose. Thus, we examined whether eccDNAs could have arisen between short stretches of microhomologies. We found that 90% of all eccDNAs contained small direct and/or inverted repeats at edges of eccDNA coordinates (≥7 nt, 200-bp windows) although these repeats were not overrepresented at eccDNA regions (Dataset S3). We finally considered the frequency of different eccDNA types. When information about the number of reads per eccDNA was taken into consideration, eccDNAs that derived from genomic regions with ≥15-nt repetitive sequences constituted 99.3%. More than 99% of these eccDNAs were [rDNAcircles] (Fig. 3). Excluding [rDNAcircles] from the analysis showed that 67% arose from regions with ≥15-nt homology. Thus, one-third of the eccDNA seemed to arise from regions in the yeast genome with short (<15 nt) or no repetitive sequences.
Discussion
In this, to our knowledge, first systematic survey of the yeast genome for eccDNA, we found more than a 1,000 unique eccDNA elements, which increases the number of reported eccDNAs in yeast by more than 100-fold. Our findings are likely an underestimate of the true set of eccDNAs because the majority of the identified eccDNAs were unique to a single population and elements smaller than 1 kb were not included in the analysis. Therefore, we expect that additional sampling would reveal a higher coverage of the genome, and we suggest that virtually any part of the yeast genome has the potential to circularize and form eccDNA.
In this study, eccDNA seemed to form throughout the S. cerevisiae genome (Fig. 3) although some eccDNAs were more abundant than others. We found that unique [LTRcircles] were significantly overrepresented at putative eccDNA recombination sites, suggesting that adjacent retrotransposons increased the likelihood of DNA circularization. However, we suggest that selection, even mild selection, is likely to be a more important factor in explaining differences in abundances, as exemplified by our results with [GAP1circles]. Also significant is the ability of the eccDNA to replicate, which likely depends on the specific type of ARS or origin it contains. We speculate that the core consensus sequence for replication (ACS) could be sufficient for DNA synthesis of circular molecules (e.g., [CDC8circle]) (Fig. 3). Alternatively, eccDNAs without active replication origins or ACS sites (e.g., [ASN2circle]) (Fig. 3) may amplify through high formation rates and/or by rolling circle replication during their formation (40, 41).
A proportion of eccDNAs (7%, 130) were confirmed by reads that spanned the circularization junctions (e.g., in Fig. 6). These reads provided information about the chromosomal loci that served as substrate for eccDNA formation and could be indicative of the mechanisms by which formation took place. Combined with our search for homologous sequences at putative recombination sites, we found in the annotated groups of 395 eccDNAs (Fig. 6B) that 15% of the chromosomal loci had <7-nt homology around junctions, 41% had 7- to 14-nt homology, and another 44% had ≥15-nt homology, suggesting that DNA circularization is mediated by homologous recombination, nonhomologous end joining, and microhomology-mediated DNA repair. When the frequency of eccDNA was taken into consideration, eccDNA that arose from regions with repeats of ≥15-nt homology constituted two-thirds of the eccDNA (67%). However, in-depth understanding of the molecular mechanisms that lead to the formation of eccDNA requires additional experimental evidence, such as eccDNA profiles of mutants depleted of factors in homologous recombination and nonhomologous end joining (e.g., rad52 and dnl4).
The most notable eccDNA that appeared in all samples was the [HXT6/7circle] (Fig. 5). This eccDNA contains a well-documented ARS sequence, and selection for chromosomal gene amplifications of HXT6 and HXT7 has been reported when yeast evolve under glucose limitation (42). Thus, it is possible that the [HXT6/7circle] is an intermediate in the generation of stable chromosomal HXT6 and HXT7 amplifications. In general, any eccDNA could contribute to stable gene amplification and gene conversion.
Besides eccDNAs’ impact on genetic variation through deletions and amplifications, self-replicating eccDNAs might also affect the rate of evolution on their own. When the unit of selection is a gene, its heritability is greatly altered when it is situated on a self-replicating eccDNA element compared with the same locus on the chromosome. Unlike chromosomes, acentric eccDNAs segregate asymmetrically, which can lead to rapid loss or amplification of eccDNA genes compared with chromosomal genes, thereby following the criteria for faster evolution (43). For instance, [GAP1circles] self-replicate and were frequently found in DNA preparations from cultures that were propagated on glutamine as the sole nitrogen source (Fig. 4D), but not in cultures grown on complete media with abundant amino acids as source of nitrogen (Fig. S6). This difference underlines that a gene on eccDNA can provide a fitness advantage (14) and presumably, once a [GAP1circle] is generated, offers a more rapid response to selection. Thus, the large array of discovered eccDNAs in the present study provide a large set of opportunities for copy number variations that may have large and so far overlooked impact on the evolution of genomes.
The Circle-Seq method should be readily adaptable to find eccDNA in other organisms. Other global screening methods, such as whole-genome sequencing and tiling arrays, have failed to identify large numbers of eccDNAs. This circumstance might be because of the low frequency of many eccDNAs, which are not expected to be detectable with conventional sequencing methods (44). De novo discovery of eccDNAs in the past has been by Giemsa staining of metaphase chromosomes (45), fluorescence in situ hybridization (15), 2D gel electrophoresis (35), or introduction of selective markers combined with Southern blotting (12, 33) and inverse PCR (14). These methods detect abundant eccDNA elements but are not suitable for genome-wide screening for novel eccDNAs or detecting eccDNA that is low in abundance. A recent study from human tissue identified thousands of 200- to 400-base-pair-long nucleotide circles, which carried parts of exons and other gene fragments (21). It remains to be investigated whether larger eccDNA can be abundantly isolated by this method.
Because yeast is a typical eukaryote in its genome organization, chromatin structure, and DNA biochemistry, eccDNA may well be a common feature of variation in other eukaryotes, including plants and animals. Genome-wide screening for eccDNA elements using Circle-Seq could facilitate studies on eukaryotic genomes that will lead to better understanding of copy number variation, evolution, and many both rare and common genetic diseases that are associated with copy number variation.
Materials and Methods
Media.
Complete yeast peptone dextrose (YPD) media contained 1% (wt/vol) yeast extract (BD Difco), 2% (wt/vol) peptone (BD Difco), and 2% (wt/vol) d-glucose. YPD containing 1,000 mg/L Geneticin (G418) (Gibco-BRL) was used to propagate strains with integrated KanMX-cassettes. Cultures treated with a DNA damaging agent were grown in YPD+G418 medium supplemented with 0.03 mM Zeocin (Invitrogen, Life Technologies). Minimal l-glutamine medium (MG) included 0.16% (wt/vol) yeast nitrogen base without amino acids and ammonium sulfate (BD Difco), 2% (wt/vol) d-glucose, and 0.35 mM l-glutamine.
Strains and Plasmids.
Reference samples (R1–R4 and Z1–Z4) consisted of a pool of 4,400 viable single-gene deletion yeast mutants with KanMX module disruptions from the yeast deletion collection in the S288C background BY4741 (MATa his3∆1 leu2∆0 met15∆0 ura3∆0) (46). M3750 served as the clonal isogenic population in the S288C background (S1 and S2, MATa ura3 Gal2) (47). Clones carrying [GAP1circles] in the CEN.PK background (G1 and G2) served as the positive control for eccDNA (14). Plasmids pBR322 (4,361 bp; New England Biolabs), pUC19 (2,686 bp; New England Biolabs), and pUG72 (originally pJJH726, 3988 bp; EUROSCARF) were maintained in Escherichia coli and purified with QIAprep Spin Miniprep Kits (Qiagen).
Cell Culturing.
Overnight cultures were used to inoculate independent starter cultures for quadruplicate cultures, except for clonal isogenic S288C and [GAP1circle] populations, which were grown in duplicate. Cells were diluted to 6 × 105 ± 3 × 105/mL in baffled flasks containing 45 mL of complete YPD medium and propagated for nearly 10 generations, to a maximum cell density after 48 h growth at 30 °C with agitation, 150 rpm. Yeast cells counts and viability were measured by propidium iodide staining, obtaining 1 × 1010 cells, of which 98% were viable (NucleoCounter NC-3000; Chemometec). Quadruplicate cultures of reference samples were grown in YPD+G418 whereas another set of quadruplicate cultures were grown in YPD+G418 with 0.03 mM Zeocin throughout the 48-h growth period. The [GAP1circle] mutant (14) was grown in 100 mL of MG medium for 72 h to 4.8 × 107 cells per mL.
Circle-Seq eccDNA Purification.
Purification of eukaryotic eccDNA was accomplished in three steps, developed and modified from a previous prokaryote plasmid DNA protocol (26, 27).
Purification and enrichment of eccDNA.
For each sample, 1 × 1010 cells were pelleted (5 min, 3,000 rpm), washed in 5 mM Tris⋅Cl, pH 8, and resuspended in 0.6 mL of L1 solution (Plasmid Mini AX; A&A Biotechnology). Suspended cells were transferred to 2-mL microcentrifuge tubes with 0.5 mL of 0.5-mm glass beads (Scientific Industries, Inc.) and disrupted by maximum vortexing for 10 min. Beads were removed by centrifugation at 270 × g, 30 s. Supernatants with cell lysates were transferred to clean tubes. Plasmid mixtures were added in a per-cell ratio of 1:1 for pBR322, 1:50 for pUC19, and 1:2,500 for pUG72, assuming that each sample contained 8 × 109 cells (genomes). Following the manufacturer’s instructions for Plasmid Mini AX kits (A&A Biotechnology), each sample was alkaline treated, followed by precipitation of proteins and separation of chromosomal DNA from circular DNA through an ion exchange membrane column. Column-bound DNA was eluted, precipitated, and dissolved in TE buffer (10 mM Tris-Cl, pH 8.0; 1 mM EDTA, pH 8.0), obtaining 6–16 µg of DNA.
Digestion of remaining linear DNA.
To increase the number of ends accessible for exonuclease digestion, the DNA from each sample was treated with 1 FastDigest Unit of NotI endonuclease (Fermentas) at 37 °C. After 17 h, samples were heat treated for 5 min at 80 °C and digested with exonuclease at 37 °C using Plasmid-Safe ATP-dependent DNase (Epicentre). To ensure complete hydrolysis of linear double-stranded DNA, additional ATP and DNase was added every 24 h (16 units per day). Samples were tested for the elimination of linear DNA by quantitative PCR (Fig. S1). After 130 h, the initial amount of linear DNA (estimated as 130 µg) was diminished between 5 × 107-fold and 4 × 108-fold. The DNase was heat inactivated for 30 min at 70 °C.
Amplification of eccDNA.
From each eccDNA-enriched sample, 1% (5 µL) was amplified by ϕ29 polymerase at 30 °C for 16 h according to the REPLI-g Mini Kit protocol (Qiagen).
PCR.
The ACT1 gene on chromosome VI was used to measure remaining linear DNA. Linear DNA content was analyzed in samples during the exonuclease time course using the following standard PCR methods and primers: ACT1 5′-TGGATTCTGGTATGTTCTAGC-3′ and 5′-GAACGACGTGAGTAACACC-3′ and quantitative PCR (qPCR) primers 5′-TCCGTCTGGATTGGTGGTTCTA-3′ and 5′-TGGACCACTTTCGTCGTATTC-3′. Reactions were in an Mx3000 (Agilent Technologies) qPCR machine in 20-µL reactions containing 2 µL of template (exonuclease-treated samples), 150 nM primers, 2% (vol/vol) DMSO, and 10 µL of Brilliant III SYBR Green PCR Master Mix (Agilent Technologies). Reaction conditions were 3 min at 95 °C followed by 45 cycles of 15 s at 95 °C and 30 s at 60 °C. Denaturing curves were generated at the ends of runs to verify reaction specificity. Purified genomic DNA from S. cerevisiae strain S288C in seven serial dilutions was included in quadruplicate for each run to produce standard curves. Copy numbers were calculated assuming a molar mass per base pair of 650 g⋅mol−1⋅bp−1 and a genome length of 1.2 × 107 bp and one copy of ACT1 per genome. Samples were run in triplicate for SD calculations. Concentrations of standards were measured using a Qubit High Sensitivity assay (ThermoFisher Scientific). PCR products were visualized by 1% agarose gel electrophoresis with 100-bp ladders from New England Biolabs.
The following inverse PCR primers were used for detection of eccDNA: [GAP1circle], 5′-CGAAGATATTCGACGAGG-3′ and 5′-GAACTTTGGGGATTCGG-3′; [HXT6/7circle], 5′-CTCCGTCAGAGGCTG-3′ and 5′-CAGTACCGAGGTGAGC-3′; [HXT3/6circle], 5′-AAACGAAATCCATCATCACG-3′ and 5′-GAAATCCGGACGGAAAACTC-3′; and [CSM3circle], 5′-GCGCCATTCATGAAGATGAT-3′ and 5′-ACCACCCGAACATGGAATTTA-3′. For detection of spiked-in plasmids, the inverse primers were pBR322, 5′-CCAACCCTTGGCAGAACATA-3′ and 5′-TTTGCGCATTCACAGTTCTC-3′; pUC19, 5′-GGTACCGAGCTCGAATTCAC-3′ and CGGGGATCCTCTAGAGTCG-3′; and pUG72, 5′-ATTGACGGGAGTGTATTGACG-3′ and 5′-TAGTTGCACCATGTCCACAAA-3′. Inverse PCR primers were designed to yield full-length products for [HXT6/7circle] (5.4 kb), [HXT3/6circle] (3.3 kb), pUC19 (2.7 kb), pUG72 (4.0 kb), and pRB322 (4.4 kb). Inverse PCR primers were designed to yield products of the recombination site [CSM3circle] (6 kb) and [GAP1circle] (0.6 kb). PCR reactions were carried out with 0.2 ng of template from ϕ29 amplified sample or 4 µL of template from 130-h exonuclease-treated sample using standard conditions. All reactions were performed at least twice.
Sequencing.
The ϕ29-amplified DNA samples were sheared by sonication to an average of 300 nucleotides (Covaris LE220). Postsonication, DNA was adjusted to 20 ng/µL. For each library preparation, 300 ng of fragmented DNA was loaded onto a robotic Apollo 324 system (IntegenX Inc.), adding adapters and barcode index labels (6-base oligos). The 12 samples were sequenced up to 100 Mb as single ends using an Illumina HiSEq. 2000 platform.
Mapping of Reads.
Reads were filtered by index barcodes, allowing 0 mismatches in the first sequence lane (>10 samples) and 1 mismatch in additional sequencing (<5 samples per lane). Adapter sequences were trimmed using Cutadapt (48), only for the longest sequence reads (215 nucleotides for S2, Z4, and R1 samples). The 141-nucleotide reads were mapped to the S288C reference genome using the Galaxy workflow system (49) and Bowtie2 mapping software, using local alignment settings and very sensitive search mode (50).
Identification of Regions with Contiguous Coverage >1 kb.
Mapped data from identical samples were merged, and a workflow was established to identify regions longer than 1 kb with contiguous read coverage (i.e., no gaps in coverage, usually translated to more than seven connected reads). Reads were allowed to map to multiple regions of the genome to capture information about repetitive regions. After mapping, Sambamba (https://zenodo.org/record/13200) was used to select only uniquely mapped reads that mapped to a single genome position. The number of fragments mapping to each contiguous region was counted, both for all mapped reads and for uniquely mapped reads. Counts were used to calculate fragments per kilobase per million mapped reads (FPKM) (51). When calculating the FPKM for regions using only uniquely mapped reads, region size was reduced to include only uniquely mappable positions within the region. In addition, the number of proposed origins (29–32) in each contiguous region >1 kb was counted. Moreover, the 17-bp conserved ARS consensus sequence (ACS) reported by Breier et al. (WWWWTTTAYRTTTWGTT; W = A or T, Y = C or T, and R = A) (29) was intersected with putative eccDNA regions (Dataset S1).
Monte Carlo Simulation.
The confidence level of each contiguous region >1 kb was assessed using a Monte Carlo simulation. The locations of putative eccDNAs were randomized throughout the genome, excluding regions known to be circular: 2µ, mtDNA, spiked plasmids, and the redundant [rDNAcircle] at chromosome XII:451323.0.491422. The FPKM of the randomized regions was calculated and used to generate a distribution. For each putative eccDNA region, an empirical P value was generated for each contiguous region >1 kb, based on the frequency of a randomly permuted location having the same or higher FPKM value. P value calculations were performed twice, first using all mapped reads (Dataset S1) and second using only uniquely mapped reads (Fig. S3 and Dataset S1). Each contiguous region >1 kb, with P < 0.1 using uniquely mapped reads was considered to originate from circular DNA and designated an unique eccDNA. In the case of P = 1 from simulations with uniquely mapped reads (i.e., no uniquely mapped reads), the P < 0.1 from simulation with all mapped reads was used as the cutoff, and each of these eccDNAs were designated an nonunique eccDNA.
An [LTRcircle] was defined as a region in which annotated LTR regions (52) overlapped within the first 100 bp of the start of the eccDNA region and the last 100 bp of the end of the eccDNA region. For each of the 10,000 iterations, the locations of unique eccDNAs were randomized across the S288C genome, excluding plasmids and nonunique [LTRcircle] regions identified in any sample. The number of [LTRcircles] for randomized regions was counted and compared with the actual count to calculate P values. To determine whether eccDNAs were enriched for replication origins and ARS regions (29–32), we performed a Monte Carlo simulation using pybedtools version 0.6.8 (53, 54). The locations of eccDNAs were randomly shuffled 10,000 times; each time we counted the number of intersections with origins/ARS regions. Using these counts, we constructed an empirical distribution and compared the number of intersections between the original, unshuffled eccDNA locations to obtain a P value.
BLAST Analysis.
Each eccDNA region was extended 200 nucleotides at each flank (if possible) and then divided into two regions of equal length. The two pieces were aligned using BLAST+ blastn (55) and the “blastn” task. Only the top hit was recorded, annotating the positions of aligned sequences, number of mismatches, gaps, and e-values (Dataset S1). EccDNAs that contained homologous sequences ≥15 nucleotide long (identified by BLAST+) were considered to had circularized by homologous recombination (Dataset S1).
Junction Read Analysis.
Each read was split into three pieces of equal length. The start and ends of each split read were mapped to the reference genome. The read fragments were mapped as paired-end reads using Bowtie2, with the expectation that each split read would map in the same orientation and within 300 bases of each other. Reads that mapped discordantly (i.e., misoriented and/or more than 300 bp from each other) were extracted and treated as potential junction reads. Junction reads that mapped uniquely to the genome and within annotated unique eccDNA regions (extended by 200 nucleotides at each flank) were counted, and the genomic coordinates mapped by more than two split reads were merged and annotated (Dataset S1). The 669 nonunique eccDNAs were excluded from this analysis due to the redundant mapping of reads.
Distribution of eccDNA Types.
All 1,756 eccDNAs (P < 0.1) were grouped (i) according to the content of homologous regions at putative eccDNA flanks or (ii) based on homologous regions at sites mapped by junction reads (see BLAST Analysis and Junction Read Analysis). The frequency of eccDNAs that arose from regions with ≥15-nt homology was calculated after the 395 annotated eccDNAs (Fig. 6B) ± the 17 recorded [rDNAcircles] on chromosome XII:451323.0.479776. The 17 recorded [rDNAcircles] were annotated to the group with ≥15-nt homology. The fraction was calculated as the total number of reads per eccDNA in the group with ≥15-nt homology junctions divided by the sum of the total number of reads per eccDNA in both groups (<15-nt homology junctions plus ≥15-nt homology junctions).
Direct and Inverted Repeat Search.
Each eccDNA region was extended by 100 nucleotides at each flank (if possible), and 200 nucleotides from each edge were aligned with BLAST+ blastn (55), using the “blastn-short” task. Each hit was recorded as either a direct or an inverted repeat. At regions where merged split reads were identified (see Junction Read Analysis), a similar BLAST+ analysis was performed adding 50 nucleotides to the edge of a merged split read region and aligning 75 nucleotides from each end.
Junction Mapping, [GAP1circle].
For the [GAP1circle], formed by homologous recombination, we mapped the recombination junction using single-nucleotide polymorphism (SNP) differences in the two long terminal repeats. We determined the recombination site between two informative SNPs. The site was located by first generating a list of coordinates of the repeat regions flanking the eccDNA plus flanking sequences inside the DNA circle. These coordinates were used to extract FASTA sequences of repeat regions using the BedTools getfasta command (53). Informative SNPs were identified by aligning the repeat regions using ClustalW (56) and identifying positions where the two repeats differed. Extracted sequences were joined into a single sequence representing the estimated circular joining region containing a single copy of the repeat. Sequencing reads were aligned to the join region using Bowtie2 with default parameters (50). Freebayes (57) was run in pooled-discrete mode with the list of informative SNPs as input. The number of reads that agreed with the genotype of each copy of the repeat was captured and used to identify the two closest SNP locations that flanked the recombination junction.
Supplementary Material
Acknowledgments
We thank David Gresham and Tom Gilbert for constructive comments. H.D.M. and B.R. received funding through a fellowship from the Faculty of Science of the University of Copenhagen and through the Blissett family. L.P. and D.B. received funding from National Institute of General Medical Sciences Center for Quantitative Biology Grant GM071508, and D.B. received funding from National Institutes of Health Grant GM046406.
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequence reported in this paper has been deposited in the European Nucleotide Archive (primary accession no. PRJEB6368 secondary accession no. ERP005892).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1508825112/-/DCSupplemental.
References
- 1.Ohno S. Evolution by Gene Duplication. Springer; Berlin: 1970. [Google Scholar]
- 2.Storlazzi CT, et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: Origin and structure. Genome Res. 2010;20(9):1198–1206. doi: 10.1101/gr.106252.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsieh JCF, Van Den Berg D, Kang H, Hsieh C-L, Lieber MR. Large chromosome deletions, duplications, and gene conversion events accumulate with age in normal human colon crypts. Aging Cell. 2013;12(2):269–279. doi: 10.1111/acel.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447(7147):932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 5.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gresham D, et al. Genome-wide detection of polymorphisms at nucleotide resolution with a single DNA microarray. Science. 2006;311(5769):1932–1936. doi: 10.1126/science.1123726. [DOI] [PubMed] [Google Scholar]
- 7.Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453(7191):56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biedler JL, Spengler BA. Metaphase chromosome anomaly: Association with drug resistance and cell-specific products. Science. 1976;191(4223):185–187. doi: 10.1126/science.942798. [DOI] [PubMed] [Google Scholar]
- 9.von Schwedler U, Jäck HM, Wabl M. Circular DNA is a product of the immunoglobulin class switch rearrangement. Nature. 1990;345(6274):452–456. doi: 10.1038/345452a0. [DOI] [PubMed] [Google Scholar]
- 10.Navrátilová A, Koblízková A, Macas J. Survey of extrachromosomal circular DNA derived from plant satellite repeats. BMC Plant Biol. 2008;8(1):90. doi: 10.1186/1471-2229-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinclair DA, Guarente L. Extrachromosomal rDNA circles: A cause of aging in yeast. Cell. 1997;91(7):1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 12.Horowitz H, Haber JE. Identification of autonomously replicating circular subtelomeric Y’ elements in Saccharomyces cerevisiae. Mol Cell Biol. 1985;5(9):2369–2380. doi: 10.1128/mcb.5.9.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Libuda DE, Winston F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature. 2006;443(7114):1003–1007. doi: 10.1038/nature05205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gresham D, et al. Adaptation to diverse nitrogen-limited environments by deletion or extrachromosomal element formation of the GAP1 locus. Proc Natl Acad Sci USA. 2010;107(43):18551–18556. doi: 10.1073/pnas.1014023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Windle B, Draper BW, Yin YX, O’Gorman S, Wahl GM. A central role for chromosome breakage in gene amplification, deletion formation, and amplicon integration. Genes Dev. 1991;5(2):160–174. doi: 10.1101/gad.5.2.160. [DOI] [PubMed] [Google Scholar]
- 16.Wolfson R, Higgins KG, Sears BB. Evidence for replication slippage in the evolution of Oenothera chloroplast DNA. Mol Biol Evol. 1991;8(5):709–720. doi: 10.1093/oxfordjournals.molbev.a040680. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, et al. Gene copy-number variation in haploid and diploid strains of the yeast Saccharomyces cerevisiae. Genetics. 2013;193(3):785–801. doi: 10.1534/genetics.112.146522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turner DJ, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40(1):90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Loon N, Miller D, Murnane JP. Formation of extrachromosomal circular DNA in HeLa cells by nonhomologous recombination. Nucleic Acids Res. 1994;22(13):2447–2452. doi: 10.1093/nar/22.13.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogt N, et al. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc Natl Acad Sci USA. 2004;101(31):11368–11373. doi: 10.1073/pnas.0402979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shibata Y, et al. Extrachromosomal microDNAs and chromosomal microdeletions in normal tissues. Science. 2012;336(6077):82–86. doi: 10.1126/science.1213307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brewer BJ, Payen C, Raghuraman MK, Dunham MJ. Origin-dependent inverted-repeat amplification: A replication-based model for generating palindromic amplicons. PLoS Genet. 2011;7(3):e1002016. doi: 10.1371/journal.pgen.1002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukherjee K, Storici F. A mechanism of gene amplification driven by small DNA fragments. PLoS Genet. 2012;8(12):e1003119. doi: 10.1371/journal.pgen.1003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kugelberg E, et al. The tandem inversion duplication in Salmonella enterica: Selection drives unstable precursors to final mutation types. Genetics. 2010;185(1):65–80. doi: 10.1534/genetics.110.114074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reams AB, Kofoid E, Savageau M, Roth JR. Duplication frequency in a population of Salmonella enterica rapidly approaches steady state with or without recombination. Genetics. 2010;184(4):1077–1094. doi: 10.1534/genetics.109.111963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li LL, Norman A, Hansen LH, Sørensen SJ. Metamobilomics: Expanding our knowledge on the pool of plasmid encoded traits in natural environments using high-throughput sequencing. Clin Microbiol Infect. 2012;18(Suppl 4):5–7. doi: 10.1111/j.1469-0691.2012.03862.x. [DOI] [PubMed] [Google Scholar]
- 27.Brown Kav A, et al. Insights into the bovine rumen plasmidome. Proc Natl Acad Sci USA. 2012;109(14):5452–5457. doi: 10.1073/pnas.1116410109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blanco L, et al. Highly efficient DNA synthesis by the phage ϕ 29 DNA polymerase: Symmetrical mode of DNA replication. J Biol Chem. 1989;264(15):8935–8940. [PubMed] [Google Scholar]
- 29.Breier AM, Chatterji S, Cozzarelli NR. Prediction of Saccharomyces cerevisiae replication origins. Genome Biol. 2004;5(4):R22. doi: 10.1186/gb-2004-5-4-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raghuraman MK, et al. Replication dynamics of the yeast genome. Science. 2001;294(5540):115–121. doi: 10.1126/science.294.5540.115. [DOI] [PubMed] [Google Scholar]
- 31.Wyrick JJ, et al. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: High-resolution mapping of replication origins. Science. 2001;294(5550):2357–2360. doi: 10.1126/science.1066101. [DOI] [PubMed] [Google Scholar]
- 32.Nieduszynski CA, Knox Y, Donaldson AD. Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev. 2006;20(14):1874–1879. doi: 10.1101/gad.385306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore IK, Martin MP, Dorsey MJ, Paquin CE. Formation of circular amplifications in Saccharomyces cerevisiae by a breakage-fusion-bridge mechanism. Environ Mol Mutagen. 2000;36(2):113–120. doi: 10.1002/1098-2280(2000)36:2<113::aid-em5>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 34.Libuda DE, Winston F. Alterations in DNA replication and histone levels promote histone gene amplification in Saccharomyces cerevisiae. Genetics. 2010;184(4):985–997. doi: 10.1534/genetics.109.113662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen S, Yacobi K, Segal D. Extrachromosomal circular DNA of tandemly repeated genomic sequences in Drosophila. Genome Res. 2003;13(6A):1133–1145. doi: 10.1101/gr.907603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stanfield S, Helinski DR. Small circular DNA in Drosophila melanogaster. Cell. 1976;9(2):333–345. doi: 10.1016/0092-8674(76)90123-9. [DOI] [PubMed] [Google Scholar]
- 37.Ruiz JC, Choi KH, von Hoff DD, Roninson IB, Wahl GM. Autonomously replicating episomes contain mdr1 genes in a multidrug-resistant human cell line. Mol Cell Biol. 1989;9(1):109–115. doi: 10.1128/mcb.9.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reifenberger E, Freidel K, Ciriacy M. Identification of novel HXT genes in Saccharomyces cerevisiae reveals the impact of individual hexose transporters on glycolytic flux. Mol Microbiol. 1995;16(1):157–167. doi: 10.1111/j.1365-2958.1995.tb02400.x. [DOI] [PubMed] [Google Scholar]
- 39.L’Abbate A, et al. Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res. 2014;42(14):9131–9145. doi: 10.1093/nar/gku590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watanabe T, Horiuchi T. A novel gene amplification system in yeast based on double rolling-circle replication. EMBO J. 2005;24(1):190–198. doi: 10.1038/sj.emboj.7600503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hourcade D, Dressler D, Wolfson J. The amplification of ribosomal RNA genes involves a rolling circle intermediate. Proc Natl Acad Sci USA. 1973;70(10):2926–2930. doi: 10.1073/pnas.70.10.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown CJ, Todd KM, Rosenzweig RF. Multiple duplications of yeast hexose transport genes in response to selection in a glucose-limited environment. Mol Biol Evol. 1998;15(8):931–942. doi: 10.1093/oxfordjournals.molbev.a026009. [DOI] [PubMed] [Google Scholar]
- 43.Lewontin RC. The units of selection. Annu Rev Ecol Syst. 1970;1(1):1–18. [Google Scholar]
- 44.Lang GI, et al. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature. 2013;500(7464):571–574. doi: 10.1038/nature12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carroll SM, et al. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Mol Cell Biol. 1988;8(4):1525–1533. doi: 10.1128/mcb.8.4.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brachmann CB, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14(2):115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 47.Grauslund M, Didion T, Kielland-Brandt MC, Andersen HA. BAP2, a gene encoding a permease for branched-chain amino acids in Saccharomyces cerevisiae. Biochim Biophys Acta. 1995;1269(3):275–280. doi: 10.1016/0167-4889(95)00138-8. [DOI] [PubMed] [Google Scholar]
- 48.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12. [Google Scholar]
- 49.Giardine B, et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005;15(10):1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carr M, Bensasson D, Bergman CM. Evolutionary genomics of transposable elements in Saccharomyces cerevisiae. PLoS ONE. 2012;7(11):e50978. doi: 10.1371/journal.pone.0050978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dale RK, Pedersen BS, Quinlan AR. Pybedtools: A flexible Python library for manipulating genomic datasets and annotations. Bioinformatics. 2011;27(24):3423–3424. doi: 10.1093/bioinformatics/btr539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Camacho C, et al. BLAST+: Architecture and applications. BMC Bioinformatics. 2009;10(1):421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 57.Garrison E, Marth G. 2012. Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.